
Abstract
In this review, we explore the concept of ‘double diabetes’, a combination of type 1 diabetes with features of insulin resistance and type 2 diabetes. After considering whether double diabetes is a useful concept, we discuss potential mechanisms of increased insulin resistance in type 1 diabetes before examining the extent to which double diabetes might increase the risk of cardiovascular disease (CVD). We then go on to consider the proposal that weight gain from intensive insulin regimens may be associated with increased CV risk factors in some patients with type 1 diabetes, and explore the complex relationships between weight gain, insulin resistance, glycaemic control and CV outcome. Important comparisons and contrasts between type 1 diabetes and type 2 diabetes are highlighted in terms of hepatic fat, fat partitioning and lipid profile, and how these may differ between type 1 diabetic patients with and without double diabetes. In so doing, we hope this work will stimulate much-needed research in this area and an improvement in clinical practice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In this review, we address the following questions: (1) What are the potential mechanisms linking type 1 diabetes with insulin resistance and how is this combination (‘double diabetes’) associated with increased cardiovascular risk? (2) Might weight gain associated with intensive insulin regimens be associated with increased insulin resistance and what implications might this have for cardiovascular risk? (3) What role does the liver play in double diabetes? (4) Might we need to consider changes in current clinical practice in the context of the issues raised in this review?
Double diabetes
The term ‘double diabetes’ was first coined in 1991 based on the observation that patients with type 1 diabetes who had a family history of type 2 diabetes were more likely to be overweight and rarely achieved adequate glycaemic control even with higher insulin doses [1]. The more extensive, or stronger, the family history, the higher the dose the patient received. The authors suggested that this might indicate the presence of increased resistance to insulin-mediated glucose disposal in this subgroup of people with type 1 diabetes and asserted that, over a lifetime, some of these individuals would likely have been diagnosed with type 2 diabetes at some point, had they not first developed beta cell destruction by an independent pathological process (i.e. type 1 diabetes). At this stage, it is important to differentiate this description of double diabetes, which considers autoimmune diabetes to be an independent process from obesity and insulin resistance, from the accelerator hypothesis [2], which describes triggering of autoimmune diabetes by factors including BMI and insulin resistance.
Other studies of people with type 1 diabetes and a family history of type 2 diabetes have supported the notion that this combination might promote both microvascular and macrovascular complications of type 1 diabetes. For example, in a prospective study of 3250 patients with type 1 diabetes recruited from 16 European countries (EURODIAB), it was demonstrated that women with a parental history of type 2 diabetes had a higher risk of developing albuminuria than those without a positive family history (HR 1.36, p = 0.04) [3]. Furthermore, in a cross-sectional study of 658 patients from the Pittsburgh Epidemiology of Diabetes Complications (EDC) cohort, 112 of whom had a confirmed family history of type 2 diabetes, and 119 of whom had experienced a CHD event, a positive family history was associated with a significant excess risk of CHD (HR 1.89, 95% CI 1.27, 2.84) [4]. Those with a greater number of affected family members had a greater risk (p = 0.001 for trend), with one family member conferring an OR of 1.62 and two family members increasing this to 5.13 [4]. These data are consistent with either genetically determined insulin resistance/double diabetes or shared parental–offspring lifestyle factors contributing to type 2 diabetes in parents and increasing the risk of complications. Given the results of genome-wide association studies of type 2 diabetes published to date [5], the latter explanation currently appears more plausible.
Measuring insulin resistance in type 1 diabetes and examining its association with complications
Clamp studies
If insulin resistance, either inherited or acquired, is implicated in the pathogenesis of double diabetes, how is it likely to manifest and can it be identified? The gold standard method of measuring insulin-mediated glucose uptake is the hyperinsulinaemic–euglycaemic clamp technique. Although this is a time-consuming and relatively invasive technique, several investigators have employed it to demonstrate increased insulin resistance in those with type 1 diabetes compared with age-, sex- and BMI-matched controls [6–16].
In one such study of 40 cases (mean age 45 years, mean diabetes duration of type 1 diabetes 23 years) and 47 age- and sex-matched control individuals, patients with type 1 diabetes not only had lower whole body insulin-mediated glucose uptake (mean ± SE: 6.19 ± 0.72 vs 12.71 ± 0.66 mg [kg fat-free mass]−1 min−1, p < 0.0001) but also had lower insulin-mediated NEFA suppression. Both measurements were correlated (p < 0.0001 within the type 1 diabetes group) with coronary artery calcification (CAC) measured by computed tomography. Of note, HbA1c did not correlate with either insulin resistance or CAC [16].
Derived measures of insulin resistance
Data from one clamp study [6], (n = 24) were used to derive estimated glucose disposal rate (eGDR) from a ‘best-fit’ model:
where hypertension status (1 or 0) is defined as BP >140/90 mmHg or on anti-hypertensive drugs; HbA1c refers to the value in %. Low eGDR was proposed as a surrogate measure of insulin resistance for use either in a clinical setting or for epidemiological analyses [6].
Prediction of CVD using eGDR
eGDR was tested prospectively in 603 type 1 diabetes Pittsburgh EDC cohort participants (mean baseline age 28 years, mean diabetes duration 19 years) followed up over 10 years [17]. Independent predictors of CVD events (n = 42) were disease duration, presence of nephropathy, non-HDL-cholesterol (HDL-C), white cell count and eGDR. HbA1c was not in itself a predictor, except as a component of eGDR [17]. However, in a subsequent analysis of the same cohort by the same group, eGDR did not appear in CHD risk prediction models (although, importantly, these incorporated three of its individual components: WHR, HDL-C and BP) [18].
In an independent examination of the DCCT/Epidemiology of Diabetes Interventions and Complications (EDIC) data, Kilpatrick et al estimated eGDR retrospectively (using BMI data instead of WHR) as a predictor of CVD events. In a Cox regression model, high eGDR as a surrogate for preserved insulin sensitivity was independently associated with a lower risk for CV events (HR 0.70; 95% CI 0.56, 0.88) [19]. In contrast, total insulin dose requirement had no predictive value. Intriguingly, high eGDR was also associated with a lower risk for microvascular events, including retinopathy and nephropathy—a finding also corroborated by data from the EURODIAB study [20].
Other derived measures related to insulin resistance: does the metabolic syndrome identify CVD risk in type 1 diabetes?
Given the mechanistic relevance of insulin resistance in the metabolic syndrome, a number of prospective studies have assessed the utility of various definitions of the metabolic syndrome in predicting risk in type 1 diabetes cohorts. However, as the glycaemia criterion of the WHO, National Cholesterol Education Program (NCEP) and International Diabetes Federation (IDF) definitions are automatically fulfilled, there is a major caveat. For example, an individual with type 1 diabetes can meet the WHO definition by virtue of microalbuminuria alone, even if non-obese and with a normal lipid profile.
In a prospective analysis of the Pittsburgh EDC cohort, the prevalence of the metabolic syndrome was 21% (WHO definition), 12% (NCEP) and 8% (IDF). Using a composite endpoint of CHD, renal failure and diabetes-related death, the metabolic syndrome conferred significantly elevated HRs (NCEP: 5.8, 95% CI 3.9, 8.6; WHO: 6.5, 95% CI 4.5, 9.4); however, these ratios were not higher than those for individual components of the syndrome (for example, microalbuminuria: 6.3, 95% CI 3.8, 10.5; BP 4.5, 95% CI 3.1, 6.6; triacylglycerol: 4.4, 95% CI 3.0, 6.6) [21].
In a similar analysis of the FinnDiane study [22], a high prevalence of the metabolic syndrome was reported (44% WHO definition; 35% NCEP; 36% IDF). The WHO definition predicted incident CV events after adjustment for traditional risk factors and nephropathy (HR 2.05, 95% CI 1.38, 3.04), even in participants with raised albumin excretion (HR 1.44, 95% CI 1.06, 1.96). However, the individual components of the metabolic syndrome were also significant predictors, with the exception of obesity. In the DCCT/EDIC study, an adapted IDF definition of the metabolic syndrome had a baseline prevalence of 22%, largely driven by low HDL-C, but did not predict either macro- or microvascular events over 17 years of follow-up [19].
Thus, in broad terms, the metabolic syndrome and its individual features are associated with more events but, as in the general population [23], dichotomous classification of individuals with type 1 diabetes according to the metabolic syndrome does not appear to add value for CV risk prediction in type 1 diabetes over and above its individual components.
Intensive insulin therapy, weight/weight gain and their relationship to insulin resistance and CV risk in type 1 diabetes
It is generally accepted that intensive insulin therapy reduces the incidence of CVD in type 1 diabetes. The most robust interventional clinical trial evidence for this is from the DCCT in which 1,441 adolescents and young adults (mean age 27 years, BMI 28 kg/m2) were randomised for 6.5 years to intensive or conventional glycaemic control and followed up post-randomisation in the EDIC study up to a total of 17 years [24]. Intensive glycaemic control was associated with a 57% reduction in RR (95% CI 12, 79%, p = 0.02) in a composite endpoint of non-fatal MI, stroke or CV death (approximate overall rate of six events per 1,000 patient-years) [24]. Of course, one of the consequences of intensive therapy is weight gain; the intensively treated group in the DCCT study gained 4.6 kg more than the conventionally treated group over the 5 year study period [25].
In this context, subgroup analyses of the DCCT/EDIC study have raised concerns about participants in whom weight gain over the course of the trial was associated with the emergence of features associated with increased CV risk (Table 1) [26]. Those in the highest quartile for weight gain exhibited higher BP and LDL-cholesterol (LDL-C) values, a higher insulin dose requirement and WHR and a more atherogenic lipid profile (raised apolipoprotein B, high VLDL, higher small, dense LDL, lower apolipoprotein A-I and lower HDL). In the intensive group, mean BMI increased from 24 to 31 kg/m2. These differences remained significant after adjustment for HbA1c [26]. A similar pattern was observed among those allocated to conventional treatment (albeit with a reduced effect size) [26], which is consistent with the existence of individual predisposing factors to insulin-induced weight gain, an area that merits further study.
Interestingly, in the DCCT/EDIC study, a family history of type 2 diabetes was also a significant independent predictor of weight gain in both the conventional and intensive groups [27], consistent with an inherited tendency to store excess fat in response to insulin, a degree of inherited peripheral insulin resistance and/or a familial effect on environmental factors (energy intake and physical activity).
There is increasing contemporary evidence that the prevalence of obesity has risen substantially among the type 1 diabetic population, as it has in the general population. For example, in the Pittsburgh EDC cohort between 1987 and 2007 the prevalence of obesity rose sevenfold (to 22.7%) and overweight increased by 47% (to 42%) [28]. While in the pre-DCCT era baseline BMI in patients with type 1 diabetes was lower than population norms, probably owing to a combination of weight loss prior to diagnosis and suboptimal glycaemic control, current BMI patterns are similar. Although much of the weight gain seen in contemporary patients with type 1 diabetes is likely to reflect cultural, societal and lifestyle changes, some of this increase may be due to use of more intensive insulin regimens. Mechanisms for the latter may include reduced excretion of urinary glucose and altered feeding behaviour (more frequent treatment of hypoglycaemic episodes, appetite stimulation), in addition to the known anabolic effects of insulin.
Adverse CV risk factors associated with weight gain in some recipients of intensive insulin therapy were also seen in the observational data from the EURODIAB cohort, in which 1,800 type 1 diabetic patients (mean age 33 years, duration of diabetes 14.8 years, HbA1c 8.2% [66 mmol/mol]) were followed up for 7.3 years [29]. Those who gained more than 5 kg over this time had better glycaemic control (mean HbA1c difference of 0.2%) than those with less or no weight gain but also had raised BP, LDL-C and triacylglycerol in association with lower HDL-C (differences that remained significant after adjustment for glycaemic control).
Insulin resistance in type 1 diabetes might relate to the route of administration of therapeutic exogenous insulin. Insulin absorbed from subcutaneous depots results in relative peripheral hyperinsulinaemia and hepatic hypoinsulinaemia compared with normal physiology. Chronic adaptation to this combination could reduce peripheral insulin-mediated glucose uptake and increase hepatic glucose production. In addition, it has been proposed that reduced hepatic insulin exposure results in a reduced level of circulating IGF-1, which, along with a parallel increase in growth hormone and IGF-binding proteins, may also contribute to increased peripheral insulin resistance [30, 31].
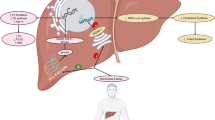
Therefore, several factors could underlie the phenotype of double diabetes (Fig. 1). First, the genetic and lifestyle factors that lead to type 2 diabetes may exist at similar frequency in those with type 1 diabetes—this would be consistent with the robust data on family history of type 2 diabetes described above. Second, weight gain caused by intensive insulin therapy may lead to insulin resistance. Third, exogenous insulin therapy might induce insulin resistance in patients with type 1 diabetes.

Mechanisms potentially contributing to insulin resistance in type 1 diabetes patients with double diabetes. T1DM, type 1 diabetes
There are several important unanswered questions:
-
1.
Are some people with type 1 diabetes (including those with a family history of type 2 diabetes or of specific ethnicities, e.g. South Asians) more susceptible to weight gain during intensive insulin treatment, perhaps owing to differences in metabolism or substrate handling – or can the observations regarding weight gain simply be explained by the fact that those who are more insulin resistant are treated with higher doses of insulin?
-
2.
What are the most important factors, e.g. inherited or familial factors, differences in feeding strategies for avoidance of hypoglycaemia, insulin resistance?
-
3.
To what extent do adverse CVD risk factors associated with higher weight gain attenuate or reverse the presumed earlier CV (and other) benefits of intensive glycaemic control in type 1 diabetes?
Lipid metabolism in type 1 diabetes
Insulin not only exerts effects on peripheral glucose uptake and suppression of hepatic glucose production, it also has profound effects on fat partitioning by promoting hepatic and peripheral lipogenesis (fat storage) as well as suppressing hepatic and peripheral lipolysis (inhibition of fat oxidation). In this section, we compare lipid handling in type 1 diabetes with non-diabetic metabolism and with type 2 diabetes.
The most obvious and striking difference between type 1 diabetes on the one hand and obesity/type 2 diabetes on the other, is low portal vein insulin concentration. This can be concluded by inference from the understanding of type 1 diabetes pathophysiology and anatomy (i.e. if beta cells are not secreting insulin, then portal insulin levels will be low, only reflecting recirculating insulin from subcutaneous injection). Further evidence for this is reviewed below and includes extrapolation from animal models. In type 1 diabetes, absent pancreatic insulin secretion is the opposite phenotype to the endogenous hyperinsulinaemia characteristic of most conditions characterised by insulin resistance. As type 1 diabetes is characterised by higher peripheral insulin concentrations and lower portal concentrations, it follows that contrasting hepatic and peripheral lipid handling might be predicted.
Reduced hepatic fat
In contrast to obesity and type 2 diabetes, in which the importance of non-alcoholic steatohepatitis (fatty liver) has been highlighted [32], there is some evidence that intra-hepatic fat content is reduced in type 1 diabetes. Comparing 19 patients with type 1 diabetes (mean age 35, BMI 23 kg/m2, HbA1c 8.7% [72 mmol/mol]) and carefully matched non-diabetic controls using 1H magnetic resonance spectroscopy, Perseghin et al demonstrated significantly reduced intra-hepatic fat content in those with type 1 diabetes (1.5 ± 0.7% vs 2.2 ± 1.0%, p < 0.03) [9]. This was associated with increased fasting lipid oxidation (1.5 ± 0.7 vs 0.8 ± 0.4 mg/kg). The estimated hepatic insulin (EHI) level was lower and the glucagon:EHI ratio (reported as an indicator of the balance between catabolism and anabolism) was higher in the type 1 diabetes group (both p < 0.05) [9].
Hyperinsulinaemia has been shown to promote the deposition of hepatic fat. Insulin stimulates sterol regulatory element-binding protein 1c (SREBP1c), which plays a crucial role in the regulation of triacylglycerol accumulation in the liver [32–35]. Moreover, two enzymes involved in de novo lipogenesis, namely, fatty acid synthase (FAS) and acetyl-CoA carboxylase, are activated by SREBP1c. In parallel with this, hepatic fatty acid oxidation is inhibited and the balance shifts to lipid storage as triacylglycerol is incorporated into VLDL [33, 36]. It has been suggested that relative under-insulinisation of the liver with preserved glucagon secretion in type 1 diabetes favours a shift in metabolism from lipid storage to oxidation [9].
Fat partitioning
Fat partitioning (the tissue distribution of fat storage) reflects the balance of lipogenesis and fat oxidation in different tissues. In type 1 diabetes, this is likely to be influenced by the absence of endogenous insulin combined with therapeutic administration of subcutaneous insulin into the peripheral (as opposed to portal) circulation. This results in reduced inhibition of hepatic lipolysis with consequent increased levels of circulating NEFAs, which, in combination with peripheral hyperinsulinaemia, may promote relatively greater lipid storage in skeletal muscle [7–9]. This is supported by recent evidence from recipients of hepatic islet cell transplants, in whom OGTT-induced NEFA suppresion was normalised [37]. The subsequent pattern of increased (ectopic) intramyocellular fat storage is similar to that found in obesity/type 2 diabetes and is associated with impaired sensitivity of muscle to glucose uptake, i.e. peripheral insulin resistance [38]. Therefore, in type 1 diabetes, this may represent an indirect mechanism of insulin resistance. Taking this idea further, anything that exaggerates these features (i.e. increased fat mass, increased lipid/NEFA flux, higher insulin dose requirements) is likely to promote further insulin resistance.
It follows that other sites of ectopic fat deposition might be influenced by abnormal fat partitioning. For example, epicardial and perivascular fat may play a role in modulating coronary function and pathophysiology, providing a potential mechanism to link double diabetes with CVD. In this regard, data have recently been published on epicardial fat thickness (measured by echocardiography) in 36 type 1 diabetic patients and 43 matched controls. Not only was epicardial fat thicker in those with type 1 diabetes (p < 0.0001), but it also correlated significantly with both WHR (r = 0.67, p = 0.003) and eGDR (r = −0.55, p = 0.0004) within the patient group [39].
HDL-C
HDL-C levels are normal or high in type 1 diabetes unless renal impairment develops, in which case HDL-C falls [7, 40–43]. Moreover, long-term survivors of type 1 diabetes are characterised by unusually high HDL-C levels (1.84 mmol/l) [44], and patients with CHD events have been shown to have lower HDL-C levels (along with higher triacylglycerol and LDL-C levels) [45]. Exogenous insulin treatment usually drives higher mean HDL-C levels in type 1 diabetes, but even those individuals with poorer glycaemic control have higher levels than healthy controls [43]. Several mechanisms have been suggested to explain this phenomenon. The most important of these appears to be lipoprotein lipase (LPL), which is highly active in adipocytes and avidly hydrolyses triacylglycerol-rich particles, resulting in high HDL-C levels. Peripheral hyperinsulinaemia associated with strict glycaemic control is associated with increased LPL activity and HDL-C, while poor glycaemic control results in lower LPL activity and HDL-C [46, 47]. There may also be a smaller contribution from a reduction in the activity of hepatic lipase (HL), an enzyme that hydrolyses triacylglycerol and phospholipids present in circulating plasma lipoproteins. Low levels of portal vein insulin in type 1 diabetes are associated with reduced HL activity and increased HDL-C [48–50]. Increasing hepatic insulin exposure by changing insulin delivery from the subcutaneous to the intraperitoneal route reverses this pattern [50]. In contrast, in type 2 diabetes, raised HL activity secondary to portal hyperinsulinaemia may contribute to low HDL-C levels. Finally, phospholipid transfer protein activity is markedly elevated in patients with type 1 diabetes, and this activity is correlated with HDL-C levels [42].
High HDL-C levels in patients with type 1 diabetes are likely to be atheroprotective, even though some compositional abnormalities may reduce the protective effect. However, if central obesity and insulin resistance then develop (double diabetes), a more atherogenic lipid profile seems to emerge. This assertion is supported by data from a subgroup of 61 DCCT participants, in whom increased HL activity accounted for most of the association between increased intra-abdominal fat stores and decreased HDL-C levels [49].
Portal insulinopenia and lipid effects
As discussed above, the liver is under-insulinised in type 1 diabetes. This raises the hypothesis that type 1 diabetes may actually confer simultaneous protective and harmful CV effects. This is a difficult phenomenon to study, but interesting insights have been gained from the field of pancreas transplantation in type 1 diabetes. When an insulin-secreting graft is attached to the portal vein, an apparently more atherogenic lipid profile is later observed than when it is attached to a systemic vein [51]. By contrast, this model has also been used to demonstrate that hepatic IGF-1 secretion is increased and growth hormone secretion is reduced by increased portal insulin levels, with the resultant systemic effects on insulin resistance mentioned above [52]. A number of other studies of patients on peritoneal dialysis (i.e. receiving insulin by the portal vein route) support these findings. In all cases, despite a more physiological method of delivering insulin, the lipid profile switched to an apparently more atherogenic pattern, with significant reductions in HDL-C and increases in LDL-C:HDL-C ratios [50, 53–55]. The pathophysiological significance of this is uncertain given Mendelian randomisation studies showing that differences in HDL-C levels do not necessarily result in harm or benefit [56]. Rather, HDL particle flux may be more important—again, a difficult process to measure. Whatever the net effect of portal insulinisation, it is of interest that new insulins have been specifically developed to have a relatively more hepatic than systemic mode of action; thus, we will shortly have new tools to help answer the specific questions raised by this review. We hypothesise that these insulins will help avoid excess weight gain, peripheral insulin resistance and cardiac fat accumulation, but may lead to greater liver fat. The net effects of these insulins on CV outcomes will therefore be of major interest.
Summary and conclusions
A summary of our current knowledge of double diabetes and areas for future research is presented in the text box. While the risks of CVD in type 1 diabetes may be falling, the relative risk of CVD, CHD, stroke and all-cause mortality continues to be unacceptably high for this patient population [57]. Our current understanding of double diabetes is insufficient to produce a rigid definition, but the literature we have summarised suggests that it has emerged as a relevant clinical concept in which there is: (1) marked weight gain over time; (2) a high daily insulin requirement; (3) a positive family history of type 2 diabetes, particularly when two or more relatives are affected; and/ or (4) a low eGDR. Affected individuals may have high normal BP (or hypertension) and relatively low HDL-C. Whether these effects associated with greater weight gain in at-risk individuals with double diabetes attenuate or reverse the vascular benefits of glycaemia reduction over time, remains to be fully established. In the meantime, avoidance of greater weight gain (while retaining glycaemic benefits) would have quality of life benefits for patients.
Summary of what is known and what remains unknown (and thus requires further research) in ‘double diabetes’ | |
|---|---|
Known | Unknown: areas for future research |
Individuals with T1DM and positive FHT2 have increased risks of albuminuria and CHD [3, 4]. | Is the effect of FHT2 on complications in T1DM mediated by genetic or familial factors (or both)? |
Peripheral insulin resistance is a consistent feature of T1DM (across the BMI range) [6–16]. | Is peripheral insulin resistance in T1DM related to increased ectopic fat stores (e.g. skeletal muscle) as a result of abnormal fat partitioning owing to non-physiological subcutaneous insulin administration? |
Clamp and derived measures of insulin resistance in T1DM predict coronary disease [16, 17, 19]. | There are no prospective studies of baseline clamp insulin resistance in a T1DM cohort with follow-up for incident CV events. |
Defining metabolic syndrome in T1DM does not add value for CV risk prediction [19, 21, 22]. | Newer risk models in contemporary populations of T1DM patients are urgently needed. |
DCCT participants in the intensive insulin therapy group in the highest quartile for weight gain developed a higher BP and a more atherogenic lipid profile [26]. | Numbers of CV events in the DCCT/EDIC study are too small to demonstrate any excess risk in those patients who gained the most weight. Further prospective cohort studies are required. It remains possible (and clinically relevant) that a beneficial CV effect of intensive glycaemic control might, in some patients, be outweighed by the adverse effects of weight gain. |
Hepatic fat stores are reduced while intramuscular and epicardial fat stores are increased in lean patients with T1DM compared with matched non-diabetic controls [9, 39]. | What is the effect of weight gain on hepatic fat stores (and ectopic fat stores) in T1DM? Any increase in perivascular or epicardial fat could have implications for CV risk. |
In T1DM following pancreas transplant, portal vein drainage results in a more atherogenic lipid profile than systemic vein drainage [51]. Similar patterns are found in patients on peritoneal dialysis (portal insulin delivery) [53–55]. | Do newer insulins that target liver rather than systemic tissues lead to more liver fat but less weight gain overall and less cardiac and intramyocellular fat accumulation? How do newer vs current insulins affect the function of HDL particles? |
FHT2, family history of type 2 diabetes; T1DM, type 1 diabetes | |
When a patient with type 1 diabetes on an intensive insulin regimen is clearly gaining a significant amount of weight, early consideration should be given to adjusting that regimen in the context of diet and lifestyle in an effort to limit weight gain. It may also be useful to ascertain a more complete family history of type 2 diabetes in such patients. We suggest that the roles of structured educational approaches, continuous subcutaneous insulin infusion and closed-loop systems are potentially useful future strategies that should be assessed for the prevention and treatment of double diabetes. Lower thresholds (BP and lipid lowering) for primary prevention of CVD should also be explored, as should the role of metformin as an ‘insulin-sparing’ agent [58] (now being evaluated in the Reducing with Metformin Vascular Adverse Lesions in Type 1 Diabetes [REMOVAL] trial [59]). Furthermore, it needs to be determined, whether newer insulins (currently in early trials) that target the liver more than the systemic tissues can lessen weight gain and, as a result, improve outcome in such patients.
From a basic science perspective, further elucidation of the complex interplay of glucose and lipid factors driving CV risk in type 1 diabetes could help to better identify at-risk patients and improve primary prevention strategies.
Abbreviations
- CAC:
-
Coronary artery calcification
- CVD:
-
Cardiovascular disease
- EDC:
-
Epidemiology of Diabetes Complications
- EDIC:
-
Epidemiology of Diabetes Interventions and Complications
- eGDR:
-
Estimated glucose disposal rate
- EHI:
-
Estimated hepatic insulin
- FAS:
-
Fatty acid synthase
- HDL-C:
-
HDL-cholesterol
- HL:
-
Hepatic lipase
- IDF:
-
International Diabetes Federation
- LPL:
-
Lipoprotein lipase
- NCEP:
-
National Cholesterol Education Program
- SREBP1c:
-
Sterol regulatory element-binding protein 1c
References
Teupe B, Bergis K (1991) Epidemiological evidence for “double diabetes”. Lancet 337:361–362
Wilkin TJ (2009) The accelerator hypothesis: a review of the evidence for insulin resistance as the basis for type 1 as well as type II diabetes. Int J Obes 33:716–726
Roglic G, Colhoun HM, Stevens LK, Lemkes HH, Manes C, Fuller JH (1998) Parental history of hypertension and parental history of diabetes and microvascular complications in insulin-dependent diabetes mellitus: the EURODIAB IDDM Complications Study. Diabet Med 15:418–426
Erbey JR, Kuller LH, Becker DJ, Orchard TJ (1998) The association between a family history of type 2 diabetes and coronary artery disease in a type 1 diabetes population. Diabetes Care 21:610–614
Petrie JR, Pearson ER, Sutherland C (2011) Implications of genome wide association studies for the understanding of type 2 diabetes pathophysiology. Biochem Pharmacol 81:471–477
Williams D, Erbey J, Becker D, Orchard TJ (2000) Can clinical factors estimate insulin resistance in type 1 diabetes? Diabetes 49:626–632
Heptulla RA, Stewart A, Enocksson S et al (2003) In situ evidence that peripheral insulin resistance in adolescents with poorly controlled type 1 diabetes is associated with impaired suppression of lipolysis: a microdialysis study. Pediatr Res 53:830–835
Perseghin G, Lattuada G, Danna M et al (2003) Insulin resistance, intramyocellular lipid content and plasma adiponectin in patients with type 1 diabetes. Am J Physiol Endocrinol Metab 285:E1174–E1181
Perseghin G, Lattuanda G, De Cobelli F et al (2005) Reduced intrahepatic fat content is associated with increased whole-body lipid oxidation in patients with type 1 diabetes. Diabetologia 48:2615–2621
Makimattila S, Virkamaki A, Malmstrom R, Urtiainen T, Yki-Jarvinen H (1996) Insulin resistance in type 1 diabetes mellitus: a major role for reduced glucose extraction. J Clin Endocrinol Metab 81:707–712
Yip J, Mattock MB, Morocutti A, Sethi M, Trevisan R, Viberti G (1993) Insulin resistance in insulin-dependent diabetic patients with microalbuminuria. Lancet 342:883–887
Kacerovsky M, Brehm A, Chmelik M et al (2011) Impaired insulin stimulation of muscular ATP production in patients with type 1 diabetes. J Intern Med 269:189–199
Nadeau KJ, Regensteiner JG, Bauer TA et al (2010) Insulin resistance in adolescents with type 1 diabetes and its relationship to cardiovascular function. J Clin Endocrinol Metab 95:513–521
Maahs DM, Hokanson JE, Wang H et al (2010) Lipoprotein subfraction cholesterol distribution is proatherogenic in women with type 1 diabetes and insulin resistance. Diabetes 59:1771–1779
Maahs DM, Nadeau K, Snell-Bergeon JK et al (2011) Association of insulin sensitivity to lipids across the lifespan in people with type 1 diabetes. Diabet Med 28:148–155
Schauer IE, Snell-Bergeon JK, Bergman BC et al (2011) Insulin resistance, defective insulin-mediated fatty acid suppression, and coronary artery calcification in subjects with and without type 1 diabetes: The CACTI study. Diabetes 60:306–314
Orchard TJ, Olson JC, Erbey JR et al (2003) Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes. Diabetes Care 26:1374–1379
Zgibor JC, Ruppert K, Orchard TJ et al (2010) Development of a coronary heart disease risk prediction model for type 1 diabetes: the Pittsburgh CHD in type 1 diabetes risk model. Diabetes Res Clin Pract 88:314–321
Kilpatrick ES, Rigby AS, Atkin SL (2007) Insulin resistance, the metabolic syndrome, and complication risk in type 1 diabetes: “double diabetes” in the Diabetes Control and Complications Trial. Diabetes Care 30:707–712
Chaturvedi N, Sjolie A-K, Porta M et al (2001) Markers of insulin resistance are strong risk factors for retinopathy incidence in type 1 diabetes. Diabetes Care 24:284–289
Pambianco G, Costacou T, Orchard TJ (2007) The prediction of major outcomes of type 1 diabetes: a 12-year prospective evaluation of three separate definitions of the metabolic syndrome and their components and estimated glucose disposal rate: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes Care 30:1248–1254
Thorn LM, Forsblom C, Waden J, Finnish Diabetic Nephropathy (FinnDiane) Study Group et al (2009) Metabolic syndrome as a risk factor for cardiovascular disease, mortality, and progression of diabetic nephropathy in type 1 diabetes. Diabetes Care 32:950–952
Sattar N, McConnachie A, Shaper AG et al (2008) Can metabolic syndrome usefully predict cardiovascular disease and diabetes? Outcome data from two prospective studies. Lancet 371:1927–1935
Nathan DM, Cleary PA, Backlund JY, The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group et al (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643–2653
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Purnell JQ, Hokanson JE, Marcovina SM, Steffes MW, Cleary PA, Brunzell JD (1998) Effect of excessive weight gain with intensive therapy of type 1 diabetes on lipid levels and blood pressure: results from the DCCT. Diabetes Control and Complications Trial. JAMA 280:140–146
Purnell JQ, Dev RK, Steffes MW et al (2003) Relationship of family history of type 2 diabetes, hypoglycemia, and autoantibodies to weight gain and lipids with intensive and conventional therapy in the Diabetes Control and Complications Trial. Diabetes 52:2623–2629
Conway B, Miller RG, Costacou T et al (2009) Adiposity and mortality in type 1 diabetes. Int J Obes 33:796–805
Ferriss JB, Webb D, Chaturvedi N, Fuller JH, Idzior-Walus B, EURODIAB Prospective Complications Group (2006) Weight gain is associated with improved glycaemic control but with adverse changes in plasma lipids and blood pressure in type 1 diabetes. Diabet Med 23:557–564
Taylor AM, Dunger DB, Grant DB, Preece MA (1988) Somatomedin-C/IGF-1 measured by radioimmunoassay and somatomedin bioactivity in adolescents with insulin dependent diabetes compared with puberty matched controls. Diabetes Res 9:177–181
Edge JA, Dunger DB, Matthews DR, Gilbert JP, Smith CP (1990) Increased overnight growth hormone concentrations in diabetic compared with normal adolescents. J Clin Endocrinol Metab 71:1356–1362
Norbert S, Konstantinos K, Hans-Ulrich H (2008) Causes and metabolic consequences of fatty liver. Endocr Rev 29:939–960
Taskinen MR (2003) Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 46:733–749
Savage DB, Semple RK (2010) Recent insights into fatty liver, metabolic dyslipidaemia and their links to insulin resistance. Curr Opin Lipidol 21:329–336
Hodson L, Frayn KN (2011) Hepatic fatty acid partitioning. Curr Opin Lipidol 22:216–224
Christ ER, Carroll PV, Albany E et al (2001) Normal VLDL metabolism despite altered lipoprotein composition in type 1 diabetes mellitus. Clin Endocrinol 55:777–787
Vethakkan SR, Walters JM, Gooley JL et al (2012) Normalized NEFA dynamics during an OGTT after islet transplantation. Transplantation 94:e49–e51
Sinha R, Dufour S, Petersen KF et al (2002) Assessment of skeletal muscle triglyceride content by 1H nuclear magnetic resonance spectroscopy in lean and obese adolescents: relationships to insulin sensitivity, total body fat, and central adiposity. Diabetes 51:1022–1027
Yazici D, Ozben B, Yavuz D et al (2011) Epicardial adipose thickness in type 1 diabetic patients. Endocrine 40:250–255
Nikkila EA, Hormila P (1978) Serum lipids and lipoproteins in insulin-treated diabetes: demonstration of increased high density lipoprotein concentrations. Diabetes 27:1078–1086
Taskinen MR (1992) Quantitative and qualitative lipoprotein abnormalities in diabetes mellitus. Diabetes 41:12–17
Colhoun HM, Taskinen MR, Otvos JD, Van Den Berg P, O’Connor J, Van Tol A (2002) Relationship of phospholipid transfer protein activity to HDL and apolipoprotein B-containing lipoproteins in subjects with and without type 1 diabetes. Diabetes 51:3300–3305
Guy J, Ogden L, Wadwa RP et al (2009) Lipid and lipoprotein profiles in youth with and without type 1 diabetes: the SEARCH for Diabetes in Youth case–control study. Diabetes Care 32:416–420
Bain SC, Gill GV, Dyer PH et al (2003) Characteristics of type 1 diabetes of over 50 years duration (the Golden Years Cohort). Diabet Med 20:808–811
Prince CT, Becker DJ, Costacou T, Miller RG, Orchard TJ (2007) Changes in glycaemic control and risk of coronary artery disease in type 1 diabetes mellitus: findings from the Pittsburgh Epidemiology of Diabetes Complications Study (EDC). Diabetologia 50:2280–2288
Taskinen MR (1987) Lipoprotein lipase in diabetes. Diabetes Metabol Rev 3:551–570
James RW, Pometta D (1990) Differences in lipoprotein subfraction composition and distribution between type I diabetic men and control subjects. Diabetes 39:1158–1164
Caixas A, Perez A, Payes A et al (1998) Effects of short-acting insulin analog (Insulin Lispro) versus regular insulin on lipid metabolism in insulin-dependent diabetes mellitus. Metabolism 47:371–376
Sibley SD, Palmer JP, Hirsch IB, Brunzell J (2003) Visceral obesity, hepatic lipase activity, and dyslipidaemia in type 1 diabetes. J Clin Endocrinol Metab 88:3379–3384
Ruotolo G, Parlavecchia M, Taskinen MR et al (1994) Normalization of lipoprotein composition by intraperitoneal insulin in IDDM. Role of increased hepatic lipase activity. Diabetes Care 17:6–12
Petruzzo P, Badet L, Lefrancois N et al (2006) Metabolic consequences of pancreatic systemic or portal venous drainage in simultaneous pancreas-kidney transplant recipients. Diabet Med 23:654–659
Frystyk J, Ritzel RA, Maubach J et al (2008) Comparison of pancreas-transplanted type 1 diabetic patients with portal-venous versus systemic-venous graft drainage: impact of glucose regulatory hormones and the growth hormone/insulin-like growth factor axis. J Clin Endocrinol Metab 93:1758–1766
Nevalainen P, Lahtela JT, Mustonen J, Pasternack A (1997) The influence of peritoneal dialysis and the use of subcutaneous and intraperitoneal insulin on glucose metabolism and serum lipids in type 1 diabetic patients. Nephrol Dial Transplant 12:145–150
Selam J-L, Kashyap M, Alberti KGMM et al (1989) Comparison of intraperitoneal and subcutaneous insulin administration on lipids, apolipoproteins, fuel metabolites, and hormones in type 1 diabetes mellitus. Metabolism 38:908–912
Lahtela JT, Mustonen J, Pasternack A (1995) Comparison of intraperitoneal and subcutaneous insulin administration on insulin sensitivity and serum lipids in type 1 diabetic patients on continuous ambulatory peritoneal dialysis. Clin Sci 88:427–432
Voight BF, Peloso GM, Orho-Melander M et al (2012) Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 380:572–580
Livingstone SJ, Looker HC, Hothersall EJ et al (2012) Risk of cardiovascular disease and total mortality in adults with type 1 diabetes: Scottish registry linkage study. PLoS Med 9:e1001321
Vella S, Buetow L, Royle P, Livingstone S, Colhoun HM, Petrie JR (2010) The use of metformin in type 1 diabetes: a systematic review of efficacy. Diabetologia 53:809–820
REMOVAL: http://clinicaltrials.gov/ct2/show/NCT01483560?term=REMOVAL&rank=1
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors were responsible for the conception and design of the manuscript, drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Cleland, S.J., Fisher, B.M., Colhoun, H.M. et al. Insulin resistance in type 1 diabetes: what is ‘double diabetes’ and what are the risks?. Diabetologia 56, 1462–1470 (2013). https://doi.org/10.1007/s00125-013-2904-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2904-2