
Abstract
Aims/hypothesis
The aim of this cohort study was to investigate the risk of malignant neoplasms and mortality in patients with diabetes treated either with human insulin or with one of three insulin analogues.
Methods
Data were provided by the largest German statutory health insurance fund (time-frame: January 1998 to June 2005 inclusive), on patients without known malignant disease who had received first-time therapy for diabetes mellitus exclusively with human insulin, aspart, lispro or glargine. The primary outcome was the diagnosis of a malignant neoplasm. Data were analysed by multiple Cox regression models adjusting for potential confounders.
Results
A total of 127,031 patients were included, with a mean follow-up time of 1.63 (median 1.41, maximum 4.41) years. A positive association between cancer incidence and insulin dose was found for all insulin types. Because patients receiving combined therapy with insulin analogues and human insulin were excluded, the mean daily dose was much lower for glargine than for human insulin, and a slightly lower cancer incidence in the glargine group was found. After adjusting for dose, a dose-dependent increase in cancer risk was found for treatment with glargine compared with human insulin (p < 0.0001): the adjusted HR was 1.09 (95% CI 1.00 to 1.19) for a daily dose of 10 IU, 1.19 (95% CI 1.10 to 1.30) for a daily dose of 30 IU, and 1.31 (95% CI 1.20 to 1.42) for a daily dose of 50 IU. No increased risk was found for aspart (p = 0.30) or lispro (p = 0.96) compared with human insulin.
Conclusions/interpretation
Considering the overall relationship between insulin dose and cancer, and the lower dose with glargine, the cancer incidence with glargine was higher than expected compared with human insulin. Our results based on observational data support safety concerns surrounding the mitogenic properties of glargine in diabetic patients. Prospective long-term studies are needed to further evaluate the safety of insulin analogues, especially glargine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although insulin has predominantly metabolic effects, it also has weak direct and indirect mitogenic properties [1, 2]. A higher risk of several types of malignant neoplasm has been found in people with diabetes in systematic reviews of epidemiological studies. In patients with diabetes mellitus, compared with individuals without this condition, Larsson et al. found an RR of 1.30 (95% CI 1.20 to 1.40) for colorectal cancer, 1.24 (95% CI 1.08 to 1.42) for bladder cancer, and 1.20 (95% CI 1.12 to 1.28) for breast cancer [3–5]. An OR of 1.82 (95% CI 1.66 to 1.89) has been reported for pancreatic cancer, and an RR of 2.10 (95% CI 1.75 to 2.53) has been calculated for endometrial cancer [6, 7]. Similar results have been reported for other cancer entities. An exposure–response relationship was reported by Yang et al., who found indications that the duration of insulin treatment was positively associated with an increase in the incidence of colorectal cancer among type 2 diabetic patients [8].
Several insulin analogues have been developed in recent years and these are increasingly being used in the treatment of diabetes. However, modifying the insulin molecule not only changes its metabolic effects, but can also alter its mitogenic potency. Although it has been shown that the differences in growth-promoting and mitogenic potencies between human insulin and insulin analogues, as well as between the analogues themselves, are related to different affinities to the IGF-I receptor, the exact mechanisms are not fully understood, and the clinical importance of this finding remains unclear [9, 10]. Comparison of the mitogenic potency of insulin analogues relative to human insulin in cell experiments showed that B10Asp was tenfold and glargine (A21Gly, B31Arg, B32Arg human insulin) was eightfold more potent, whereas lispro (B28Lys, B29Pro human insulin) and aspart (B28Asp human insulin) were slightly less potent [10].
The proliferative effects of insulin analogues on mammary epithelial cells relative to human insulin have recently been analysed; at physiologically relevant concentrations, a significantly higher proliferative effect on MCF-7 breast cancer cells was found for glargine, but not for aspart or lispro [11]. In addition, recent experiments with colorectal, prostate and breast cancer cell lines showed that glargine and lispro stimulated cancer cell proliferation significantly more than human insulin did [12].
In vivo carcinogenicity was first demonstrated for the B10Asp insulin analogue, which, in contrast to human insulin, induced mammary tumours in rats [13]. Animal experiments using high-dose glargine have been inconclusive because of the excessive short-term mortality from repeated severe hypoglycaemia and subsequent neuronal necrosis or cardiovascular failure [14].
Because of safety concerns, long-term studies to evaluate the safety of insulin analogues have been demanded [15]. However, to our knowledge, there have not been any randomised or non-randomised trials conducted in humans that have been designed and sufficiently powered to detect differences in carcinogenic safety between insulin analogues and human insulin [16–18].
Our objective was to conduct a cohort study to investigate the risk of malignant neoplasms in patients with diabetes treated either with human insulin or an insulin analogue, taking into consideration important potential confounders and effect modifiers.
Methods
Data
Pseudonymous data on all insurants of Germany’s largest statutory health insurance fund, the Allgemeine Ortskrankenkasse (AOK; local healthcare fund), were provided by Wissenschaftliches Institut der AOK (AOK Research Institute) for 13 of 16 federal states, representing 17.9 million people. The following data were available for the total period between 1 January 1998 and 30 June 2005: (1) basic data (patient ID, age, sex); (2) data from primary care prescriptions for insulin billed to the AOK (including information on the insulin type and dose); (3) data from primary care prescriptions for concomitant medications billed to the AOK (classified with the German version of the Anatomical Therapeutic Chemical [ATC] Classification System [19]); (4) data from hospital claims billed to the AOK (date of admission, date of discharge, diagnoses at admission and discharge according to the International Classification of Diseases, 10th revision, German Modification (ICD 10; available at www.dimdi.de/dynamic/de/klassi/downloadcenter/icd-10-gm/vorgaenger/version2005/systematik/xlgbp2005.zip, accessed 29 May 2009); (5) data on the survival status of AOK members. For one of the federal states (Bavaria), data were not available for the whole time period.
Study population and definition of variables
The study population comprised adult (≥18 years) patients without known malignant disease who had received first-time insulin therapy for diabetes mellitus exclusively with human insulin or with only one type of insulin analogue. We restricted the type of insulin analogue to the short-acting analogues lispro and aspart and the long-acting analogue glargine, as other analogues had not been on the German market long enough for evaluation. The diagnosis ‘diabetes mellitus’ was derived from prescription data, as human insulin and insulin analogues are only prescribed to patients with diabetes. No definite distinction between diabetes types 1 and 2 could be made on the basis of these data and no information on insulin treatment modalities (e.g. number of insulin injections, insulin pump) was available; however, it is very likely that patients treated with insulin and oral glucose-lowering agents had type 2 diabetes mellitus. We classified participants as insulin-naive if they had not received any prescription for human insulin or insulin analogues within 1 year before inclusion in the study. We considered participants to be without known malignant disease if they had not received a corresponding diagnosis within 3 years prior to inclusion in the study. To ensure that only event-free patients were included, we excluded participants with the slightest suspicion of a malignant disease (e.g. patients with the ICD-10 diagnosis Z03.1—observation for suspected malignant neoplasm). Data on the following potential confounders were available at baseline: age, sex, year of start of treatment, AOK branch (name of federal state), number of hospital stays, duration of hospitalisation and concomitant medications. Data on hospitalisation and concomitant medication refer to the time period 3 years prior to the study entry date. Individual doses of human insulin and insulin analogues were available as the cumulative dose over time. We divided the cumulative dose for each participant by the time at risk, i.e. the days until an event or until censoring, to obtain a mean daily dose for each participant. We excluded participants with implausibly high daily doses (>1000 IU), as well as participants with an event date that was equal to the study inclusion date.
Definition of cohorts
We defined four study groups according to the treatment received: human insulin, aspart, lispro and glargine. Eligible participants were those exposed to only one of these agents during follow-up. Patients who received any concomitant insulin prescriptions were excluded, as were patients who received porcine or bovine insulin.
Outcomes
The predefined primary outcome was the diagnosis of a malignant neoplasm, which was defined according to ICD-10 (diagnoses C00-C97 and D00-D09) or the corresponding diagnoses in ICD-9. The secondary outcome was all-cause mortality.
Follow-up
Because of the required preceding period of 3 years without known malignant disease, the earliest possible study inclusion date (start of treatment with human insulin or an insulin analogue) was 1 January 2001. Follow-up ended when an event occurred or on 30 June 2005 (study end). Follow-up also ended if the patient cancelled his or her membership of the AOK. Continuous follow-up was ensured, as we only analysed data for patients with at least one insulin prescription every 6 months until an event occurred or until the end of the observation period.
Statistical analysis
We used the χ² test to compare categorical variables between the treatment groups at baseline. We applied analysis of variance (ANOVA, F test) for comparison of continuous covariates between the treatment groups at baseline. We applied survival time methods for time-to-event data [20]. Patients who did not experience an event during the study period were censored at study end. For the analysis of the time until the occurrence of a malignant neoplasm, patients who died without a prior diagnosis of malignant neoplasm were censored at the last observation date before death. We used HRs as effect measures, calculated by means of Cox proportional hazards regression models [21]. We applied multiple Cox regression models to adjust for potential confounders. Starting with a Cox model including all given potential confounders as main effects, we performed a backward selection procedure with a significance level of α = 0.01 to identify covariates significantly associated with the time to event. The federal states and the year of treatment were selected ‘en bloc’, i.e. either all or none of the categories of the covariate were included in the model. The scale of the continuous covariates was assessed using a graphical procedure proposed by Hosmer and Lemeshow [20]. In this procedure, a Cox model is fitted after categorising the continuous covariates into groups formed by the quartiles of the covariates. The estimated coefficients of the groups are then plotted against the midpoints of the groups. If the effect of the covariates is linear in the logarithmically transformed hazard, the polygon connecting these points forms an approximately straight line. We checked the proportional hazard assumption graphically by plotting log(-log survival time) versus log survival time and by a formal test based on Schoenfeld residuals [22]. If the proportional hazard assumption was violated, a stratified Cox model was fitted. We assessed possible interactions between the most relevant covariates—insulin group, sex, age, and dose—by performing a backward selection procedure based on a Cox model including all significant main effects and interaction terms at level α = 0.01. As this study was intended to generate hypotheses, we made no adjustments for multiple testing [23]. All analyses were performed with the SAS statistical package, Version 9.1 (SAS Institute, Cary, NC, USA).
Results
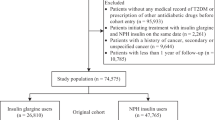
Out of nearly 18 million insurants and 322,732 insulin-treated diabetic patients, we included a total of 127,031 eligible patients in the analysis. Figure 1 shows a flow chart of the data and Table 1 shows the baseline characteristics of the patients analysed. The baseline characteristics of patients within different dose ranges for human insulin and glargine are presented in Electronic supplementary material (ESM) Table 1.

Flow diagram of the data analysed
The mean follow-up time was 1.63 years (median 1.41, maximum 4.41) for malignant neoplasms and 1.67 years (median 1.50, maximum 4.41) for mortality after starting human insulin or insulin analogue therapy. The time under observation for malignant neoplasms was 1.70 years for human insulin, 1.44 years for aspart, 2.10 years for lispro and 1.31 years for glargine; the difference between the groups was significant (p < 0.0001 by ANOVA). The corresponding values for mortality were only slightly higher. The mean duration of follow-up was shorter for those patients prescribed glargine or aspart owing to the later introduction of these analogues to the German market (1999 and 2000, respectively).
Among the 127,031 insulin-treated diabetic patients included in the study, malignant neoplasms were observed in 5,009 patients and 18,253 patients died. The crude incidence rate (per 100 patient-years) for malignant neoplasms was 2.50 in patients treated with human insulin, 2.16 in those treated with aspart, 2.13 in those treated with lispro and 2.14 in those treated with glargine. The crude incidence rate for the secondary endpoint, mortality, was 9.24, 5.75, 6.91 and 6.30, respectively.
The mean (SD) insulin dose until the occurrence of a malignant neoplasm was 43.8 (37.4) IU for human insulin, 38.9 (33.7) IU for aspart, 36.2 (32.7) IU for lispro and 25.9 (22.5) IU for glargine (p < 0.0001 by ANOVA). The median (and lower quartile, upper quartile and 95% quantile) was 37.0 (21.1, 56.2, 100) IU for human insulin, 33.0 (20.0, 49.3, 98.4) IU for aspart, 31.0 (15.2, 49.2, 91.6) IU for lispro and 21.8 (14.8, 30.0, 59.3) IU for glargine. The corresponding values for mortality differed only marginally.
ESM Tables 2 and 3 summarise the final multiple Cox models for the primary outcome, a diagnosis of a malignant neoplasm, and the secondary outcome, all-cause mortality, respectively, including significant interactions and interactions between insulin group and dose.
Significance levels other than α = 0.01 (α = 0.05 or α = 0.15) in the backward selection procedure essentially led to the same results for the parameter estimates of the treatment groups. The assessment of the scale of the continuous covariates suggested a linear association for all continuous covariates. Thus, all continuous covariates were included as a linear term in the final model. Graphical checks indicated a possible deviation from the proportional hazard assumption for the factor ‘dose’ for both outcomes. Thus, a stratified Cox model was fitted, using quantiles of the dose as the strata. This model essentially yielded the same HRs for the insulin groups as the unstratified Cox model.
Tables 2 and 3 show the results of intermediate models with different degrees of complexity to enable a better assessment of the influence of the different covariates on the primary and secondary outcomes, respectively. The results of a model including age, sex and dose in addition to treatment group as the only covariates are consistent with the main results of the final Cox model. In a model adjusting for age and sex, dose had a significant overall effect on the incidence of malignant neoplasms (p < 0.0001). The significant dose effect remained when each insulin type was analysed separately.
Because the final multiple Cox models contain interaction terms between the insulin analogues and dose, the adjusted HRs for insulin analogues compared with human insulin change when the dose is varied. Therefore, in Tables 2 and 3 the adjusted HRs from the final model are given for different dose levels. Figure 2 further illustrates the adjusted HRs for the insulin analogues compared with human insulin regarding malignant neoplasms and mortality in relation to the insulin dose, based on the final multiple Cox model.

HRs (solid line) and 95% confidence limits (dotted lines) for malignant neoplasms (a–c) and mortality (d–f) for aspart (a, d), lispro (b, e) and glargine (c, f) based on the final models shown in ESM 2 and ESM 3. Because of the interaction with dose, the HRs change with varying dose levels. For mortality, the final model includes an additional interaction of glargine with age. Therefore, the HR curve shown for glargine refers to a fixed age of 70 years. The curves are plotted between the 5% and the 95% quantiles of the observed data for each insulin analogue
To check the robustness of the results, we conducted several sensitivity analyses. To assess detection bias, we analysed the potential influence of particular tumour entities to identify potential bias caused by malignant neoplasms that were possibly already prevalent but undiagnosed at study entry; skin cancer (C44), precancerous lesions and in situ carcinoma (D00–D09) were not treated as malignant neoplasms in further analyses. Furthermore, we considered a subclassification of treatment with oral glucose-lowering agents. Table 4 summarises the results of the sensitivity analyses. To further illustrate and analyse the dose-dependent risk increase with glargine, we performed additional analyses. Detailed analyses of subgroups of patients treated with different doses of glargine or human insulin are given in Table 5. Table 6 shows the change in incidence rates of malignant neoplasms and mortality per dose increase of 1 SD (within each treatment group).
Discussion
The results of our study indicate that, in patients with diabetes, the insulin dose is positively associated with the risk of malignant neoplasms; this applies both to human insulin and to insulin analogues. Compared with human insulin, this dose-dependent risk increase was higher for glargine (p < 0.0001) but not for lispro (p = 0.96) or aspart (p = 0.30).
In this pragmatic cohort study there were notable differences between the baseline characteristics of the treatment groups. This underlines the importance of accounting for confounding factors by applying a careful model-building process including relevant covariates and interaction terms.
At baseline, the patients in the glargine group seemed to be healthier than those in the human insulin group (fewer and shorter hospital stays), and the mean daily dose was significantly lower in the glargine group, leading to a significantly lower risk in the unadjusted analysis as well as in the analysis adjusted for age and sex, in which the modifying effects of dose were not taken into account. After also adjusting for dose, a significantly higher risk was observed for glargine relative to human insulin (Table 2). This means that glargine is associated with a higher risk of malignant neoplasms when the risk is adjusted for patients’ characteristics and the insulin dosage. Our findings are in line with the previously demonstrated in vitro mitogenic potency of glargine [10–12].
Our study has both strengths and limitations. The major strength of our study is the evaluation of a comprehensive data set from the largest German statutory health insurance fund, which is most likely to be representative of insulin-treated diabetic patients in Germany. Our access to these data enabled the analysis of almost 130,000 diabetic patients treated with insulin who were observed over a mean period of 1.63 years for malignant neoplasms under real-life conditions. For four of the 16 federal states in Germany, data were either missing or only partly available. This was due to technical reasons and, in our view, did not influence the representative nature of the database.
One major limitation of the study is the fact that patients were not randomised to treatment groups. Although the results were adjusted for all known and available confounders, potentially relevant factors such as insulin resistance, body mass index, smoking, social status and duration of diabetes were not available and therefore could not be considered in the analyses. However, to explain the observed dose-dependent risk increase in the glargine group, these potential confounders would have to be associated with both a higher cancer risk and a higher glargine dosage. For example, we have no evidence that the glargine group included more smokers or people from a lower socioeconomic class; in fact, at baseline, the glargine group generally seemed to be healthier than the human insulin group, and these findings were consistent when comparing subgroups of patients within the same dose range.
Although we tried to consider bias as much as the available data allowed, we cannot entirely exclude the possibility that some known or unknown factors could have influenced both the dose of human insulin and insulin analogues and the risk of cancer, especially given that the groups being compared were clinically dissimilar. Ultimately, only a randomised controlled trial could dispel these concerns. Such an analysis would also require the follow-up of large patient groups. In our study, the patient numbers were highest for human insulin and glargine. Hence, the conclusions are less robust for aspart and lispro.
Detection bias may have occurred because of the more frequent and longer hospitalisation periods of patients in the human insulin group compared with those in the glargine group. This could have resulted in relatively higher detection rates of neoplasms, particularly skin cancer (C44), as well as of precancerous lesions and in situ carcinoma (D00–D09). We took great care to avoid bias through prevalent but undiagnosed neoplasms when designing this study; hence, we excluded patients with the slightest suspicion of a malignant disease. We performed sensitivity analyses including and excluding these particular entities, and the main results remained unchanged. Furthermore, a higher detection rate of malignant neoplasms in the human insulin group would lead to an underestimation of the true effect, and the real difference between the incidence rates of the insulin and the glargine groups could be even greater than the one observed.
Another disparity is that more patients in the glargine group were treated with oral glucose-lowering agents. To take the effect of these agents into account, we adjusted for the factor ‘concomitant treatment with any oral glucose-lowering agent’ in the final Cox regression model and additionally for the specific subclasses of oral glucose-lowering agents in sensitivity analyses. In particular, we analysed a potential influence of metformin therapy, for which potential effects on cancer incidence have been previously discussed [24]. However, we found no evidence in our analyses of a substantial influence of metformin or any other oral glucose-lowering agent on the risk of morbidity or mortality.
A further potential limitation is prescription bias. Members of the human insulin group may have been treated with both short- and long-acting agents, whereas patients in the glargine group were treated only with glargine, and presumably had residual insulin secretion or received prescriptions for oral agents. Prescription bias could have affected the results if patients at a higher risk of malignant neoplasms had been prescribed glargine (at a higher dosage); however, the glargine group generally seemed to be healthier at baseline than the human insulin group (in all dose ranges).
Furthermore, we have no evidence that patients who still have residual insulin secretion are at a higher risk of malignant neoplasms than those who require treatment with human insulin with both short- and long-acting agents.
The fact that the members of the glargine group were treated only with glargine, i.e. did not receive additional treatment with a short-acting insulin, contributes to the observed higher average daily insulin dose in the human insulin group. This was not unexpected and was accounted for by the study design: we conducted an analysis based on real-life data representing the usual care of patients treated with glargine or human insulin. In addition, to avoid confounding, we explicitly aimed to compare exposure to human insulin and insulin analogues without overlapping or crossover effects. Therefore, we included patients who had been exposed to only one of the types of insulin investigated.
Taking these aspects into account, the slightly lower crude incidence rate of malignant neoplasms associated with glargine compared with human insulin seems to be at least partly a result of the different doses in the treatment groups.
One also has to take into account that glargine is being actively marketed promoting the added advantage of once-daily application in patients who have not reached their treatment goals with oral glucose-lowering agents alone. This might have led to an increased use of comparatively smaller doses of glargine.
We considered these dose differences in subgroup analyses, and even in these analyses (which only considered the most relevant covariates) and despite the lower statistical power, a significantly higher relative risk of cancer was observed for glargine in the high-dose range. Considering that the lower number of patients and events reduced the statistical power, it is not unexpected that we only found statistically significant differences in the subgroup that received doses in the highest range.
The observed results for the secondary outcome (mortality) require closer examination. We observed lower crude mortality rates in patients treated with glargine doses of <40 IU than in those treated with human insulin at the same doses (dose range <20 IU: 5.81 vs 8.83 per 100 patient-years; dose range 20 to 40 IU: 5.58 vs 9.67 per 100 patient-years).
However, we observed higher mortality rates in patients treated with high doses of glargine (>40 IU) than in patients treated with equivalent doses of human insulin (14.79 vs 9.17 per 100 patient-years). We carefully adjusted for the available covariates, took dose effects into account, and found a treatment–dose interaction caused by the higher mortality rate in patients treated with high doses. We have no indication that patients who were at high risk of dying were more likely to be treated with high doses of glargine than with human insulin, and the demographic data do not provide any plausible explanations for this finding. We are therefore cautious in interpreting these results and we would need further data to draw any valid conclusions.
Unfortunately, data on cause-specific mortality were not available and we do not know if causes of death differed between the groups. In this regard we would like to point out that an analysis of patients treated over a longer period of time would of course have been desirable; however, the available data were limited because of the framework of this study. We performed an additional analysis including only participants with a follow-up time (time between study entry and end of study) of at least 1.5 years, and found consistent results regarding the dose-dependent risk of malignant neoplasms and mortality.
One of the strengths of our study is that the patients were treated with a single type of insulin, which avoids confounding between treatments. We think that it would be difficult to treat diabetic patients over several years with only one insulin analogue. Moreover, such a study protocol would in itself create additional confounders.
In our opinion, this issue should be investigated using a prospective study design, and a reliable assessment of causes of death should be performed by a clinical review committee [25]. We plan to conduct additional analyses to further explore the effects of human insulin and insulin analogues on specific cancer entities.
To our knowledge, the risk of cancer in patients with diabetes has so far never been compared between patients treated with human insulin and those treated with insulin analogues. Despite the relatively fragile nature of the data, the results of our cohort study support safety concerns surrounding the mitogenic and potential tumour growth-promoting properties of glargine. The findings underline the necessity for a prospective, randomised, controlled, long-term study that is designed and sufficiently powered to evaluate insulin analogues with regard to their effects on morbidity and mortality in patients with diabetes. Taking into account the fact that a non-randomised study cannot prove that the association between glargine and cancer is causal, we still think that our results may have practical implications. In our view, the decision for or against treatment with glargine has to be made on an individual basis after providing comprehensive information to patients about the limited long-term data available and the uncertain potential benefits and harms. We think that, especially in type 2 diabetes mellitus, the potential harm of glargine must be weighed against rather minor potential therapeutic advantages [18, 26, 27]. In general, before the widespread introduction of a new drug that is to be taken for decades, we think that an evaluation of its patient-relevant beneficial and harmful effects should be conducted under controlled conditions for at least several years. Because such studies are rare and not required for market approval, patients and physicians will still often have to make treatment decisions in the face of uncertainty.
Abbreviations
- AOK:
-
Allgemeine Ortskrankenkasse (local healthcare fund)
- ATC:
-
Anatomical Therapeutic Chemical
- ICD:
-
International Classification of Diseases
References
Sandhu MS, Dunger DB, Giovannucci EL (2002) Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J Natl Cancer Inst 94:972–980
Tran TT, Medline A, Bruce WR (1996) Insulin promotion of colon tumors in rats. Cancer Epidemiol Biomarkers Prev 5:1013–1015
Larsson SC, Mantzoros CS, Wolk A (2007) Diabetes mellitus and risk of breast cancer: a meta-analysis. Int J Cancer 121:856–862
Larsson SC, Orsini N, Brismar K, Wolk A (2006) Diabetes mellitus and risk of bladder cancer: a meta-analysis. Diabetologia 49:2819–2823
Larsson SC, Orsini N, Wolk A (2005) Diabetes mellitus and risk of colorectal cancer: a meta-analysis. J Natl Cancer Inst 97:1679–1687
Huxley R, Ansary-Moghaddam A, Berrington de González A, Barzi F, Woodward M (2005) Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer 92:2076–2083
Friberg E, Orsini N, Mantzoros CS, Wolk A (2007) Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia 50:1365–1374
Yang YX, Hennessy S, Lewis JD (2004) Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients. Gastroenterology 127:1044–1050
Bornfeldt KE, Gidlöf RA, Wasteson A, Lake M, Skottner A, Arnqvist HJ (1991) Binding and biological effects of insulin, insulin analogues and insulin-like growth factors in rat aortic smooth muscle cells. Comparison of maximal growth promoting activities. Diabetologia 34:307–313
Kurtzhals P, Schäffer L, Sørensen A et al (2000) Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 49:999–1005
Mayer D, Shukla A, Enzmann H (2008) Proliferative effects of insulin analogues on mammary epithelial cells. Arch Physiol Biochem 114:38–44
Weinstein D, Simon M, Yehezkel E, Laron Z, Werner H (2009) Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab Res Rev 25:41–49
Drejer KK, Sørensen AR, Wiberg FC, Klein HH, Lundemose AG (1992) The bioactivity of insulin analogues from in vitro receptor binding to in vivo glucose uptake. Diabetes Metab Rev 8:259–285
Stammberger I, Bube A, Durchfeld-Meyer B, Donaubauer H, Troschau G (2002) Evaluation of the carcinogenic potential of insulin glargine (LANTUS) in rats and mice. Int J Toxicol 21:171–179
Owens DR, Zinman B, Bolli GB (2001) Insulins today and beyond. Lancet 358:739–746
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (2007) Kurzwirksame Insulinanaloga zur Behandlung des Diabetes mellitus Typ 1: Abschlussbericht; Auftrag A05-02. [Rapid-acting insulin analogues for the treatment of diabetes mellitus type 1: final report A05-02.] Available from www.iqwig.de/download/A05-02_Abschlussbericht_Kurzwirksame_Insulinanaloga_bei_Diabetes_mellitus_Typ_1.pdf. Accessed 20 March 2009 (article in German)
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (2005) Kurzwirksame Insulinanaloga zur Behandlung des Diabetes mellitus Typ 2: Abschlussbericht; Auftrag A05-04. [Rapid-acting insulin analogues for the treatment of diabetes mellitus type 2: final report A05-04.] Available from www.iqwig.de/download/A05-04_Abschlussbericht_Kurzwirksame_Insulinanaloga_bei_Typ_2_Diabetes_mellitus.pdf. Accessed 20 March 2009 (article in German)
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (2009) Langwirksame Insulinanaloga zur Behandlung des Diabetes mellitus Typ 2: Abschlussbericht; Auftrag A05-03. [Long-acting insulin analogues for the treatment of diabetes mellitus type 2: final report A05-03.] Available from www.iqwig.de/download/A05-03_Abschlussbericht_Langwirksame_Insulinanaloga_bei_Diabetes_mellitus_Typ_2_V1.1.pdf. Accessed 20 March 2009 (article in German)
Fricke U, Günther J, Zawinell A (2007) Anatomisch-therapeutisch-chemische Klassifikation mit Tagesdosen für den deutschen Arzneimittelmarkt. Methodik der ATC-Klassifikation und der DDD-Festlegung. ATC-Index mit DDD-Angaben. [The Anatomical Therapeutic Classification with daily doses for the German pharmaceutical market. Methodology of the ATC classification and DDD specification. ATC index with DDD information.] Available from http://wido.de/fileadmin/wido/downloads/pdf_arzneimittel/wido_arz_atc-index_0406.pdf. Accessed 20 March 2009 (article in German)
Hosmer DW, Lemeshow S (1999) Applied survival analysis. Wiley, New York
Cox DR (1972) Regression models and life tables (with discussion). J R Stat Soc Ser B 34:187–220
Kleinbaum DG, Klein M (2005) Survival analysis. A self-learning text, 2nd edn. Springer, New York
Bender R, Lange S (2001) Adjusting for multiple testing—when and how? J Clin Epidemiol 54:343–349
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330:1304–1305
Mühlhauser I, Sawicki PT, Blank M, Overmann H, Richter B, Berger M (2002) Reliability of causes of death in persons with type I diabetes. Diabetologia 45:1490–1497
Horvath K, Jeitler K, Berghold A et al (2007) Long-acting insulin analogues versus NPH insulin (human isophane insulin) for type 2 diabetes mellitus. Cochrane Database Syst Rev (2):CD005613. doi:10.1002/14651858.CD005613.p
Singh SR, Ahmad F, Lal A, Yu C, Bai Z, Bennett H (2009) Efficacy and safety of insulin analogues for the management of diabetes mellitus: a meta-analysis. CMAJ 180:385–397
Acknowledgements
We thank N. McGauran (Institute for Quality and Efficiency in Health Care, Cologne, Germany) for editorial support.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
L. G. Hemkens and U. Grouven contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Baseline characteristics of patients who received human insulin or glargine, categorised according to daily dose received (PDF 73.1 kb)
ESM Table 2
Results of the final Cox regression model for malignant neoplasms (reference group: human insulin) based on data for 127,031 patients (PDF 34 kb)
ESM Table 3
Results of the final Cox regression model for all-cause mortality (reference group: human insulin) based on data for 127,031 patients (PDF 36 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hemkens, L.G., Grouven, U., Bender, R. et al. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia 52, 1732–1744 (2009). https://doi.org/10.1007/s00125-009-1418-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1418-4