
Abstract
Aims/hypothesis
Obesity is closely associated with muscle insulin resistance and is a major risk factor for the pathogenesis of type 2 diabetes. Regular physical activity not only prevents obesity, but also considerably improves insulin sensitivity and skeletal muscle metabolism. We sought to establish and characterise an in vitro model of human skeletal muscle contraction, with a view to directly studying the signalling pathways and mechanisms that are involved in the beneficial effects of muscle activity.
Methods
Contracting human skeletal muscle cell cultures were established by applying electrical pulse stimulation. To induce insulin resistance, skeletal muscle cells were incubated with human adipocyte-derived conditioned medium, monocyte chemotactic protein (MCP)-1 and chemerin.
Results
Similarly to in exercising skeletal muscle in vivo, electrical pulse stimulation induced contractile activity in human skeletal muscle cells, combined with the formation of sarcomeres, activation of AMP-activated protein kinase (AMPK) and increased IL-6 secretion. Insulin-stimulated glucose uptake was substantially elevated in contracting cells compared with control. The incubation of skeletal muscle cells with adipocyte-conditioned media, chemerin and MCP-1 significantly reduced the insulin-stimulated phosphorylation of Akt. This effect was abrogated by concomitant pulse stimulation of the cells. Additionally, pro-inflammatory signalling by adipocyte-derived factors was completely prevented by electrical pulse stimulation of the myotubes.
Conclusions/interpretation
We showed that the effects of electrical pulse stimulation on skeletal muscle cells were similar to the effect of exercise on skeletal muscle in vivo in terms of enhanced AMPK activation and IL-6 secretion. In our model, muscle contractile activity eliminates insulin resistance by blocking pro-inflammatory signalling pathways. This novel model therefore provides a unique tool for investigating the molecular mechanisms that mediate the beneficial effects of muscle contraction.
Similar content being viewed by others

Introduction
Obesity in combination with a lack of exercise is a strong risk factor for the development of type 2 diabetes. It is well established that physical inactivity causes accumulation of visceral fat and that the health consequences of both are related to systemic low-grade inflammation [1, 2]. Importantly, the visceral fat compartment is a major secretory and endocrine-active tissue producing numerous cytokines that regulate energy metabolism and insulin sensitivity [3–5]. Adipocytes from obese persons are characterised by altered endocrine function, leading to increased secretion of pro-inflammatory adipokines, such as TNFα, chemerin, monocyte chemotactic protein (MCP)-1 and resistin [6–9]. The activation of inflammatory pathways leads to insulin resistance [10] in peripheral tissues such as skeletal muscle and adipose tissue, constituting an early defect in the pathogenesis of type 2 diabetes [11]. Insulin-resistant and type 2 diabetic patients display impaired insulin action on whole-body glucose uptake, in part due to impaired insulin-stimulated glucose uptake in skeletal muscle [11].
It is well accepted that physical activity exerts major beneficial effects on the prevention of chronic diseases like type 2 diabetes, cardiovascular disease, dementia and depression [12, 13]. Regular physical activity not only prevents obesity and reduces adipose tissue mass, but is also known to increase insulin-stimulated glucose uptake in the immediate post-exercise period [14], while chronic physical activity enhances insulin sensitivity in human skeletal muscle [15, 16]. Acute exercise increases glucose uptake in skeletal muscle by an insulin-independent mechanism that bypasses the insulin signalling defects associated with pathophysiological conditions [17]. Additionally, exercise activates AMP-activated protein kinase (AMPK), which phosphorylates and thereby inhibits acetyl-CoA carboxylase, resulting in reduced malonyl CoA content and hence enhanced fatty acid oxidation [18].
At present, the molecular mechanisms mediating the health-promoting effects of physical activity are not entirely understood. In the last decade, it became evident that skeletal muscle is an endocrine organ that produces and releases myokines in response to contraction [19]; these myokines probably mediate the health-promoting effects of physical activity. Myokines such as IL-6 and brain derived neurotrophic factor are released by skeletal muscle cells after exercise and lead to enhanced fatty acid oxidation in an AMPK-dependent fashion [20, 21].
We have previously shown that primary human skeletal muscle cells (hSkMC) incubated with adipocyte-conditioned medium (CM) from primary human adipocytes or treated with adipokines like MCP-1 and chemerin are characterised by impaired insulin signalling and glucose uptake [8, 9, 22]. Our aim here was to establish and characterise an in vitro contraction model of hSkMC, which mimics exercise; the overall goal was to analyse the signalling pathways and mechanisms involved in the beneficial effects of muscle activity. We also combined this contraction model with the insulin resistance model to analyse the cross-talk between adipocytes and contracting hSkMC. We show here that the contractile activity of hSkMC exerts an anti-inflammatory action, which prevents the induction of insulin resistance.
Methods
Materials
Reagents for SDS-PAGE were supplied by GE Healthcare (Munich, Germany) and Sigma (Munich, Germany), and rotiphorese was supplied by Carl Roth (Karlsruhe, Germany). The following antibodies were used: anti-phospho glycogen synthase kinase (GSK) 3α/β (Ser21/9), anti-phospho Akt (Ser473, Thr308), anti-Akt, anti-phospho nuclear factor ‘kappa-light-chain-enhancer’ of activated B cells (NFκB) (Ser536), anti-NFκB, anti-IκB kinase (IKK)α/ß, anti-IκBα, anti-phospho AMPKα (Thr172) and anti-AMPKα (Cell Signalling Technology, Frankfurt, Germany); anti-tubulin (Calbiochem, Merck Biosciences, Schmalbach, Germany); sarcomeric α-actinin (Sigma); anti-myosin heavy chain (MHC) (Upstate, San Diego, CA, USA); and mitochondria oxidative phosphorylation antibody cocktail and anti-GLUT12 antibody (Acris, Herford, Germany). Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse IgG were purchased from Promega (Mannheim, Germany). Collagenase NB4 standard grade was obtained from Serva (Heidelberg, Germany) and culture media from Gibco (Berlin, Germany). Recombinant human chemerin was supplied by R&D Systems (Wiesbaden-Nordenstadt, Germany) and MCP-1 by PeproTech (Hamburg, Germany). Primary hSkMC and the supplement pack for growth medium were obtained from PromoCell (Heidelberg, Germany). Horse serum for the differentiation medium was provided by Gibco. All other chemicals were of the highest analytical grade commercially available and purchased from Sigma.
Culture of hSkMC
Primary hSkMC from five healthy white donors (three males, 16, 21 and 47 years old; two females, 33 and 37 years old) were supplied as proliferating myoblasts and cultured as described earlier [23]. For an individual experiment, myoblasts were seeded in six-well culture dishes at a density of 1 × 105 cells/well and cultured to near-confluence in α-modified Eagle’s medium (αMEM)/Ham’s F-12 medium containing skeletal muscle cell growth medium supplement. The cells were then differentiated in αMEM containing 2% (vol./vol.) horse serum until day 5 of differentiation, followed by overnight starvation in αMEM without serum. Differentiated cells were electrically stimulated and incubated as indicated with CM, chemerin, MCP-1 and TNFα, respectively. Afterwards, cells were stimulated with 100 nmol/l insulin for 10 min.
Electrical pulse stimulation
Electrical pulse stimulation (EPS) was applied to fully differentiated myotubes in six-well dishes using a C-Dish combined with a pulse generator (C-Pace 100; IonOptix, Milton, MA, USA). The instrument emits bipolar stimuli to the carbon electrodes of the C-dish, which are placed in the cell culture medium. The myotubes were stimulated at 1 Hz, 2 ms and 11.5 V for 2 to 24 h. The medium was changed directly before stimulation. To document the contraction of the stimulated myotubes, a series of two images per s was taken using a microscope (DM RBE; Leica, Heidelberg, Germany) and camera (HV-C20; Hitachi, Tokyo, Japan). The images, shown at a rate of two images per s, can be viewed in the electronic supplementary material [ESM] Video 1.
Adipocyte isolation and culture
Adipose tissue samples were obtained from subcutaneous fat of normal or moderately overweight women (BMI 27.9 ± 0.9 kg/m2 [mean ± SEM], age 26–44 years). The procedure for obtaining adipose tissue was approved by the Ethics Committee of Heinrich-Heine-University, Duesseldorf, Germany. All tissue donors were healthy, free of medication and had no evidence of diabetes according to routine laboratory tests. Pre-adipocytes were isolated by collagenase digestion and differentiated as previously described [9, 24]. After 15 days, 70–90% of the seeded pre-adipocytes developed to differentiated adipocytes, as defined by accumulation of lipid droplets. These mature adipocytes were then used to generate CM by incubation with αMEM for 48 h, as previously described [25].
Immunofluorescence staining
hSkMC were seeded on glass coverslips, differentiated and stimulated by EPS. Afterwards cells were fixed with 2% (wt/vol.) paraformaldehyde dissolved in PBS for 15 min at room temperature. Cells were washed twice and permeabilised on ice for 5 min with 0.2% (vol./vol.) Triton-X in buffer containing 20 mmol/l HEPES, 300 mmol/l saccharose, 50 mmol/l NaCl and 3 mmol/l MgCl2. After blocking with 5% (wt/vol.) non-fat dry milk in PBS, myotubes were incubated with anti-sarcomeric α-actinin, washed and incubated with a secondary rhodamine-conjugated antibody. Myotubes were washed with 2× saline-sodium citrate (SSC) buffer (0.3 mol/l NaCl, 0.03 mol/l sodium citrate, pH 7.0) and incubated with 100 μg/ml RNAse in 2× SSC buffer for 20 min at 37°C. After washing with 2× SSC buffer, the nuclei were stained with 5 μmol/l Syto13 green for 5 min at room temperature.
Electron microscopy
Embedding of hSkMC in Epon 812 was performed as described by Luft [26] and modified by Reale [27]. In brief, cells were fixed in 2.5% (vol./vol.) glutaraldehyde/190 mmol/l cacodylate buffer, pH 7.4, and post-fixed in 1% (wt/vol.) osmium tetroxide. We used 1% (wt/vol.) uranyl acetate and lead citrate [28] to stain ultra-thin sections. Sections were investigated using a transmission electron microscope (TEM910; Zeiss, Oberkochem, Germany).
Measurement of IL-6 and vascular endothelial growth factor
The cytokine concentration in the supernatant fractions was determined by IL-6 and vascular endothelial growth factor (VEGF) ELISA (Biovendor, Heidelberg, Germany), respectively, both assays used according to the manufacturer’s protocol.
Cell viability assays
hSkMC were differentiated and electrically stimulated for 2–24 h.
ATP assay
Changes of relative ATP level were analysed using an ATP cell viability assay kit (ApoSENSOR; BioVision, Heidelberg, Germany) according to the instructions.
Lactate assay
l-(+)-lactate was detected in the supernatant fraction with a kit (Lactate Assay Kit II; BioVision).
MTT assay
The NADH content in the cells was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Myotubes were incubated for 2 h at 37°C with 1.2 mmol/l MTT solution in medium. Afterwards, cells were washed with PBS and lysed with 500 μl DMSO. Absorption at 540 nm was determined using a plate reader (InfiniteM200; Tecan, Maennedorf, Switzerland).
Cytotoxicity assay
The supernatant fraction was collected and lactate dehydrogenase release into the medium measured with a kit (Cytotoxicity Detection Kit Plus; Roche Applied Science, Mannheim, Germany) used according to the manufacturer’s protocol.
Marker of mitochondrial function
hSkMC were incubated with 1 μmol/l JC-1 dye for 30 min under culture conditions after the indicated time points. Afterwards, hSkMC were washed and analysed using a plate reader (InfiniteM200; Tecan). JC-1 monomers were assessed using excitation/emission wavelengths of 485/530 nm, while J-aggregates were measured at 560/595 nm.
RNA-isolation and quantitative real-time PCR
Total RNA was isolated and reverse-transcribed using kits (RNeasy Mini, Omniscript Reverse Transcription; Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Gene expression was determined by quantitative real-time PCR using QuantiTect primer assays and SYBR green reagents (Qiagen) with 0.016 to 20.00 ng cDNA on a cycler (Step One Plus; Applied Biosystems, Carlsbad, CA, USA). Expression of the genes investigated was normalised to actin. Gene expression was analysed via the ΔΔCt method.
Glucose uptake
hSkMC were electrically stimulated for 7.5 h on day 6 of differentiation. During the last 30 min of EPS, cells were treated with insulin (100 nmol/l) and uptake of 2-deoxy-glucose was measured for 2 h as described above [9, 29].
Fatty acid and glucose oxidation
hSkMC were seeded on 10 mm coverslips in six-well culture dishes and electrically stimulated as described above. Subsequently, coverslips were transferred to 48 well culture dishes, and 11.1 kBq/well of [14C]oleic acid and [14C]palmitic acid supplemented with 1 μmol/l l-carnitine, or 7.4 kBq/well [U-14C]glucose supplemented with 0.35 mmol/l glucose were added to hSkMC. Culture dishes were incubated for 4 h in an oxidation chamber, which allows gas exchange between two neighbouring wells. Filter papers soaked with NaOH were placed in the empty neighbouring wells. Oxidation was stopped, and CO2 was liberated via acidification of culture media by injecting 1 mol/l HCl and trapped in filter paper. Radioactivity was counted in a liquid scintillation counter (Beckman, Munich, Germany).
Immunoblotting
hSkMC were treated as indicated and lysed in a buffer containing 50 mmol/l HEPES (pH 7.4), 1% (vol./vol.) Triton-X, PhosStop and complete protease inhibitor cocktail (Roche). Western blot analysis was performed as described before [9]. Signals were visualised and evaluated on a work station (VersaDoc 4000 MP; BioRad, Munich, Germany) and analysed with an analysis software package (Quantity One, version 4.6.7, BioRad, Munich, Germany).
Presentation of data and statistics
Data are presented as means ± SEM. Unpaired two-tailed Student’s t test or one-way ANOVA (post hoc test Bonferroni’s multiple comparison test) were used to determine statistical significance. All statistical analyses were performed using Prism5 (GraphPad, La Jolla, CA, USA), with a value of p < 0.05 considered to be statistically significant. Corresponding significance levels are as indicated.
Results
EPS induces de novo sarcomere structure assembly in human skeletal myotubes
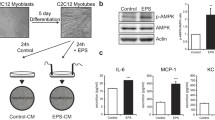
After 6 days of differentiation, most of the hSkMC fused and formed typical multinucleated myotubes. As reported in our earlier study [22], differentiating hSkMC display increased protein abundance of myogenin, myoblast determination protein and MHC, which are typical markers of myogenesis, but cells did not contract spontaneously. The conditions of 1 Hz frequency, 2 ms pulse duration and 11.5 V intensity were selected with regard to most marked AMPK activation and IL-6 secretion after differentiation with 2% horse serum (Fig. 1). Using this protocol, we observed that after a few hours of continuous EPS, a subset of myotubes showed noticeable, vigorous contraction, with most of the myotubes contracting after 8 h EPS (ESM Video 1). Immunofluorescence staining of sarcomeric α-actinin showed uniform distribution of the protein in the cytoplasm of unstimulated cells. After EPS, a reorganisation of the cytoskeleton and de novo formation of sarcomeric structures with their typical striated pattern were observed (Fig. 2a–c). These findings were confirmed by electron microscopy (Fig. 2d–f). The typical Z-lines appeared as a series of dark lines as described for cross-striated muscles.

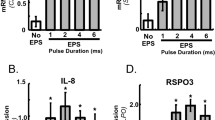
Process of selecting the conditions of EPS protocol. Based on previously published conditions of EPS, frequencies of 0.1, 1 and 10 Hz were tested [21, 30, 42]. The conditions were selected with regard to optimised AMPK activation and IL-6 secretion. Effects of variation of frequency (a, b) and test of differentiation with horse serum (c, d) were quantified. a, b Myotubes were differentiated in αMEM serum-free medium and subjected to EPS at 0.1, 1 and 10 Hz, 2 ms and 11.5 V. Total cell lysates (a) were obtained, resolved by SDS-PAGE and immunoblotted with phospho-specific (p) AMPK (Thr172) antibody; n = 3. b IL-6 secretion was determined in supernatant fractions; n = 4–5; **p < 0.01 vs control. c Myotubes were differentiated in αMEM serum-free medium containing 2% (vol./vol.) horse serum during differentiation, in combination with overnight starvation. Total cell lysates were obtained and treated as above (a); n ≥ 3; *p < 0.05. White symbols, control; black symbols, EPS. d IL-6 secretion was determined in supernatant fractions; n ≥ 3; ***p < 0.001 vs control

Effect of EPS on sarcomere structure assembly in human skeletal myotubes. a The cells were fixed and analysed for localisation of sarcomeric α-actinin without (control), and after 8 (b) and 24 h (c) of EPS (1 Hz, 2 ms, 11.5 V) by immunofluorescence staining, as described. Red, sarcomeric α-actinin; green, sytogreen. Magnification × 100. Sarcomeric α-actinin was evenly distributed in the cytoplasm without EPS (a). Sarcomeres became rapidly visible after EPS, with (b, c) Z-lines appearing as a series of red lines. These findings were confirmed by electron microscopy (d–f). Magnification × 10,000. MT, mitochondria; N, nucleus
EPS increases IL-6 and VEGF secretion, as well as AMPK activation
EPS did not influence the abundance of motor proteins like MHC and sarcomeric α-actinin, or that of the glucose transporters GLUT1 and GLUT4 (Fig. 3a). However, in response to contraction, we observed an increase of MYH1 mRNA, while no changes were measured for MYH2 mRNA (Fig. 3b). EPS-induced contraction upregulated secretion of IL-6 and VEGF, two known myokines. EPS significantly upregulated IL-6 secretion after 8 and 24 h compared with controls, reaching a concentration of 35 ± 8 pg/ml (mean±SEM) after 24 h EPS, compared with 14 ± 4 pg/ml in control (Fig. 3c). EPS immediately upregulated VEGF secretion, reaching a maximum after 24 h of 274 ± 40 pg/ml (fivefold increase vs control) (Fig. 3d).

Effect of EPS on the abundance of muscle proteins, secretion of IL-6, and on VEGF and AMPK activation. hSkMC were differentiated and exposed to EPS for the indicated times at 1 Hz, 2 ms and 11.5 V. a Total cell lysates were resolved by SDS-PAGE; representative blots of proteins as labelled are shown. b Relative gene expression of MYH1 and MYH2 was measured by real-time PCR as described; n = 4–5; ***p < 0.001. c IL-6 and (d) VEGF secretion of human myotubes with and without EPS was measured after indicated time points by ELISA; n ≥ 3; *p < 0.05. White symbols, control; black symbols, EPS. e Relative ATP concentration of myotubes was measured after indicated time points of EPS and in control, as described; n = 8, *p < 0.05. f Total cell lysates were obtained and analysed by SDS-PAGE and western blot using phospho-specific (p) AMPKα antibody. Quantified data were normalised to tubulin; n ≥ 3; **p < 0.01 vs corresponding basal level. Data (b–f) are mean ± SEM
ATP content in hSkMC significantly increased during the first 2 h of EPS (1.5-fold). Afterwards, the ATP level in hSkMC decreased significantly (Fig. 3e). After 8 h, EPS induced a 3.8-fold increase in AMPK (Thr172) phosphorylation compared with non-stimulated hSkMC (Fig. 3f). This effect was diminished, but still significant after 24 h. However, significant changes in acetyl-CoA carboxylase phosphorylation or protein abundance were not observed upon EPS (data not shown).
EPS exerts no damaging effect on myotubes
Decreased levels of ATP and increased levels of ADP are recognised in apoptotic cells. To rule out the possibility that EPS may cause cell damage, several cell-based assays were performed. Cytotoxicity, as assessed by l-lactate dehydrogenase release, did not differ significantly between cells with or without EPS (Fig. 4a). The NADH level in cells, as assessed by MTT assay, was not changed by EPS compared with control cells (Fig. 4b). The concentration of lactate in the medium significantly increased from 8 to 24 h of culture (Fig. 4c) from 187 ± 7 nmol/ml (mean ± SEM) to 336 ± 10 nmol/ml. EPS induced significant, higher lactate concentrations in the medium compared with controls. The polarisation of mitochondrial membranes was not disturbed, since the ratio of JC-1 aggregates to monomers was not changed during culture, either in unstimulated or in EPS-treated cells (Fig. 4d). Protein abundance of the complexes of the electron transport chain showed a significant increase for complexes II, IV and V (Fig. 4e). In addition UCP3, PGC1α (also known as PPARGC1A) and COX2 mRNA were significantly increased compared with non-stimulated cells (Fig. 4f).

Determination of metabolic activity of skeletal muscle cells after EPS. hSkMC were differentiated and stimulated for 8 and 24 h at 1 Hz, 2 ms and 11.5 V. a Lactate dehydrogenase (LDH) abundance in the medium; n ≥ 5, ***p < 0.001. b The quantity of NADH in the cells was measured using an MTT assay; n ≥ 3, **p < 0.01. c Lactate concentrations in the supernatant fractions of EPS-treated and untreated cells. As positive control, lysis reagent (supplied with the kit) was used. n ≥ 3, ***p < 0.001. d The ratio of JC-1 aggregates to monomers. As a positive control, exposure to 100 μmol/l CCCP (carbonyl-cyanide m-chlorophenyl hydrazone) for 45 min was used prior to JC-1 staining. e Total cell lysates were obtained and analysed by SDS-PAGE and western blot using an oxidative phosphorylation antibody cocktail. Data are normalised to tubulin; n = 4, *p < 0.05. f Relative gene expression of UCP3, PGC1α and COX2 as measured by real-time PCR; n = 4–5; **p < 0.01 for UCP3 and PGC1α, *p > 0.05 for COX2. Values (a–f) are mean ± SEM
EPS augments insulin-stimulated glucose uptake, but does not change fatty acid oxidation
To test whether EPS induces changes in glucose homeostasis in hSkMC, we measured glucose uptake. Insulin-stimulated glucose uptake was significantly increased over the basal level (2.6-fold; Fig. 5a), as reported in our earlier studies [22, 29]. Contraction of hSkMC led to a significant 2.4-fold increase compared with basal control. Importantly, hSkMC showed a marked increase (fivefold) in insulin-stimulated glucose uptake after EPS compared with basal control. The incremental increase in insulin-stimulated glucose uptake, which reflects the efficiency of insulin action, was profoundly augmented by contractile activity of myotubes (Fig. 5b). However, levels of insulin-sensitive glucose transporters, namely GLUT4 and GLUT12, were not affected (Fig. 5c). Oxidation of the fatty acids, oleic acid and palmitic acid, and glucose oxidation were assessed by 14CO2 production. Glucose oxidation was increased in response to insulin and EPS alone, while the combined effect was additive (Fig. 5d). However, oxidation of both fatty acids was not changed after EPS-induced contraction (Fig. 5e).

Effect of EPS on glucose uptake and fatty acid oxidation in hSkMC. a In the last 30 min of 7.5 h of EPS (1 Hz, 2 ms, 11.5 V) cells were stimulated with 100 nmol/l insulin. Glucose uptake was assessed for 2 h after acute insulin stimulation. n = 5; *p < 0.05 vs basal control; †p < 0.05 vs basal control and EPS control. White bars, basal; black bars, insulin-stimulated. b The increment of insulin-stimulated glucose uptake was calculated as the difference between insulin-stimulated glucose uptake (2-deoxy-glucose, 2-DOG) and basal control; n = 5; *p < 0.05. c Total cell lysates were obtained and analysed by SDS-PAGE and western blot. Representative blots for proteins as indicated are shown. d Glucose oxidation. Cells were treated with 100 nmol/l insulin as indicated during incubation in the oxidation chamber; n = 4–5; *p < 0.05 vs basal control; †p < 0.05 vs basal control and EPS control. White bars, basal; black bars, insulin-stimulated e Fatty acid oxidation as assessed after 24 h EPS; n ≥ 3; ‡p = 0.4. Data (a, b, d, e) are means ± SEM. OA, oleic acid; PA, palmitic acid
EPS protects hSkMC from impaired insulin signalling induced by CM, MCP-1 and chemerin
Under control conditions, insulin induced a significant increase of Akt phosphorylation (Ser473) in hSkMC, with a significant reduction of this response after incubation with CM (Fig. 6a). When the cells were EPS-stimulated during incubation with CM, this effect on insulin-stimulated Akt phosphorylation was abrogated. Comparable effects were observed at the level of GSK3α (Ser21) phosphorylation, with very marked inhibition by CM and complete prevention by EPS. Chemerin and MCP-1 induced a significant reduction of insulin-stimulated Akt phosphorylation at Ser473 and Thr308 sites, respectively, as reported in our earlier studies [8, 9, 23]. The application of EPS during treatment with chemerin and MCP-1, respectively, restored insulin signalling in hSkMC (Fig. 6b).

Effect of CM, chemerin and MCP-1 on insulin signalling in control and electrically stimulated hSkMC. a Myotubes were simultaneously incubated with CM for 8 h and exposed to EPS. After acute stimulation with insulin, total cell lysates were obtained, resolved by SDS-PAGE and immunoblotted with phospho-specific (p)Akt and GSK3 antibodies. All data were normalised to the level of tubulin and are expressed relative to insulin-stimulated control values. Data are means ± SEM; n ≥ 5; *p < 0.05. b Myotubes were incubated with 2 μg/ml chemerin (Chem) and 2 ng/ml MCP-1, respectively, and simultaneously exposed to EPS. After acute stimulation with insulin, total cell lysates were obtained, resolved by SDS-PAGE and immunoblotted with phospho-specific Akt antibodies for Ser473 and Thr308. All data were normalised to the level of tubulin and are expressed relative to the insulin-stimulated control value. Data are presented as means ± SEM; n ≥ 4, *p < 0.05 vs insulin-stimulated control. White bars, basal; black bars, 100 nmol/l insulin
EPS prevents activation of NFκB and p42/44 mitogen-activated protein kinase in hSkMC
NFκB and IKKβ protein abundance was significantly reduced in EPS-treated cells, whereas IKKα and IκBα protein abundance were not affected (Fig. 7a). Incubation of cells with TNFα led to activation of NFκB, reaching a maximum after 10 min (3.2-fold; Fig. 7c). As shown by application of the selective inhibitor of IKK, I229 (Fig. 7b), this effect is specifically mediated by IKK downstream signalling, After EPS, NFκB activation was diminished due to reduced NFκB protein abundance. Most importantly, after EPS, maximal NFκB activation in response to TNFα was substantially reduced (1.7-fold after 10 min). Thus contractile activity exerts an anti-inflammatory effect and interferes with TNFα-induced NFκB signalling.

Prevention of TNFα-induced inflammatory signalling by EPS. a Skeletal muscle cells were stimulated by EPS for 8 h, after which total cell lysates were resolved by SDS-PAGE, and NFκB, IKKα, IKKβ and IκBα levels determined by western blotting. White bar, control; black bars, EPS. b Skeletal muscle cells were pre-incubated with 10 μmol/l specific IKK inhibitor I229 and afterwards stimulated with 50 pg/ml TNFα for 10 min. c–e Cells were treated with 50 pg/ml TNFα for times as indicated after 8 h EPS. Total cell lysates were resolved by SDS-PAGE and immunoblotted with (c) phospho-specific (p) antibody for NFκB, and (d) specific antibodies for IKKß and (e) IκBα. All data were normalised to the level of tubulin and are presented as means ± SEM; n ≥ 3; *p < 0.05 vs basal level. White symbols, control; black symbols, EPS
Incubation of hSkMC with TNFα led to a significant decrease (59% after 20 min) of IKKβ protein levels (Fig. 7d). In contrast, IKKβ protein abundance was significantly reduced after EPS, compared with quiescent controls (Fig. 7a), but was not altered after TNFα treatment (Fig. 7d). The incubation of myotubes with TNFα diminished IκBα protein abundance by more than 60% after 20 min, while TNFα had no effect on IκBα levels in contracting cells (Fig. 7e). CM and chemerin induced NFκB activation (1.8-fold and 1.5-fold; Fig. 8a), while MCP-1 induced significant activation of p44/p42 mitogen-activated protein kinase (MAPK) (1.5-fold; Fig. 8c). Both effects on NFκB and p44/p42 MAPK activation were diminished to control level after EPS (Fig. 8b, c).

Prevention of CM-, MCP-1- and chemerin-induced inflammatory signalling by EPS. a Skeletal muscle cells were pre-incubated with or without 10 μmol/l of the specific IKK inhibitor I229. Afterwards, cells were treated with CM and 2 μg/ml chemerin for 30 min. Total cell lysates were resolved by SDS-PAGE and immunoblotted with a phospho-specific (p) antibody for NFκB. b, c Skeletal muscle cells were exposed to EPS for 8 h at 1 Hz, 2 ms and 11.5 V. Cells were treated with CM and 2 μg/ml chemerin (b) or with 2 ng/ml MCP-1 (c) for the last 30 min of stimulation. Total cell lysates were resolved by SDS-PAGE and immunoblotted with a phospho-specific antibody for NFκB (b) or MAPK (c). Blots are representative. All data were normalised to the level of tubulin and are means ± SEM; n ≥ 4; *p < 0.05 vs basal level. b, c White bars, control (non-EPS); black bars, EPS
Discussion
Exercise has been shown to have a positive impact on a number of diseases in humans, including obesity and type 2 diabetes [1, 2, 13]. The pathogenesis of type 2 diabetes has been intensively studied and is characterised by chronic hyperglycaemia, resulting in defects in insulin secretion, insulin action or both. However, knowledge of the interplay between different molecular signalling pathways during exercise is still incomplete and experimentally adequate models of exercise remain elusive. In this study, we established and validated a model of contracting hSkMC and used it to further analyse the beneficial effect of exercise in the context of insulin resistance.
It is well known that hSkMC display increased levels of typical myogenesis markers like myogenin, myoblast determination protein, MHC and GLUT4 during differentiation in vitro [20]. One limitation of the models used is the lack of contraction, a central characteristic of muscle cells. Therefore, we stimulated hSkMC with electrical pulses, resulting in vigorous contraction and formation of striation patterns of sarcomeric structures, as visualised by immunofluorescence staining of α-actinin and electron microscopy. In line with this, Fujita et al. observed de novo formation of sarcomeres after EPS in murine C2C12 cells, whereas calcium channel blockers like verapamil and BAPTA-AM suppressed sarcomere structure development [30]. Thus it can be speculated that manipulated Ca2+ transients, achieved by applying an appropriate EPS to differentiated hSkMC, are primarily necessary to accelerate de novo sarcomere assembly and to rapidly develop contractile activity. In response to contraction, we observed an increase of MYHI mRNA expression after EPS, in combination with an increase of mitochondrial marker proteins and mitochondrial mRNA expression. MYHI fibres are more efficient at using oxygen to generate ATP for continuous, extended muscle contractions over a long time, reflecting the experimental setup used by us with its rather long EPS time of 24 h.
In skeletal muscle, three energy systems function to replenish ATP, namely creatine kinase, glycolysis and mitochondrial respiration. All systems contribute to different degrees to the replenishment of ATP on the basis of an interaction between the intensity and duration of exercise [31]. Using EPS, we observed enhanced glucose uptake and increased lactate concentrations. During high-performance sports, glucose is catabolised and pyruvate is substantially generated. When the mitochondrial capacity is exceeded, pyruvate is reduced to lactate, resulting in oxidation of NADH/H+ to NAD+ [31]. As we did not observe increased fatty acid oxidation after 24 h of EPS, it is likely that cells mainly catabolise glucose for ATP regeneration under the applied conditions. However, after depletion of muscle glycogen by contraction, cells may replenish glycogen stores, as well.
Human studies using the one-legged exercise model followed by a euglycaemic–hyperinsulinaemic clamp have shown increased insulin sensitivity after exercise [32]. C2C12 cells derived from mouse skeletal muscle are mostly used to investigate muscle cell differentiation, sarcomere development, myotube contraction and glucose uptake [30, 33]. However, these cells do not produce sufficient levels of GLUT4 and their insulin responsiveness is reportedly minimal, even after differentiation [34]. In our model, GLUT12 and GLUT4 protein abundance was not altered in cells during EPS, but human cells exhibited a profound insulin-stimulated glucose uptake (2.6-fold greater), compared with C2C12 cells (1.4-fold) [33]. We observed increased insulin efficiency after EPS, resulting in substantially elevated glucose uptake in contracting myotubes. As insulin signalling, GLUT4 and GLUT12 protein abundance remained unaltered after EPS, it may be that contractile activity affects the GLUT4 trafficking machinery, mediating a more efficient mobilisation of the transporter by insulin. Future studies will be needed to address these issues.
One of the major achievements in obesity research is the finding that adipose tissue is a major endocrine organ, which secretes numerous adipokines. Indeed, several hundred adipokines have now been identified [35]. The secretion of adipokines is changed dramatically in the obese state, affecting a wide range of physiological functions and leading to insulin resistance in skeletal muscle, among other tissues [6, 7, 36]. A main finding of the present study was the improved insulin sensitivity of contracting hSkMC in conditions of insulin resistance. As previously shown by our group, CM (which contains the whole secretory output of mature adipocytes, including adipokines like chemerin, pigment epithelium-derived factor and MCP-1) induced insulin resistance in hSkMC at the level of Akt and GSK3, and reduced insulin-stimulated glucose uptake by activating inflammatory signalling pathways [8, 9, 23, 37]. Incubation of hSkMC with CM or adipokines is a suitable model to dissect different mechanisms leading to muscle insulin resistance. In this study, we showed that EPS completely prevented insulin resistance in hSkMC at the level of Akt and GSK3 during incubation with CM, MCP-1 or chemerin. To date, we have only analysed the effects of EPS with regard to insulin signalling. Future work will be required to assess its effects at effector systems located further downstream from insulin.
One potential reason for the observation described above might be the blocking effect of EPS on NFκB activation by CM and several adipokines. Some adipokines involved in the development of insulin resistance are known to activate inflammatory signalling pathways by activating IKK and its downstream effector NFκB [38]. These proteins belong to a family of transcription factors that controls production of pro-inflammatory proteins. In our model, CM, chemerin and TNFα induced activation of NFκB, which is consistent with the elevated NFκB activity observed in muscle of insulin-resistant participants in comparison with lean control participants under basal conditions [39]. EPS, which mimics contraction of an active muscle, diminished NFκB activity and prevented activation of NFκB by CM and chemerin. Additionally, the increment of NFκB activation by TNFα treatment after EPS was profoundly diminished.
Thus contractile activity of hSkMC appears to directly inhibit TNFα signalling and activation of NFκB. This may involve: (1) downregulation of pro-inflammatory signalling components like IKK and NFκB, as shown here; (2) Ca2+-mediated activation of anti-inflammatory pathways; and (3) probably the release of myokines by contracting cells. The anti-inflammatory effect of exercise is well established [40] and muscular IL-6 is thought to play a key role in this process [41]. However, the effect of IL-6 is related to its profound increase in the circulation after exercise, as well as to the inhibition of TNFα and IL-1 receptor alpha production [19]. Thus additional myokines exerting an autocrine action may be involved in the anti-inflammatory effect of muscle contractile activity. Very recent work in our laboratory suggests that several hundred myokines are released from contracting hSkMC (S. Lambernd, unpublished observations). Identification of these factors will be instrumental to understanding the beneficial effects of muscle contraction.
In conclusion, using this model of contracting myotubes, we observed that the risk of insulin resistance is directly diminished, as insulin signalling was not disturbed after incubation with CM and inflammatory signalling was not activated. This model provides a unique tool for investigation of the mechanisms and underlying signalling pathways that mediate the beneficial effects of muscle contraction, and will help further clarify the potential of exercise as a way of combating insulin resistance.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- CM:
-
Adipocyte-conditioned medium
- EPS:
-
Electrical pulse stimulation
- GSK:
-
Glycogen synthase kinase
- hSkMC:
-
Human skeletal muscle cells
- IKK:
-
IκB kinase
- MAPK:
-
Mitogen-activated protein kinase
- MCP:
-
Monocyte chemotactic protein
- αMEM:
-
α-Modified Eagle’s medium
- MHC:
-
Myosin heavy chain
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NFκB:
-
Nuclear factor ‘kappa-light-chain-enhancer’ of activated B cells
- SSC:
-
Saline-sodium citrate
- VEGF:
-
Vascular endothelial growth factor
References
Handschin C, Spiegelman BM (2008) The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 454:463–469
Yudkin JS (2007) Inflammation, obesity, and the metabolic syndrome. Horm Metab Res 39:707–709
Bloomgarden ZT (2000) Obesity and diabetes. Diabetes Care 23:1584–1590
Felber JP, Golay A (2002) Pathways from obesity to diabetes. Int J Obes Relat Metab Disord 26(Suppl 2):S39–S45
Finegood DT (2003) Obesity, inflammation and type II diabetes. Int J Obes Relat Metab Disord 27(Suppl 3):S4–S5
Arner P (2001) Regional differences in protein production by human adipose tissue. Biochem Soc Trans 29:72–75
Trayhurn P, Beattie JH (2001) Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc 60:329–339
Sell H, Dietze-Schroeder D, Kaiser U, Eckel J (2006) Monocyte chemotactic protein-1 is a potential player in the negative cross-talk between adipose tissue and skeletal muscle. Endocrinology 147:2458–2467
Sell H, Laurencikiene J, Taube A et al (2009) Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes 58:2731–2740
Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW (1999) C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol 19:972–978
DeFronzo RA, Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(Suppl 2):S157–S163
Pedersen BK (2011) Muscles and their myokines. J Exp Biol 214:337–346
Pedersen BK (2009) The diseasome of physical inactivity and the role of myokines in muscle fat cross talk. J Physiol 587:5559–5568
Richter EA, Garetto LP, Goodman MN, Ruderman NB (1982) Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest 69:785–793
Wojtaszewski JF, Hansen BF, Gade et al (2000) Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 49:325–331
King DS, Dalsky GP, Clutter WE et al (1988) Effects of exercise and lack of exercise on insulin sensitivity and responsiveness. J Appl Physiol 64:1942–1946
Zierath JR, Krook A, Wallberg-Henriksson H (2000) Insulin action and insulin resistance in human skeletal muscle. Diabetologia 43:821–835
Kahn BB, Alquier T, Carling D, Hardie DG (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1:15–25
Pedersen BK, Febbraio MA (2008) Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev 88:1379–1406
Pedersen BK, Steensberg A, Fischer C et al (2004) The metabolic role of IL-6 produced during exercise: is IL-6 an exercise factor? Proc Nutr Soc 63:263–267
Matthews VB, Astrom MB, Chan MH et al (2009) Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 52:1409–1418
Sell H, Eckardt K, Taube A et al (2008) Skeletal muscle insulin resistance induced by adipocyte-conditioned medium: underlying mechanisms and reversibility. Am J Physiol Endocrinol Metab 294:E1070–E1077
Dietze D, Koenen M, Rohrig K, Horikoshi H, Hauner H, Eckel J (2002) Impairment of insulin signaling in human skeletal muscle cells by co-culture with human adipocytes. Diabetes 51:2369–2376
Hauner H, Petruschke T, Russ M, Rohrig K, Eckel J (1995) Effects of tumour necrosis factor alpha (TNF alpha) on glucose transport and lipid metabolism of newly-differentiated human fat cells in cell culture. Diabetologia 38:764–771
Dietze-Schroeder D, Sell H, Uhlig M, Koenen M, Eckel J (2005) Autocrine action of adiponectin on human fat cells prevents the release of insulin resistance-inducing factors. Diabetes 54:2003–2011
Luft JH (1961) Improvements in epoxy resin embedding methods. J Biophys Biochem Cytol 9:409–414
Reale E, Luciano L, Brandes G (1992) Alterations in the morphology of glycoconjugate molecules caused by histochemical procedures: comparison of renal glomeruli and articular cartilage. Histochem J 24:153–165
Reynolds ES (1963) The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol 17:208–212
Eckardt K, Sell H, Taube A et al (2009) Cannabinoid type 1 receptors in human skeletal muscle cells participate in the negative crosstalk between fat and muscle. Diabetologia 52:664–674
Fujita H, Nedachi T, Kanzaki M (2007) Accelerated de novo sarcomere assembly by electric pulse stimulation in C2C12 myotubes. Exp Cell Res 313:1853–1865
Baker JS, McCormick MC, Robergs RA (2010) Interaction among skeletal muscle metabolic energy systems during intense exercise. J Nutr Metab 2010:905612
Frosig C, Rose AJ, Treebak JT, Kiens B, Richter EA, Wojtaszewski JF (2007) Effects of endurance exercise training on insulin signaling in human skeletal muscle: interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes 56:2093–2102
Nedachi T, Kanzaki M (2006) Regulation of glucose transporters by insulin and extracellular glucose in C2C12 myotubes. Am J Physiol Endocrinol Metab 291:E817–E828
Sargeant R, Mitsumoto Y, Sarabia V, Shillabeer G, Klip A (1993) Hormonal regulation of glucose transporters in muscle cells in culture. J Endocrinol Invest 16:147–162
Lehr S, Hartwig S, Lamers D et al (2011) Identification and validation of novel adipokines released from primary human adipocytes. Mol Cell Proteomics. doi:10:1074/mcp.M111.010504
Sell H, Eckel J, Dietze-Schroeder D (2006) Pathways leading to muscle insulin resistance—the muscle–fat connection. Arch Physiol Biochem 112:105–113
Famulla S, Lamers D, Hartwig S et al (2010) Pigment epithelium-derived factor is one of the most abundant proteins secreted by human adipocytes and induces insulin resistance and inflammatory signaling in muscle and fat cells. Int J Obes (Lond) 35:762–772
Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 18:621–663
Tantiwong P, Shanmugasundaram K, Monroy A et al (2010) NF-kappaB activity in muscle from obese and type 2 diabetic subjects under basal and exercise-stimulated conditions. Am J Physiol Endocrinol Metab 299:E794–E801
Petersen AM, Pedersen BK (2005) The anti-inflammatory effect of exercise. J Appl Physiol 98:1154–1162
Pedersen BK (2011) Exercise-induced myokines and their role in chronic diseases. Brain Behav Immun 25:811–816
Nedachi T, Fujita H, Kanzaki M (2008) Contractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscle. Am J Physiol Endocrinol Metab 295:E1191–E1204
Acknowledgements
We thank J. Liebau (Department of Plastic Surgery, Florence-Nightingale-Hospital, Duesseldorf, Germany) and C. Andree (Department of Plastic Surgery, Sana-Hospital, Duesseldorf-Gerresheim, Germany) for support in obtaining adipose tissue samples. The secretarial assistance of B. Hurow and the technical help of A. Cramer, A. Horrighs and D. Herzfeld de Wiza are gratefully acknowledged.
Funding
This work was supported by the Ministerium für Wissenschaft und Forschung des Landes Nordrhein-Westfalen (Ministry of Science and Research of the State of North Rhine-Westphalia), the Bundesministerium für Gesundheit (Federal Ministry of Health), the Commission of the European Communities (Collaborative Project ADAPT, contract no. HEALTH-F2-2008-201100), the European Union COST Action (BM0602) and the Jühling Foundation.
Contribution statement
SL contributed to the concept, acquired, analysed and interpreted data, wrote the manuscript and had the main responsibility together with JE. AT, AS, BP, SWG, RS, KJ and JW performed research and contributed to analysis and interpretation of data. KE and JE contributed to the concept, analysis of the data, and the discussion and revision of the manuscript. All authors approved the final version of the manuscript.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Differentiated skeletal muscle cells were stimulated with EPS at 1 Hz, 2 ms and 11.5 V. After 24 h of EPS, contraction of human myotubes was observed and documented as described in Methods section (AVI 55.8 MB)
Rights and permissions
About this article
Cite this article
Lambernd, S., Taube, A., Schober, A. et al. Contractile activity of human skeletal muscle cells prevents insulin resistance by inhibiting pro-inflammatory signalling pathways. Diabetologia 55, 1128–1139 (2012). https://doi.org/10.1007/s00125-012-2454-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2454-z