
Abstract
Aims/hypothesis
Membrane phospholipids are the major intracellular source for fatty acid-derived mediators, which regulate myriad cell functions. We showed previously that high glucose levels triggered the hydrolysis of polyunsaturated fatty acids from beta cell phospholipids. These fatty acids were subjected to free radical-catalysed peroxidation to generate the bioactive aldehyde 4-hydroxy-2E-nonenal (4-HNE). The latter activated the nuclear peroxisome proliferator-activated receptor-δ (PPARδ), which in turn augmented glucose-stimulated insulin secretion. The present study aimed at investigating the combined effects of glucose and fatty acid overload on phospholipid turnover and the subsequent generation of lipid mediators, which affect insulin secretion and beta cell viability.
Methods
INS-1E cells were incubated with increasing glucose concentrations (5–25 mmol/l) without or with palmitic acid (PA; 50–500 μmol/l) and taken for fatty acid-based lipidomic analysis and functional assays. Rat isolated islets of Langerhans were used similarly.
Results
PA was incorporated into membrane phospholipids in a concentration- and time-dependent manner; incorporation was highest at 25 mmol/l glucose. This was coupled to a rapid exchange with saturated, mono-unsaturated and polyunsaturated fatty acids. Importantly, released arachidonic acid and linoleic acid were subjected to peroxidation, resulting in the generation of 4-HNE, which further augmented insulin secretion by activating PPARδ in beta cells. However, this adaptive increase in insulin secretion was abolished at high glucose and PA levels, which induced endoplasmic reticulum stress, apoptosis and cell death.
Conclusions/interpretation
These findings highlight a key role for phospholipid remodelling and fatty acid peroxidation in mediating adaptive and cytotoxic interactions induced by nutrient overload in beta cells.
Similar content being viewed by others


Introduction
NEFA that are incorporated into phospholipids and triacylglycerols also serve as important regulators of insulin secretion [1–3]. Beta cells respond to increased circulating levels of NEFA by augmenting the secretion of insulin, which facilitates their peripheral disposal [4–7]. Long-term exposure of beta cells to high levels of glucose and NEFA reduces this response in a phenomenon termed ‘glucolipotoxicity’, which is characterised by altered membrane fluidity, protein palmitoylation, generation of ceramides, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, autophagy and apoptosis [8–11].
Previous studies have shown that arachidonic acid (AA) and linoleic acid (LA) and their metabolites are involved in the regulation of beta cell function [12]. We have shown in INS-1E cells that high glucose levels induced remodelling of membrane phospholipids and released AA and LA. Reactive oxygen species (ROS) prompted non-enzymatic peroxidation of AA and LA to produce 4-hydroxy-2E-nonenal (4-HNE), which augments glucose-stimulated insulin secretion (GSIS) via activation of the nuclear receptor peroxisome proliferator-activated receptor-δ (PPARδ) [13].
Previous lipidomic analyses have been performed predominantly on beta cells exposed to glucolipotoxic conditions [14]. The incorporation of high levels of palmitic acid (PA) into phospholipids was associated with beta cell damage [15, 16]. Similarly, beta cell failure in animal models of diabetes was linked to changes in the level of lipid metabolites such as phosphatidylethanolamine [17]. Hitherto, no comprehensive analysis of fatty acid turnover in membrane phospholipid of beta cells exposed to physiological and increasing concentrations of glucose and PA has been reported. Our study was undertaken with the following aims: (1) to determine fatty acid turnover in beta cell phospholipids following exposure to increasing levels of glucose and/or PA; (2) to investigate the impact of these changes on beta cell function and (3) to ascertain whether 4-HNE mediates the adaptive responses.
Methods
Materials
A list of chemicals, reagents, antibodies, vectors and suppliers is given in electronic supplementary material [ESM] Methods.
Animals, islet isolation and INS-1E beta cell culture
The ethics committee for animal welfare (IACUC) of the Hebrew University approved the study protocol. Islets of Langerhans were isolated from male Wistar rats (150–175 g; Harlan, Jerusalem, Israel) following collagenase digestion of pancreases, as described [13]. INS-1E cells (passages 65–85) were grown and maintained as described [18]. GSIS assays in INS-1E cell cultures and rat isolated islets and insulin RIA (Merck Millipore, Billerica, MA, USA) were performed as described [13].
Solubilisation of PA
Sodium palmitate (10 mmol/l) was dissolved in 11% (wt/vol.) fatty acid-free BSA in RPMI 1640 medium (pH 7.3), heated to 56°C for 1 h and sterilised by filtration. The final BSA concentration in INS-1E or islet cultures was 0.5% (wt/vol.).
Fatty acid-based lipidomic analysis
The lipidomic analysis of INS-1E cells was performed as described [13]. Fatty acids were identified by G. Cohen and quantified by comparison with standard methylated references.
Western blot and dot blot analyses
Cell lysates were prepared as described [19] and used for western blot analyses of PPARδ, p-Thr980-protein kinase RNA-like ER kinase (PERK), p-Ser724-inositol-requiring enzyme-1 (IRE), total IRE and α-tubulin, according to the suppliers’ protocols. Dot blot analysis was used to detect 4-HNE-histidine protein adducts.
Real-time PCR analysis
The RNeasy kit (Qiagen, Valencia, CA, USA) was used for RNA isolation. The RevetAid kit (Fermentas, Glen Burnie, MD, USA) was used for cDNA synthesis, using oligodT primers. Real-time PCR was performed using the Stratagene MX3000P system (La Jolla, CA, USA) according to the manufacturer’s guidelines. Oligonucleotide primers, designed using Primer Express program (Applied Biosystems, Foster City, CA, USA), were synthesised by Sigma-Aldrich (Rehovot, Israel). The PCR protocols have been described previously [13, 20]. Primer sequences are given in ESM Table 1.
PPARδ–luciferase reporter assay
Subconfluent INS-1E cultures were co-transfected with the hPPARδ expression vector along with the hRXR-, green fluorescent protein (GFP)- and Renilla luciferase expression vectors and the 3XPPRE-TK-luciferase reporter, as described [19]. The yield of the transfection, assessed by GFP fluorescence, was >85%. Firefly luciferase-induced luminescence was determined in cell lysates by means of the dual luciferase reporter assay using the Mithras LB-940 luminometer (Berthold Technologies, Bad Wildbad, Germany). Results were normalised to the Renilla luciferase activity, used as an internal control according to the kit’s instructions.
PPAR silencing with siRNA
The Pparδ (also known as Ppard) siRNA sequences were 5′-ACGAGAAGUGCGAUCGGAU-3′, 5′-CCUCAAGUACGGCGUGCAU-3′, 5′-CCACAACGCUACCGCUUU-3′ and 5′-CAUGAGUUCUUGCGCAGUA-3′. Scrambled RNA sequences were as follows: sense 5′-GAGGUAACGUCGAACUAUA-3′; antisense 5′-UAUAGUUCGACGUUACCUC-3′. The silencing procedure in INS-1E cells was performed as described previously [13].
Extraction of polar lipids and HPLC analysis
Polar lipid extraction from culture media (20 ml) and HPLC analysis of 4-HNE in the extracts were performed as described previously [13].
Cell viability and apoptosis assays
The methylthiazol tetrazolium (MTT) cell viability assay was performed as described previously [13]. Apoptosis was determined with the annexin V–propidium iodide fluorescent detection assay (MBL-Bion, Tel-Aviv, Israel) in a FACScan System and quantified with the CellQuest Software (version 7.5.3, BD Biosciences, San Jose, CA, USA).
Statistical analysis
Results are given as the means ± SEM of separate experiments in INS-1E cells or in islets pooled from five rats. Statistical analyses were performed using Student’s t test. Multiple comparisons were performed by one-way ANOVA followed by the Dunnett’s test using GraphPad Prism 5.0 software (La Jolla, CA, USA); p < 0.05 was considered significant.
Results
Effects of PA on function and viability of INS-1E cells
INS-1E cell cultures were maintained at 5-, 11- and 25 mmol/l glucose for 48 h. Serum-free RPMI 1640 medium, supplemented with 0.5% fatty acid-free BSA without or with 50–500 μmol/l PA, was present during the last 16 h of incubation. Figure 1 (a, b; at zero PA) confirms previous reports on glucose-dependent beta cell proliferation [18]. This effect persisted in the presence of 50–125 μmol/l PA; yet, at PA concentrations of 250 μmol/l or above cell death was intensified in a glucose-dependent manner (Fig. 1a, b). Cell loss was coupled to PA-induced apoptosis and ROS generation, while increasing glucose concentrations exacerbated this effect (Fig. 1c, d). The minimum PA concentration required to initiate apoptosis was 500, 350 and 250 μmol/l at 5, 11 and 25 mmol/l glucose, respectively. Significant cell death occurred in the presence of 500 μmol/l PA at 11 and 25 mmol/l glucose (Fig. 1e, f).

Effects of glucose and PA on INS-1E cells. (a–d) In the dose–response analysis, cells were exposed to 5 (white bars and circles), 11 (grey bars and circles) or 25 mmol/l glucose (black bars and circles) for 48 h. During the last 16 h they were exposed to serum-free medium containing 0.5% (wt/vol.) BSA, without or with PA. This was followed by cell counting (a), MTT assay (b) and annexin–propidium iodide FACS analysis (c). (d) ROS was measured with 2′,7′-dichlorofluorescein diacetate (DCFDA) dye 1 h after PA addition. (e, f) In the time course analysis, the cells were similarly treated without or with 500 μmol/l PA for the indicated times, followed by cell counting (e) and MTT assay (f). Data are means ± SEM, n = 3; *p < 0.05 for differences from the corresponding 5 mmol/l glucose control; † p < 0.05 for differences from the respective PA-untreated cells. RU, relative units
Figure 2 shows the results of GSIS assays in similarly treated cells: PA augmented significantly the GSIS already occurring at 50 μmol/l in cells exposed to 5 and 11 mmol/l glucose, and at 125 μmol/l in cells incubated with 25 mmol/l glucose. The vertical broken lines in Fig. 2 indicate PA concentrations above which insulin secretion and/or content were significantly reduced in a glucose-dependent manner. Another observed abnormal effect was the increased basal insulin secretion (at 3.3 mmol/l glucose) in cells exposed to 11 and 25 mmol/l glucose (but not to 5 mmol/l glucose) and high PA levels (Fig. 2a, c, e). It is also worth noting that at 25 mmol/l glucose and increasing PA levels the augmentation of basal insulin secretion preceded the reduction in cell number.

Effects of glucose and PA on insulin secretion and content of INS-1E cells. INS-1E cells were maintained at 5 (a, b), 11 (c, d) or 25 mmol/l glucose (e, f) without or with the indicated PA concentrations, as described in Fig. 1, and then taken for the standard GSIS assay (1 h in KRB-HEPES containing 3.3 mmol/l glucose [white bars], followed by 1 h at 16.7 mmol/l glucose [black bars]). The vertical broken lines indicate PA concentrations above which insulin secretion and/or content were significantly reduced in a glucose-dependent manner. Data are means ± SEM, n = 3; *p < 0.05 for differences from the respective 5 mmol/l glucose control or PA-untreated cells; † p < 0.05 for differences from the respective untreated cells or cells treated with stimulatory levels of PA
PA-induced phospholipid remodelling in INS-1E cells
Table 1 shows data from lipidomic analyses of INS-1E cells maintained at 5, 11 or 25 mmol/l glucose without or with increasing PA concentrations. As shown previously [13, 21], increased glucose levels affected the abundance of saturated fatty acids (SFA), mono-unsaturated fatty acids (MUFA) and polyunsaturated fatty acids (PUFA) in membrane phospholipids. SFA incorporation following PA treatment was correlated positively to ambient glucose; the incremental SFA incorporation following treatment with 500 μmol/l PA increased from 24.0 ± 0.6 ng/106 cells at 5 mmol/l glucose to 33.2 ± 3.5 and 41.9 ± 1.6 ng/106 cells at 11 and 25 mmol/l glucose, respectively. The respective gain in PA incorporation was 36.0 ± 0.7, 43.5 ± 6.2 and 46.6 ± 1.9 ng/106 cells. Yet, total fatty acid content in phospholipids remained unaltered at all PA levels (Table 1), indicating that the incorporation of PA into phospholipids was coupled to a comparable exchange with other fatty acids. PA treatment also markedly increased the incorporation of palmitoleic acid (16:1,cis-9) into phospholipids (Fig. 3b), while the abundance of stearic acid (18:0) and oleic acid (18:1,cis-9) was significantly reduced in a PA concentration-dependent manner at all glucose levels (Fig. 3c, d). Finally, PA further intensified the depletion of AA and LA at all glucose concentrations (Fig. 3e, f).

Effects of glucose and PA on abundance of fatty acids in phospholipids in INS-1E cells. The data in Table 1 were used to calculate the relative abundance of PA (a), palmitoleic acid (b), stearic acid (c), oleic acid (d), AA (e) and LA (f) in cells incubated at 5 (white) circles, 11 (grey circles) or 25 mmol/l glucose (black circles). The results are shown as fold of the levels obtained for the respective 5 mmol/l glucose controls. Data are means ± SEM, n = 4. The statistical analyses are given in Table 1
Time-dependent lipidomic analysis of cells exposed to a non-cytotoxic concentration of PA (200 μmol/l) is presented in ESM Table 2. Maximal PA incorporation and the concomitant decline in the abundance of stearic acid, oleic acid, AA and LA were observed within 2 h of incubation (Fig. 4a, c–f) whereas the incorporation of palmitoleic acid continued throughout the incubation period (Fig. 4b).

Time course analysis of the effects of PA on the abundance of fatty acids in phospholipids. INS-1E cells were incubated for the indicated times with 200 μmol/l PA, processed and taken for fatty acid-based lipidomic analysis. (a–f) Relative abundance of the indicated fatty acids was calculated from the data in ESM Table 2. (g) Lysates prepared from cells exposed to 5, 11 or 25 mmol/l glucose without (white bars) or with 200 μmol/l PA (black bars) were used for western blot analyses of total cPLA2 and p-Ser505-cPLA2. (h) Similar western blot analyses were performed on cells exposed to 5 mmol/l glucose and the indicated concentrations of PA, added during the last hour of incubation. Representative western blots are shown in (g) and (h). (i) INS-1E cells treated as in (h) were incubated without or with 10 μmol/l GW1100 followed by western blot analysis. A representative western blot is shown. (j) Rat pooled islets were treated similarly and processed for western blot analysis. α-Tubulin served as a protein loading control. Data are means ± SEM, n = 3 or 4; *p < 0.05 for differences from the untreated cells
Activated phospholipase A2 (PLA2) hydrolyses PUFA from the sn-2 position in phospholipids [22]. Figure 4(g, h) shows that PA dose-dependently induced Ser505-phosphorylation in cPLA2 in cells at 5 mmol/l glucose [13, 22], while the enzyme was maximally phosphorylated at 11 and 25 mmol/l glucose The finding that GW1100, a specific GPR40 antagonist [23], blocked PA-induced cPLA2 phosphorylation (Fig. 4i, j) links this receptor to the phospholipid remodelling process.
PA increased insulin secretion in a PPARδ-dependent manner
We have shown before that 4-HNE, the peroxidation product of AA and LA, amplifies insulin secretion by activating PPARδ [13]. To test whether PA could enhance GSIS by activating PPARδ we measured the activity of a PPAR response element (PPRE)–luciferase reporter vector in cells exposed to non-cytotoxic PA concentrations at various glucose levels. As shown before [13], 11 and 25 mmol/l glucose increased PPRE–luciferase reporter activity (2.06-fold and 2.74-fold, respectively, in comparison with cells at 5 mmol/l; Fig. 5a). Importantly, PA significantly increased luciferase activity in a concentration-dependent manner at all glucose concentrations. Worthy of note, PPARδ protein levels in the cells remained similar under these conditions (ESM Fig. 1).

Role of PPARδ in PA-induced insulin secretion. (a) INS-1E cells were transfected with the reporter system, as described under ‘Methods’, and incubated at 5 (white bars), 11 (grey bars) and 25 mmol/l glucose (black bars) with the indicated PA concentrations, followed by the luciferase activity assay. A relative light unit value of 1 was assigned to the respective untreated control group. (b) Similarly transfected cells were incubated at 5 mmol/l glucose for 48 h, without or with 200 μmol/l PA, as described for Fig. 1, without or with GSK0660 (GSK) during the last 16 h. Luciferase activity was then measured; a relative light unit value of 1 was assigned to vehicle-treated cells. (c) The cells were maintained at 5 mmol/l glucose for 48 h; PA and GSK0660 were present during the last 16 h, after which a standard GSIS assay (3.3 mmol/l glucose [white bars] and 16.7 mmol/l glucose [black bars]) was performed. The vehicle DMSO (1:1,000 dilution) had no significant effect on GSIS. (d) siRNA sequences (si) were used to silence PPARδ expression. Untreated cells (C) and cells transfected with scrambled RNA sequences (sc) served as controls. Pparδ mRNA and PPARδ protein levels were measured by quantitative real-time PCR and western blot analyses. (e) Similarly treated cells were incubated at 5 mmol/l glucose without or with 200 μmol/l PA and taken for the standard GSIS assay. Data are means ± SEM, n = 3; *p < 0.05 for differences from the respective untreated controls (a–e); † p < 0.05 for differences from the respective control at the same glucose concentration (a), from PA-treated cells (b, d) or from the respective C and sc cells (e). RU, relative units
The role of 4-HNE in PA-induced PPARδ activation and insulin secretion was further investigated under non-cytotoxic conditions (5 mmol/l glucose without or with 200 μmol/l PA). The selective PPARδ antagonist GSK0660 blocked PA-enhanced luciferase activity (Fig. 5b) and GSIS (Fig. 5c). Furthermore, siRNA-mediated silencing of PPARδ expression markedly reduced the capacity of PA to amplify GSIS. Control GSIS was also reduced in vehicle-treated cells. Naive cells or cells transfected with scrambled RNA sequences served as controls (Fig. 5d, e).
Previous studies suggest that 4-HNE activates PPARδ [13, 24]. Figure 6a shows that cells exposed to 200 μmol/l PA for 16 h generated 5.6-fold higher amounts of 4-HNE than vehicle-treated cells. l-Carnosine, a potent scavenger of α,β-unsaturated aldehydes [25], reduced the amount of free 4-HNE in the culture medium to the control level, while eliminating PPARδ activation and GSIS amplification (Fig. 6b, c). Similarly, l-carnosine abolished the ability of PA to induce luciferase activity and augment insulin secretion (Fig. 6d, e). Similar effects were also observed in rat isolated islets exposed to PA without or with GSK0660 or l-carnosine (Fig. 6f, g). The concentration-dependent effects of PA on GSIS in rat isolated islets are shown in ESM Fig 2a, b.

PA augments 4-HNE generation and activates PPARδ. (a) INS-1E cells were maintained at 5 mmol/l glucose for 48 h without or with 200 μmol/l PA, in the absence or presence of l-carnosine (Car), during the last 16 h of incubation. The culture media were then collected, extracted and taken for HPLC analysis of 4-HNE. (b) Cells at 5 mmol/l glucose were transfected, as described in the legend to Fig. 4, treated with the indicated concentrations of 4-HNE and l-carnosine and taken for the luciferase activity assay. A relative light unit value of 1 was assigned to the respective untreated control groups. (c) INS-1E cells were similarly treated with glucose, 4-HNE and l-carnosine and taken for the standard GSIS assay. (d, e) Transfected (d) or control cells (e) were treated with PA and taken for the luciferase activity and the GSIS assays, respectively. (f, g) Rat isolated islets, maintained at 5 mmol/l glucose, were treated with 200 μmol/l PA without or with GSK0600 (f) or l-carnosine (g) for 16 h and taken for the GSIS assay (3.3 mmol/l glucose [white bars] and 16.7 mmol/l glucose [black bars]). Data are means ± SEM, n = 3; *p < 0.05 for differences from the respective untreated cells; † p < 0.05 for differences from the respective PA-treated cells
Ravnskjaeret el al [20] and Jiang et al [26] proposed that pyruvate dehydrogenase kinase 4 (PDK4), carnitine palmitoyltransferase-1 (CPT-1α) and fatty acid translocase (CD36) were PPARδ downstream targets involved in the regulation of insulin secretion. We measured their expression in 4-HNE-treated cells (Fig. 7a–f) and observed a significant increase in mRNA levels of Cd36 and Pdk4 (but not Cpt-1α [also known as Cpt1a] mRNA) in both INS-1E cells and rat isolated islets.

4-HNE increases the expression of Pdk4 and Cd36 in INS-1E cells. INS-1E cells (a, c, e) and rat isolated islets (pooled from five rats; b, d, f) were treated with the indicated 4-HNE concentrations as described above and processed for quantitative real-time PCR analysis of Cd36 (a, b), Pdk4 (c, d) and Cpt-1α mRNA (e, f). Data are means ± SEM, n = 3; *p < 0.05 for differences from the respective untreated controls
PA induced beta cell death in a 4-HNE-independent manner
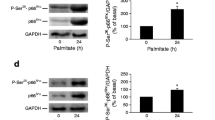
High exogenous levels of 4-HNE cause beta cell apoptosis and death, primarily due to excessive covalent protein modifications [13, 27]. ESM Fig. 3a shows that exogenously added 4-HNE formed protein adducts in INS-1E cells in a concentration-dependent manner. PA increased the formation of 4-HNE-protein adducts in a concentration- and time-dependent manner in cells maintained at 25 mmol/l glucose. Maximal formation of adducts was observed with 500 μmol/l PA following 16 h of incubation (ESM Fig. 3b). Strikingly, l-carnosine, which effectively diminished the accumulation of 4-HNE-protein adducts in INS-1E cells and rat isolated islets exposed to PA, failed to rescue the cells from death (Fig. 8a, b). ER stress is considered a major contributor to beta cell dysfunction under glucolipotoxic conditions [8, 28]. However, l-carnosine had no noticeable lowering effect on the expression and phosphorylation of the key ER stress markers IRE, pSer724-IRE (p-IRE) and pThr980-PERK (p-PERK), which were activated by PA (Fig. 8c–f).

Glucolipotoxicity is not mediated by 4-HNE. INS-1E cells, maintained at 25 mmol/l glucose for 48 h, in the absence or presence of 500 μmol/l PA, without or with l-carnosine (Car) during the last 16 h of incubation, were processed and taken for a dot blot analysis of 4-HNE-protein adducts (a), cell number counting (b) and western blot analyses of p-Thr980-PERK, p-Ser724-IRE and total IRE (c). Corresponding densitometric analysis of the western blots are shown (d–f). Dot blot analysis of lysates prepared from rat pooled isolated islets that were incubated with 500 μmol/l PA is shown in (a). Data are means ± SEM, n = 3; *p < 0.05 for differences from the respective untreated cells; † p < 0.05 for differences from the respective PA-treated cells. RU, relative units
Discussion
This study shows that the combination of increasing levels of PA and glucose enhanced phospholipid remodelling and the release of AA and LA in beta cells. These conditions triggered the peroxidation of these PUFA to generate 4-HNE, which amplified insulin secretion in a PPARδ-dependent manner. However, this effect was lost at high glucose and PA concentrations due to cytotoxic effects of the combination.
The present lipidomic analysis revealed fast and profound changes in the abundance of fatty acids in phospholipids of PA-treated cells. The extent of incorporation and exchange of PA with other fatty acids increased in a glucose- and PA-dependent manner, while the total content of phospholipid-associated fatty acids was comparable at all PA concentrations. Further studies are required to determine whether phospholipid remodelling occurs similarly in isolated islets. PA-induced reorganisation of the phospholipid compartment was completed within 2 h and was associated with Ser505 phosphorylation in cPLA2. Worthy of note, a two-step activation of cPLA2 by Ser505 and Ser515 phosphorylation has been proposed [29]. Plausibly, the maximal Ser505 phosphorylation at high glucose concentration (Fig. 4g) may not reflect full activation of the enzyme unless Ser515 is co-phosphorylated. The inhibition of PA-induced Ser505 phosphorylation in cPLA2 by GW1100 (Fig. 4i, j) suggests a role for free fatty acid receptor 1 (GPR40) in PA-induced activation of this enzyme [2, 30]. Interestingly, previous findings [31] showing that PA could alter the abundance of lyso-phosphatidylethanolamine species in islets from Gpr40 (also known as Ffar1) knockout mice suggest that other GPR subtypes could be involved the regulation of phospholipid turnover in beta cells.
The abundance of stearic-, oleic-, arachidonic- and linoleic acid in phospholipids was reduced maximally when PA incorporation reached peak levels. Concomitantly, palmitoleic acid incorporation increased linearly and was maximal at 250 μmol/l PA. This reflects Δ9-desaturase (SCD1)-mediated transformation of PA to its mono-unsaturated form. Previous reports assigned protective roles for SCD1 and oleic acid against palmitate-induced damage in beta cells [32, 33]. The reciprocal alterations in PA and oleic acid content in phospholipids result from competition for SCD1 between endogenous oleic acid and high levels of exogenously added PA. Previous findings demonstrating the protective role of oleic acid in PA-treated beta cells [34] could be explained by the replenishment of the former when SCD1 predominately transforms PA to palmitoleic acid.
PUFA are transformed by cyclooxygenases, lipoxygenases and cytochrome P450 epoxidase/ω-hydrolase to a large number of mediators, which regulate various beta cell functions [12, 35, 36]. The increased cyclooxygenase-mediated production of PGE2 in PA-treated cells (ESM Fig. 3c and [37]) corroborates our findings regarding increased availability of AA to intracellular metabolism. It is worth noting that PGE2, as well as other PUFA metabolites, can also regulate beta cell function by activating PPARα, PPARδ or PPARγ nuclear receptors. In addition, free radical-induced peroxidation of PUFA generates 4-hydroxyalkenals [35, 36]. Being saturated, PA is not subjected to peroxidation, yet it effectively augmented 4-HNE production in INS-1E cells by increasing the availability of AA and LA to peroxidation. We, and others, have reported that 4-hydroxyalkenals activate the PPARδ nuclear receptor in various cells, including beta cells [13, 19, 24, 35, 38]. The findings that 4-HNE scavenging with l-carnosine and PPARδ inhibition or its silencing attenuated PA-induced GSIS further support the role of the 4-HNE–PPARδ axis in beta cell function [13, 39]. However, additional strategies aimed at reducing endogenous 4-HNE levels (i.e. modulation of the expression of fatty aldehyde dehydrogenases) may further be employed.
Our findings complement well previous reports on ameliorating effects of PPARδ agonists on PA-induced dysfunction in beta cells [26, 40]. We have found that 4-HNE augmented the expression of the PPARδ-sensitive genes Pdk4 and Cd36 (but not Cpt-1α) in beta cells. These proteins have been linked to enhanced mitochondrial fatty acid oxidation and augmented insulin secretion [20, 41]. The reason for a lack of effect on CPT-1α in our study is unclear. The different sensitivity of the INS-1E cell line and isolated islets to 4-HNE has been previously observed and discussed [13]. Collectively, our findings support the hypothesis that non-cytotoxic combinations of glucose and PA have a hormetic role in response to nutrient overload by increasing insulin secretion to maintain homeostasis of both nutrients. The in vitro cytotoxic threshold level of PA in beta cells was correlated conversely with the ambient glucose. However, it is unclear whether similar glucolipotoxic threshold levels are attained in vivo. There is a wide range of plasma PA levels in healthy individuals and the levels are altered significantly in various diseases but barely reach the in vitro threshold levels reported here [42–44]. Therefore, clinical and population-based studies are needed to ascertain the relevant in vivo glucolipotoxic threshold levels in relation to beta cell pathophysiology. Similarly, the idea that other fatty acids in the plasma attenuate these threshold levels should be carefully evaluated.
ER stress and enhanced ROS production have been implicated in beta cell glucolipotoxicity [11, 28, 45], although others contest this theory [37]. The contribution made by beta cell-generated 4-hydroxyalkenals to this process is unclear. High levels of 4-hydroxyalkenals and their metabolites have been linked to the development of diabetic complications [25, 35, 38, 46–48]. Indeed, exposure of beta cell cultures and rat isolated islets to high levels of 4-HNE induced apoptosis and cell death [13, 27]. Nonetheless, this study shows that the neutralisation of 4-HNE with l-carnosine failed to rescue the cells from ER stress and demise. Therefore, endogenous 4-HNE may not be a major glucolipotoxic factor. However, PA-induced stress could result from the excessive generation of palmitoyl-CoA [11], which is a substrate for ceramide biosynthesis [49]. Ceramides are pro-apoptotic and can blunt the stimulatory effect of glucose on proinsulin gene transcription [49]. Thus, PA-derived metabolites other than 4-HNE appear to contribute to beta cell dysfunction and death. Our study focused on the rapid effects of PA on phospholipid remodelling and its consequences. Further studies are required to investigate whether this mechanism remains operative after prolonged incubation of beta cells with non-toxic PA and glucose levels.
In summary, this study shows that glucose- and PA-induced phospholipid turnover generates intracellular metabolites that regulate beta cell function and survival. The adaptive GSIS response was mediated via the 4-HNE-activated PPARδ pathway. Notably, the abundance of fatty acids in phospholipids may vary considerably among individuals due to diet, nutrient availability and metabolism [50]. Therefore, these factors may differentially influence phospholipid remodelling resulting in variable beta cell response to nutritionally challenging conditions. Importantly, this study points to an important role for physiological levels of PA in the maintenance normal beta cell function.
Abbreviations
- AA:
-
Arachidonic acid
- CPT-1α:
-
Carnitine palmitoyltransferase-1α
- ER:
-
Endoplasmic reticulum
- GFP:
-
Green fluorescent protein
- GSIS:
-
Glucose-stimulated insulin secretion
- 4-HNE:
-
4-hydroxy-2E-nonenal
- IRE:
-
Inositol-requiring enzyme-1
- LA:
-
Linoleic acid
- MTT:
-
Methylthiazol tetrazolium
- MUFA:
-
Mono-unsaturated fatty acids
- PA:
-
Palmitic acid
- PERK:
-
Protein kinase RNA-like ER kinase
- PLA2 :
-
Phospholipase A2
- PPARδ:
-
Peroxisome proliferator-activated receptor-δ
- PPRE:
-
PPAR response element
- PUFA:
-
Polyunsaturated fatty acids
- PDK4:
-
Pyruvate dehydrogenase kinase 4
- ROS:
-
Reactive oxygen species
- SCD1:
-
Δ9-Desaturase
- SFA:
-
Saturated fatty acids
References
Metz SA (1988) Membrane phospholipid turnover as an intermediary step in insulin secretion. Putative roles of phospholipases in cell signaling. Am J Med 85:9–21
Nolan CJ, Madiraju MS, Delghingaro-Augusto V, Peyot ML, Prentki M (2006) Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 55(Suppl 2):S16–S23
Yaney GC, Corkey BE (2003) Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 46:1297–1312
Carpentier AC, Frisch F, Brassard P et al (2007) Mechanism of insulin-stimulated clearance of plasma nonesterified fatty acids in humans. Am J Endocrinol Metab 292:E693–E701
Nestel PJ, Ishikawa T, Goldrick RB (1978) Diminished plasma free fatty acid clearance in obese subjects. Metabolism 27:589–597
Chow CC, Periwal V, Csako G et al (2011) Higher acute insulin response to glucose may determine greater free fatty acid clearance in African-American women. J Clin Endocrinol Metab 96:2456–2463
Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS (2007) Regulation of lipolysis in adipocytes. Annu Rev Nutr 27:79–101
Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL (2004) Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 145:5087–5096
Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY (2001) Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50:69–76
Stein DT, Stevenson BE, Chester MW et al (1997) The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J Clin Invest 100:398–403
Elsner M, Gehrmann W, Lenzen S (2011) Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 60:200–208
Luo P, Wang MH (2011) Eicosanoids, β-cell function, and diabetes. Prostaglandins Other Lipid Mediat 95:1–10
Cohen G, Riahi Y, Shamni O et al (2011) Role of lipid peroxidation and PPAR-delta in amplifying glucose-stimulated insulin secretion. Diabetes 60:2830–2842
Boslem E, Weir JM, MacIntosh G et al (2013) Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic β-cells. J Biol Chem 288:26569–26582
Hoppa MB, Collins S, Ramracheya R et al (2009) Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules. Cell Metab 10:455–465
Busch AK, Gurisik E, Cordery DV et al (2005) Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic beta-cells from lipoapoptosis. Diabetes 54:2917–2924
Medina-Gomez G, Yetukuri L, Velagapudi V et al (2009) Adaptation and failure of pancreatic beta cells in murine models with different degrees of metabolic syndrome. Dis Models Mech 2:582–592
Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P (2004) Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145:667–678
Riahi Y, Sin-Malia Y, Cohen G et al (2010) The natural protective mechanism against hyperglycemia in vascular endothelial cells: roles of the lipid peroxidation product 4-hydroxydodecadienal and peroxisome proliferator-activated receptor delta. Diabetes 59:808–818
Ravnskjaer K, Frigerio F, Boergesen M, Nielsen T, Maechler P, Mandrup S (2010) PPARdelta is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J Lipid Res 51:1370–1379
Wallace M, Whelan H, Brennan L (2013) Metabolomic analysis of pancreatic beta cells following exposure to high glucose. Biochim Biophys Acta 1830:2583–2590
Jolly YC, Major C, Wolf BA (1993) Transient activation of calcium-dependent phospholipase A2 by insulin secretagogues in isolated pancreatic islets. Biochemistry 32:12209–12217
Briscoe CP, Peat AJ, McKeown SC et al (2006) Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol 148:619–628
Coleman JD, Prabhu KS, Thompson JT et al (2007) The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-β/δ(PPARβ/δ). Free Radic Biol Med 42:1155–1164
Aldini G, Orioli M, Rossoni G et al (2011) The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J Cell Mol Med 15:1339–1354
Jiang L, Wan J, Ke LQ, Lu QG, Tong NW (2010) Activation of PPARδ promotes mitochondrial energy metabolism and decreases basal insulin secretion in palmitate-treated beta-cells. Mol Cell Biochem 343:249–256
Suarez-Pinzon WL, Strynadka K, Rabinovitch A (1996) Destruction of rat pancreatic islet beta-cells by cytokines involves the production of cytotoxic aldehydes. Endocrinology 137:5290–5296
Bachar E, Ariav Y, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G (2009) Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic β-cells via activation of mTORC1. PLoS One 4:e4954
Pavicevic Z, Leslie CC, Malik KU (2008) cPLA2 phosphorylation at serine-515 and serine-505 is required for arachidonic acid release in vascular smooth muscle cells. J Lipid Res 49:724–737
Latour MG, Alquier T, Oseid E et al (2007) GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes 56:1087–1094
Alquier T, Peyot ML, Latour MG et al (2009) Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes 58:2607–2615
Thorn K, Hovsepyan M, Bergsten P (2010) Reduced levels of SCD1 accentuate palmitate-induced stress in insulin-producing β-cells. Lipids Health Dis 9:108
Lai E, Bikopoulos G, Wheeler MB, Rozakis-Adcock M, Volchuk A (2008) Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic β-cells. Am J Physiol Endocrinol Metab 294:E540–E550
Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY (2003) Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic β-cell turnover and function. Diabetes 52:726–733
Cohen G, Riahi Y, Sunda V et al (2013) Signaling properties of 4-hydroxyalkenals formed by lipid peroxidation in diabetes. Free Radic Biol Med 65:978–987
Riahi Y, Cohen G, Shamni O, Sasson S (2010) Signaling and cytotoxic functions of 4-hydroxyalkenals. Am J Physiol Endocrinol Metab 299:E879–E886
Moore PC, Ugas MA, Hagman DK, Parazzoli SD, Poitout V (2004) Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes 53:2610–2616
Jaganjac M, Tirosh O, Cohen G, Sasson S, Zarkovic N (2013) Reactive aldehydes–second messengers of free radicals in diabetes mellitus. Free Radic Res 47(Suppl 1):39–48
Poulsen L, Siersbaek M, Mandrup S (2012) PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol 23:631–639
Cao M, Tong Y, Lv Q et al (2012) PPARδ activation rescues pancreatic β-cell line INS-1E from palmitate-induced endoplasmic reticulum stress through enhanced fatty acid oxidation. PPAR Res 2012:680–684
Wan J, Jiang L, Lu Q, Ke L, Li X, Tong N (2010) Activation of PPARδ up-regulates fatty acid oxidation and energy uncoupling genes of mitochondria and reduces palmitate-induced apoptosis in pancreatic β-cells. Biochem Biophys Res Commun 391:1567–1572
Fraser DA, Thoen J, Rustan AC, Forre O, Kjeldsen-Kragh J (1999) Changes in plasma free fatty acid concentrations in rheumatoid arthritis patients during fasting and their effects upon T-lymphocyte proliferation. Rheumatology 38:948–952
Carlsten A, Hallgren B, Jagenburg R, Svanborg A, Werko L (1966) Amino acids and free fatty acids in plasma in diabetes. I. The effect of insulin on the arterial levels. Acta Med Scand 179:361–370
Shen F, Qi J, Xu F et al (2013) Age-related distributions of nine fasting plasma free fatty acids in a population of Chinese adults. Clin Chim Acta 415:81–87
Bachar-Wikstrom E, Wikstrom JD, Ariav Y et al (2013) Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes 62:1227–1237
Bacot S, Bernoud-Hubac N, Baddas N et al (2003) Covalent binding of hydroxy-alkenals 4-HDDE, 4-HHE, and 4-HNE to ethanolamine phospholipid subclasses. J Lipid Res 44:917–926
Toyokuni S, Yamada S, Kashima M et al (2000) Serum 4-hydroxy-2-nonenal-modified albumin is elevated in patients with type 2 diabetes mellitus. Antioxid Redox Signal 2:681–685
Negre-Salvayre A, Auge N, Ayala V et al (2010) Pathological aspects of lipid peroxidation. Free Radic Res 44:1125–1171
Boslem E, Meikle PJ, Biden TJ (2012) Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets 4:177–187
Ferreri C, Chatgilialoglu C (2012) Role of fatty acid-based functional lipidomics in the development of molecular diagnostic tools. Expert Rev Mol Diagn 12:767–780
Acknowledgements
We thank V. Sunda and S. Deplano (Lipinutragen, Lipidomic Laboratory Consiglio Nazionale delle Ricerche, Bologna, Italy) for conducting the lipidomic analyses and D. Faham (The Hebrew University) for performing the flow cytometric analysis.
Funding
The study was supported by grants from the Israel Science Foundation (44/10), the Legacy Heritage Biomedical Science Partnership of the Israel Academy of Sciences and Humanities (1429/13), the Brettler Center for Research of Molecular Pharmacology and Therapeutics in the Hebrew University and by COST (European Cooperation in Science and Technology) Action CM1201 on ‘Biomimetic Radical Chemistry’. GC and OS received fellowships from the Hebrew University Center for Diabetes Research. SS holds the Adolf D. and Horty Storch Chair in Pharmaceutical Sciences at the Faculty of Medicine, the Hebrew University of Jerusalem, Israel and is affiliated with the David R. Bloom Center for Pharmacy and the Dr Adolf and Klara Brettler Center for Research of Molecular Pharmacology and Therapeutics in the Hebrew University.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
GC researched data, contributed to the discussion and wrote and edited the manuscript. OS researched data (HPLC analysis). CC and CF researched data (lipidomic analysis), contributed to the discussion and reviewed the manuscript. YA, OC ECB and NFL performed the studies with GW1100, PPAR isoform expression and the quantitative real-time PCR measurements. NK contributed to the research design and discussion and reviewed and edited the manuscript. SS designed the study and wrote and edited the manuscript. All authors contributed to the study conception and design, acquisition of data and analysis and interpretation of data. All authors contributed to the drafting of the article and approved the final version of the manuscript. SS is the guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 150 kb)
ESM Table 1
(PDF 208 kb)
ESM Table 2
(PDF 80 kb)
ESM Figure 1
(PDF 182 kb)
ESM Figure 2
(PDF 142 kb)
ESM Figure 3
(PDF 162 kb)
Rights and permissions
About this article

Cite this article
Cohen, G., Shamni, O., Avrahami, Y. et al. Beta cell response to nutrient overload involves phospholipid remodelling and lipid peroxidation. Diabetologia 58, 1333–1343 (2015). https://doi.org/10.1007/s00125-015-3566-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3566-z