
Abstract
Aims/hypothesis
We assessed whether per-arnt-sim (PAS) domain-containing protein kinase (PASK) is involved in the regulation of glucagon secretion.
Methods
mRNA levels were measured in islets by quantitative PCR and in pancreatic beta cells obtained by laser capture microdissection. Glucose tolerance, plasma hormone levels and islet hormone secretion were analysed in C57BL/6 Pask homozygote knockout mice (Pask −/−) and control littermates. Alpha-TC1-9 cells, human islets or cultured E13.5 rat pancreatic epithelia were transduced with anti-Pask or control small interfering RNAs, or with adenoviruses encoding enhanced green fluorescent protein or PASK.
Results
PASK expression was significantly lower in islets from human type 2 diabetic than control participants. PASK mRNA was present in alpha and beta cells from mouse islets. In Pask−/− mice, fasted blood glucose and plasma glucagon levels were 25 ± 5% and 50 ± 8% (mean ± SE) higher, respectively, than in control mice. At inhibitory glucose concentrations (10 mmol/l), islets from Pask−/− mice secreted 2.04 ± 0.2-fold (p < 0.01) more glucagon and 2.63 ± 0.3-fold (p < 0.01) less insulin than wild-type islets. Glucose failed to inhibit glucagon secretion from PASK-depleted alpha-TC1-9 cells, whereas PASK overexpression inhibited glucagon secretion from these cells and human islets. Extracellular insulin (20 nmol/l) inhibited glucagon secretion from control and PASK-deficient alpha-TC1-9 cells. PASK-depleted alpha-TC1-9 cells and pancreatic embryonic explants displayed increased expression of the preproglucagon (Gcg) and AMP-activated protein kinase (AMPK)-alpha2 (Prkaa2) genes, implying a possible role for AMPK-alpha2 downstream of PASK in the control of glucagon gene expression and release.
Conclusions/interpretation
PASK is involved in the regulation of glucagon secretion by glucose and may be a useful target for the treatment of type 2 diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Elevated glucose concentrations (>3.5 mmol/l) normally suppress the release of glucagon from pancreatic alpha cells; dysregulation of this process is a feature of type 1 and type 2 diabetes [1, 2]. Both direct [3] and indirect [4] effects of glucose on the release of glucagon have been described. Whereas the latter appear to involve the release of secretory products, including insulin [3, 5], gamma-aminobutyric acid [6–8] or zinc ions [4, 9] from neighbouring beta cells, as well as somatostatin from delta cells [10, 11], the signalling events involved in the direct sensing of glucose are more controversial [12]. These may involve enhanced metabolism of glucose and the closure of ATP-sensitive K+ channels, followed by limited membrane depolarisation [13, 14]. AMP-activated protein kinase (AMPK) appears also to be involved in the regulation of glucagon release [15].
Per-arnt-sim (PAS) domain-containing protein kinases (PASKs) are related to AMPK and are common in prokaryotes. However, there is currently only one known mammalian counterpart [16, 17]. We [18] and others [19, 20] have shown that PASK is important for energy sensing and maintenance of normal cellular energy balance in mammalian systems.
In pancreatic beta cells, PASK activity is regulated by glucose and is involved in the regulation of glucose-induced preproinsulin and pancreatic duodenum homeobox-1 (Pdx1) gene expression [18, 20]. Expression of the Pask gene in rodent islets and beta cell lines [18, 20] is also glucose-sensitive. Recently, PASK has been implicated in regulation of lipogenic gene expression [19] and might, therefore, influence glucose signalling through lipid intermediates as proposed for glucose-induced insulin secretion [21].
Pask homozygote knockout mice (Pask−/−), which are globally inactivated for Pask, have previously been reported to display lower plasma insulin levels than control littermates [19], but normal glucose tolerance [19, 22], reflecting enhanced insulin sensitivity [19]. Insulin secretion from Pask−/− islets has variously been shown to be not different [22] or lower [19] than in control islets. It has also been previously reported that total insulin content and/or beta cell mass were not altered in Pask−/− mice [19, 22]. However, data from these earlier studies are difficult to interpret for a number of reasons. In the first instance, total islet insulin was not always measured, making it difficult to evaluate the insulin secretory capacity of Pask−/− islets. Moreover quantification of beta cell mass using pancreatic sections is prone to substantial variability [19]. Due to these limitations [19, 22], the role of PASK in the regulation of pancreatic hormone release and glucose homeostasis remains to be clarified.
A recent report [20] showed that inhibition of insulin gene expression by palmitate was reversed by Pask overexpression in MIN6 beta cells. The loss of PASK, and hence the loss of regulation by Pdx1 and Mafa gene expression [18], was proposed to be a mechanism by which insulin gene expression in the beta cell might be lost on exposure to palmitate [20]. Thus, PASK appears to exert a protective effect in mature beta cells. Moreover, aberrant PASK expression and/or function may play a significant role in the development of diabetes and, interestingly, Pask −/− mice develop glucose intolerance on a high-fat diet [18].
Although glucose homeostasis reflects the release of multiple islet hormones in addition to insulin, there are currently no published data on the role of PASK in other islet cell types. The present study demonstrates that PASK is involved in the regulation of glucagon secretion from pancreatic alpha cells. We show that PASK expression is decreased in the islets of human type 2 diabetic patients and that Pask is at least as strongly expressed in highly purified mouse alpha cells as in beta cells. Silencing or ablation of Pask in clonal alpha cells or islets, respectively, drastically blunted the inhibition of glucagon secretion by glucose. Whereas the insulin content of PASK-deficient islets was dramatically reduced, the acute regulation of insulin secretion by glucose was normal. Thus, PASK regulates hormone release reciprocally from pancreatic alpha and beta cells. Given that dysregulation of insulin and glucagon secretion are characteristics of type 2 diabetes, we propose that PASK may be a potential drug target to modulate glucagon release in vivo.
Methods
Materials
Adenoviruses encoding for human PASK have been previously described [16]. All general chemicals and tissue culture reagents were purchased from Sigma (Poole, UK) or Invitrogen (Paisley, UK), unless otherwise stated in the text.
Isolation and culture of islets
Studies on human islets were conducted with local ethics committee approval at all sites (Charing Cross Research Ethics Committee Ref. 07/H0711/114). Human islets were isolated as previously described in Oxford, UK [23], or in Pisa, Italy [24], and maintained in medium containing 11 mmol/l glucose for 10 days to allow the loss of exocrine tissue [25]. Human type 2 diabetic donors were selected according to established criteria [23, 26]. Mouse pancreatic islets were isolated and cultured as described in [3]. Alpha-TC1-9 cells (passage 35–45; American Type Culture Collection, Manassas, VA, USA) were cultured as previously described [7].
Laser capture microdissection and microarray analysis
These procedures were performed as described in Electronic supplementary material (ESM) Methods.
RNA sequencing
We prepared 300 ng of total RNA with a kit (mRNA-Seq 8; Illumina, Little Chesterford, UK). For clustering and sequencing we used Illumina cluster generation and sequencing kits v4. 9pM were loaded to the flowcell (one sample per lane) and sequenced on a sequencer (GAIIx) with RTA software version 2.6 (Little Chesterford, UK). On average we obtained 28 million reads per lane, passing Illumina’s quality filtering. Short reads (35 bp) were aligned to the mouse genome (Ensembl v54) using a mapping station (Genomatix) [27] and to a database of known and potential splice junctions using Bowtie [28]. Up to three mismatches and no insertions or deletions were allowed, and, on average, more than 85% of the reads in a sample could be mapped to the genome. Gene expression was measured as proposed by Mortazavi et al. [29] and reads per kilobase per million (RPKM) values were computed for every gene.
Mice Pask−/− mice (kindly provided by R. Wenger, Institute of Physiology and ZIHP, Zurich, Switzerland) [30] were back-crossed for ten generations with C57BL/6 mice prior to use. Mice were housed with two to five animals per cage in a pathogen-free facility on a 12 h light–dark cycle with free access to standard mouse chow diet, unless otherwise stated. All in vivo procedures were performed in the Imperial College Central Biomedical Service in accordance with the Principles of Laboratory Care and the UK Home Office (Animals Scientific Procedures Act, 1986), and approved by the local ethics committee. Genotyping was performed as previously described [30].
Intraperitoneal glucose tolerance test
An intraperitoneal glucose tolerance test was performed on 8-week-old mice as described in [31], at 09:00 hours on each experimental day.
Measurement of total pancreatic insulin and glucagon
Pancreases were excised from 8-week-old mice and suspended in ice-cold acid–ethanol (75.0% ethanol–23.4% molecular grade water–1.5% HCl–0.1% Triton X-100, vol./vol.) prior to disruption by sonication (microsonicator; Misonix, Farmingdale, NY, USA) at 4°C. Total protein was measured by Bradford’s assay [32], and insulin and glucagon content were measured by radioimmunoassay (Linco, Watford, UK).
Measurement of plasma glucagon
At 8 weeks of age mice were starved overnight prior to being killed by cervical dislocation. Blood (200 μl) was immediately removed by cardiac puncture. Plasma was collected using high-speed centrifugation (2,000 g, 5 min) in heparin-coated tubes (Microvette; Sarstedt, Leicester, UK) and plasma glucagon assessed by radioimmunoassay (Linco, Watford, UK).
Manipulation of PASK content in alpha-TC1-9 cells and cultured islets of Langerhans
Alpha-TC1-9 cells and islets were infected with adenoviruses encoding for PASK or enhanced green fluorescent protein at a multiplicity of infection of 100 and cultured for 48 h prior to use. Alpha-TC1-9 cells were transfected with anti-Pask small interfering RNA (siRNA) or control siRNA (1 nmol/l) [18]. Protein content was assessed by western (immuno-)blot analysis.
Culture of E13.5 rat pancreatic epithelia
E13.5 rat pancreatic epithelia were isolated and cultured as previously described [33]. Epithelia were cultured in the presence of anti-Pask siRNA or control siRNA [18] (1 nmol/l) for 10 days prior to RNA isolation for real-time quantitative PCR analysis.
Real-time quantitative PCR analysis
Highly purified primary mouse islet beta and alpha cells were obtained by fluorescence-activated cell sorting of transgenic mice expressing the fluorescent protein Venus selectively in the alpha cell under the preproglucagon promoter [34, 35]. RNA was extracted from alpha-TC1-9 cells and cultured rat E13.5 explants using Trizol (Invitrogen) according to the manufacturer’s guidelines. Total RNA was subjected to DNAse treatment (Ambion, Warrington, UK), followed by cDNA conversion (high-capacity cDNA conversion kit; Applied Biosystems, Warrington, UK) and real-time quantitative PCR using SYBR green (Applied Biosystems) in a 7500 Real-Time PCR System (ABI, Warrington, UK).
Western (immuno-)blot analysis
Western (immuno-)blot analysis was performed as previously described [18].
Statistical analysis
Data are the means ± SE for the number of observations indicated. Statistical significance and differences between means were assessed by Student’s t test with Bonferroni correction for multiple analyses.
Results
PASK gene expression is regulated by glucose in human pancreatic islets of Langerhans and is lowered in type 2 diabetes
We have previously shown that Pask mRNA and protein levels are increased by high glucose in rat islets and clonal MIN6 beta cells [18]. Here, we first determined whether islet PASK expression may also be regulated by glucose or by diabetes in humans. Measured in human islets of Langerhans by quantitative PCR, PASK mRNA was elevated after 24 h culture at 11 mmol/l glucose (2.7 ± 0.2-fold vs culture at 3 mmol/l glucose [means ± SE]; Fig. 1a). Measured after culture at 11 mmol/l glucose, PASK gene expression was significantly lower in islets from patients with type 2 diabetes than in control islets (Fig. 1b).

PASK expression is regulated by glucose in human islets and is lowered in type 2 diabetic islets. Real-time quantitative PCR on non-diabetic (a, b) or type 2 diabetic (T2D) (b) human islet RNA, following culture at the indicated glucose concentrations for 24 h (a) or at 11 mmol/l glucose (b); n = 3; *p < 0.05, **p < 0.01. cPask mRNA content in FACS-purified mouse alpha and beta cells; †p = 0.07
mRNA expression data obtained using whole islets may be confounded by variable degrees of contamination with other pancreatic cell types, as well as by other factors. Therefore, we sought to further support the above results by using laser capture microdissection and array analysis to quantify mRNA levels selectively in beta cell-enriched samples [26]. A clear tendency for decreased PASK expression in islets in type 2 diabetic pancreases vs controls was also observed using this approach (ESM Fig. 1). In contrast, consistent changes in levels of the AMPK-alpha1 and -alpha2 subunit (encoded by PRKAA1 and PRKAA2, respectively) were not observed with array data on samples obtained by laser capture microdissection. However, with quantitative PCR done on the same samples with enough RNA (10 vs 10), a small but statistically significant decrease in PRKAA1 expression was apparent (18%; p < 0.019), using RPL32 as an internal control.
Pask is expressed in murine pancreatic beta and alpha cells
In the mouse, Pask mRNA has previously been shown to be most abundant in testes as well as in haemopoietic tissues including thymus and spleen [30]. Levels in other tissues, including islets, are much lower [30] (G. A. Rutter and G. Sun, unpublished observations). Indeed, the expression of Pask in adult mouse islets and its induction by glucose have recently been questioned, since expression in these cells of the lacZ gene, expressed in mice null for Pask alleles under the Pask promoter, was barely detectable by beta-Gal staining [22]. By contrast, we have previously detected Pask mRNA in mouse islets and MIN6 cells [18], data recently confirmed by others [20]. Examined here in islets from mice on a mixed C57BL/6/sv129 background cultured at 11 mmol/l glucose, massive parallel sequencing (RNAseq) confirmed the presence of Pask mRNA at low but detectable levels, lying in the lower 30th percentile of all mRNAs at approximately 0.2 ± 0.3% (n = 3 female mice) of β-actin mRNA levels (ESM Fig. 2).
To determine in which cell types Pask was expressed in mouse islets, we next compared the expression of Pask mRNA in highly purified primary mouse alpha and beta cells (Fig. 1c). These were obtained by fluorescence-activated cell sorting of transgenic mice selectively expressing the fluorescent protein, Venus, in the alpha cell [34, 35] and by real-time quantitative PCR analysis. This approach confirmed the presence of Pask mRNA in both cell types and indicated that levels in purified alpha cells (1.45 ± 0.46% of beta-actin mRNA) tended to be higher than in beta cells (0.54 ± 0.044%; n = 3 preparations; p = 0.07 by two-tailed Student’s t test).
Pask−/− mice display normal glucose tolerance but impaired plasma glucagon concentration
Given the greater expression of Pask in alpha than in beta cells and the dysregulation of glucagon secretion observed in type 2 diabetes, as reviewed by others [2], we next analysed the potential contribution of this enzyme to the regulation of glucagon secretion using Pask −/− mice. At 8 weeks of age, Pask −/− male (Fig. 2a, c, e) and female (Fig. 2b, d) mice displayed 25 ± 5% higher plasma glucose levels after 16 h of fasting than wild-type littermate control mice, but normal glucose tolerance after intraperitoneal injection of the sugar (Fig. 2a–d). Measured after fasting, plasma glucagon was significantly higher in Pask −/− male mice than in littermate controls (91 ± 5.4 vs 58 ± 3 pg/ml; Fig. 2e).

Pask−/− mice display higher fasted glucose levels than littermate controls. At 8 weeks of age, male (a, c) and female (b, d) Pask−/− mice were starved for 16 h prior to an intraperitoneal glucose tolerance test (1 g glucose per kg body weight). c, d Blood glucose at 0 min, as indicated by dotted squares (a, b), for male and female mice, respectively. e Plasma glucagon concentrations from male mice following 16 h of fasting. n = 8–10 mice per group; *p < 0.05, **p < 0.01. Black squares and solid lines, Pask+/+, wild-type; rhombuses and dotted lines, Pask+/−, heterozygote; black triangles and dashed lines, Pask−/−, homozygous knockout
Islets of Langerhans from Pask−/− mice display impaired glucose-regulated glucagon secretion
We next examined whether the elevated plasma glucagon concentration was due to dysregulation of alpha cell function. Supporting this view, the inhibition of glucagon secretion in response to elevated glucose was impaired in islets from Pask −/− mice (Fig. 3a). Thus, Pask −/− islets, in which Pask gene expression was undetectable (Fig. 3e), secreted 2.04 ± 0.2-fold more glucagon at inhibitory (10 mmol/l) glucose concentrations than did islets from wild-type littermates (Fig. 3a), whereas release at stimulatory (0.5 mmol/l) glucose was unaltered. Interestingly, a slight inhibitory effect on glucagon secretion in Pask −/− islets was still observed at 10 mmol/l glucose (Fig. 3a). Although glucose-stimulated insulin release when normalised to total islet insulin content was not affected by Pask deletion (Fig. 3b), the total amount of insulin per islet was lower (0.38 ± 0.3-fold) in Pask −/− than in wild-type islets (Fig. 3d), i.e. the amount of insulin release was compromised in Pask −/− islets, in agreement with published data [19].

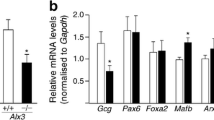
Islets from Pask −/− mice display abnormal inhibition of glucagon secretion in response to glucose. a Glucagon and (b) insulin secretion in response to glucose was assessed in groups of ten mouse islets, as previously described [7]. Data presented ar e% secreted hormone/total hormone content in 10 islets. White bars, islets exposed to 0.5 mmol/l glucose; black bars, islets exposed to 10 mmol/l glucose. c Total glucagon and (d) insulin from mouse islets of each genotype. n = 3 separate preparations of islets. e Total RNA was extracted from groups of 100 islets using Tri-reagent (Invitrogen) and gene expression analysed by real-time quantitative PCR analysis using SYBR-green (Applied Biosystems). Cyclophilin gene expression was used as the internal calibrator. n = 3 Nd, not detected; black bars, Pask +/+ wild-type islets; grey bars, Pask +/− heterozygote islets; white bars, Pask −/− homozygous knockout mice islets. *p < 0.05, **p < 0.01
The expression of mouse preproinsulin 2 (Ins2) and Pdx1 genes was strongly impaired in Pask −/− mouse islets (Fig. 3e), as previously reported [18, 20]. Moreover, the insulin content of whole Pask −/− mouse pancreases was 1.98 ± 0.3-fold lower than that of wild-type pancreases (67 ± 15 vs 133 ± 12 ng/mg protein for Pask −/− and wild-type mice, respectively; n = 3 mice for each genotype). While glucagon protein content in Pask −/− islets (Fig. 3c) was unaffected, preproglucagon (Gcg) gene expression was slightly but significantly increased, as was Prkaa2 mRNA expression (Fig. 3e).
Glucagon release is activated in the absence of Pask in alpha-TC1-9 cells
Since the above experiments using intact islets did not allow ready discrimination between an action of PASK cell-autonomously in the alpha cell and an effect mediated by changes in the release of beta cell-derived factors, we next explored the role of the enzyme in the clonal alpha-TC1-9 cell line [3]. Culture of alpha-TC1-9 cells in the presence of an siRNA against Pask led to near complete ablation of Pask gene expression and protein content (Fig. 4a). Glucagon release from Pask-deficient alpha-TC1-9 cells at inhibitory (10 mmol/l) glucose concentrations was comparable to that observed in control cells at stimulatory (0.5 mmol/l) glucose concentrations (Fig. 4b). By contrast, addition of extracellular insulin (20 nmol/l) led to inhibition of glucagon secretion in control and Pask-silenced alpha-TC1-9 cells, indicating (1) the presence of distinct glucose and insulin signalling pathways in the alpha cell and (2) that the insulin signalling pathway in Pask-silenced alpha-TC1-9 cells was intact (Fig. 4b). Total glucagon protein content, as assessed by radioimmunoassay, was not different between alpha-TC1-9 cells treated with control or anti-Pask siRNA (data not shown).

PASK silencing activates glucagon secretion in alpha-TC1-9 cells. a Transfection of alpha-TC1-9 cells with 1 nmol/l Pask siRNA led to decreased Pask expression and PASK protein content, as assessed by real-time quantitative PCR and western (immuno-)blot analysis. The migration of molecular weight markers (BioRad, Hemel Hempstead, UK) is indicated on the western (immuno-)blot. b Glucagon secretion in response to elevated glucose and insulin was assessed as described in Fig. 3, n = 3. Data presented are % secreted hormone/total hormone content per sample. White bars, transfected with control; grey bars, transfected with anti-Pask siRNA. *p < 0.05, **p < 0.01
Glucagon release is inhibited by PASK overexpression in alpha-TC1-9 cells and human islets of Langerhans
The above findings indicated that PASK may be an inhibitor of glucagon release from alpha cells. To test this hypothesis, we next explored the impact of forced activation of the enzyme in these cells. Adenovirus-mediated overexpression of PASK in alpha-TC1-9 cells (Fig. 5a) led to inhibition of glucagon secretion at normally stimulatory (1 mmol/l) glucose concentrations (0.36 ± 0.1-fold vs control).

Overexpression of PASK inhibits glucagon secretion in alpha-TC1-9 cells and human islets of Langerhans. a Alpha-TC1-9 cells were infected with adenovirus encoding for PASK (grey bars) or enhanced green fluorescent protein (EGFP; control virus, white bars) at multiplicity of infection of 100 and cultured for 24 h prior to glucagon secretion as described in Fig. 3, n = 3. b Human islets of Langerhans were infected with adenovirus for human PASK or EGFP at multiplicity of infection of 100 and cultured for 48 h prior to glucagon secretion assay [3], n = 3. Data presented are % secreted hormone/total hormone content per sample. c Western (immuno-)blot analysis of protein extracts from alpha-TC1-9 cells and human pancreatic islets after 48 h transduction with adenoviruses encoding for PASK or EGFP. PASK protein content was normalised to tubulin protein content as assessed by densitometry using ImageJ (http://rsbweb.nih.gov/ij/index.html; n = 3). Representative images from at least three separate preparations of each are shown. *p < 0.05, **p < 0.01
Similarly, overexpression of PASK in human islets of Langerhans (Fig. 5b) inhibited glucagon secretion at all glucose concentrations tested, while the stimulatory effects of KCl were largely maintained. Interestingly, there was still an apparent effect of glucose on glucagon secretion in human islets overexpressing PASK (Fig. 5b), consistent with a maintained effect on glucagon release mediated by beta cell-derived factors.
Silencing of Pask gene expression in alpha-TC1-9 cells and E13.5 rat pancreatic epithelial explants causes increased AMPK-alpha2 and glucagon gene expression
In an effort to identify the mechanism(s) through which PASK may regulate glucagon secretion, we measured the expression of a number of potential target genes. Pask gene expression was inhibited in alpha-TC1-9 cells treated with a siRNA against Pask (Fig. 6). In line with the above findings in Pask −/− mouse islets (Fig. 3e), preproglucagon (Gcg) gene expression was increased substantially by Pask ablation in alpha-TC1-9 cells (7.4 ± 1.3-fold vs control; Fig. 6a), although total glucagon protein content was unaltered (Fig. 2c), consistent with the relatively slow turnover of mature glucagon. Prkaa2 (Fig. 6a), but not Prkaa1 (data not shown) gene expression was also increased (4.3 ± 1.5-fold vs control) by Pask silencing.

Pask silencing in alpha-TC1-9 cells leads to increased expression of Gcg and Prkaa2. Real-time quantitative PCR analysis, using SYBR green (Applied Biosystems), of RNA from alpha-TC1-9 cells treated with control or Pask siRNA, n = 3. Cyclophilin gene expression was used as the internal calibrator for real-time quantitative PCR. White bars, treated with control; grey bars, treated with anti-Pask siRNA. **p < 0.01
To determine whether the dysregulation of glucagon gene expression and release in Pask −/− mice was due to a role of the enzyme in the development of pancreatic alpha and beta cells, we next examined the impact of Pask silencing in developing pancreatic epithelia. E13.5 rat pancreatic epithelial explants [33], in which Pask gene expression was silenced with an anti-Pask siRNA, developed endocrine buds to an extent indistinguishable from that observed in explants treated with a control (scrambled) siRNA (ESM Fig. 3). However, explants in which Pask was silenced displayed similar increases in Gcg and Prkaa2 gene expression (ESM Fig. 3) to those observed in alpha-TC1-9 cells (Fig. 6). Explants overexpressing Pask did not survive the 10 day culture period.
Discussion
The present data indicate that PASK/Pask is expressed in human and mouse pancreatic islets, respectively, and regulated by glucose. Moreover, in the mouse, Pask mRNA levels are at least as high in alpha cells as in beta cells. Correspondingly, we provide evidence that changes in Pask gene expression within the islet regulate both insulin content [18, 20] and glucagon release, i.e. that PASK mediates a ‘high glucose’ signal in alpha and beta cells. In the alpha cell, this might be achieved either via a cell-autonomous action on cellular activity and/or through changes in the release of beta cell-derived factors (Fig. 7). Either of these mechanisms are likely to modulate intracellular signalling within the alpha cell through changes in membrane excitability, levels of unbound Ca2+, cAMP, etc.

Proposed mechanism for PASK action in alpha cells. Glucagon secretion is regulated by changes in extracellular glucose and other factors, including insulin and possibly other molecules released from pancreatic beta cells. We propose (dashed lines) that PASK may be involved in cell-autonomous regulation of glucagon secretion, possibly through the regulation of Prkaa2 gene expression (AMPK-alpha2) (1) and/or an as yet unidentified pathway (2). PASK may also regulate glucagon secretion through its action on beta cell function and/or beta cell mass (3)
PASK is expressed in pancreatic islets
An important finding of the present study is that Pask mRNA is clearly present in mouse islets maintained at permissive glucose concentrations (11 mmol/l), in contrast to a recent suggestion [22]. These results and those from another recent publication [20] support our own previous findings [18]. We would stress, however, that the level of gene expression, at least at the mRNA level, was relatively low compared with tissues in which Pask is strongly expressed (notably in the testes) [22], an observation that probably underlies the failure to detect significant expression using alternative approaches [22].
PASK regulates insulin and glucagon secretion
It has previously been shown that islet architecture and beta cell mass in Pask −/− mice are intact [19, 22]. Thus, it was hypothesised in earlier studies that the observed decrease in insulin release may have been due to defective glucose-signalling in the beta cell [19]. In contrast, our data show that glucose-regulated insulin secretion was intact in Pask −/− islets (Fig. 3b), but that the insulin content of the islet (Fig. 3d) and pancreas (see Results section) were lowered by Pask deletion. This apparent discrepancy between the present and earlier [19, 22] studies may reflect differences in the genetic background, age and sex of the mice used. We would, however, note that Pask −/− beta cell mass was previously assessed by immunohistochemical analysis of fixed pancreatic sections, which may be prone to error.
Nevertheless, it was previously reported that plasma insulin content was compromised in Pask −/− mice [19], a result consistent with the present findings. In our hands, glucagon release from Pask −/− mouse islets (Fig. 3a) and human islets in which PASK was overexpressed (Fig. 5b) still displayed some degree of glucose-responsiveness, even though this was markedly decreased compared with the control condition. We hypothesise that this ‘residual’ regulation of release may be due, at least in part, to the release of insulin or other factors (Zn2+, gamma-aminobutyric acid, etc.) from neighbouring pancreatic beta cells and is consistent with the maintained effect of exogenous insulin on glucagon release from alpha-TC1-9 cells (Fig. 4b).
Close inspection of the data of Fig. 3a vs Fig. 4 reveals that whereas loss of Pask enhanced glucagon secretion from islets only at high (inhibitory) glucose levels (Fig. 3a), stimulation of glucagon release was apparent at all glucose concentrations examined in alpha-TC1-9 cells (Fig. 4). The mechanisms responsible for this difference are unclear. However, we note that while insulin mRNA was present at low but detectable levels in control alpha-TC-1-9 cells (Ct values ≥ 29; data not shown), silencing of Pask led to decreased insulin gene expression such that this was below the limit of detection. Hence an action of Pask silencing on endogenous insulin secretion from alpha-TC-1-9 cells, and thus loss of tonic inhibition of glucagon secretion by this hormone, may contribute to the above mechanisms. Further studies are needed to resolve this question.
Interestingly, the increase in plasma glucagon levels measured after 16 h fasting in 8-week-old Pask −/− mice was similar to that observed in alpha cell-specific insulin receptor knockout mice of the same age [5]. We have previously shown that PASK activity is not regulated by insulin, but that PASK may regulate insulin gene expression in pancreatic beta cells [18]. Thus, we propose that the regulation of insulin content by Pask is one of the mechanisms through which PASK may indirectly regulate glucagon release.
A role for AMPK alpha2 downstream of PASK?
Our gene-expression studies revealed that expression of the catalytic subunit of another glucose-responsive fuel gauge, Prkaa2 (encoding AMPK-alpha2), is upregulated when Pask gene expression is compromised (Fig. 6). This is an interesting observation, since AMPK activity has been shown to be glucose-responsive in pancreatic beta cells [36] and to regulate insulin release [37, 38]; in addition, we have recently shown that activation of AMPK in the alpha cell stimulates glucagon secretion [15]. However, as also observed here, Prkaa2 mRNA levels are normally much lower (more than tenfold) than Prkaa1 in purified mouse beta and alpha cells [31], suggesting that the inappropriate activation of this isoform and an increase in AMPK activity in the nucleus (from which AMPK alpha1 is excluded in mature pancreatic endocrine cells) [36] may impact on the transcription of key genes, including Gcg, to reprogram the alpha cell. Interestingly, induction of Prkaa2 expression was also observed in E13.5 pancreatic epithelia [33] in which Pask was silenced (Fig. 6c), indicating that defects during pancreatic development may contribute to the dysregulation of glucagon release in Pask −/− mice.
PASK may have a role in the pathophysiology of type 2 diabetes
The observation that pancreatic islets from patients with type 2 diabetes display lower PASK mRNA levels than islets from non-diabetic individuals (Fig. 1b) suggests that lowered PASK activity may contribute to decreased insulin release and to enhanced glucagon secretion in this condition [1]. PASK may therefore represent a potential therapeutic target, the activation of which might favourably affect the secretion of both hormones in patients with type 2 diabetes.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- PAS:
-
Per-arnt-sim
- PASK:
-
PAS domain-containing protein kinase
- Pask −/− :
-
Pask homozygote knockout mice
- siRNA:
-
Small interfering RNA
References
Unger RH (1985) Glucagon physiology and pathophysiology in the light of new advances. Diabetologia 28:574–578
Gromada J, Franklin I, Wollheim CB (2007) Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 28:84–116
Ravier MA, Rutter GA (2005) Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes 54:1789–1797
Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB (2003) Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol 5:330–335
Kawamori D, Kurpad AJ, Hu J et al (2009) Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 9:350–361
Rorsman P, Berggren PO, Bokvist K et al (1989) Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 341:233–236
Bailey SJ, Ravier MA, Rutter GA (2007) Glucose-dependent regulation of gamma-aminobutyric acid (GABA A) receptor expression in mouse pancreatic islet alpha-cells. Diabetes 56:320–327
Gilon P, Bertrand G, Loubatieres-Mariani MM, Remacle C, Henquin JC (1991) The influence of gamma-aminobutyric acid on hormone release by the mouse and rat endocrine pancreas. Endocrinology 129:2521–2529
Zhou H, Zhang T, Harmon JS, Bryan J, Robertson RP (2007) Zinc, not insulin, regulates the rat alpha-cell response to hypoglycemia in vivo. Diabetes 56:1107–1112
Gromada J, Hoy M, Buschard K, Salehi A, Rorsman P (2001) Somatostatin inhibits exocytosis in rat pancreatic alpha-cells by G(i2)-dependent activation of calcineurin and depriming of secretory granules. J Physiol 535:519–532
Hauge-Evans AC, King AJ, Carmignac D et al (2009) Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 58:403–411
Rutter GA (2009) Regulating glucagon secretion: somatostatin in the spotlight. Diabetes 58:299–301
Gopel SO, Kanno T, Barg S, Weng XG, Gromada J, Rorsman P (2000) Regulation of glucagon release in mouse -cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J Physiol 528:509–520
MacDonald PE, de Marinis YZ, Ramracheya R et al (2007) A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol 5:e143
Leclerc I, Sun G, Morris C, Fernandez-Milan E, Nyirenda M, Rutter GA (2011) AMP-activated protein kinase regulates glucagon secretion from mouse pancreatic alpha cells. Diabetologia 54:125–134
Rutter J, Michnoff CH, Harper SM, Gardner KH, McKnight SL (2001) PAS kinase: an evolutionarily conserved PAS domain-regulated serine/threonine kinase. Proc Natl Acad Sci USA 98:8991–8996
Hofer T, Spielmann P, Stengel P et al (2001) Mammalian PASKIN, a PAS-serine/threonine kinase related to bacterial oxygen sensors. Biochem Biophys Res Commun 288:757–764
da Silva XG, Rutter J, Rutter GA (2004) Involvement of Per-Arnt-Sim (PAS) kinase in the stimulation of preproinsulin and pancreatic duodenum homeobox 1 gene expression by glucose. Proc Natl Acad Sci USA 101:8319–8324
Hao HX, Cardon CM, Swiatek W et al (2007) PAS kinase is required for normal cellular energy balance. Proc Natl Acad Sci USA 104:15466–15471
Fontes G, Semache M, Hagman DK et al (2009) Involvement of Per-Arnt-Sim kinase and extracellular-regulated kinases-1/2 in palmitate inhibition of insulin gene expression in pancreatic beta-cells. Diabetes 58:2048–2058
Corkey BE, Deeney JT, Yaney GC, Tornheim K, Prentki M (2000) The role of long-chain fatty acyl-CoA esters in beta-cell signal transduction. J Nutr 130:299S–304S
Borter E, Niessen M, Zuellig R et al (2007) Glucose-stimulated insulin production in mice deficient for the PAS kinase PASKIN. Diabetes 56:113–117
Cross SE, Hughes SJ, Partridge CJ, Clark A, Gray DW, Johnson PR (2008) Collagenase penetrates human pancreatic islets following standard intraductal administration. Transplantation 86:907–911
Marchetti P, Bugliani M, Lupi R et al (2007) The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 50:2486–2494
Leclerc I, Woltersdorf WW, da Silva XG et al (2004) Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: impact on glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 286:E1023–E1031
Marselli L, Thorne J, Dahiya S et al (2010) Gene expression profiles of beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS ONE 5:e11499
Sultan M, Schulz MH, Richard H et al (2008) A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321:956–960
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Katschinski DM, Marti HH, Wagner KF et al (2003) Targeted disruption of the mouse PAS domain serine/threonine kinase PASKIN. Mol Cell Biol 23:6780–6789
Sun G, Tarasov AI, McGinty J et al (2010) Ablation of AMP-activated protein kinase alpha1 and alpha2 from mouse pancreatic beta cells and RIP2.Cre neurons suppresses insulin release in vivo. Diabetologia 53:924–936
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Miralles F, Serup P, Cluzeaud F, Vandewalle A, Czernichow P, Scharfmann R (1999) Characterization of beta cells developed in vitro from rat embryonic pancreatic epithelium. Dev Dyn 214:116–126
Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM (2008) Glucose sensing in L cells: a primary cell study. Cell Metab 8:532–539
Nicolson TJ, Bellomo EA, Wijesekara N et al (2009) Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 58:2070–2083
da Silva Xavier G, Leclerc I, Salt IP et al (2000) Role of AMP-activated protein kinase in the regulation by glucose of islet beta cell gene expression. Proc Natl Acad Sci USA 97:4023–4028
da Silva XG, Leclerc I, Varadi A, Tsuboi T, Moule SK, Rutter GA (2003) Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem J 371:761–774
Tsuboi T, da Silva XG, Leclerc I, Rutter GA (2003) 5′-AMP-activated protein kinase controls insulin-containing secretory vesicle dynamics. J Biol Chem 278:52042–52051
Acknowledgements
The authors thank S. Vakhshouri for technical assistance, and F. Semplici and M. K. Loder for scientific input during manuscript preparation. This work was supported by a Juvenile Diabetes Research Foundation post-doctoral fellowship and an European Foundation for the Study of Diabetes Albert Renold travelling fellowship to G. da Silva Xavier, and by grants to G. A. Rutter from the Wellcome Trust (081958/2/07/2) and the Medical Research Council (G0401641). H. Farhan, H. Kim and S. Caxaria were supported by Imperial College London BSc and MSc project funds. The Diabetes Research and Wellness Foundation supported P. Johnson and S. Hughes.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
H. Farhan and H. Kim contributed equally to this study.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM 1
(PDF 37.8 kb)
ESM Fig. 1
(PDF 11.0 kb)
ESM Fig. 2
(PDF 20.9 kb)
ESM Fig. 3
(PDF 48.5 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
da Silva Xavier, G., Farhan, H., Kim, H. et al. Per-arnt-sim (PAS) domain-containing protein kinase is downregulated in human islets in type 2 diabetes and regulates glucagon secretion. Diabetologia 54, 819–827 (2011). https://doi.org/10.1007/s00125-010-2010-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-2010-7