
Abstract
Aims/hypothesis
We designed a chemically modified, enzyme-resistant peptide with triple-acting properties based on human glucagon with amino acid substitutions aligned to strategic positions in the sequence of glucose-dependent insulinotropic polypeptide (GIP).
Methods
Y1-dA2-I12-N17-V18-I27-G28,29-glucagon (termed YAG-glucagon) was incubated with dipeptidylpeptidase IV (DPP-IV) to assess stability, BRIN-BD11 cells to evaluate insulin secretion, and receptor-transfected cells to examine cAMP production. Acute glucose-lowering and insulinotropic properties of YAG-glucagon were assessed in National Institutes of Health (NIH) Swiss mice, while longer-term actions on glucose homeostasis, insulin secretion, food intake and body weight were examined in high-fat-fed mice.
Results
YAG-glucagon was resistant to DPP-IV, increased in vitro insulin secretion (1.5–3-fold; p < 0.001) and stimulated cAMP production in GIP receptor-, glucagon-like peptide-1 (GLP-1) receptor- and glucagon receptor-transfected cells. Plasma glucose levels were significantly reduced (by 51%; p < 0.01) and insulin concentrations increased (1.2-fold; p < 0.01) after acute injection of YAG-glucagon in NIH Swiss mice. Acute actions were countered by established GIP, GLP-1 and glucagon antagonists. In high-fat-fed mice, twice-daily administration of YAG-glucagon for 14 days reduced plasma glucose (40% reduction; p < 0.01) and increased plasma insulin concentrations (1.8-fold; p < 0.05). Glycaemic responses were markedly improved (19–48% reduction; p < 0.05) and insulin secretion enhanced (1.5-fold; p < 0.05) after a glucose load, which were independent of changes in insulin sensitivity, food intake and body weight.
Conclusions/interpretation
YAG-glucagon is a DPP-IV-resistant triple agonist of GIP, GLP-1 and glucagon receptors and exhibits beneficial biological properties suggesting that it may hold promise for treatment of type 2 diabetes.
Similar content being viewed by others


Introduction
Glucagon is a 29-amino-acid polypeptide which is processed via pro-glucagon cleavage by prohormone convertase 2 in the pancreatic alpha cells [1]. It is the counter-regulatory hormone to insulin, which is secreted when plasma glucose concentrations become low, acting as a key regulator of metabolism in the fasting state. The metabolic actions of glucagon are triggered through binding to G-protein-coupled glucagon receptors to stimulate lipolysis, suppress hepatic glycogen synthesis and stimulate glycogenolysis and gluconeogenesis, thereby securing sufficient energy supply to the central nervous system and peripheral tissue [2].
Patients with type 2 diabetes often present with excessive fasting glucagon levels and poor suppression of postprandial glucagon secretion resulting in increased hepatic glucose output, which raises fasting glucose levels and contributes to hyperglycaemia [3]. In order to combat this, peptide and non-peptide glucagon receptor antagonists have been developed [4, 5]. The efficacy of these agents after chronic administration has been questioned amidst concerns that hepatic glucose output might rebound if glucagon receptor antagonism is not maintained [6]. Consistent with this, animals with a null mutation of the glucagon receptor have been shown to exhibit promising anti-diabetic potential [7]. Moreover, studies in transgenic mice overexpressing the glucagon receptor gene in pancreatic beta cells demonstrated increased insulin secretion, pancreatic insulin content and beta cell mass, partial protection against hyperglycaemia, and impaired glucose tolerance under conditions of high-fat feeding [8, 9].
Despite the classical role of glucagon as a hyperglycaemic hormone, some studies have shown that dual agonism of glucagon and glucagon-like peptide-1 (GLP-1) receptors with modified glucagon-like peptides represents a novel approach to treatment of obesity-diabetes [10, 11]. Thus oxyntomodulin analogues and peptides designed to trigger both glucagon and GLP-1 receptors represent unexpected therapeutic agents [12–17]. Glucose-dependent insulinotropic polypeptide (GIP), also from the glucagon/secretin family of peptides [2], is the major physiological incretin with powerful insulin-releasing and glucose-lowering properties, which may also have therapeutic potential [18]. In the present research, we chose to chemically modify the amino acid sequence of human glucagon by substituting key amino acids with those that are known to be important in the biological function of GIP. It was hypothesised that such chemically modified peptides would exhibit glucagon- and GIP-like properties to act on beta cells, with the possibility of additional GLP-1 receptor effects. The novel triple-acting peptide analogue (Y1-dA2-I12-N17-V18-I27-G28,29-glucagon, referred to as YAG-glucagon) described in this paper was initially assessed for dipeptidylpeptidase IV (DPP-IV) resistance, in vitro insulin secretion and cAMP production, and acute glucose-lowering and insulinotropic actions in NIH Swiss mice. After these acute studies, YAG-glucagon was further evaluated to reveal powerful glucose-lowering and insulinotropic actions in a longer-term study in mice with high-fat-diet-induced obesity and insulin resistance.
Methods
Peptide synthesis, purification and identification
Peptides (see Table 1 for amino acid sequence information) were purchased from GL Biochem (Shanghai, China; 20% purity). All peptides were purified by reversed phase (rp)-HPLC, and structural identities confirmed in-house using matrix-assisted laser desorption ionisation-time of flight mass spectrometry (MALDI-ToF MS). For rp-HPLC, a Phenomenex Jupiter semi-preparative column (C-18, 5 μ, 300 A; Phenomenex, Macclesfield, UK) was equilibrated with 0.12% (vol./vol.) trifluoroacetic acid/water at a flow rate of 1 ml/min using 0.1% (vol./vol.) trifluoroacetic acid in 70% acetonitrile/water. The concentration of the eluting solvent was raised from 0 to 80/85% over 18–20 min and to 90/95% over the next 10–15 min. For MALDI-ToF MS, samples (1 μl) were mixed with matrix solution (1 μl of a 10 mg/ml solution of α-cyano-4-hydroxycinnamic acid; Sigma-Aldrich, Dorset, UK) in acetonitrile/ethanol (1/1, vol./vol.) placed on one well of a 100-well stainless-steel sample plate and allowed to dry at room temperature. Mass spectra were recorded using a Voyager-DE BioSpectrometry Workstation (PerSeptive BioSystems, Framingham, MA, USA) [19]. Masses were recorded as mass-to-charge (m/z) ratio vs relative peak intensity and compared with theoretical values [19].
DPP-IV stability studies
Peptides were incubated at 37°C in 50 mmol/l triethanolamine/HCl (pH 7.8; Sigma-Aldrich) with purified porcine DPP-IV (5 mU; Sigma-Aldrich) for 0, 2, 4 and 8 h. Degradation profiles were obtained using rp-HPLC analysis as described above, and HPLC peak area data were used to calculate the percentage of intact peptide remaining at time points during the incubation as described previously [19].
In vitro insulin secretion
Effects of modified peptides on in vitro insulin secretion were examined using clonal pancreatic BRIN-BD11 cells, the characteristics of which have been reported previously [20]. Briefly, BRIN-BD11 cells were seeded (100,000 cells per well) into 24-well plates (Nunc, Roskilde, Denmark) and allowed to attach overnight at 37°C. After a 40 min preincubation (1.1 mmol/l glucose; 37°C), cells were incubated (20 min; 37°C) in the presence of 5.6 or 16.7 mmol/l glucose with a range of peptide concentrations (10−12–10−6 mol/l). In addition, YAG-glucagon (10−6 mol/l) was incubated in the presence of antagonists of GIP, GLP-1 and glucagon, namely 10−6 mol/l (Pro3)GIP, 10−6 mol/l exendin(9–39) and 10−6 mol/l DesHis1DesPhe6glucagon-amide, respectively (at 5.6 mmol/l glucose). After 20 min incubation, buffer was removed from each well, and aliquots (200 μl) were stored at −20°C before determination of insulin.
GIP receptor, GLP-1 receptor and glucagon receptor gene expression in BRIN-BD11 cells
Total RNA was isolated and purified using QIAzol lysis reagent (Qiagen, Crawley, West Sussex, UK), and RNA concentration determined from the absorbance at 260 nm. First-strand cDNA was synthesised using 2 μg total RNA at 42°C for 50 min in the presence of 0.5 μg oligo-dT(12–18) primer, 10 mmol/l dNTP and 200 U Superscript II reverse transcriptase (Invitrogen, Paisley, UK) in a final volume of 20 μl using a GeneStorm GS1 Thermal Cycler (Gene Technologies, Braintree, Essex, UK). Primers used for PCR are provided in electronic supplementary material (ESM) Table 1. The DNA-denaturing step was carried out at 95°C for 5 min using Bio-Rad MJ Mini personal Thermal cycler (Bio-Rad Laboratories, Hemel Hempstead, UK). cDNA amplification was then started for 40 cycles with 95°C denaturation for 30 s, 58°C annealing for 30 s and 72°C elongation for 30 s, with SYBR Green fluorescence being read after each cycle and recorded by Bio-Rad CFX Manager software (Version 1.5; Bio-Rad Laboratories) to construct an amplification curve. Gene expression was calculated from \( {2^{{-\Delta {{\mathrm{C}}_{\mathrm{t}}}}}} \) values normalised to rat β-actin control primer.
In vitro cAMP production
Chinese Hamster Lung (CHL) cells transfected with either the human GIP receptor (GIP-R) [21] or GLP-1 receptor (GLP-1-R) [22] and glucagon receptor (glucagon-R)-transfected HEK293 cells [23] were used to assess effects of YAG-glucagon on cAMP production. Cells were seeded (200,000 cells per well) into 96-well plates (Nunc) and washed with Hank’s Balanced Salt Solution (HBSS) buffer before incubation with YAG-glucagon (10−6 to 10−12 mol/l) or respective control peptides (in the presence of 200 μmol/l 3-isobutyl-1-methylxanthine (IBMX) for 20 min at 37°C). After incubation, medium was removed and cells lysed before measurement of cAMP using Parameter cAMP assay (R&D Systems, Abingdon, UK) according to the manufacturer’s instructions.
Animals
Acute animal studies were carried out in male NIH Swiss mice (Harlan, Oxon, UK; 10–16 weeks old). Longer-term experiments were performed in NIH Swiss mice previously fed a high-fat diet for 4 months composed of 45% fat, 20% protein and 35% carbohydrate (total energy 26.15 kJ/g; Special Diet Services, Witham, UK) for 140 days. This diet resulted in progressive body weight gain (56.2 ± 4.0 vs 47.4 ± 3.5 g; p < 0.05) and hyperglycaemia (9.8 ± 1.0 vs 4.3 ± 0.5 mmol/l; p < 0.05) compared with age-matched controls on normal laboratory chow (data not shown). Animals were housed in a 12:12 h light/dark cycle (08:00–20:00 hours) and had free access to drinking water and food. All animal experiments were conducted according to UK Home Office Regulations (UK Animals Scientific Procedures Act 1986) and the Principles of Laboratory Animal Care (NIH Publication Number 86-23, revised 1985). No adverse effects were observed after administration of any of the peptides.
Acute actions of YAG-glucagon on plasma glucose and insulin concentrations in vivo
Normal NIH Swiss mice were used to assess acute actions of peptide on plasma glucose and insulin. Briefly, plasma glucose and insulin responses were evaluated after an i.p. injection of glucose alone (18 mmol/kg body weight) or in combination with test peptide (25 nmol/kg body weight). In some experiments, (Pro3)GIP, exendin(9–39) or DesHis1DesPhe6glucagon-amide (each at 25 nmol/kg body weight) were co-administered with YAG-glucagon to evaluate the extent to which the effects were mediated through GIP-R, GLP-1-R or glucagon-R. All test solutions were administered in a final volume of 8 ml/kg body weight.
Effects of twice-daily administration of YAG-glucagon in high-fat-fed mice
Twice-daily injections of YAG-glucagon (25 nmol/kg body weight; i.p.) or saline vehicle (0.9% (wt/vol.) NaCl) were administered at 09:00 and 16:00 hours over 14 days to high-fat-fed mice. Food intake, body weight, non-fasting plasma glucose and insulin concentrations were monitored at 2–4 day intervals. Glucose tolerance (18 mmol/kg body weight; i.p.) and insulin sensitivity (50 U/kg body weight; i.p.) tests were performed after 14 days of treatment.
Biochemical analyses
Blood samples were collected from the cut tip on the tail vein of conscious mice into chilled fluoride/heparin glucose micro-centrifuge tubes (Sarstedt, Numbrecht, Germany) at the time points indicated in the figures. Samples were immediately centrifuged using a Beckman microcentrifuge (Beckman Instruments, Galway, Ireland) for 30 s at 13,000 g. Plasma glucose was assayed by an automated glucose oxidase procedure using a Beckman Glucose Analyzer II (Beckman Instruments, Galway, Ireland). Plasma insulin was assayed by a modified dextran-coated charcoal RIA [24].
Statistical analysis
Results are expressed as means ± SEM, and data were compared using the unpaired Student’s t test. Where appropriate, data were compared using repeated-measures ANOVA or one-way ANOVA, followed by the Student–Newman–Keuls post-hoc test. Incremental AUC analyses for plasma glucose and insulin were calculated using GraphPad Prism version 5.0. Groups of data were considered to be significantly different if p < 0.05.
Results
Peptide characterisation and DPP-IV stability
Peptides were purified using repeated runs of rp-HPLC and this resulted in sharp, well-resolved peaks with retention times shown in Table 1. Experimental masses obtained for all peptides after MALDI-ToF MS corresponded very closely to the theoretical masses, confirming successful peptide synthesis (Table 1). Moreover, YAG-glucagon and modified analogues were completely stable to the actions of DPP-IV up to and including 8 h, whereas native peptides were degraded with estimated half-lives of approximately 2.0–4.5 h (Table 1).
In vitro insulin secretion and cAMP production
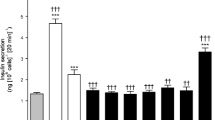
Gene receptor expression analysis revealed that receptors for GIP, GLP-1 and glucagon were expressed in BRIN-BD11 cells (relative expression 1.0 ± 0.3, 5.0 ± 0.5 and 0.05 ± 0.3 arbitrary units, respectively). Figure 1 illustrates effects of YAG-glucagon on insulin secretion from BRIN-BD11 cells at 5.6 mmol/l (Fig. 1a) and 16.7 mmol/l glucose (Fig. 1b). At both glucose concentrations, YAG-glucagon significantly increased insulin secretion at all peptide concentrations compared with glucose alone (1.5–3.0-fold; p < 0.001) and glucagon (1.4–1.8-fold; p < 0.001) (Fig. 1a, b). Native GIP and GLP-1 (10−6 mol/l) included as positive controls similarly stimulated insulin secretion (p < 0.001; Fig. 1a, b). Analogues 1 and 2 were 30–70% less potent (p < 0.01) at stimulating insulin secretion (1.3 ± 0.1 and 1.1 ± 0.04 ng/106 cells/20 min, respectively) compared with YAG-glucagon (3.0 ± 0.3 ng/106 cells/20 min) at 10−6 mol/l. (Pro3)GIP, exendin(9–39) and DesHis1DesPhe6glucagon-amide (each tested at 10−6 mol/l) inhibited maximal YAG-glucagon-induced insulin secretion by 19% (p < 0.05), 24% (p < 0.01) and 29% (p < 0.001), respectively (Fig. 1c). YAG-glucagon (10−12 and 10−6 mol/l) stimulated cAMP production by GIP-R-, GLP-1-R- and glucagon-R-transfected cells equivalent to 93–109%, 50–85% and 70–90% of that elicited by the respective native peptide (p < 0.01 to p < 0.001; Fig. 1d). At a high concentration of 10−6 mol/l, native GIP, GLP-1 and glucagon peptides (n = 3) stimulated cAMP production from GIP-R-, GLP-1-R- and glucagon-R-transfected cells, but the responses were less than or comparable to those induced by a 106 times lower concentration of the native peptides (10−12 mol/l). Thus, compared with 10−12 mol/l, native peptide cross-talk at 10−6 mol/l was: for GIP-R, GLP-1 75.7 ± 0.4% and glucagon 65.1 ± 0.2%; for GLP-1-R, GIP 83.5 ± 0.4% and glucagon 76.6 ± 0.2%; and for glucagon-R, GLP-1 125.7 ± 1.4% and GIP 128.5 ± 1.0%.

Effects of YAG-glucagon on insulin secretion in BRIN-BD11 cells and cAMP production in receptor-transfected cells. Various concentrations of YAG-glucagon (10−12 to 10−6 mol/l) were incubated with BRIN-BD11 cells in the presence of (a) 5.6 and (b) 16.7 mmol/l glucose for 20 min (n = 8), and insulin release was measured using RIA. (c) YAG-glucagon (10−6 mol/l) was incubated in the presence of (Pro3)GIP, exendin(9–39) and DesHis1DesPhe6glucagon-amide (each at 10−6 mol/l), respectively (at 5.6 mmol/l glucose). (d) YAG-glucagon was incubated in CHL cells transfected with human GIP-R or human GLP-1-R and HEK-293 cells transfected with glucagon-R (20 min; n = 3), and cAMP production was measured and represented as % of control (native peptide). (a) and (b): white bars, glucose control; black bars, glucagon; light grey bars, YAG-glucagon; striped bars, GLP-1; dark grey bars, GIP. (c): white bars, YAG-glucagon; light grey bars, YAG-glucagon plus exendin(9–39); dark grey bars, YAG-glucagon plus DesHis1DesPhe6glucagon-amide; black bars, YAG-glucagon plus (Pro3)GIP. (d): white bars, 10−12 mol/l peptide concentrations; black bars, 10−6 mol/l peptide concentrations. Data are expressed as means ± SEM. ***p < 0.001 compared with glucose control. ††† p < 0.001 compared with glucagon. ‡ p < 0.05, ‡‡ p < 0.01 and ‡‡‡ p < 0.001 compared with YAG-glucagon. §§ p < 0.01 and §§§ p < 0.001 compared with stimulation by respective native peptide (100%)
Acute glucose-lowering and insulinotropic actions of YAG-glucagon in NIH Swiss mice
As shown in Fig. 2, injection of glucose alone resulted in a sharp increase in plasma glucose concentrations, which gradually increased over the whole 60 min observation period. YAG-glucagon significantly lowered (51% reduction; p < 0.01; Fig. 2a, c) plasma glucose concentrations compared with controls. In agreement with the marked reduction in plasma glucose, YAG-glucagon significantly (1.2-fold increase; p < 0.01) increased plasma insulin concentrations (Fig. 2b, d). When administered conjointly with YAG-glucagon, (Pro3)GIP, exendin(9–39) and DesHis1DesPhe6glucagon-amide each significantly inhibited YAG-glucagon-induced insulin secretion (9%, 19% and 27%, respectively; p < 0.01 to p < 0.001; Fig. 2b, d). Consistent with this, the glucose-lowering actions of YAG-glucagon were abolished (Fig. 2a, c). Indeed, the glucagon antagonist, DesHis1DesPhe6glucagon-amide, actually elicited a marked hyperglycaemic response.

Acute actions of YAG-glucagon on plasma glucose and insulin concentrations in NIH Swiss mice. (a) Plasma glucose and (b) insulin concentrations were measured before and after i.p. administration of glucose alone (18 mmol/kg body weight) and YAG-glucagon (25 nmol/kg body weight) alone or in combination with (Pro3)GIP, exendin(9–39) or DesHis1DesPhe6glucagon-amide (each at 25 nmol/kg body weight). (c) Plasma glucose and (d) plasma insulin AUC graphs also included. White triangles and white bars, glucose alone; black circles and black bars, YAG-glucagon; white circles and light grey bars, YAG-glucagon plus exendin (9–39); white squares and striped bars, YAG-glucagon plus DesHis1DesPhe6glucagon-amide; black triangles and dark grey bars, YAG-glucagon plus (Pro3)GIP. Data are expressed as means ± SEM for six mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with glucose alone. †† p < 0.01 and ††† p < 0.001 compared with YAG-glucagon
Effects of twice-daily administration of YAG-glucagon on body weight, food intake, non-fasting plasma glucose and insulin concentrations in high-fat-fed mice
Twice-daily administration of YAG-glucagon for 14 days had no effect on body weight or food intake compared with saline-treated controls (Fig. 3a, b). Non-fasting plasma glucose concentrations were significantly decreased (40% decrease; p < 0.01; Fig. 3c), and plasma insulin concentrations were significantly increased (1.8-fold increase; p < 0.05; Fig. 3d) on days 7, 10 and 13 in YAG-glucagon-treated mice compared with saline-treated controls.

Effects of twice-daily administration of YAG-glucagon on (a) body weight, (b) food intake, (c) non-fasting plasma glucose and (d) non-fasting plasma insulin in high-fat-fed mice. Variables were measured before and during daily treatment (14 days) with saline vehicle (0.9% wt/vol. NaCl) or in combination with YAG-glucagon (25 nmol/kg body weight). White squares, saline vehicle; black circles, YAG-glucagon. Data are expressed as means ± SEM for eight mice. *p < 0.05 and **p < 0.01 compared with saline-treated controls
Effects of twice-daily administration of YAG-glucagon on glucose tolerance, plasma insulin response to glucose and insulin sensitivity in high-fat-fed mice
As shown in Fig. 4a, twice-daily administration of YAG-glucagon for 14 days resulted in significantly decreased plasma glucose concentrations (19–48% decrease; p < 0.05) compared with saline-treated controls (Fig. 4a) after an exogenous glucose load. These beneficial effects of YAG-glucagon were particularly evident from plasma glucose AUC values, which were significantly decreased (47% reduction; p < 0.05; Fig. 4c). In line with this, glucose-mediated plasma insulin concentrations were significantly elevated (1.5-fold increase; p < 0.05; Fig. 4b) 30 min after injection for YAG-glucagon-treated mice, and this was further highlighted by significantly increased plasma insulin AUC values (1.5-fold increase; p < 0.05; Fig. 4d). No significant effects on insulin sensitivity were observed after 14 days of treatment with YAG-glucagon compared with saline controls (4,155 ± 286 vs 4,523 ± 351 mmol/l × min, respectively) (data not shown).

Effects of twice-daily administration of YAG-glucagon on (a) glucose tolerance and (b) plasma insulin response to glucose in high-fat-fed mice. Tests were conducted after daily treatment with YAG-glucagon (25 nmol/kg body weight) or with saline vehicle (0.9% wt/vol. NaCl) for 14 days. (c) Plasma glucose and (d) plasma insulin AUC graphs also included. White squares and white bars, saline vehicle; black circles and black bars, YAG-glucagon. Data are expressed as means ± SEM for eight mice. *p < 0.05 compared with saline-treated controls
Discussion
Increasing attention has focused on agents based on the structure of glucagon that bind to and activate both the glucagon and GLP-1 receptors in an attempt to improve glucose homeostasis in type 2 diabetes [4, 10, 11]. Thus it appears that the strong insulinotropic actions of GLP-1 overcome the hyperglycaemic actions of glucagon, while not affecting potential beneficial actions on lipid metabolism and energy balance [10, 11]. The present study examined the antihyperglycaemic and insulinotropic properties of a chemically modified glucagon peptide, Y1-dA2-I12-N17-V18-I27-G28,29-glucagon (referred to as YAG-glucagon), engineered to combine the actions of glucagon with the powerful insulinotropic actions of GIP and possibly GLP-1.
YAG-glucagon was engineered to comprise eight amino acid substitutions/modifications along the backbone amino acid sequence of human glucagon including: Y1H, [dA]2S, I12K, N17R, V18R, I27M, G28N and G29T. These particular modifications were selected to align with key amino acid residues that are important in the biological action of GIP [18]. This rationale was chosen, as it was hypothesised that such strategic amino acid modifications would result in a chemically modified peptide that would exhibit enhanced beta cell insulin secretion by specifically harnessing the insulinotropic actions of GIP receptor activation. Synthetic peptides were generated by conventional Fmoc chemistry [25], purified by repeated runs of rp-HPLC, and molecular mass confirmed by MALDI-ToF MS. Importantly, experimental peptide masses correlated well with theoretical masses, thus confirming successful peptide synthesis.
Native peptides were rapidly degraded by DPP-IV, with in vitro half-lives in the range 2–4.5 h, whereas YAG-glucagon was stable up to and including the 8 h incubation time point. This resistance to DPP-IV is most likely due to substitution of naturally occurring l-Ala2 with the d-isomer, as studies with related peptides such as GIP have shown that DPP-IV cannot cleave the d-isomeric form [26]. YAG-glucagon markedly increased in vitro insulin secretion in BRIN-BD11 cells under both physiological and supraphysiological glucose conditions. This is an interesting finding, as potency was better than glucagon per se, suggesting that the actions of YAG-glucagon to enhance insulin production may be as a result of a stimulatory effect on related receptors including GIP and possibly GLP-1, as is the case with other proglucagon-derived peptides [10–17]. Indeed, the insulin-releasing actions of YAG-glucagon were comparable to the potent insulin-releasing actions of the established native incretin peptides GIP and GLP-1. Consistent with this view, established antagonists of receptors of GIP, GLP-1 and glucagon, namely (Pro3)GIP, exendin(9–39) and DesHis1DesPhe6glucagon-amide, inhibited YAG-glucagon-induced insulin release from BRIN-BD11 cells. Novel analogues 1 and 2 with sequence modifications between that of glucagon and YAG-glucagon were significantly less potent at stimulating in vitro insulin secretion compared with YAG-glucagon and thus were not investigated any further. Interestingly, when we incubated 10−6 mol/l native GIP, GLP-1 and glucagon peptides with each of the receptor-transfected cell lines, cAMP responses indicated that there was cross-talk at each of the receptors, but these effects were less than those evoked by 106 times lower concentration of the native peptides. YAG-glucagon stimulated cAMP production by GIP-R-, GLP-1-R- and glucagon-R- transfected cells equivalent to ∼100%, 68% and 80% of the responses evoked by native GIP, GLP-1 and glucagon, respectively. Moreover, YAG-glucagon exhibited greater cAMP responses at both GIP-R and GLP-1-R, even at 10−12 mol/l, compared with any of the native peptides tested. Thus, our data clearly illustrate that YAG-glucagon acts as a triple agonist and contrasts with oxyntomodulin and related analogues, which evoke relatively weak receptor responses compared with the native peptide ligands [12–16].
To assess the acute antihyperglycaemic and insulinotropic actions of YAG-glucagon, we used normal NIH Swiss mice. YAG-glucagon significantly lowered plasma glucose concentrations after acute i.p. administration, and corresponding plasma insulin concentrations were increased. The enhanced insulinotropic actions of YAG-glucagon suggest that the chemical modifications present in this peptide offset proteolytic degradation and also enhance its ability to stimulate beta cell insulin secretion. These effects are reminiscent of other related modified incretin hormones [13, 18, 27]. As with in vitro studies, chemical antagonists of GIP-R, GLP-1-R and glucagon-R abolished both the insulin-releasing and glucose-lowering actions of YAG-glucagon in normal mice. Thus, rather than exhibiting dual action, YAG-glucagon acts as a triple-acting agonist of GIP-R, GLP-1-R and glucagon-R. A substantial hyperglycaemic rebound was observed with DesHis1DesPhe6glucagon-amide consistent with potential problems of therapeutic glucagon-R blockade [6].
Results of acute in vivo studies provided a strong basis for the subsequent 14-day study in high-fat-fed mice. Consistent with earlier observations, mice on a high-fat diet exhibited increased food intake, progressive body weight gain, hyperglycaemia and hyperinsulinaemia [13]. Twice-daily administration of YAG-glucagon for 14 days significantly decreased non-fasting plasma glucose concentrations and increased plasma insulin levels. Furthermore, glucose tolerance was significantly improved after 14 days of treatment with YAG-glucagon, and this was accompanied by a marked improvement in glucose-mediated insulin secretion. This confirms the long-acting insulinotropic effects of YAG-glucagon and its ability to overcome any associated pancreatic beta cell defects in this high-fat model [13]. Interestingly, improvements in glucose homeostasis over this relatively short 14-day study period were independent of any change in insulin sensitivity, food intake or body weight. Although not measured in the present study, future experiments could be conducted to ascertain if long-term treatment with YAG-glucagon affects lipid variables.
In conclusion, this study demonstrates that selective chemical modification of native glucagon generated a novel peptide, YAG-glucagon, which is a DPP-IV-resistant triple agonist of GIP-R, GLP-1-R and glucagon-R, with powerful insulin-releasing and glucose-lowering properties. This combined approach can be anticipated to overcome any diabetes-induced defect at any individual receptors, as illustrated by improvement in the actions of GIP after glucose-lowering with other therapies [28, 29]. This research represents an important step in the progression of glucagon-based peptides as a potential therapy for type 2 diabetes mellitus; however, further studies are required to fully answer questions of mechanism of action and whether or not these analogues will be useful in a clinical setting.
Abbreviations
- DPP-IV:
-
Dipeptidylpeptidase IV
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- MALDI-ToF MS:
-
Matrix-assisted laser desorption ionisation-time of flight mass spectrometry
- NIH:
-
National Institutes of Health
- rp-HPLC:
-
Reversed-phase-HPLC
- YAG-glucagon:
-
Y1-dA2-I12-N17-V18-I27-G28,29-glucagon
References
Cryer PE (2012) Glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology 153:1039–1048
Drucker DJ (2005) Biologic actions and therapeutic potential of the proglucagon-derived peptides. Nat Clin Pract Endocrinol Metabol 1:22–31
Del Prato S, Marchetti P (2004) Beta- and alpha-cell dysfunction in type 2 diabetes. Horm Metab Res 36:775–781
Christensen M, Bagger JI, Vilsbøll T, Knop FK (2011) The alpha-cell as target for type 2 diabetes. Rev Diabet Stud 8:369–381
Bagger JI, Knop FK, Holst JJ, Vilsbøll T (2011) Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes Metabol 13:965–971
Agius L (2007) New hepatic targets for glycaemic control in diabetes. Best Pract Res Clin Endocrinol Metabol 21:587–605
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha-cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA 100:1438–1443
Gelling RE, Vuguin PM, Du XQ et al (2009) Pancreatic beta-cell overexpression of the glucagon receptor gene results in enhanced beta-cell function and mass. Am J Physiol Endocrinol Metab 297:E695–E707
Vuguin PM, Charron MJ (2011) Novel insights into glucagon receptor action: lessons from knockout and transgenic mouse models. Diabetes Obes Metabol 13:144–150
Pocai A, Carrington PE, Adams JR et al (2009) Glucagon-like peptide-1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 58:2258–2266
Day JW, Ottaway N, Patterson JT et al (2009) A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol 5:749–757
Maida A, Lovshin JA, Baggio LL, Drucker DJ (2008) The glucagon-like peptide-1 receptor agonist oxyntomodulin enhances beta-cell function but does not inhibit gastric emptying in mice. Endocrinology 149:5670–5678
Kerr BD, Flatt PR, Gault VA (2010) (d-Ser2)Oxm[mPEG-PAL]: a novel chemically modified analogue of oxyntomodulin with antihyperglycaemia, insulinotropic and anorexigenic actions. Biochem Pharmacol 80:1727–1735
Druce MR, Minnion JS, Field BC et al (2008) Investigation of structure-activity relationships of oxyntomodulin (Oxm) using Oxm analogs. Endocrinology 150:1712–1722
Du X, Kosinski JR, Lao J et al (2012) Differential effects of oxyntomodulin and GLP-1 on glucose metabolism. Am J Physiol Endocrinol Metab 303:E265–E271
Santoprete A, Capito E, Carrington PE et al (2011) DPP-IV-resistant, long-acting oxyntomodulin derivatives. J Pept Sci 17:270–280
Liu YL, Ford HE, Druce MR et al (2010) Subcutaneous oxyntomodulin analogue administration reduces body weight in lean and obese rodents. Int J Obes (Lond) 34:1715–1725
Gault VA, Flatt PR, O’Harte FP Glucose-dependent insulinotropic polypeptide analogues and their therapeutic potential for the treatment of obesity-diabetes. Biochem Biophys Res Commun 308:207–213
Irwin N, Green BD, Gault VA et al (2005) Degradation, insulin secretion, and antihyperglycemic actions of two palmitate-derivatized N-terminal pyroglutamyl analogues of glucose-dependent insulinotropic polypeptide. J Med Chem 48:1244–1250
McClenaghan NH, Barnett CR, Ah-Sing E et al (1996) Characterization of a novel glucose-responsive insulin-secreting cell line, BRIN-BD11, produced by electro-fusion. Diabetes 45:1132–1140
Gremlich S, Porret A, Hani EH et al (1995) Cloning, functional expression and chromosomal localization of the human pancreatic islet glucose-dependent insulinotropic polypeptide receptor. Diabetes 44:1202–1208
Thorens B, Porret A, Buhler L et al (1993) Cloning and functional expression of the human islet GLP-1 receptor: demonstration that exendin-4 is an agonist and exendin-(9–39) an antagonist of the receptor. Diabetes 42:1678–1682
Ikegami T, Cypess AM, Bouscarel B (2001) Modulation of glucagon receptor expression and response in transfected human embryonic kidney cells. Am J Physiol Cell Physiol 281:1396–1402
Flatt PR, Bailey CJ (1981) Abnormal plasma glucose and insulin responses in heterozygous lean (ob/+) mice. Diabetologia 20:573–577
Fields GB, Noble RL (1990) Solid-phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int J Pept Protein Res 35:161–214
Hinke SA, Gelling RW, Pederson RA et al (2002) Dipeptidyl peptidase IV-resistant [d-Ala2]glucose-dependent insulinotropic polypeptide polypeptide (GIP) improves glucose tolerance in normal and obese diabetic rats. Diabetes 51:652–661
Gallwitz B (2011) Glucagon-like peptide-1 analogues for type 2 diabetes mellitus: current and emerging agents. Drugs 71:1675–1688
Aaboe K, Knop FK, Vilsboll T et al (2009) KATP channel closure ameliorates the impaired insulinotropic effect of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. J Clin Endocrinol Metab 4:603–608
Hojberg PV, Vilsboll T, Rabol R et al (2009) Four weeks of near-normalization of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 52:199–207
Acknowledgements
The authors wish to thank B. Thorens (University of Lausanne, Switzerland) for GLP-1-R- and GIP-R-transfected CHL cells, and C. Unson (The Rockefeller University, USA) for glucagon-R-transfected HEK293 cells.
Funding
These studies were supported by University of Ulster Strategy Research Funding and a Vice-Chancellors Research Scholarship to V. Bhat.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
VKB, BDK and SV contributed to acquisition of data and analysis and interpretation of data, and helped draft the manuscript. VAG and PRF contributed to conception and design of the study and data interpretation and critically revised the manuscript for important intellectual content. All authors approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 9 kb)
Rights and permissions
About this article
Cite this article
Bhat, V.K., Kerr, B.D., Vasu, S. et al. A DPP-IV-resistant triple-acting agonist of GIP, GLP-1 and glucagon receptors with potent glucose-lowering and insulinotropic actions in high-fat-fed mice. Diabetologia 56, 1417–1424 (2013). https://doi.org/10.1007/s00125-013-2892-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2892-2