
Abstract
Aims/hypothesis
Insulin resistance is characterised by impaired glucose utilisation when measured by a euglycaemic–hyperinsulinaemic clamp. We hypothesised that, in response to postprandial conditions, non-diabetic individuals would have similar intracellular glycolytic and oxidative glucose metabolism independent of the degree of insulin resistance.
Methods
Fourteen (seven male) sedentary, insulin-sensitive participants (mean±SD: BMI 25 ± 4 kg/m2; age 39 ± 10 years; glucose disposal rate 9.4 ± 2.1 mg [kg estimated metabolic body size]−1 min−1) and 14 (six male) sedentary, non-diabetic, insulin-resistant volunteers (29 ± 4 kg/m2; 34 ± 13 years; 5.3 ± 1.2 mg [kg estimated metabolic body size]−1 min−1) received after a 10 h fast 60 g glucose plus 15 g [6,6-2H2]glucose. Serum glucose and insulin concentrations, plasma 2H enrichment and whole-body gas exchange were determined before glucose ingestion and hourly thereafter for 4 h. Plasma 2H2O production is an index of glycolytic disposal. On day 2, participants received a weight-maintenance diet. On day 3, a euglycaemic–hyperinsulinaemic clamp was performed.
Results
Insulin-resistant individuals had about a twofold higher postprandial insulin response than insulin-sensitive individuals (p = 0.003). Resting metabolic rate was similar in the two groups before (p = 0.29) and after (p = 0.33–0.99 over time) glucose ingestion, whereas a trend for blunted glucose-induced thermogenesis was observed in insulin-resistant vs insulin-sensitive individuals (p = 0.06). However, over the 4 h after the 75 g glucose ingestion, glycolytic glucose disposal was the same in insulin-sensitive and insulin-resistant individuals (36.5 ± 3.7 and 36.2 ± 6.4 mmol, respectively; p = 0.99). Similarly, whole-body carbohydrate oxidation did not differ between the groups either before or after glucose ingestion (p = 0.41).
Conclusions/interpretation
Postprandial hyperinsulinaemia and modest hyperglycaemia overcome insulin resistance by enhancing tissue glucose uptake and intracellular glucose utilisation.
Similar content being viewed by others
Introduction
Insulin resistance is defined as a decreased sensitivity to the metabolic actions of insulin on glucose uptake by tissues such as skeletal muscle and adipose tissue or the inhibition of hepatic glucose production in the liver [1]. The glucose clamp technique is widely accepted as the reference method to determine insulin-stimulated glucose disposal rate [2]. This is, however, a non-physiological method during which serum insulin and glucose concentrations are kept constant. At similar insulin concentrations, insulin-resistant individuals require lower glucose infusion rates to ‘clamp’ serum glucose concentration than insulin-sensitive individuals. As a consequence, insulin resistance is characterised by decreased insulin-stimulated oxidative and non-oxidative glucose disposal rates [3–6].
The impaired insulin-stimulated glucose utilisation during the clamp may not necessarily be present under physiological conditions such as in the postabsorptive and/or postprandial state. Two factors play a role in promoting postprandial tissue glucose uptake, i.e. hyperinsulinaemia and hyperglycaemia. First, insulin-resistant individuals with a well-preserved pancreatic beta cell function release more insulin (hyperinsulinaemia) to compensate for the impaired insulin action. Second, postprandial hyperglycaemia itself is also known to promote tissue glucose uptake by a mass action effect. Therefore the fact that insulin-stimulated glucose utilisation is impaired in insulin-resistant individuals during a euglycaemic–hyperinsulinaemic clamp may not be valid under physiological postprandial conditions.
A novel method for measuring insulin sensitivity under physiological conditions was proposed by Beysen et al. [7] and confirmed by us [8]. The method is based on the assessment of glycolytic metabolism using 15 g [6,6-2H2]glucose in a 75 g glucose load. The 2H label is metabolised into water by carboxylation–decarboxylation between pyruvate and dicarboxylic acids (i.e. oxaloacetate, malate and fumarate) and in the liver via the action of glutamate–pyruvate transaminase [9]. This metabolic water is then incorporated into the body water pool. Thus release of 2H2O after a 75 g glucose load normalised for serum insulin concentration is an estimate of the ability of insulin to facilitate tissue glucose transport, phosphorylation and glycolytic pathways [7, 8]. Using this novel technique to assess intracellular glucose metabolism, we hypothesised that non-diabetic insulin-resistant vs insulin-sensitive individuals (by a euglycaemic–hyperinsulinaemic clamp) will have similar postprandial whole-body glycolytic metabolism.
Methods
Subjects
Twenty-eight healthy (physical examination and routine medical laboratory tests), non-smoking and non-diabetic participants (13 male and 15 female) were recruited by advertising. They all had a stable body weight (change <2 kg) over the preceding 3 months and none performed regular physical activity (>60 min/week) or took medications. After determination of insulin sensitivity by a 120 mU/m2 euglycaemic–hyperinsulinaemic clamp, all participants were classified as insulin-resistant or insulin-sensitive by using the median value as a cut-off (Table 1). The protocol was approved by the Pennington Biomedical Research Center Institutional Review Board, and all participants provided written informed consent. Body fat mass was measured on a Hologic dual-energy x-ray absorptiometer in the fan beam mode (QDR 4500; Hologic, Waltham, MA, USA). Fat-free mass (FFM) was calculated as the difference between body mass and fat mass. Whole-body water content was calculated as FFM times 0.73 [10].
Experimental design
Participants were instructed to avoid any vigorous physical activity for the 2 days preceding metabolic testing. On day −1, they were admitted to our inpatient unit to eat a three-meal standardised diet (50% carbohydrate, 20% protein and 30% fat). After a 12 h overnight fast, an [2H]glucose disposal test (2H-GDT) and gas exchange measurements were performed on day 1. On day 2, participants received a weight-maintenance diet, and on day 3 a euglycaemic–hyperinsulinaemic clamp was performed after a 12 h overnight fast.
2H glucose disposal test
The 2H-GDT consists of a 75 g glucose load containing 15 g [6,6-2H2]glucose dissolved in 300 ml water. Complete glycolytic disposal of 15 g [6,6-2H2]glucose results in the release of 0.0824 mol 2H2O. Dilution in the body water pool (~2,200 mol in a 70 kg man) results in body 2H enrichment of about 0.0037 atom% excess (250 δ units). The limit for 2H detection by isotope-ratio mass spectrometry is about 1 δ. Blood samples were taken at −30, −20 and −10 min before glucose ingestion, and every 30 min for 4 h thereafter to determine serum glucose and insulin concentrations, as well as blood 2H2O enrichments.
From plasma samples, 100 μl aliquots in the cap of an inverted vial were placed in a 70°C glass-bead-filled heating block overnight. Water distillate inside the vial was then collected, and analysis was performed in triplicate. The 2H content of the samples was determined using a ThermoFinnigan High Temperature Conversion/Elemental Analyzer coupled with a ThermoFinnigan MAT 253 isotope-ratio mass spectrometer via a Conflo-III Interface (Thermo Fisher Scientific, Waltham, MA, USA). 2H isotope abundance was first calculated in 2H values relative to the International Vienna Standard Mean Ocean Water and then transformed to atom% excess. The net plasma 2H2O enrichment was calculated as the difference between 2H2O at a given time minus baseline 2H2O (mean of three samples). Then this value was converted into mmol by multiplying 2H2O enrichment by whole-body water content divided by 20 (relative molecular mass of 2H2O).
Indirect calorimetry
Gas exchange was measured while volunteers remained in a supine position, awake, and in a room at 22°C by using a Deltatrac II metabolic cart (Datex-Ohmeda, Helsinki, Finland) for 30 min before and 20 min at the end of each hour after glucose ingestion. The analysers were calibrated before each study with standardised gases containing 5% CO2 and 95% O2. A transparent plastic hood connected to the device was placed over the head of the participant. \( \dot{V}{{\text{O}}_{{2}}} \) and \( \dot{V}{\text{C}}{{\text{O}}_{{2}}} \) were calculated at 1 min intervals from continuous measurements of CO2 and O2 concentrations, and energy expenditure calculated after standardisation for temperature, pressure and moisture. Energy substrate oxidation was calculated taking into account urinary nitrogen excretion rate [11]. Metabolic rate and carbohydrate oxidation were adjusted for estimated metabolic body size (EMBS = FFM [kg] + 17.7) [12]. The resting metabolic rate response to the oral glucose was evaluated as the net incremental AUC (iAUC) [13].
Euglycaemic–hyperinsulinaemic clamp
After an overnight fast, insulin (120 mU m−2 min−1) was infused for 2 h, while a 20% glucose solution was infused to maintain glycaemia at 5 mmol/l [2]. Serum insulin concentration was measured at 10 min intervals at baseline and during steady-state from 90 to 120 min. A high insulin dose was selected in order to fully suppress hepatic glucose production and better represent skeletal muscle insulin sensitivity [14]. The glucose disposal rate, a measure of insulin sensitivity, was adjusted for EMBS (EMBS = FFM [kg] + 17.7) [12].
Blood analysis
Serum glucose concentration was measured by the glucose oxidase method on a Beckman Coulter DXC 600 Pro, and insulin was analysed using immunoassays on a Siemen’s 2000 instrument. The serum glucose and insulin responses to the oral glucose were evaluated as the net iAUC [13].
Statistical analysis
Data are presented as means±SD, except in figures in which SEs are shown. Analyses were performed using SAS version 9.2 (SAS Institute, Cary, NC, USA). Frequency of male and female participants in each group was compared by χ 2. Differences in the participants’ characteristics were assessed by t test. Other data were analysed using repeated-measures ANOVA (SAS PROC MIXED) with group, time and group × time interaction as fixed effects and individual as a random effect. The statistical significance for multiple comparisons was adjusted by the Tukey–Kramer method to control for type I errors. Stepwise multiple regression analysis was used to identify the factors that best predicted postprandial thermogenesis. A regression analysis was then derived to generate a predictive equation with postprandial thermogenesis as the dependent variable. Associations between variables were assessed by Pearson correlation. p < 0.05 was considered significant.
Results
Subject characteristics
The groups of insulin-resistant and insulin-sensitive individuals included similar proportions of males and females (p = 0.70) and had a similar age (p = 0.26) and body composition (p = 0.33) despite a higher BMI (p = 0.01) in insulin-resistant individuals. On average and by design, insulin sensitivity by clamp or by HOMA-IR was almost twofold different between the groups (Table 1).
Oral glucose tolerance
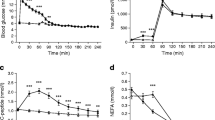
In response to glucose ingestion, serum glucose concentration reached maximal values after 60 min and returned to baseline after 150 and 180 min in insulin-sensitive and insulin-resistant participants, respectively (Fig. 1a). The integrated serum glucose response over 4 h was increased in insulin-resistant vs insulin-sensitive participants (59 ± 9 vs 50 ± 5 mmol/l × min, respectively; p = 0.002).

Serum glucose (a), serum insulin (b), whole-body resting metabolic rate (c) and whole-body carbohydrate oxidation (d) before and after a 75 g oral glucose dose in insulin-resistant (continuous line) and insulin-sensitive (dashed line) individuals. Insert: 4 h AUC for the increase in resting metabolic rate relative to fasting values after a 75 g oral glucose dose in insulin-resistant (black bar) and insulin-sensitive (white bar) individuals. *p < 0.05 and † p = 0.06 for comparison between groups. **p < 0.01 compared with its respective fasting value in each group
Serum insulin concentration reached maximal values at 60–90 min in both groups. In the insulin-sensitive group, serum insulin concentration returned to baseline at 150 min after glucose ingestion whereas it remained high for the entire period in the insulin-resistant group (Fig. 1b). As expected, the integrated serum insulin response over 4 h was larger in insulin-resistant than insulin-sensitive participants (3703 ± 1920 vs 1918 ± 763 pmol/l × min; p = 0.003).
Whole-body metabolic rate and carbohydrate oxidation
Resting metabolic rate assessed during fasting conditions was similar in insulin-resistant and insulin-sensitive participants (89 ± 9 vs 82 ± 6 kJ [kg EMBS]−1 day−1, respectively; p = 0.29). After glucose ingestion, an increase in metabolic rate was noted at 60 and 120 min (p < 0.0001; Fig. 1c), with no differences over the entire period between groups (p = 0.33–0.99; Fig. 1c). We also calculated the glucose-induced change in resting metabolic rate relative to the fasting metabolic rate (in kJ/h) at each time point using the AUC. On average, insulin-sensitive vs insulin-resistant individuals had a borderline higher postprandial thermogenesis (75 ± 28 vs 38 ± 64 kJ/h × min; p = 0.06; Fig. 1c, insert). In order to better compare the latter values, we searched for potential confounder factors such as the glucose dose relative to body size, FFM, fat mass, sex and age. Only fat mass was a significant determinant of the postprandial increase in resting metabolic rate (adjusted R 2 = 0.11; p = 0.05; slope = −2.2 ± 1.1 kJ/h × min). Based on this model, residual values between insulin-sensitive and insulin-resistant participants were not different (12 ± 30 and –14 ± 59 kJ/h × min, respectively; p = 0.15).
Regarding carbohydrate oxidation, there was no difference between groups either before or after glucose ingestion (p = 0.41; Fig. 1d). In both groups, carbohydrate oxidation increased after the glucose load and remained high for the entire period (p < 0.01; Fig. 1d).
Postprandial glycolytic disposal
Glycolytic disposal assessed by net 2H2O production measured in plasma showed a linear increase throughout the 4 h assessment period, with no differences between the two groups (p = 0.82, Fig. 2a). At the end of the assessment period, net 2H2O production was 36.5 ± 3.7 and 36.2 ± 6.4 mmol (p = 0.99) in insulin-sensitive and insulin-resistant participants, respectively. Consistently, there was no association between insulin-stimulated glucose disposal rate and 4 h net 2H2O production (r = −0.07; p = 0.72; Fig. 2b).

a Plasma net 2H2O production before and after a 75 g oral glucose dose containing 15 g [6,6-2H2]glucose in insulin-resistant (black line/circles) and insulin-sensitive (dashed line/white circles) participants. b Relationship between insulin-stimulated glucose disposal rate measured by hyperinsulinaemic clamp and 4 h net 2H2O production. c Ratio between 4 h net 2H2O production and 4 h glucose AUC in insulin-resistant (black bar) and insulin-sensitive (white bar) participants. Pearson value, −0.07; p = 0.72. **p = 0.01 for comparison between groups
In order to assess the influence of postprandial glycaemia on glycolytic glucose disposal, the ratio between 4 h net 2H2O production and 4 h glucose AUC was calculated. A higher ratio was observed in insulin-sensitive than insulin-resistant participants (p = 0.01; Fig. 2c).
Discussion
In this study we found that higher postprandial insulinaemia and glycaemia in insulin-resistant individuals did compensate for impaired insulin signalling leading to similar whole-body glycolytic and oxidative glucose disposal to that in insulin-sensitive individuals. Our findings are in contrast with the pattern noted during the non-physiological euglycaemic–hyperinsulinaemic clamp conditions, at least when oxidative glucose disposal is analysed. On the one hand, such a difference may be explained because, during a clamp, serum insulin concentration is kept constant among individuals and glucose infusion rate is adjusted to maintain euglycaemia. As a consequence, glucose utilisation will always be lower in insulin-resistant than insulin-sensitive individuals [3–6]. On the other hand, tissue-specific glucose utilisation may differ between insulin-resistant and insulin-sensitive individuals. For instance, splanchnic glucose disposal is mostly driven by hyperglycaemia [15], which will increase the proportion of glucose taken up by this tissue. Then insulin-resistant individuals, having elevated glycaemia, may have increased splanchnic glucose disposal compared with insulin-sensitive individuals.
Our finding of similar whole-body postprandial glycolytic glucose disposal in insulin-resistant and insulin-sensitive individuals was previously established by Woerle et al. in type 2 diabetic vs non-diabetic people [16]. Such concordance between study results highlights the combined role of hyperinsulinaemia and hyperglycaemia in the regulation of cellular glucose uptake. In the case of insulin resistance, hyperinsulinaemia will have a predominant role, since glycaemia is moderately increased compared with that in insulin-sensitive individuals. However, when type 2 diabetes becomes associated with moderate to low serum insulin concentration, frank hyperglycaemia will be a main driver for tissue glucose uptake. In order to take into account the effect of moderate hyperglycaemia on glycolytic glucose disposal, we divided whole-body 2H-labelled water production by serum glucose concentration. In line with our results, insulin-sensitive participants had increased glycolytic glucose disposal per unit of glycaemia compared with insulin-resistant participants.
Baseline resting metabolic rate was similar in the two groups. However, in response to glucose ingestion, insulin-resistant individuals tended to have a blunted postprandial thermogenic response (p = 0.06). Such a finding is in line with a previous report in which meal-induced thermogenesis was compared between insulin-resistant and insulin-sensitive individuals matched for age, body size and body composition [5]. However, in the present study insulin-resistant vs insulin-sensitive participants had a higher BMI and a tendency for a higher fat mass, which may have affected our comparison. Since we provided a fixed amount of glucose (75 g) to all the participants, the reduced postprandial thermogenesis in insulin-resistant individuals may be due to a lower glucose dose relative to body size. To perform a better comparison of glucose-induced thermogenesis between groups, we searched for potential confounding variables including the size of the glucose dose relative to body mass, fat mass, FFM, age and sex. Only fat mass was significantly associated with thermogenesis. When postprandial thermogenesis was adjusted for fat mass, there was no more difference between groups. The discrepancy between the study of Segal et al. [5] and the present work may be accounted for by some obvious differences, such as the larger liquid mixed meal (3,012 kJ), the use of a threefold higher insulin dose for the glucose clamp, and the difference in insulin sensitivity between groups. In contrast with the groups studied by Segal et al. [5], all the above factors may explain why our insulin-resistant participants had higher postprandial glycaemia than the insulin-sensitive participants. Alternatively, our study may well be underpowered to detect such a difference in postprandial thermogenesis, as suggested by our post hoc power analysis (not shown). Finally, our model for correcting for differences in body composition had a rather poor predictive power for postprandial thermogenesis (R 2 = 0.11).
In summary, the results of the present study indicate that insulin resistance did not affect whole-body postprandial glycolysis and glucose oxidation. These data indicate that the relative postprandial hyperinsulinaemia and hyperglycaemia overcome insulin resistance by enhancing tissue glucose uptake and intracellular glucose utilisation.
Abbreviations
- EMBS:
-
Estimated metabolic body size
- FFM:
-
Fat-free mass
- GDT:
-
Glucose disposal test
- HOMA-IR:
-
HOMA-insulin resistance
- iAUC:
-
Net incremental AUC
References
McGarry JD (1992) What if Minkowski had been ageusic? An alternative angle on diabetes. Science 258:766–770
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
Galgani JE, Heilbronn LK, Azuma K et al (2008) Metabolic flexibility in response to glucose is not impaired in people with type 2 diabetes after controlling for glucose disposal rate. Diabetes 57:841–845
Kelley DE, Goodpaster B, Wing RR, Simoneau JA (1999) Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol 277:E1130–E1141
Segal KR, Albu J, Chun A, Edano A, Legaspi B, Pi-Sunyer F (1992) Independent effects of obesity and insulin resistance on postprandial thermogenesis in men. J Clin Invest 89:824–833
Thorburn AW, Gumbiner B, Bulacan F, Wallace P, Henry RR (1990) Intracellular glucose oxidation and glycogen synthase activity are reduced in non-insulin-dependent (type II) diabetes independent of impaired glucose uptake. J Clin Invest 85:522–529
Beysen C, Murphy EJ, McLaughlin T et al (2007) Whole-body glycolysis measured by the deuterated-glucose disposal test correlates highly with insulin resistance in vivo. Diabetes Care 30:1143–1149
Galgani JE, de Jonge L, Rood JC, Smith SR, Young AA, Ravussin E (2010) Urinary C-peptide excretion: a novel alternate measure of insulin sensitivity in physiological conditions. Obesity (Silver Spring) 18:1852–1857
Katz J, Rognstad R (1966) The metabolism of tritiated glucose by rat adipose tissue. J Biol Chem 241:3600–3610
Wang Z, Deurenberg P, Wang W, Pietrobelli A, Baumgartner RN, Heymsfield S (1999) Hydration of fat-free body mass: new physiological modeling approach. Am J Physiol 276:E995–E1003
Jequier E, Acheson K, Schutz Y (1987) Assessment of energy expenditure and fuel utilization in man. Annu Rev Nutr 7:187–208
Lillioja S, Bogardus C (1988) Obesity and insulin resistance: lessons learned from the Pima Indians. Diabetes Metab Rev 4:517–540
Gannon MC, Nuttall FQ, Westphal SA, Neil BJ, Seaquist ER (1989) Effects of dose of ingested glucose on plasma metabolite and hormone responses in type II diabetic subjects. Diabetes Care 12:544–552
Campbell PJ, Mandarino LJ, Gerich JE (1988) Quantification of the relative impairment in actions of insulin on hepatic glucose production and peripheral glucose uptake in non-insulin-dependent diabetes mellitus. Metabolism 37:15–21
Ferrannini E, Bjorkman O, Reichard GA Jr et al (1985) The disposal of an oral glucose load in healthy subjects. A quantitative study. Diabetes 34:580–588
Woerle HJ, Szoke E, Meyer C et al (2006) Mechanisms for abnormal postprandial glucose metabolism in type 2 diabetes. Am J Physiol Endocrinol Metab 290:E67–E77
Funding
This work was funded by a CNRU P30 grant DK072476. J.E. Galgani was supported by a fellowship from The International Nutrition Foundation/Ellison Medical Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
JEG designed the study, analysed and interpreted the data, and drafted/revised and approved the final version of the manuscript. ER designed the study, interpreted the data, and revised and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Galgani, J.E., Ravussin, E. Postprandial whole-body glycolysis is similar in insulin-resistant and insulin-sensitive non-diabetic humans. Diabetologia 55, 737–742 (2012). https://doi.org/10.1007/s00125-011-2413-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2413-0