
Abstract
Aims/hypothesis
The activation of NADPH oxidase has been implicated in NEFA-induced beta cell dysfunction. However, the causal role of this activation in vivo remains unclear. Here, using rodents, we investigated whether pharmacological or genetic inhibition of NADPH oxidase could prevent NEFA-induced beta cell dysfunction in vivo.
Methods
Normal rats were infused for 48 h with saline or oleate with or without the NADPH oxidase inhibitor apocynin. In addition, NADPH oxidase subunit p47phox-null mice and wild-type littermate controls were infused with saline or oleate for 48 h. This was followed by measurement of NADPH oxidase activity, reactive oxygen species (ROS) and superoxide imaging and assessment of beta cell function in isolated islets and hyperglycaemic clamps.
Results
Oleate infusion in rats increased NADPH oxidase activity, consistent with increased total but not mitochondrial superoxide in islets and impaired beta cell function in isolated islets and during hyperglycaemic clamps. Co-infusion of apocynin with oleate normalised NADPH oxidase activity and total superoxide levels and prevented beta cell dysfunction. Similarly, 48 h NEFA elevation in wild-type mice increased total but not mitochondrial superoxide and impaired beta cell function in isolated islets. p47phox-null mice were protected against these effects when subjected to 48 h oleate infusion. Finally, oleate increased the levels of total ROS, in both models, whereas inhibition of NADPH oxidase prevented this increase, suggesting that NADPH oxidase is the main source of ROS in this model.
Conclusions/interpretation
These data show that NADPH-oxidase-derived cytosolic superoxide is increased in islets upon oleate infusion in vivo; and whole-body NADPH-oxidase inhibition decreases superoxide in concert with restoration of islet function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic exposure of pancreatic beta cells to NEFA impairs beta cell function [1]. Although not all studies are concordant [2], a growing body of evidence implicates oxidative stress as a mechanism of NEFA-induced beta cell dysfunction [3–7]. Pancreatic beta cells have low antioxidant defences [8] and are thus susceptible to decrease in function induced by reactive oxygen species (ROS) [9]. We have previously demonstrated that: (1) prolonged elevation of plasma NEFA in rats impairs glucose-stimulated insulin secretion (GSIS) in vivo during hyperglycaemic clamps and ex vivo in freshly isolated islets and (2) treatment with antioxidants (N-acetylcysteine or taurine or tempol), which decreases islet ROS measured with dihydro-dichlorofluorescein diacetate (H2DCF-DA), prevents the impairing effects of NEFA on beta cell function [10]. Zhang et al confirmed our findings in rats [10] in a different model of prolonged NEFA elevation [11]. Our group has also shown that the antioxidant taurine alleviated NEFA-induced impairment in beta cell function in humans [12].
The type and cellular localisation of ROS involved in NEFA-induced beta cell dysfunction are still unclear. Although the mitochondrial electron transport chain was traditionally regarded as the main source of ROS generation, a growing body of evidence now suggests that NADPH oxidase is also a source of ROS production in beta cells. In fact, beta cells express NADPH oxidase (NOX1, NOX2 and NOX4 isoforms) [13, 14] which generates superoxide in the cytosol/plasma membrane and components of this enzyme are elevated in islets of animal models of type 2 diabetes [15]. NOX1 and NOX2 are multi-subunit enzymes, comprising cytosolic (p40phox, p47phox and p67phox and the small protein rho rac) and membrane-bound (gp91phox and p22phox) subunits. Upon activation (e.g. by protein kinase C [PKC]), the cytosolic subunits migrate to the membrane, and the complex assembles its components, leading to production of superoxide radical [16]. Therefore, we hypothesised that NEFA activate NADPH oxidase and that this enzyme is a possible site of ROS generation in NEFA-induced beta cell dysfunction.
To investigate the role of NADPH oxidase in fat-induced beta cell dysfunction, the beta cell function was evaluated ex vivo in isolated islets and in vivo during hyperglycaemic clamps after prolonged elevation of plasma NEFA with or without NADPH oxidase inhibition. We used two models of NADPH oxidase inhibition: a pharmacological model (apocynin treatment) and a genetic model (p47phox-knockout mouse). Oleate is the most prevalent circulating fatty acid and was used instead of the standard infusion of Intralipid and heparin, because we have previously shown that the prolonged infusion of oleate impairs GSIS to a greater degree than Intralipid and heparin [17].
Here we show that pharmacological or genetic inhibition of NADPH oxidase prevents the increase in superoxide in islets of oleate-treated rodents. In addition, this inhibition prevents the impairing effect of oleate on beta cell function assessed both ex vivo and in vivo. Therefore, NADPH oxidase-derived superoxide plays a causal role in NEFA-induced beta cell dysfunction in vivo, and is thus a potential target for prevention of beta cell dysfunction in type 2 diabetes.
Methods
Animals
All procedures were in accordance with the Canadian Council of Animal Care Standards and were approved by the Animal Care Committee of the University of Toronto. Female Wistar rats (250–300 g; Charles River, Senneville, QC, Canada) and male mice (25–30 g) with knockout of the p47phox subunit of NADPH oxidase, as previously described [18] or their littermate wild-type controls (WT) were used for experiments. Animals were housed in the University of Toronto’s Department of Comparative Medicine. They were exposed to a 12 h light/dark cycle and were fed rodent chow (Teklad Global 2018, Madison, WI, USA).
Surgery and intravenous infusion
All surgical procedures were performed under general anaesthesia. The left jugular vein and the right carotid artery of the rats were cannulated as previously described [10]. After 3 days of recovery from surgery, rats were randomised and infused for 48 h with either: (1) NaCl (saline, 154 mmol/l) as control; (2) oleate (OLE, 1.3 μmol/min; Sigma, St Louis, MO, USA), prepared as described previously [10, 17], to elevate plasma NEFA by 1.5- to twofold; (3) oleate and apocynin (Calbiochem, La Jolla, CA, USA); or (4) apocynin alone. Apocynin dissolved in saline at pH 7.4 was given at a dose of 0.5 μmol kg−1 min−1 [19].
In mice, an indwelling catheter was inserted into the right internal jugular vein for infusion. After a minimum 3–4 day period of post-surgery recovery, knockout and WT mice were infused for 48 h with either saline or oleate to elevate plasma NEFA by 1.5- to twofold. Oleate was prepared as for rats and administered to mice at a dose of 0.4 μmol/min.
Pancreatic islet isolation
After the 48 h infusion, islets were isolated from the overnight-fasted rats using the Ficoll/Histopaque method, as described previously [10]. Pancreatic islets of mice were isolated after a 5 h fast, following 48 h infusion, as previously described [20].
NADPH oxidase activity assay
The chemiluminescence lucigenin assay was carried out using a single-tube luminometer (Berthold FB12; Berthold Technologies, Bad Wildbad, Germany) modified to maintain the sample temperature at 37°C [18]. Islets of rats were isolated as above. Following 1 h of pre-incubation at 37°C in Krebs–Ringer buffer with 10 mmol/l HEPES (KRBH) containing 2.8 mmol/l glucose, islets were washed in PBS and suspended in 1 ml PBS. Lucigenin (5 μmol/l) and NADPH (100 μmol/l) were added to the samples and light emission was recorded every 2 min over a 15 min period. The results were normalised per 1 mg protein. Islet protein was measured using the Bradford assay.
Islet NADPH/NADP+ measurement
Islet NADPH and NADP+ levels were measured from 200 islets of rats using an NADPH/NADP+ quantification kit (Sigma). The levels were normalised by mg of protein.
ROS measurement
Islet ROS were measured using H2DCF-DA (Sigma) [21], which detects most ROS, including hydrogen peroxide, lipid peroxides, peroxyl radical and peroxynitrite anion [22]. However, H2DCF-DA does not directly detect superoxide [23]. Total superoxide was measured by hydroethidine (HEt; Invitrogen, Burlington, ON, Canada), and mitochondrial superoxide was measured by MitoSOX (MS; Invitrogen) as described previously [21].
Thiobarbituric acid reacting substances (TBARS) assay
Lipid peroxidation in islets of rats was measured indirectly by assay of the secondary product malondialdehyde (MDA) using the TBARS assay kit (Cayman Chemical Company, Ann Arbor, MI, USA). The levels of MDA were expressed as μmol/mg of islet protein.
Ex vivo evaluation of GSIS
Isolated islets of rats and mice were pre-incubated for 1 h at 37°C in KRBH supplemented with 2.8 mmol/l glucose. Thereafter, five rat islets of approximately the same size were incubated in triplicate at 2.8, 6.5, 13 and 22 mmol/l glucose for 2 h at 37°C. Similarly, ten mouse islets of approximately the same size were incubated in duplicate at 6.5 and 22 mmol/l glucose for 2 h at 37°C. Insulin concentration in the medium was analysed using a radioimmunoassay kit specific for rat/mouse insulin (Linco, St Charles, MO, USA). The islets used for secretion were then subjected to acid ethanol extraction [21] for measurement of insulin content.
In vivo evaluation of GSIS
GSIS in vivo was determined by measuring plasma insulin and C-peptide levels in rats during two-step hyperglycaemic clamps. An infusion of 37.5% glucose was started at time t = 0 min. Plasma glucose was maintained at 13 mmol/l by adjusting the rate of the glucose infusion according to frequent (every 5–10 min) glucose determinations. At 120 min, the glucose infusion was raised to achieve and maintain plasma glucose levels of 22 mmol/l until the end of the experiment (t = 240 min). During the clamp, blood samples were frequently drawn to measure plasma NEFA, insulin and C-peptide levels.
Plasma assays
Plasma NEFA were measured with an enzymatic colorimetric kit (Wako Industries, Neuss, Germany). Radioimmunoassay kits specific for rat/mouse insulin and C-peptide (Linco) were used to determine plasma concentrations (interassay CV <10%). Plasma TBARS assay was carried out as described above. The results are expressed in nmol/l. The ratio of reduced glutathione to oxidised glutathione (GSH/GSSG) in plasma was measured using the GSH/GSSG EnzyChrom kit (BioAssays Systems, Hayward, CA, USA).
Statistical analysis
Data are means ± SEM. One-way nonparametric ANOVA for repeated measurements followed by Tukey’s t test was used to compare treatments. Calculations were performed using SAS (Cary, NC, USA).
Results
Plasma determinations in rats
During the 48 h infusions, as expected, the oleate-treated rats had higher plasma levels of NEFA than the rats treated with saline or apocynin alone (Table 1). The infusion of oleate and/or apocynin did not affect plasma glucose or insulin (data not shown). Oleate decreased the plasma GSH/GSSG and tended to increase the plasma levels of MDA (a lipid peroxidation product) (Table 2). The effects of oleate were prevented by the co-infusion of apocynin.
Beta cell function ex vivo and determinations in rat islets
Forty-eight-hour oleate infusion increased the activity of NADPH oxidase (p < 0.05 vs SAL; Fig. 1a). This increase was prevented by the co-infusion of apocynin. Oleate also tended to increase the islet NADPH/NADP+ ratio (Fig. 1b). As expected, this tendency was accentuated by apocynin. Oleate elevated total islet superoxide (p < 0.01 vs SAL; Fig. 1c) but not mitochondrial superoxide (Fig. 1d). The increase in total superoxide was reduced by apocynin (p < 0.01 oleate vs oleate + apocynin; NS oleate + apocynin vs saline; Fig. 1c). In addition, oleate elevated total ROS (p < 0.01 vs saline; Fig. 1e); this elevation was attenuated by apocynin (p < 0.05 oleate vs oleate + apocynin; NS saline vs oleate + apocynin). Islets of rats treated with oleate showed an increase in MDA (p < 0.05 oleate vs saline; Fig. 1f), which was completely prevented by apocynin (p < 0.01 oleate vs oleate + apocynin).

The NADPH oxidase inhibitor apocynin normalises NADPH oxidase activity, total superoxide, total ROS and MDA levels increased by oleate in rat islets. Rats were treated for 48 h with saline (SAL, 5 μl/min); oleate, to elevate plasma NEFA 1.5- to twofold (OLE, 1.3 μmol/min); oleate + apocynin (OLE + APO, 1.3 μmol/min + 0.5 μmol kg−1 min−1, respectively) or apocynin (APO, 0.5 μmol kg−1 min−1). (a) NADPH oxidase activity in freshly isolated islets of the rats treated as above. Data are means ± SEM (SAL, n = 7; OLE, n = 9; OLE + APO, n = 6; APO, n = 6). (b) NADPH/NADP+ in freshly isolated islets of the rats treated as above. Data are means ± SEM (n = 6 per group). Total (c) and mitochondrial (d) superoxide levels (n = 6 rats per group) and total ROS (e) levels (n = 4 rats per group) in freshly isolated islets of rats treated as above. Data are expressed as mean % of SAL ± SEM. The representative fluorescent images of islets stained with hydroethidine, MitoSOX and H2DCF-DA are also shown. (f) MDA levels in freshly isolated islets of rats treated as described above. Data are means ± SEM (SAL, n = 7; OLE, n = 8; OLE + APO, n = 9; APO, n = 9); *p < 0.05 OLE vs SAL, **p < 0.01 OLE vs SAL, † p < 0.05 OLE vs OLE + APO and APO, †† p < 0.01 OLE vs OLE + APO and APO, ‡‡ p < 0.01 OLE vs all
After the 48 h infusions, we evaluated GSIS ex vivo in freshly isolated islets. At 2.8 and 6.5 mmol/l of glucose, insulin secretion did not differ among groups. The insulin secretory response of islets isolated from oleate-infused rats was markedly decreased compared with the response of islets from rats infused with saline at both 13 (p < 0.05 vs saline) and 22 mmol/l (p < 0.05 vs saline) glucose (Fig. 2a). Apocynin prevented the oleate-induced decrease in insulin secretion at both of these glucose concentrations. Likewise, the islet insulin content was significantly decreased by oleate (p < 0.01 vs saline), but not in the presence of apocynin (Fig. 2b).

Apocynin prevents oleate-induced beta cell dysfunction ex vivo in islets isolated from rats. (a) Insulin secretory response to glucose and (b) islet insulin content in freshly isolated islets of rats treated as described in the Fig. 1 legend. Data are means ± SEM (saline [SAL], n = 7; oleate [OLE], n = 7; OLE + apocynin [APO], n = 7; APO, n = 8); **p < 0.01 OLE vs SAL; ‡ p < 0.05 OLE vs all
Rat beta cell function in vivo
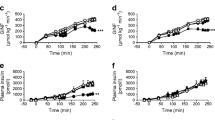
As shown in Fig. 3a, basal plasma NEFA levels before the hyperglycaemic clamps were higher in oleate-treated rats than in control rats or rats treated with apocynin alone (p < 0.05). NEFA levels declined during the clamp because of hyperglycaemia and hyperinsulinaemia. However, NEFA remained highest in oleate-treated rats.

Apocynin prevents the decrease in beta cell function induced by oleate in vivo during two-step hyperglycaemic clamp in rats. Rats were treated as described in the Fig. 1 legend. Plasma levels of NEFA (a), glucose (b), insulin (d) and C-peptide (e) and glucose infusion rate (Ginf) (c) were measured during the two-step hyperglycaemic clamp. Data are means ± SEM (saline [SAL], n = 8; oleate [OLE], n = 8; OLE + apocynin [APO], n = 5; APO, n = 7); **p < 0.01 OLE vs SAL, ***p < 0.001 OLE vs SAL, † p < 0.05 OLE vs OLE + APO, ‡‡ p < 0.01 OLE vs all. White circles, SAL; black circles, OLE; black squares, OLE + APO; white squares, APO
Basal plasma glucose was similar in all groups (Fig. 3b). During the first step of the clamp, glucose levels rose to 13 mmol/l and during the second step, to 22 mmol/l, with no differences among groups (Fig. 3b). The glucose infusion rate (Ginf) required to maintain the target glucose level was lower in the oleate-treated group than in the saline-treated group, consistent with decreased insulin secretion, decreased insulin sensitivity, or both (p < 0.01 oleate vs saline; Fig. 3c). Apocynin, in combination with oleate, prevented the lowering of Ginf by oleate (p < 0.01 oleate vs oleate + apocynin; Fig. 3c). Apocynin alone had no effect on Ginf.
Basal plasma insulin and C-peptide levels were similar in all groups of rats. As expected, plasma insulin rose in response to increasing glucose levels (Fig. 3d). Plasma C-peptide also rose, indicating that the rise in insulin was due to increased secretion (Fig. 3e). Plasma insulin and C-peptide were lower in oleate-treated rats than in those treated with saline. In the oleate + apocynin group plasma insulin and C-peptide were similar to control levels (Fig. 3d and e). Apocynin alone had no effect on insulin or C-peptide levels.
Ginf and insulin levels were proportionally decreased in the oleate vs saline group; the sensitivity index (glucose metabolism divided by plasma insulin [M/I] = Ginf/insulin [24]) was not significantly different during either the first step (0.42 ± 0.027 μmol kg−1 min−1 [pmol/l]−1 for saline and 0.43 ± 0.016 μmol kg−1 min−1 [pmol/l]−1 for oleate, NS) or the second step (0.31 ± 0.027 μmol kg−1 min−1 [pmol/l]−1 for saline and 0.34 ± 0.01 μmol kg−1 min−1 [pmol/l]−1 for oleate, NS) of the clamp. Apocynin, either alone or in combination with oleate, had no effect on M/I.
Studies in mice
During the 48 h infusions, as expected, the mice treated with oleate had higher plasma NEFA levels than the mice treated with saline (Table 3). The infusion of saline or oleate did not affect plasma glucose or insulin (data not shown).
Forty-eight hours of oleate infusion elevated total islet superoxide (p < 0.01; Fig. 4a) but not mitochondrial superoxide (Fig. 4b) in WT mice. The increase in total superoxide did not occur in islets of oleate-infused p47phox-knockout mice. Forty-eight hours of oleate infusion elevated total ROS in the islets of WT mice (p < 0.01 vs WT-SAL; Fig. 4c). There was no significant increase in total ROS in the islets of p47phox-knockout mice infused with oleate. The evaluation of GSIS ex vivo in freshly isolated mouse islets (Fig. 5a) showed that at 6.5 mmol/l of glucose, there was no significant difference among the different groups. However, at 22 mmol/l glucose, 48 h oleate infusion in WT mice impaired GSIS compared with saline infusions (p < 0.01). In contrast, p47phox-knockout mice subjected to 48 h elevation of NEFA had no significant impairment in GSIS (p < 0.01 WT treated with oleate vs knockout treated with oleate). Likewise, the islet insulin content was significantly decreased in WT mice infused with oleate as compared with saline (p < 0.01 vs SAL; Fig. 5b). p47phox-knockout mice were protected from the oleate-induced decrease in islet insulin content.

The genetic deletion of p47phox in mice normalises total superoxide and total ROS levels increased by oleate in islets. p47phox-null mice (KO) and their WT littermate controls were treated for 48 h with saline (SAL, 0.5 μl/min) or oleate (OLE, 0.4 μmol/min) to elevate plasma NEFA 1.5- to twofold. Total (a) and mitochondrial (b) superoxide levels (WT-saline [SAL], n = 7; WT-oleate [OLE], n = 6; KO-OLE, n = 6; KO-SAL, n = 4) and total ROS (c) levels (WT-SAL, n = 6; WT-OLE, n = 6; KO-OLE, n = 7; KO-SAL, n = 3) in freshly isolated islets of the mice treated as above. Data are expressed as mean % of SAL ± SEM; ‡‡ p < 0.01 WT-OLE vs all. The representative fluorescent images of islets imaged with HEt, MitoSOX and H2DCF-DA are also shown

The genetic deletion of p47phox in mice prevents oleate-induced beta cell dysfunction ex vivo in isolated islets. (a) Insulin secretory response to glucose and (b) islet insulin content in freshly isolated islets of p47phox-null mice (KO) and their WT littermate controls treated as described in the Fig. 4 legend. Data are means ± SEM (WT-saline [SAL], n = 8; WT-oleate [OLE], n = 7; KO-OLE, n = 7; KO-SAL, n = 6); **p < 0.01 KO-OLE vs WT-SAL, ‡‡ p < 0.01 WT-OLE vs all
Discussion
We herein investigated whether NADPH oxidase is a source of ROS production in beta cells and whether NADPH oxidase activation is causally linked to oleate-induced beta cell dysfunction in vivo. As demonstrated previously by our group [10, 17], 48 h oleate infusion impaired beta cell function ex vivo in isolated islets and in vivo during hyperglycaemic clamps in association with an increase in markers of oxidative stress. Although a transient increase in ROS generation is required for GSIS in beta cells [25–28], chronic oxidative stress leads to beta cell dysfunction [10, 21, 29–32]. In the present study, both genetic and pharmacological inhibition of NADPH oxidase protected against the impairing effects of oleate. Beta cells express NADPH oxidase [13] and components of this enzyme are elevated in islets of animal models of type 2 diabetes [15]. In fact, the inhibition of NADPH oxidase in islets of db/db mice partially restored islet insulin content [15], suggesting that this enzyme plays a role in beta cell dysfunction.
Several in vitro studies have reported the causal effect of the NEFA activation of NADPH oxidase in inducing beta cell dysfunction [33–35]. One in vivo study investigated the effects of high-fat diet on NADPH oxidase in beta cells of rats and found decreased subunit content and ROS concentrations in islets, with elevated GSIS in order to overcome the high-fat-diet-induced insulin resistance [36]. The findings of the latter study conflict with those of other studies showing that high-fat diet induces oxidative stress in islets of rats [29, 30]. Other in vivo studies using apocynin demonstrate a role for NADPH oxidase in high-fat-diet-induced insulin resistance [37, 38]. However, this is the first in vivo study that has investigated the causal effect of NADPH oxidase in fat-induced beta cell dysfunction. We have shown that 48 h infusion of oleate activated NADPH oxidase and increased total, but not mitochondrial, superoxide in islets. A similar finding was reported in myocytes by Yuzefovych et al [39]. Pharmacological or genetic inhibition of NADPH oxidase prevented the NEFA-induced increase in cytosolic superoxide in islets and decrease in secretory function of beta cells.
NADPH is a substrate for NADPH oxidase. Oleate increased the NADPH/NADP+ ratio in accordance with the findings of previous studies [40, 41], despite increasing NADPH oxidase activity. The effect of oleate on NADPH/NADP+ ratio is likely a result of increased provision of acetyl-CoA to the Krebs cycle (increased production) [40] in association with some impairment in electron transport system flux (increased redox ratio) [42]. When NADPH oxidase activity is inhibited, the NADPH already generated from fat accumulates. NADPH is a substrate for thioredoxin and glutaredoxin [43], which are antioxidants involved in mediating insulin secretion and can also increase insulin secretion via inhibition of Kv channels [44]. Therefore, we cannot rule out the possibility that the increase in NADPH contributes to the beneficial effects of inhibition of GSIS by NADPH oxidase, via reduced oxidative stress and/or independent effects of NADPH.
Here, we showed that NADPH oxidase is the source of superoxide production in a model of 48 h oleate-induced beta cell dysfunction, as oleate did not elevate the levels of mitochondrial superoxide. The latter finding is consistent with our previous result in MIN6 cells where superoxide dismutase (Sod2) mRNA was unchanged after 48 h exposure to oleate [10]; however, we cannot exclude the possibility that mitochondrial superoxide is increased at other time points.
Superoxides are short-lived; thus, their impairing effects could be due to their conversion into other types of ROS that are known to impair beta cell function [9]. To assess this conversion, islets were isolated and imaged with H2DCF-DA, which detects total ROS except superoxide [23]. We found that pharmacological or genetic inhibition of NADPH oxidase prevents the increase in ROS caused by oleate, implying that extramitochondrial superoxide is converted to other ROS. Moreover, in our pharmacological inhibition model, co-infusion of apocynin normalised the levels of MDA (an indication of lipid peroxidation), suggesting that lipid peroxides are one of the ROS generated from extramitochondrial superoxide. These observations are in accordance with previous studies wherein NADPH oxidase inhibition also reduced total ROS [45] and lipid peroxides [4].
During the in vivo hyperglycaemic clamp studies, oleate-treated rats showed lower insulin and C-peptide responses to glucose compared with saline-treated rats. These results indicate reduced insulin secretion, an effect consistent with that observed ex vivo in isolated islets. Insulin secretion during hyperglycaemic clamps should be interpreted in the context of insulin sensitivity because in vivo normal beta cells compensate for insulin resistance by increasing insulin secretion independently of plasma glucose. Although NEFA induce insulin resistance, this effect was not observed in our rat model, consistent with our previous studies performed using the same oleate infusion protocol in the same animal model [10, 17]. Insulin resistance could not be detected likely due to a number of factors, including the moderate elevation of NEFA, their monounsaturation, the female sex of the rats and the use of the hyperglycaemic clamp rather than the gold-standard hyperinsulinaemic–euglycaemic clamp. Since insulin sensitivity was not changed, the beta cell did not need to secrete more insulin to compensate for insulin resistance; therefore, our in vivo GSIS results are similar to the results obtained ex vivo.
The reason for using a genetic mouse model of NADPH oxidase inhibition is that all pharmacological inhibitors may have nonspecific effects. It has been argued that apocynin does not act as an NADPH oxidase inhibitor but as an antioxidant [46]. Several mouse models of NADPH oxidase subunit knockout exist; however, special attention has been paid to the p47phox subunit [3, 33, 35] when investigating beta cell function. We used a model that targets the p47phox subunit because: (1) the NADPH oxidase inhibitor used in rats, apocynin, acts by inhibiting the translocation of the p47phox subunit to the plasma membrane subunits [47], therefore, the use of this knockout mouse model allows us to draw a parallel with our studies in rats; and (2) activation of PKC by NEFA leads to serine phosphorylation of the p47phox subunit, thus allowing it to translocate to the membrane subunits and activate the enzyme complex [48]. Indeed we have shown that NEFA tends to increase the phosphorylation of p47phox at serine 328, which is a direct phosphorylation site by PKC (electronic supplementary material [ESM] Fig. 1). Therefore, the p47phox subunit is fundamental in the mechanism of fat-induced beta cell dysfunction. Recently, some of us have shown that knockout of p47phox subunit also protects against diabetes in Akita mice [18].
The contribution of the systemic effect of NADPH oxidase inhibition towards beta cell function cannot be ruled out in our model. Markers of circulating whole-body redox, such as reduced-to-oxidised glutathione ratio (GSH/GSSG), were decreased and the levels of the lipid peroxidation product MDA tended to be increased by oleate in the plasma of rats. Such changes were prevented by the co-infusion of apocynin (Table 2). Hence, these results suggest that the inhibition of systemic NADPH oxidase could contribute to the preservation of islet function by elevating the systemic redox ratio as well as by reducing systemic oxidative stress.
The precise mechanism by which NADPH oxidase-derived ROS impair beta cell function is not completely clear but several mechanisms have been proposed. First, oxidative stress can inhibit glucose metabolism in the beta cell [49]. Although cytosolic glycolysis may be affected, extramitochondrial ROS can also induce mitochondrial dysfunction [4, 45]. Second, oxidative stress may affect insulin biosynthesis, as suggested by the decrease in islet insulin content we found in both rats and mice infused with oleate, in accordance with Nakayama et al [15]. In summary, our study demonstrates that prolonged exposure to oleate, which increases ROS in islets via NADPH oxidase, decreases GSIS both ex vivo and in vivo, and that NADPH oxidase inhibition prevents the impairing effects of oleate. These findings are the first direct in vivo demonstration of a causal role for NADPH oxidase in the NEFA-induced decrease in beta cell function. Therefore, NADPH oxidase inhibition may be considered among the therapeutic targets aimed at ameliorating beta cell function in type 2 diabetes.
Abbreviations
- Ginf:
-
Glucose infusion rate
- GSH/GSSG:
-
Reduced-to-oxidised glutathione ratio
- GSIS:
-
Glucose-stimulated insulin secretion
- H2DCF-DA:
-
Dihydro-dichlorofluorescein diacetate
- HEt:
-
Hydroethidine
- IKKβ:
-
Inhibitor of (nuclear factor) kappa B kinase beta
- JNK:
-
c-Jun-N-terminal kinase
- KRBH:
-
Krebs–Ringer buffer containing 10 mmol/l HEPES
- MDA:
-
Malonedialdehyde
- M/I:
-
Glucose metabolism (M) divided by plasma insulin (I)
- MS:
-
MitoSOX
- NFκB:
-
Nuclear factor kappa B
- PKC:
-
Protein kinase C
- RLU:
-
Relative luminescence units
- ROS:
-
Reactive oxygen species
- TBARS:
-
Thiobarbituric acid reacting substances
- WT:
-
Wild-type
References
Giacca A, Xiao C, Oprescu AI, Carpentier AC, Lewis GF (2011) Lipid-induced pancreatic beta-cell dysfunction: focus on in vivo studies. Am J Physiol Endocrinol Metab 300:E255–E262
Moore PC, Ugas MA, Hagman DK, Parazzoli SD, Poitout V (2004) Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes 53:2610–2616
Morgan D, Oliveira-Emilio HR, Keane D et al (2007) Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 50:359–369
Syed I, Jayaram B, Subasinghe W, Kowluru A (2010) Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol 80:874–883
Carlsson C, Borg LA, Welsh N (1999) Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology 140:3422–3428
Maestre I, Jordan J, Calvo S et al (2003) Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology 144:335–345
Wang X, Li H, de Leo D et al (2004) Gene and protein kinase expression profiling of reactive oxygen species-associated lipotoxicity in the pancreatic beta-cell line MIN6. Diabetes 53:129–140
Lenzen S, Drinkgern J, Tiedge M (1996) Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med 20:463–466
Maechler P, Jornot L, Wollheim CB (1999) Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem 274:27905–27913
Oprescu AI, Bikopoulos G, Naassan A et al (2007) Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes 56:2927–2937
Zhang X, Bao Y, Ke L, Yu Y (2010) Elevated circulating free fatty acids levels causing pancreatic islet cell dysfunction through oxidative stress. J Endocrinol Investig 33:388–394
Xiao C, Giacca A, Lewis GF (2008) Oral taurine but not N-acetylcysteine ameliorates NEFA-induced impairment in insulin sensitivity and beta cell function in obese and overweight, non-diabetic men. Diabetologia 51:139–146
Oliveira HR, Verlengia R, Carvalho CR, Britto LR, Curi R, Carpinelli AR (2003) Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 52:1457–1463
Uchizono Y, Takeya R, Iwase M et al (2006) Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci 80:133–139
Nakayama M, Inoguchi T, Sonta T et al (2005) Increased expression of NAD(P)H oxidase in islets of animal models of Type 2 diabetes and its improvement by an AT1 receptor antagonist. Biochem Biophys Res Commun 332:927–933
Babior BM, Lambeth JD, Nauseef W (2002) The neutrophil NADPH oxidase. Arch Biochem Biophys 397:342–344
Mason TM, Goh T, Tchipashvili V et al (1999) Prolonged elevation of plasma free fatty acids desensitizes the insulin secretory response to glucose in vivo in rats. Diabetes 48:524–530
Liu GC, Fang F, Zhou J et al (2012) Deletion of p47phox attenuates the progression of diabetic nephropathy and reduces the severity of diabetes in the Akita mouse. Diabetologia 55:2522–2532
Kimura S, Zhang GX, Nishiyama A et al (2005) Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 45:860–866
Diao J, Allister EM, Koshkin V et al (2008) UCP2 is highly expressed in pancreatic alpha-cells and influences secretion and survival. Proc Natl Acad Sci USA 105:12057–12062
Tang C, Han P, Oprescu AI et al (2007) Evidence for a role of superoxide generation in glucose-induced beta-cell dysfunction in vivo. Diabetes 56:2722–2731
LeBel CP, Ischiropoulos H, Bondy SC (1992) Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5:227–231
Bilski P, Belanger AG, Chignell CF (2002) Photosensitized oxidation of 2′,7′-dichlorofluorescin: singlet oxygen does not contribute to the formation of fluorescent oxidation product 2′,7′-dichlorofluorescein. Free Radic Biol Med 33:938–946
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H (2005) Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal 7:1021–1031
Morgan D, Rebelato E, Abdulkader F et al (2009) Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology 150:2197–2201
Newsholme P, Morgan D, Rebelato E et al (2009) Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 52:2489–2498
Pi J, Bai Y, Zhang Q et al (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56:1783–1791
Yuan L, Li X, Xu GL, Qi CJ (2010) Effects of renin-angiotensin system blockade on islet function in diabetic rats. J Endocrinol Investig 33:13–19
Zhu CF, Peng HB, Liu GQ, Zhang F, Li Y (2010) Beneficial effects of oligopeptides from marine salmon skin in a rat model of type 2 diabetes. Nutrition 26:1014–1020
Harmon JS, Stein R, Robertson RP (2005) Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem 280:11107–11113
Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC (2002) Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 277:30010–30018
Michalska M, Wolf G, Walther R, Newsholme P (2010) Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic beta-cell dysfunction. Biosci Rep 30:445–453
Newsholme P, Haber EP, Hirabara SM et al (2007) Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol 583:9–24
Santos LR, Rebelato E, Graciano MF, Abdulkader F, Curi R, Carpinelli AR (2011) Oleic acid modulates metabolic substrate channeling during glucose-stimulated insulin secretion via NAD(P)H oxidase. Endocrinology 152:3614–3621
Valle MM, Graciano MF, Lopes de Oliveira ER et al (2011) Alterations of NADPH oxidase activity in rat pancreatic islets induced by a high-fat diet. Pancreas 40:390–395
Meng R, Zhu DL, Bi Y, Yang DH, Wang YP (2010) Apocynin improves insulin resistance through suppressing inflammation in high-fat diet-induced obese mice. Mediat Inflamm 2010:858735
Meng R, Zhu DL, Bi Y, Yang DH, Wang YP (2011) Anti-oxidative effect of apocynin on insulin resistance in high-fat diet mice. Ann Clin Lab Sci 41:236–243
Yuzefovych L, Wilson G, Rachek L (2010) Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am J Physiol Endocrinol Metab 299:E1096–E1105
Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M (2011) Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta 1807:726–734
Iizuka K, Nakajima H, Namba M et al (2002) Metabolic consequence of long-term exposure of pancreatic beta cells to free fatty acid with special reference to glucose insensitivity. Biochim Biophys Acta 1586:23–31
Schonfeld P, Wojtczak L (2008) Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med 45:231–241
Ivarsson R, Quintens R, Dejonghe S et al (2005) Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54:2132–2142
MacDonald PE, Salapatek AM, Wheeler MB (2003) Temperature and redox state dependence of native Kv2.1 currents in rat pancreatic beta-cells. J Physiol 546:647–653
Subasinghe W, Syed I, Kowluru A (2011) Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic beta-cells: evidence for regulation by Rac1. Am J Physiol Regul Integr Comp Physiol 300:R12–R20
Heumuller S, Wind S, Barbosa-Sicard E et al (2008) Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51:211–217
Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ (1994) Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11:95–102
Fontayne A, Dang PM, Gougerot-Pocidalo MA, El-Benna J (2002) Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 41:7743–7750
Miwa I, Ichimura N, Sugiura M, Hamada Y, Taniguchi S (2000) Inhibition of glucose-induced insulin secretion by 4-hydroxy-2-nonenal and other lipid peroxidation products. Endocrinology 141:2767–2772
Acknowledgements
The authors thank L. Lam (Department of Physiology, University of Toronto) for her excellent technical assistance.
This work was supported by Canadian Institutes of Health Research grant MOP-69018 (A. Giacca). K. Koulajian was supported by scholarships from the Banting and Best Diabetes Centre (University of Toronto), Ontario Graduate Scholarship and Ontario Graduate Scholarship for Science and Technology.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
KK researched and analysed data and drafted the manuscript. TD, GCL, AI, CT and JNP researched and analysed data and contributed to revising the manuscript. JWS, JWJ and JEB contributed to data analysis, discussion and revision of the manuscript. AG designed the study, contributed to the discussion and reviewed and finalised the manuscript. All the authors gave final approval to the submission of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 126 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Koulajian, K., Desai, T., Liu, G.C. et al. NADPH oxidase inhibition prevents beta cell dysfunction induced by prolonged elevation of oleate in rodents. Diabetologia 56, 1078–1087 (2013). https://doi.org/10.1007/s00125-013-2858-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2858-4