
Abstract
It is now widely accepted that reactive oxygen species (ROS) contribute to cell and tissue dysfunction and damage in diabetes. The source of ROS in the insulin secreting pancreatic beta cells has traditionally been considered to be the mitochondrial electron transport chain. While this source is undoubtedly important, we fully describe in this article recent information and evidence of NADPH oxidase-dependent generation of ROS in pancreatic beta cells and identify the various isoforms that contribute to O •−2 and H2O2 production in various conditions. While glucose-stimulated ROS generation may be important for acute regulation of insulin secretion, at higher levels ROS may disrupt mitochondrial energy metabolism. However, ROS may alter other cellular processes such as signal transduction, ion fluxes and/or cell proliferation/death. The various beta cell isoforms of NADPH oxidase (described in this review) may, via differences in the kinetics and species of ROS generated, positively and negatively regulate insulin secretion and cell survival.
Similar content being viewed by others


Regulation of insulin secretion
The combination of glucose transport and phosphorylation at the glucokinase step determines a major part of metabolic flux through glycolysis in the beta cell. Glucose may be further metabolised by pyruvate dehydrogenase and pyruvate carboxylase, resulting in enhanced tricarboxylic acid cycle activity and subsequent ATP generation [1]. This results in closure of the ATP-sensitive K+ channels, decreasing the hyperpolarising outward K+ flux, depolarising the plasma membrane and opening voltage-gated Ca2+ channels to increase the influx of extracellular Ca2+, which is a primary driver of the mechanism of glucose-stimulated insulin secretion (GSIS). Glucose metabolism also results in formation of metabolic stimulus–secretion coupling factors [2–8]. Amino acids may potentiate the generation of metabolic stimulus–secretion coupling factors [1]. The glucose-dependent increase and oscillations in intracellular Ca2+ concentration associated with the mechanism of GSIS can stimulate mitochondrial generation of reactive oxygen species (ROS), whereas Ca2+, via protein kinase C activation, may enhance NADPH oxidase (NOX)-dependent generation of ROS [9–11]. The subsequent formation of H2O2, unless rapidly removed, can suppress beta cell metabolic activity resulting in inhibition of insulin secretion (Fig. 1). Rodent pancreatic beta cells have particularly low levels of H2O2-detoxifying and redox-regulating enzymes, such as catalase, glutathione peroxidase, glutathione reductase and thioredoxin, although this is less pronounced in human islets. The superoxide radical inactivating superoxide dismutase (SOD) isozymes are produced at greater levels resulting in an imbalance between H2O2-generating and -inactivating enzymes [12].

The negative impact of excessive ROS generation on insulin secretion. An imbalance of ROS formation and removal may lead to an excessive concentration of ROS. This can result in inhibition of glycolysis and tricarboxylic acid cycle, reducing stimulus–secretion coupling factor generation (ATP, citrate, glutamate) and resulting in K +ATP channel opening, membrane hyperpolarisation and impaired insulin secretion
The consequence of limited scavenging systems is that upon Ca2+ stimulation of mitochondrial and NOX systems, H2O2 concentrations in beta cells may increase rapidly and so easily reach damaging levels. The beta cell must thus strictly limit ROS production to ensure that appropriate levels are maintained, which are essential to and supportive of stimulation of insulin production and release, but not harmful to the cells.
The NADPH oxidase complex
To better explain the role of some specific isoforms in the beta cell, we will now describe the structure and organisation of the NOX family. The NOX family is a group of plasma or sub-cellular membrane-associated superoxide (O •−2 )-generating enzymes found in a wide variety of eukaryotic cells. These enzymes are members of a superfamily of flavocytochromes, which transport electrons through biological membranes and catalyse the cytosolic NADPH-dependent reduction of molecular oxygen to O •−2 [13]. The NOX family members are related through their common ability to produce O •−2 but differ in their membrane and cytosolic protein components.
The phagocyte NOX is a multicomponent enzyme complex, which plays an essential role in host defence. It is composed of two membrane components, three cytosolic proteins and a small GTPase ras-related C3 botulinum toxin substrate (Rac)2 [14]. The membrane components are the catalytic gp91phox (alternatively termed NOX2) and the regulatory p22phox. The catalytic subunit of the respiratory burst oxidase is located in phagocytic cell intracellular phagosome membrane and in the plasma membrane [15, 16]. The membrane component gp91phox has six transmembrane domains and is unstable in the absence of p22phox. The two components gp91phox and p22phox stabilise one another in a tightly associated heterodimer complex, referred to as flavocytochrome b 558 [17]. Activation of NOX2 requires translocation of cytosolic factors to the gp91phox/p22phox complex [18]. The cytosolic components are the proteins p40phox, p47phox and p67phox. Translocation of cytosolic regulatory factors to the membrane is required for enzyme activation and O •−2 production. When the cell is exposed to stimuli such as various growth factors, cytokines, hormones or bacterial components (such as fMetLeuPhe), the subsequent activation of various isoforms of protein kinase C leads to phosphorylation of the cytosolic component p47phox, which, along with p67phox and p40phox, interacts with the flavocytochrome b 558 at the membrane. Concomitantly, Rac dissociates from a RhoGDP dissociation inhibitor and is activated by exchange of GDP for GTP. Rac regulates the activity of the oxidase by a two-step process, directly via contact with gp91phox and by interaction with p67phox [19, 20]. The electron transfer from NADPH to flavin adenine dinucleotide (FAD) and then to molecular oxygen is regulated by the activation domain of p67phox [21, 22]. NOX2 production has been reported in non-phagocytic cells [18, 23].
The homologues of the phagocytic NOX are now known as the NOX family, which comprises seven members: NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1 and DUOX2 [14]. According to structural and functional characteristics, the animal NOX family enzymes can be classified into three groups: (1) the NOX1 to NOX4 group, characterised by the requirement for interaction with p22phox; (2) the NOX5 group, characterised by two calcium-binding EF-hand motifs in the cytosolic N-terminal region; and (3) the DUOX group (DUOX1 and DUOX2), characterised by EF-hand calcium-binding domain and NH2-terminal peroxidase domain [24, 25].
Recently, novel cytoplasmic members of the NOX family enzymes with significant homology to p47phox and p67phox were identified, including an isoform of p47phox, designated NOX organiser 1 (NOXO1) [26, 27], and a homologue of p67phox termed NOX activator 1 (NOXA1) [28].
p40phox was originally detected by co-immunoprecipitation with p47phox and p67phox, and is a non-glycosylated cytosolic protein with an apparent molecular mass of 40 kDa. p40phox has been shown to interact with p47phox and p67phox with a 1:1:1 stoichiometry [29].
NOX1 was the first homologue of gp91phox (NOX2) to be described [30]. The small GTPase Rac may be involved in the regulation of NOX1 activity [31, 32]. NOX4 is a gp91phox homologue, which exhibits about 39% homology to NOX2 [33]. A particular feature of NOX4 is the high level of constitutive production of hydrogen peroxide, which is partially dependent on the presence of p22phox [34, 35]. Unlike NOX1, 2 and 3, NOX4 is a constitutively active enzyme and is activated without the need for the GTPase Rac or the cytosolic components [35], and regulated by protein expression level [36]. Recent studies have demonstrated the participation of NOX4 in the regulation of cell differentiation such as in human and mouse pre-adipocytes [37–39], thus suggesting a role for H2O2 production in adipocyte differentiation and maturation. While NOX4 expression has been reported in pancreatic islets [18], the role of NOX4 in beta cells has yet to be determined.
NADPH oxidase isoforms and components in pancreatic islets
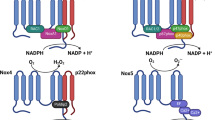
Pancreatic islets produce a phagocyte-like NOX complex (the NOX2 isoform). Expression of gp91 phox (also known as Cybb), p22 phox (also known as Cyba) and p47 phox (also known as Ncf1) mRNA by using RT-PCR and of p47phox and p67phox proteins via western blotting was demonstrated in isolated rat pancreatic islets [23]. Immunohistochemical techniques have demonstrated that p47phox is produced in the central core of pancreatic islets, an area mainly populated by beta cells [23]. Isolated rat islets constitutively express mRNAs for NOX1 and NOX2 isoforms (as membrane-associated components) and NOXO1 (homologue of p47phox), NOXA1 (homologue of p67phox) and p40phox as cytosolic components [28]. The expression and organisation of the various components of NOX1/NOX4 and NOX2 in pancreatic beta cells are described in Fig. 2. Remarkably the level of glucose-stimulated H2O2 production in beta cells was shown to reach a similar magnitude, although with slower kinetics, compared with rat neutrophils (a cell type recognised for its ability to generate large amounts of ROS) [23]. While rat neutrophils generated approximately 50% of their maximal H2O2 response within 5 min exposure to a 5.6 mmol/l glucose stimulus, rat islets required 45 min to reach 50% of their maximal H2O2 response [23]. However, both cell types exhibited a similar maximum level of ROS generation in response to glucose. While we appreciate that the kinetics of O •−2 and H2O2 generation are substantially different in these cell types, this probably reflects the different function of ROS generation, i.e. bacterial killing in the neutrophil vs signalling, control of ion fluxes and energy metabolism in the pancreatic beta cell.

NADPH oxidase isoforms in pancreatic beta cells. The prototypical NOX, NOX2, is an enzyme system that catalyses the NADPH-dependent reduction of oxygen to O •−2 . In non-stimulated beta cells, this enzyme system is inactive and its components are dispersed between the cytosol and the membranes. The flavocytochrome b558 component, which is composed of two subunits, gp91phox (which defines the NOX isoform) and p22phox, is located in the plasma membrane and in specific granules in phagocytic cells. The other components of the NADPH complex (p47phox, p67phox, p40phox and small G-protein Rac 1/2) are cytosolic proteins. The activation of beta cells by nutrients or other factors triggers phosphorylation of the p47phox, p67phox and p40phox cytosolic components, resulting in their translocation to the plasma membrane, where they interact with gp91phox and p22phox. In addition, Rac2 dissociates from its inhibitor, which allows it to interact with flavocytochrome b558 to form a binding partner for p67phox. A family of NADPH-dependent oxidases, the NOX family, is known to be widely distributed in various cells and tissues. Its members share homology with the well defined gp91phox/NOX2 catalytic subunit of phagocyte cell NOX. To date, two of these homologues, NOX1 and NOX2, have been identified and characterised in pancreatic beta cells. NOXO1 and NOXA1, which may interact with NOX1, are homologues of p47phox and p67phox respectively. FAD, Flavin adenine dinucleotide; Rap, a small molecular weight GTPase of the Ras family
We have recently shown that in rat pancreatic islets p47phox levels were acutely increased by high glucose, palmitic acid and the pro-inflammatory cytokine IL-1β, but chronically reduced in the presence of saturated fatty acid or IL-1β. Reduction of p47phox abundance in rat islets after 24 h exposure to sub-lethal levels of palmitic acid or IL-1β may reflect activation of a cellular protective mechanism that would reduce levels of apoptosis in vivo [11]. As discussed above, NOX is quantitatively an important source of ROS, either O •−2 or H2O2, in pancreatic islets. Using a hydroethidine assay, ROS production was demonstrated to significantly and dose dependently increase with respect to glucose or palmitic acid concentration, or IL-1β addition [11]. Hydroethidine operates effectively as a probe for measurement of O •−2 . The dye can freely enter a cell and is dehydrogenated to ethidium bromide. A major advantage of this probe is its ability to distinguish between the two major ROS species, O •−2 and H2O2. The normal function of ROS generation in the beta cell is related to metabolism, ion flux and transcriptional control of insulin secretion. While undoubtedly an oversimplification, it may generally be stated that ROS are acutely positively correlated with GSIS but chronically negatively correlated with insulin secretion [11, 40, 41]. Indeed specific pharmacological NOX enzyme inhibition decreased H2O2 production and impaired static and dynamic insulin secretion and glucose oxidation [42]. NOX inhibition also inhibited glucose-induced [Ca2+]i fluxes [42]. Glucose metabolism may stimulate phospholipase C (PLC) activation [43] resulting in: (1) diacylglycerol and inositol triphosphate (IP3) formation; and (2) subsequent Ca2+ release from intracellular stores, which together with diacylglycerol, can lead to ‘classic’ protein kinase C isoform activation, phosphorylation and activation of p47phox, and translocation of p47phox to the plasma membrane, followed by activation of the complete NOX complex. The rapid generation of O •−2 and H2O2 may mediate a further increase in cytosolic Ca2+ [41] so elevating glucose metabolism via activation of specific Ca2+-sensitive metabolic enzymes (especially those located in the mitochondria), which results in generation of mitochondrial-derived metabolic stimulus–secretion coupling factors (such as glutamate, citrate, NADPH and mitochondrially derived ROS) and promotion of insulin secretion (Fig. 3). Results from the laboratories of the authors of this review have demonstrated that glucose induces an acute increase of Cu/Zn SOD activity, which increases H2O2 generation in pancreatic islets (D. Morgan, E. Rebelato and A. Carpinelli, unpublished data), and that SOD activity is closely correlated with the rate of insulin secretion (r = 0.96) [44]. While we have made a strong case for the involvement of H2O2 in the promotion of insulin secretion, this is not a fully established fact with respect to all species studied, but does appear to be the case for mouse, rat and human beta cells [40, 41, 45] (D. Morgan, E. Rebelato and A. Carpinelli, unpublished data).

The positive role of NOX in insulin secretion. Glucose metabolism may stimulate phospholipase C (PLC) activation [43], which results in phosphatidyl-inositol 4,5 bisphosphate (PIP2) hydrolysis producing diacylglycerol (DAG) and inositol triphosphate (IP3) formation. Ca2+ is released from intracellular stores and in conjunction with diacylglycerol can lead to protein kinase C (PKC) activation, phosphorylation and activation of p47phox, and translocation of p47phox to the plasma membrane, followed by activation of the complete NOX complex. The O •−2 . and H2O2 thus formed may mediate a further increase in cytosolic Ca2+, elevating glucose metabolism via activation of specific metabolic enzymes. This results in generation of stimulus–secretion coupling factors and promotion of insulin secretion. GPx, Glutathione peroxidase
Site(s) of ROS generation in the pancreatic beta cell
A recent paper has described the importance of mitochondrial ROS production for GSIS [45]. The authors described experiments in which rat islets were incubated in the presence of glucose and inhibitors of mitochondrial electron transfer (rotenone or antimycin) or a mitochondrial uncoupler, CCCP. In conditions in which mitochondrial function had been disrupted by the latter pharmacological agents, so enhancing ROS production in conditions of reduced or halted ATP generation, insulin secretion was increased. The authors concluded that mitochondrial ROS production is a necessary stimulus for glucose-induced insulin secretion. However, in all conditions tested in the latter paper, NOX-generated ROS would also be present and would also be attenuated by the use of antioxidants. Thus while it is possible to conclude that ROS are essential for glucose-induced insulin secretion, the key site(s) of generation and action are yet to be conclusively determined.
A more widely held view is that mitochondrial dysfunction and ROS overproduction are involved in molecular mechanisms of cell dysfunction in diabetes (see also below) [46, 47]. Mild uncoupling can substantially decrease mitochondrial ROS generation and reduce oxidative stress. The uncoupling mechanism involves activation of uncoupling protein (UCP) 2, which protects cells and enhances GSIS. However, substantial overactivation of UCP 2 disrupts cellular energy metabolism, thus reducing insulin secretion. The critical role of UCP 2 in promotion of pancreatic beta cell insulin secretion was recently illustrated in a study making use of Ucp2 −/− mice, in which impaired beta cell function due to enhanced oxidative stress was demonstrated [48].
Superoxide in solution dismutates to H2O2 with a rate constant of 105 mol (l/s)−1 at pH 7 and this rate can be increased (by approximately four orders of magnitude) due to catalysis by SOD [49]. The biological action of NOX derived-ROS (which can be released extracellularly) may be dependent on the action of SOD and on subsequent diffusion of H2O2 through the plasma membrane or possible transport through selective membrane channels.
Although transmembrane flux of ROS has been proposed, it is tempting to speculate that NOX1/NOX4 or NOX2 could also deliver electrons to intracellular oxygen, so forming intracellular O •−2 , which could immediately impact on signalling (Fig. 2). Considering the short half-life of ROS, its participation as a signalling molecule is probably dependent on the site of its generation, which would support the hypothesis of a key role for membrane-associated NOX in signal transduction at the plasma membrane [46]. Regarding the importance of H2O2, previous work has demonstrated a H2O2 dependency of platelet-derived growth factor signalling in smooth muscle cells [50]. In addition, an improvement in insulin signalling was reported to be due to NOX-derived H2O2, which caused protein-tyrosine phosphatase inhibition in adipocytes [51]. Recently, the authors of this review demonstrated that GSIS in rat pancreatic islets was dependent on NOX activity and formation of H2O2 [41].
Considering the chemical differences between various ROS (e.g. O •−2 , H2O2) and indeed reactive nitrogen species, there are likely to be different redox targets for each to transmit its effect. For example NO• binds to the haem group of various enzymes to modulate their activity; O •−2 reacts with iron-sulphur clusters of key cellular proteins to alter their activity; and H2O2 can modify proteins primarily through oxidation of thiol groups, which is favoured when these groups are in the deprotonated form. Thiols are important biological targets of oxidation, as they form part of essential antioxidant defences (e.g. glutathione, thioredoxin and glutaredoxin) and are critical for the chemical action of many enzymes, transcription factors and regulatory proteins [52]. While it is difficult to define the relative contribution of sub-cellular structures and associated functions to ROS-associated positive or negative effects on insulin secretion, we would speculate that mitochondrial generation of metabolic stimulus–secretion coupling factors and plasma membrane-associated signal transduction events (including ion-channel activity) are the major sites of interaction (Fig. 3)
Beta cell redox regulation, metabolism and insulin secretion
Chronic exposure to high glucose levels and/or high lipid levels in vivo and in vitro results in glucotoxicity or glucolipotoxicity, whereby beta cell insulin secretion is reduced and the cell is more susceptible to apoptotic stimuli [4, 46, 53]. Under glucotoxic conditions elevated generation of mitochondrial O •−2 will occur over an extended period, resulting in overstimulation of UCP 2, mitochondrial proton gradient uncoupling and reduced ATP generation. Transcription factors have been reported to be down-regulated by H2O2, due to decreased expression and/or DNA binding. Duodenal homeobox-1 (PDX-1), a beta cell-specific transcription factor that functions as a potent activator of insulin gene transcription, and MafA are two targets of H2O2 regulation [54]. These transcriptional factors play a crucial role in pancreas development and both beta cell differentiation and function. A recent report described that adenoviral overexpression of dominant negative type c-Jun N-terminal kinase-1 preserved PDX-1 DNA-binding activity in face of oxidative stress and protected insulin gene expression and secretion [55]. Under glucolipotoxic or indeed lipotoxic conditions, elevations in fatty acid oxidation cause extended elevation of mitochondrial generation of O •−2 and H2O2 with the associated negative consequences, as described above. In addition generation of diacylglycerol or monoacylglycerol from the elevated intracellular fatty acids associated with the lipotoxic condition may chronically stimulate NOX generation of O •−2 and H2O2.
Diabetic C57BL/KsJ-db/db mice were reported to have a significantly greater beta cell mass after antioxidant treatment, possibly due to suppression of apoptosis. They had significantly higher levels of insulin content and insulin mRNA, which was correlated with higher levels of PDX-1 and associated translocation, clearly visible in the nuclei of islet cells after the antioxidant treatment [56].
With respect to pancreatic beta cell lines, RINm5F cells overexpressing catalase, glutathione peroxidase and Cu/Zn SOD were protected from a lethal pro-inflammatory cytokine mixture (IL-1β, TNF-α and interferon-γ) and exhibited a significantly lower level of ROS-dependent protein damage [57]. Indeed earlier reports, using primary rat islets, described the importance of redox enzymes to the regulation of beta cell metabolism, including a study describing the inhibition of glutathione reductase (a GSSG-reducing enzyme), which impaired glucose metabolism and insulin secretion [58]. GSIS was reported to be dependent on the level of intracellular reduced glutathione [59]. Using a patch-clamp technique, investigators reported a reduction in ATP-sensitive K+ channel activity, suppression of plasma membrane electrical excitability and inhibition of metabolism after H2O2 addition [60–62].
More recent studies have identified that mitochondrial aconitase, mitochondrial adenine-nucleotide translocase [63] and cytosolic glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are susceptible to oxidative modification [64, 65]. One of four cysteine side chains of GAPDH is especially important to the catalytic reaction process (Cys 149) and is located near to the NAD coenzyme in the active site of the enzyme [66]. Mitochondrial aconitase belongs to a family of iron/sulphur-containing dehydratases, whose activities depend on the redox state of the [4Fe–4S]2+ cluster. It has been shown that exposure of purified mitochondria to oxidants, particularly O •−2 and H2O2, resulted in enzyme inactivation [67]. Inactivation appeared to result from interactions between aconitase and a mitochondrial membrane component responsive to H2O2 [67]. GAPDH inhibition may divert metabolites to the hexosamine pathway [68] or possibly to polyol and advanced glycation end-product formation [69]. The possible mechanisms by which high levels of ROS inhibit beta cell metabolism and insulin secretion have been summarised in Fig. 1. Under specific conditions, high rates of glucose metabolism prevented the accumulation of superoxide-derived ROS in primary beta cells [70].
ROS-associated beta cell damage in diabetes
Insulin resistance and pancreatic beta cell insufficiency are major features in the progression of type 2 diabetes mellitus [71, 72]. The molecular basis for excessive mitochondrial oxidative damage in diabetes has been expertly reviewed elsewhere [47].
Interestingly, compared with many other cell types, the beta cell may be at high risk of oxidative damage with an increased sensitivity for apoptosis. This high risk may be due to: (1) excessive levels of mitochondrial O •−2 generation; (2) additional O •−2 and H2O2 generation through elevated beta cell NOX activity (as described here); and (3) limited antioxidant defences. With respect to type 2 diabetes, beta cell dysfunction and associated depressed insulin secretion must be evident before hyperglycaemia develops [72]. It is clear that excessive levels of glucose, lipid, endocrine and various inflammatory factors interact at the level of the pancreatic islet to promote beta cell dysfunction. Thus any explanation of molecular integration at the level of the beta cell must include common mechanisms of dysfunction. One possible candidate mechanism is the activation of NOX and consequent ROS production. The beta cell NOX is activated by glucose, saturated fatty acids (such as palmitic acid), endocrine factors (angiotensin II) and some proinflammatory cytokines [46]. The evidence for cytokine involvement in type 2 diabetes mellitus is still under investigation, but it is widely accepted that cytokines play a major role in beta cell dysfunction and destruction in type 1 diabetes mellitus.
Impairment of beta cell function in vivo may involve excessive generation of O •−2 and H2O2 through increased NOX activity as discussed here and elsewhere [11]. This would subsequently affect mitochondrial function, reducing ATP production and insulin secretion [11, 69]. Additionally palmitate metabolism may give rise to ceramide synthesis, ceramide being a key component of the signal transduction pathway for ROS-induced apoptosis [73]. Ceramide may also induce apoptosis by inactivating pro-survival pathways. Thus this lipid can inhibit phosphatidyl inositol 3-kinase (PI3K), which in turn results in blockade of protein kinase B (PKB, also known as Akt/PKB) activation [74]. Downstream targets of the PI3K/PKB pathway involved in survival include glycogen synthase kinase-3 (GSK-3) [75], caspase-9 [76] and the Bcl-2 family member Bad [77]. The mechanism for the effect of high glucose on pancreatic beta cell apoptosis remains to be elucidated, but several studies have been conducted on this subject. Thus exposure of islets from diabetes-prone Psammomys obesus (obese sand rat, an animal model of type 2 diabetes) to high glucose levels resulted in a dose-dependent increase in beta cell DNA fragmentation suggesting caspase activation [78]. Addition of high glucose resulted in apoptosis of pancreatic beta cells from ob/ob mice and normal Wistar rats via a Ca2+-dependent process in vitro [79].
The proinflammatory cytokine IL1-β is known to induce apoptosis of islet beta cells (associated with type 1 diabetes) by stimulating cytochrome c release from the mitochondria with subsequent activation of downstream caspases. Proinflammatory cytokine mixtures (especially IL-1β, TNF-α and IFN-γ) are typically required to induce death in rodent cell lines. The molecular mechanisms of cytokine-induced cell death appear to involve upregulation of the enzyme inducible nitric oxide synthase (iNOS) and thus nitric oxide generation and also NOX subunit expression resulting in ROS production [11]. O •−2 and NO can combine to generate peroxynitrite (ONOO−) [80], which can induce mitochondrial dysfunction, endoplasmic reticulum stress and apoptosis [81]. Oxidative stress due to elevated O •−2 and H2O2 and subsequent peroxynitrite generation may damage DNA by promoting single-strand DNA break formation, leading to initiation of the apoptotic cascade. Indeed, evidence has recently been obtained from the laboratory of one of the authors of this review that pharmacological inhibition of NOX or iNOS will attenuate cytokine-induced inhibition of insulin secretion from mouse islets (M. Michalska, G. Wolf, R. Walther and P. Newsholme, unpublished data) suggesting a potential role for peroxynitrite formation in inhibition of insulin secretion.
Experimental overexpression of free radical dissipating enzymes such as catalase, glutathione peroxidase and the cytoplasmic copper–zinc SOD protects cultured rodent beta cells from deleterious combinations of cytokines (IFN-γ, TNF-α and IL-1β) [82]. The alloxan-induced diabetes-susceptible (ALR/Lt) mouse, originally selected for resistance to alloxan (a beta cell-selective toxin that generates hydroxyl radicals [83]), provides a rare example of systemically upregulated antioxidant defences that include beta cells. The molecular basis for this resistance was a systemic upregulation of molecules and enzymes associated with free radical dissipation [84, 85]. The lower oxidative burden in ALR/Lt mice correlated with significantly higher specific activities for a battery of antioxidant enzymes not only in comparison with control mice, but also in comparison with the closely related NOD/Lt strain, which develops spontaneous T cell-mediated type 1 diabetes [85, 86]. ALR pancreatic beta cells, both in vivo and in vitro, were resistant to alloxan and also resistant in vitro to the cytotoxic combination of IFN-γ, TNF-α and IL-1β. However, a recent review has highlighted that the mechanisms of alloxan-induced cellular cytotoxicity are complex and therefore results obtained from the ALR/Lt mouse model must be evaluated in the context of the complexity of action of alloxan [87].
Evidence has been published that impaired insulin and insulin like growth factor signalling pathways contribute to the cellular mechanisms underlying the pathology of Alzheimer’s disease [88]. Recent work has shown that ROS generation in the brain of patients with Alzheimer’s disease is primarily via amyloid-β-activated microglial NOX. Melatonin provided significant protective effects due to inhibition of phosphorylation of the p47phox subunit of NOX via a PI3K/PKB-dependent signalling pathway, also blocking the translocation of p47phox and p67phox subunit to the membrane, as well as downregulating the binding of p47phox to gp91phox, thereby impairing the assembly of the functional NOX enzyme [89]. Conflicting reports exist on the effect of melatonin on insulin secretion, but it is tempting to speculate that it may alter NOX assembly and activity as described for microglial cells. This is of importance as genetic studies have demonstrated a link between variations in MTNR1B gene, hyperglycaemia, impaired early-phase insulin secretion and beta cell function [90].
Concluding remarks
While ROS appear to be beneficial for the redox-dependent regulation of insulin secretion, chronic production and elevation of ROS above a critical threshold level may lead to activation of oxidative stress concomitantly with reduced insulin secretion. As NOX is expressed at relatively high levels in the islet beta cell, relative to other islet cells, potential therapies based on targeting NOX in the islet may be beneficial for maintaining beta cell function and integrity in conditions of high glucose, lipid or inflammatory challenge, and therefore could be effective in type 2 diabetes treatment.
Abbreviations
- ALR/Lt:
-
Alloxan-induced diabetes-susceptible
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GSIS:
-
Glucose-stimulated insulin secretion
- iNOS:
-
Inducible nitric oxide synthase
- NOX:
-
NADPH oxidase
- NOXA1:
-
NOX activator 1
- NOXO1:
-
NOX organiser 1
- PDX-1:
-
Duodenal homeobox-1
- PI3K:
-
Phosphatidyl inositol 3-kinase
- PKB:
-
Protein kinase B
- Rac:
-
Ras-related C3 botulinum toxin substrate
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- UCP:
-
Uncoupling protein
References
Newsholme P, Brennan L, Bender K (2006) Amino acid metabolism, β-cell function, and diabetes. Diabetes 55(Suppl 2):S39–S47
Persaud SJ, Jones PM (1993) The involvement of protein kinase C in glucose stimulated insulin secretion. Biochem Soc Trans 21:428S
Schrey MP, Montague W (1983) Phosphatidylinositol hydrolysis in isolated guinea-pig islets of Langerhans. Biochem J 216:433–441
Newsholme P, Keane D, Welters HJ, Morgan NG (2007) Life and death decisions of the pancreatic β-cell: the role of fatty acids. Clin Sci (Lond) 112:27–42
Metz SA (1988) Membrane phospholipid turnover as an intermediary step in insulin secretion. Putative roles of phospholipases in cell signaling. Am J Med 85:9–21
Jones PM, Persaud SJ (1993) Arachidonic acid as a second messenger in glucose-induced insulin secretion from pancreatic beta-cells. J Endocrinol 137:7–14
Keane D, Newsholme P (2008) Saturated and unsaturated (including arachidonic acid) non-esterified fatty acid modulation of insulin secretion from pancreatic beta cells. Biochem Soc Trans 36:955–958
Metz SA (1988) Exogenous arachidonic acid promotes insulin release from intact or permeabilized rat islets by dual mechanisms. Putative activation of Ca2+ mobilization and protein kinase C. Diabetes 37:1453–1469
Kruman I, Guo Q, Mattson MP (1998) Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res 51:293–308
Yu JH, Kim KH, Kim H (2006) Role of NADPH oxidase and calcium in cerulein-induced apoptosis: involvement of apoptosis-inducing factor. Ann N Y Acad Sci 1090:292–297
Morgan D, Oliveira-Emilio HR, Keane D et al (2007) Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 50:359–369
Lortz S, Gurgul-Convey E, Lenzen S, Tiedge M (2005) Importance of mitochondrial superoxide dismutase expression in insulin-producing cells for the toxicity of reactive oxygen species and proinflammatory cytokines. Diabetologia 48:1541–1548
Krause KH (2004) Tissue distribution and putative physiological function of NOX family NADPH oxidases. Jpn J Infect Dis 57:S28–S29
Geiszt M (2006) NADPH oxidases: new kids on the block. Cardiovasc Res 71:289–299
Borregaard N, Tauber AI (1984) Subcellular localization of the human neutrophil NADPH oxidase b-cytochrome and associated flavoprotein. J Biol Chem 259:47–52
Vignais PV (2002) The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci 59:1428–1459
Lambeth JD, Kawahara T, Diebold B (2007) Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med 43:319–331
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Koga H, Terasawa H, Nunoi H, Takeshige K, Inagaki F, Sumimoto H (1999) Tetratricopeptide repeat (TPR) motifs of p67(phox) participate in interaction with the small GTPase Rac and activation of the phagocyte NADPH oxidase. J Biol Chem 274:25051–25060
Kawahara T, Quinn MT, Lambeth JD (2007) Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol 7:109
Nisimoto Y, Motalebi S, Han CH, Lambeth JD (1999) The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b(558). J Biol Chem 274:22999–23005
Bedard K, Lardy B, Krause KH (2007) NOX family NADPH oxidases: not just in mammals. Biochimie 89:1107–1112
Oliveira HR, Verlengia R, Carvalho CR, Britto LR, Curi R, Carpinelli AR (2003) Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 52:1457–1463
Banfi B, Molnar G, Maturana A et al (2001) A Ca(2+)-activated NADPH oxidase in testis, spleen, lymph nodes. J Biol Chem 276:37594–37601
Sumimoto H (2008) Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J 275:3249–3277
Geiszt M, Leto TL (2004) The NOX family of NAD(P)H oxidases: host defense and beyond. J Biol Chem 279:51715–51718
Takeya R, Ueno N, Kami K et al (2003) Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J Biol Chem 278:25234–25246
Uchizono Y, Takeya R, Iwase M et al (2006) Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci 80:133–139
Lapouge K, Smith SJ, Groemping Y, Rittinger K (2002) Architecture of the p40–p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox. J Biol Chem 277:10121–10128
Banfi B, Maturana A, Jaconi S et al (2000) A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science 287:138–142
Sumimoto H, Miyano K, Takeya R (2005) Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun 338:677–686
Cheng G, Diebold BA, Hughes Y, Lambeth JD (2006) Nox1-dependent reactive oxygen generation is regulated by Rac1. J Biol Chem 281:17718–17726
Shiose A, Kuroda J, Tsuruya K et al (2001) A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem 276:1417–1423
Kawahara T, Ritsick D, Cheng G, Lambeth JD (2005) Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem 280:31859–31869
Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG (2006) Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18:69–82
Brandes RP, Schröder K (2008) Composition and functions of vascular nicotinamide adenine dinucleotide phosphate oxidases. Trends Cardiovasc Med 18:15–19
Guichard C, Moreau R, Pessayre D, Epperson TK, Krause KH (2008) NOX family NADPH oxidases in liver and in pancreatic islets: a role in the metabolic syndrome and diabetes. Biochem Soc Trans 36:920–929
Schröder K, Wandzioch K, Helmcke I, Brandes RP (2009) Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol 29:239–245
Mahadev K, Motoshima H, Wu X et al (2004) The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol 24:1844–1854
Pi J, Bai Y, Zhang Q et al (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56:1783–1791
Morgan D, Rebelato E, Abdulkader F et al (2009) Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta cells. Endocrinology 150:2197–2201
Imoto H, Sasaki N, Iwase M et al (2008) Impaired insulin secretion by diphenyleneiodium associated with perturbation of cytosolic Ca2+ dynamics in pancreatic beta-cells. Endocrinology 149:5391–5400
Zawalich WS, Zawalich KC (2008) Enhanced activation of phospholipase C and insulin secretion from islets incubated in fatty acid-free bovine serum albumin. Metabolism 57:290–298
Oliveira HR, Curi R, Carpinelli AR (1999) Glucose induces an acute increase of superoxide dismutase activity in incubated rat pancreatic islets. Am J Physiol 276:C507–C510
Leloup C, Tourrel-Cuzin C, Magnan C et al (2009) Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 58:673–681
Newsholme P, Haber EP, Hirabara SM et al (2007) Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol 583:9–24
Green K, Brand MD, Murphy MP (2004) Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 53(Suppl 1):S110–S118
Pi J, Bai Y, Daniel K et al (2009) Persistent oxidative stress due to absence of uncoupling protein 2 is associated with impaired pancreatic beta cell function. Endocrinology 150:3040–3048
Halliwell B, Gutteridge JMC (2007) Free radicals in biology and medicine. Oxford University Press, Oxford
Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T (1995) Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270:296–299
Mahadev K, Zilbering A, Zhu L, Goldstein BJ (2001) Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem 276:21938–21942
Peskin AV, Winterbourn CC (2006) Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phosphate dehydrogenase. Free Radic Biol Med 40:45–53
Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF (1992) Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest 90:320–325
Kaneto H, Katakami N, Kawamori D et al (2007) Involvement of oxidative stress in the pathogenesis of diabetes. Antioxid Redox Signal 9:355–366
Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC (2002) Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 277:30010–30018
Kaneto H, Kajimoto Y, Miyagawa J et al (1999) Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 48:2398–2406
Lortz S, Tiedge M, Nachtwey T, Karlsen AE, Nerup J, Lenzen S (2000) Protection of insulin-producing RINm5F cells against cytokine-mediated toxicity through overexpression of antioxidant enzymes. Diabetes 49:1123–1130
Malaisse WJ, Dufrane SP, Mathias PC et al (1985) The coupling of metabolic to secretory events in pancreatic islets. The possible role of glutathione reductase. Biochim Biophys Acta 844:256–264
Ammon HP, Mark M (1985) Thiols and pancreatic beta-cell function: a review. Cell Biochem Funct 3:157–171
Avshalumov MV, Chen BT, Koos T, Tepper JM, Rice ME (2005) Endogenous hydrogen peroxide regulates the excitability of midbrain dopamine neurons via ATP-sensitive potassium channels. J Neurosci 25:4222–4231
Krippeit-Drews P, Kramer C, Welker S, Lang F, Ammon HP, Drews G (1999) Interference of H2O2 with stimulus-secretion coupling in mouse pancreatic beta-cells. J Physiol 514:471–481
Maechler P, Jornot L, Wollheim CB (1999) Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem 274:27905–27913
Yan LJ, Levine RL, Sohal RS (1997) Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci U S A 94:11168–11172
Brodie AE, Reed DJ (1987) Reversible oxidation of glyceraldehyde 3-phosphate dehydrogenase thiols in human lung carcinoma cells by hydrogen peroxide. Biochem Biophys Res Commun 148:120–125
Du XL, Edelstein D, Rossetti L et al (2000) Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A 97:12222–12226
Molina y Vedia L, McDonald B, Reep B et al (1992) Nitric oxide-induced S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP-ribosylation. J Biol Chem 267:24929–24932
Bulteau AL, Ikeda-Saito M, Szweda LI (2003) Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry 42:14846–14855
Sakai K, Matsumoto K, Nishikawa T et al (2003) Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem Biophys Res Commun 300:216–222
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414:813–820
Martens GA, Cai Y, Hinke S, Stange G, van de Casteele M, Pipeleers D (2005) Glucose suppresses superoxide generation in metabolically responsive pancreatic beta cells. J Biol Chem 280:20389–20396
Bell GI, Polonsky KS (2001) Diabetes mellitus and genetically programmed defects in β-cell function. Nature 414:788–791
Kahn SE (2003) The relative contributions of insulin resistance and β-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 46:3–19
Cacicedo JM, Benjachareowong S, Chou E, Ruderman NB, Ido Y (2005) Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes 54:1838–1845
Beeharry N, Chambers JA, Green IC (2004) Fatty acid protection from palmitic acid-induced apoptosis is lost following PI3-kinase inhibition. Apoptosis 9:599–607
Pap M, Cooper GM (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 273:19929–19932
Cardone MH, Roy N, Stennicke HR et al (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282:1318–1321
Datta SR, Dudek H, Tao X et al (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241
Donath MY, Gross DJ, Cerasi E, Kaiser N (1999) Hyperglycemia-induced β-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 48:738–744
Efanova IB, Zaitsev SV, Zhivotovsky B et al (1998) Glucose and tolbutamide induce apoptosis in pancreatic β-cells. A process dependent on intracellular Ca2+ concentration. J Biol Chem 273:33501–33507
Lenzen S (2008) Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans 36:343–347
Grover AK, Kwan CY, Samson CE (2003) Effects of peroxynitrite on sarco/endoplasmic reticulum Ca2+ pump isoforms SERCS2b and SERCA3a. Am J Physiol Cell Physiol 285:C1537–C1543
Azevedo-Martins AK, Lortz S, Lenzen S, Curi R, Eizirik DL, Tiedge M (2003) Improvement of the mitochondrial antioxidant defense status prevents cytokine-induced nuclear factor-kappaB activation in insulin-producing cells. Diabetes 52:93–101
Sekiguchi F, Ishibashi K, Katoh H, Kawamoto Y, Ino T (1990) Genetic profile of alloxan-induced diabetes-susceptible mice (ALS) and resistant mice (ALR). Exp Anim 39:269–272
Mathews CE, Leiter EH (1999) Constitutive differences in antioxidant defense status distinguishes alloxan resistant (ALR/Lt) and alloxan susceptible (ALS/Lt) mice. Free Radical Biol Med 27:449–455
Mathews CE, Leiter EH (1999) Resistance of ALR/Lt islets to free radical-mediated diabetogenic stress is inherited as a dominant trait. Diabetes 48:2189–2196
Mathews CE, Graser R, Savinov A, Serreze DV, Leiter EH (2001) The NOD/Lt-related ALR/Lt strain: unusual resistance of beta cells to autoimmune killing uncovers a role for beta-cell expressed resistance determinants. Proc Natl Acad Sci U S A 98:235–240
Lenzen S (2008) The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 51:216–226
Carro E, Torres-Aleman I (2004) The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur J Pharmacol 490:127–133
Zhou J, Zhang S, Zhao X, Wei T (2008) Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid beta 1–42. J Pineal Res 45:157–165
Mulder H, Nagorny CLF, Lyssenko V, Groop L (2009) Melatonin receptors in pancreatic islets: good morning to a novel type 2 diabetes gene. Diabetologia 52:1240–1249
Acknowledgements
This work was funded by The Health Research Board of Ireland and Enterprise Ireland, as well as by The State of São Paulo Research Foundation (FAPESP), the Programme for support for centres of excellence PRONEX and the National Council for Scientific and Technological Development (CNPq), Brazil.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Newsholme, P., Morgan, D., Rebelato, E. et al. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 52, 2489–2498 (2009). https://doi.org/10.1007/s00125-009-1536-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1536-z