
Abstract
Aims/hypothesis
Type 2 diabetes is characterised by excessive hepatic glucose production and frequently leads to systemic vascular complications. We therefore analysed the relationship between the gene expression profile in the liver and the pathophysiology of Type 2 diabetes.
Methods
Liver biopsy samples were obtained from twelve patients with Type 2 diabetes and from nine non-diabetic patients. To assay gene expression globally in the livers of both groups, we made complementary DNA (cDNA) microarrays consisting of 1080 human cDNAs. Relative expression ratios of individual genes were obtained by comparing cyanine 5-labelled cDNA from the patients with cyanine 3-labelled cDNA from reference RNA from the liver of a non-diabetic patient.
Results
On assessing the similarities of differentially expressed genes, the gene expression profiles of the twelve diabetic patients formed a separate cluster from those of the non-diabetic patients. Of the 1080 genes assayed, 105 (9.7%) were up-regulated and 134 (12%) were down-regulated in the diabetic livers (p<0.005). The genes up-regulated in the diabetic patients included those encoding angiogenic factors such as vascular endothelial growth factor, endothelin and platelet-derived growth factor. They also included TGF superfamily genes such as TGFA and TGFB1 as well as bone morphogenetic proteins. Among the down-regulated genes in the diabetic patients were molecules defending against stress, e.g. flavin-containing monooxygenase and superoxide dismutase.
Conclusions/interpretation
These findings suggest that livers of patients with Type 2 diabetes have gene expression profiles indicative of an increased risk of systemic vascular complications.
Similar content being viewed by others

Introduction
Type 2 diabetes, the most common form of diabetes mellitus, results from the interaction of genetic and environmental factors. Although a few cases of Type 2 diabetes involve monogenic syndrome, in which the onset of disease in young patients is predictable, the majority of cases involve multiple genetic alterations [1]. The liver plays a central role in glucose homeostasis, and Type 2 diabetes is characterised by excessive hepatic glucose production. Due to its long duration, the disease causes systemic vascular complications such as micro- and macroangiopathy. Indeed, the liver is also a major source of angiogenic factors and cytokines involved in the development of atherosclerosis, e.g. vascular endothelial growth factor (VEGF) and TGF [2, 3]. For this reason, a comprehensive analysis of gene expression in the liver could deepen understanding of Type 2 diabetes and its related complications.
The recently developed complementary DNA (cDNA) microarray technology allows simultaneous, parallel analysis of the expression of hundreds to thousands of genes in cell lines or tissues, as well as the investigation of drug-induced changes in gene expression [4]. We have already used cDNA microarrays, consisting of 1080 human cDNAs, to characterise alterations that occur in various liver diseases [5, 6]. More recently, microarray profiling of skeletal muscle tissue of Type 2 diabetic patients [7, 8] and of healthy humans treated with insulin [9] has been reported. Although the liver is central for glucose homeostasis, microarray profiling of diabetic liver has been done only in Ob/Ob mice, a genetically obese rodent model of diabetes [10, 11, 12]. We have now used it to clarify the transcriptional alterations in the liver that are associated with the pathophysiology of Type 2 diabetes. We have also used this methodology to determine differentially expressed liver genes that are representative of increased risk for systemic diabetic vascular complications.
Subjects and methods
Patients
The subjects were twelve patients with Type 2 diabetes and nine non-diabetic patients serially recruited between 1998 and 2001 at Kanazawa University Hospital, Japan. There were no significant differences in age, BMI and liver function tests between diabetic and non diabetic patients (Table 1). Informed consent was obtained from all subjects, and the experimental protocol was approved by our institution.
All subjects tested negative for hepatitis B and C viruses. The diabetic patients were diagnosed according to criteria established by an expert committee on the diagnosis and classification of diabetes mellitus [1]. The diabetic patients (DM 1 to DM 12) were treated either with diet therapy alone (DM 7, 10 and 11), or with an α-glucosidase inhibitor (DM 6 and 8) or with insulin (DM 1 to 5, 9 and 12). None of them was taking any other oral hypoglycaemic agent. Pharmacological treatments other than anti-diabetic treatments included diltiazem hydrochloride (DM 3), nicorandil (DM 6) and nifedipine (DM 6) for angina pectoris. No statins, angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers were being taken.
Informed consent was obtained from the diabetic patients both for this study and for the histolological examination of liver diseases, including non-alcoholic steatohepatitis (NASH), which is often associated with diabetes [13]. Liver biopsy specimens were obtained from all diabetic patients, immediately frozen in liquid nitrogen and stored at −80 °C until use. The nine non-diabetic subjects (Non-DM 1 to 9) were patients undergoing resection for colon cancer; for each, a tissue specimen was surgically removed from the non-cancerous part of the liver. The non-diabetic subjects were not taking medication, except anti-indigestion or anti-ulcer agents. Informed consent was also obtained from them for this study.
The study was approved by the relevant ethics committee and was carried out in accordance with the Declaration of Helsinki.
Pathology of the liver
All liver biopsy specimens were examined using haematoxylin-eosin and silver reticulin stain. A pathologist who was blinded to the patients’ clinical condition and biochemical data scored each biopsy for steatosis, inflammation and fibrosis, using previously reported criteria [14, 15]. Steatosis was scored as 0 (no steatosis), 1 (in less than 5% of the lobular parenchyma), 2 (in 5–25% of the lobular parenchyma), 3 (in 25–75% of the lobular parenchyma) or 4 (in more than 75% of the lobular parenchyma). Inflammation was scored as 0 (no hepatocyte injury or inflammation), 1 (sparse injury and/or inflammation), 2 (mild focal injury and/or inflammation), 3 (noticeable injury and/or inflammation) or 4 (severe zone 3 hepatocyte injury and/or inflammation). Fibrosis was scored as 0 (normal connective tissue), 1 (focal pericellular fibrosis in zone 3), 2 (perivenular and pericellular fibrosis confined to zones 2 and 3 with or without portal and/or periportal fibrosis), 3 (bridging or extensive fibrosis with architectural distortion) or 4 (cirrhosis). No subjects had severely inflamed or fibrotic livers (score of 3 or higher).
Preparation of cDNA microarray slides
The protocol for preparing microarray slides containing 1080 cDNA clones has been described [5, 6]. The genes to be spotted were selected from the Clontech atlas membrane array and obtained from IMAGE Consortium libraries through its distributor, Research Genetics (Huntsville, Ala., USA). Each sequence-verified clone was repeatedly sequenced in our laboratory to avoid using cross-contaminated clones. Sequence homology was confirmed using the advanced BLAST search program (http://www.ncbi.nlm.nih.gov/BLAST/). When sequencing failed to verify the purity of IMAGE clones for key genes, corresponding clones were obtained from RIKEN Gene Bank (Takaodai, Tukuba, Japan) or through personal communication. Of the 1080 cDNA clones used for analysis, 1001 were sequence-verified and 79 were sequence-unverified human expressed sequence tag clones. As negative controls for each assay, we used Firefly luciferase (Promega, Madison, Wis., USA) sequences, which have no homology to sequences in the human genome. Polymerase chain reaction products prepared from these clones were spotted on to glass slides with 16 pins using SPBIO 2000 (Hitachi Software, Fukuoka, Japan). To normalise varying efficiencies of labelling and detection, a series of housekeeping genes (encoding β-actin, ribosomal protein L32, GAPDH and albumin), as well as firefly luciferase sequences, were spotted in each of the 16 rectangles of DNA spots.
RNA isolation and antisense RNA amplification
Total RNA was isolated from frozen liver tissue samples using an RNA extraction kit [5], and first-strand cDNA was synthesised with Superscript II reverse transcriptase (GIBCO-BRL, Gaithersburg, Md., USA). Second-strand cDNA was synthesised with Escherichia coli DNA ligase and E. coli DNA polymerase (both from New England Biolabs, Beverly, Mass., USA) as described [5], and double-stranded cDNA was purified by phase lock gel (Eppendorf, Westbury, N.Y., USA) with phenol/chloroform extraction. We subsequently used double-stranded cDNA as a template for in vitro antisense RNA (aRNA) transcription and amplification using a MEGA script T7 kit (Ambion, Austin, Tex., USA) according to the manufacturer’s protocol. The resulting aRNA was purified on an affinity resin column (RNeasy Mini Kit, Qiagen, Chatsworth, Calif., USA). All aRNAs were stored at −80 °C until use.
Preparation of fluorescence-labelled cDNA and microarray hybridisation
To label the probes, approximately 2 to 3 µg of aRNA was used as a template for first-strand cDNA synthesis with cyanine (Cy) 3- or Cy5-deoxyuridine triphosphate (Amersham Biosciences, Piscataway, N.J., USA) and Superscript II reverse transcriptase [5]. As a reference for each microarray analysis, we used aRNA samples prepared from the normal liver tissue of a 72-year-old man with a single liver tumour. Reference aRNAs were labelled with Cy3 and test sample aRNAs with Cy5. The labelled probes were purified on Microcon 30 columns (Millipore, Bedford, Mass., USA), and then the blocking reagents 10 µg yeast transfer RNA, 4 µg poly(dA) and 15 µg human Cot DNA were added to each probe and each mixture was concentrated to 12 µl. We then added 2.55 µl 20× standard saline citrate (SSC) and 0.45 µl 10% sodium dodecyl sulfate to each mixture, and each 15 µl aliquot was used as a hybridisation probe for each cDNA-spotted slide. The slides were covered with glass coverslips and fixed in a hybridisation cassette (TeleChem, Sunnyvale, Calif., USA), then hybridisation was performed for 12 hours at 65 °C. Slides were washed in 2× SSC and 0.03% sodium dodecyl sulfate, in 1× SSC and in 0.2× SSC; each washing lasted 5 minutes.
Image analysis
Quantitative assessment of the signals on the slides was done by scanning on a ScanArray 5000 (General Scanning, Watertown, Mass., USA), followed by image analysis using ImaGene 3.0 software (Bio Discovery, Los Angeles, Calif., USA). The signal intensity of each spot was corrected by subtracting adjacent background signals. To normalise the data, we averaged the intensities of all spots obtained with Cy3 and Cy5 in each of the 16 rectangles and adjusted the intensity of each corrected DNA spot by the average intensity ratio of Cy5:Cy3 (=1.0). This global normalisation of intensity provided a smaller variance of the Cy5:Cy3 ratio and almost the same results as normalisation using the housekeeping genes. Because signal values of approximately 500 to 600 were obtained for luciferase genes, which have no homology to any human gene sequence, all values below 600 were set as background values.
Hierarchical clustering of the gene expression of the patients was assessed by calculating Pearson’s product–moment correlation coefficient using the program Cluster, and by visualisation using ‘Tree View’ (http://genexpress.stanford.edu). The data were log-transferred, normalised, mean-centred and applied to the average linkage clustering. The resulting dendrogram indicated the order in which patients were grouped according to similarities in their gene expression patterns. The gene cluster data were presented graphically as coloured images, and the genes that had been analysed were arranged as ordered by the clustering algorithm, so that genes with the most similar expression patterns were adjacent to each other.
Real-time quantitative PCR
Double-strand cDNA was used as a template for real-time quantitative PCR of bone morphogenetic protein-4 (BMP4), TGFB1, endothelin and glutathione S-transferase theta 1. To do this we used the ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, Calif., USA). The sets of primers and TaqMan probes were proprietary to Applied Biosystems (Assays-on-Demand gene expression product). To control for variation in the amount of DNA available for PCR in the different samples, gene expression of the target sequence was normalised relative to expression of an endogenous control, 18S ribosomal RNA (18S rRNA TaqMan Control Reagent Kit, Applied Biosystems). The PCR conditions were 1 cycle at 50 °C for 2 min and at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and at 60 °C for 1 min.
Statistical analysis
All data are expressed as means ± SEM. To test the significance of numbers and frequencies of genes or Cy5:Cy3 ratios, we used supervised analyses with the permutation-based method (BRB-ArrayTools, http://linus.nci.nih.gov/BRB-ArrayTools.html) [16]. This software for the statistical analysis of cDNA microarray gene expression data was developed by the Biometric Research Branch of the National Cancer Institute (USA). It contains a class comparison tool based on univariate F tests to find genes differentially expressed between predefined clinical groups. The permutation distribution of the F statistic, based on 2000 random permutations, was also used to confirm statistical significance. A p value of less than 0.005 was considered significant.
Results
Histological findings in the livers of Type 2 diabetic patients
The clinical characteristics of the diabetic and non-diabetic patients were similar, except for diabetic markers (Table 1). Histological examination of the livers of Type 2 diabetic patients revealed various degrees of steatosis, inflammation and fibrosis (Table 2), although all had normal liver function (Table 1). We did not identify any patients with advanced NASH, or whose livers revealed severe inflammation or fibrosis (score of 3 or more). The livers of all non-diabetic patients were histologically normal (Table 2).
Hierarchical clustering of gene expression
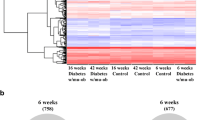
Hierarchical clustering of gene expression in the livers of all 21 patients was assessed by calculating Pearson’s product–moment correlation coefficient (Fig. 1). For differentially expressed genes, the patients clustered into two groups, with one cluster being the twelve diabetic patients and the second cluster being the nine non-diabetic patients. The presence of diabetes was the only clinical determinant of gene expression contributing to clustering. In contrast, age, sex and BMI were not clinical determinants of gene expression profiling (data not shown). Differences in the clustering of diabetic and non-diabetic patients was observed using three subsets of genes: (i) transcription factors (95 genes); (ii) cytokines and growth factors (154 genes); and (iii) apoptosis-related genes (114 genes) (data not shown). These results suggest that the livers of diabetic patients have different patterns of gene expression than livers of non-diabetic persons.

Hierarchical clustering analysis of gene expression profiles in the livers of twelve diabetic and nine non-diabetic patients. Genes up-regulated in diabetic livers are shown in red; genes down-regulated in diabetic livers are in green. DM, Type 2 diabetic patient; Non-DM, non-diabetic patient
Identification of differentially expressed genes in the livers of Type 2 diabetic patients
In characterising the differentially expressed genes in the livers of patients with Type 2 diabetes, we found that of the 1080 sequences assayed 105 (9.7%) were significantly up-regulated and 134 (12%) were significantly down-regulated, compared with those of non-diabetic patients (p<0.005 by univariate F tests) (Tables 3 and 4). Although our cDNA microarrays did not contain many genes associated with glucose and lipid metabolism, expression of other genes was differentially altered in patients with Type 2 diabetes.
Interestingly, the genes significantly up-regulated included: (i) those encoding angiogenic factors, such as VEGF, endothelin and platelet-derived growth factor (PDGF); (ii) members of the TGF superfamily, including TGF-α and BMP-4 and −7; and (iii) collagens IV and VIII (Table 3). Also up-regulated were genes coding for pro-inflammatory cytokines or their receptors, e.g. interleukin 1 alpha, fibroblast growth factor and tumour necrosis factor receptor, and stress-response proteins, including glutathione S-transferase T1 and 8-oxoguanine DNA glycosylase (Table 3).
In contrast, the following genes were down-regulated in diabetic compared with non-diabetic livers: genes coding for general transcription factors, such as TFII and Jun; genes coding for lipogenic enzymes, including fatty acid desaturase and CD36 (fatty acid translocase); and genes coding for the ketogenic enzyme, acetoacetyl coenzyme A thiolase (Table 4). Also down-regulated in the livers of diabetic patients were genes encoding cell–cell interaction molecules, such as fibronectin, vascular cell adhesion molecule (VCAM)-1, selectin L, ficolin, decorin and MHC classes I and II, as well as stress defence molecules like flavin containing monooxygenase (FMO) 5 and superoxide dismutase 2 (SOD2) (Table 4).
Real-time PCR analyses
Real-time PCR confirmed that expression of BMP4 was significantly up-regulated in the livers of diabetic patients (Fig. 2a), with expression levels of BMP4 as assessed by real-time PCR and cDNA microarray well correlated with each other (r=0.58, p=0.0053) (Fig. 2b). Real-time PCR also confirmed that the expression of endothelin and glutathione S-transferase T1 were up-regulated in the livers of diabetic patients (Fig. 2c, d). Because the microarray analysis was not sufficiently sensitive to detect TGFB1 gene expression, we used real-time PCR to assay expression of this gene. As with other members of the TGF superfamily, expression of TGFB1 message was up-regulated in the liver of diabetic patients, relative to that of non-diabetic patients (Fig. 2e).

Expression of gene transcripts by real-time quantitative PCR (a–e) and (b) gene expression levels assessed by cDNA microarray and real-time quantitative PCR. Double-strand complementary DNAs (cDNA) were used as templates for expression of bone morphogenetic protein 4 (BMP4) (a, b), endothelin (c), glutathione S-transferase theta 1 (d) and TGFB1 (e) mRNAs. Expression of each target sequence was normalised relative to the expression of 18S ribosomal RNA. Each bar represents means ± SD. The correlation (b) between BMP4 gene expression levels assessed by cDNA microarray and real-time quantitative PCR was significant (r=0.58, p=0.0053). ●, Type 2 diabetic patients; ○, non-diabetic patients. Non-DM, liver of non-diabetic patients; DM, liver of Type 2 diabetic patients; Cy, cyanine. * p<0.05 vs Non-DM
Discussion
In assessing gene expression profiles in the livers of patients with and without Type 2 diabetes using cDNA microarray analyses and real-time quantitative PCR, we found that the twelve diabetic patients formed a separate cluster from the nine non-diabetic patients. This was especially evident in three subsets of differentially expressed genes encoding (i) transcription factors, (ii) cytokines and growth factors, and (iii) proteins associated with apoptosis. Diabetes is known to cause histological changes in the liver including NASH [13], and the difference in expression levels between diabetic and non-diabetic livers could therefore be related to these histological changes.
In the diabetes group, expression of 105 genes was significantly up-regulated, while expression of 134 genes was significantly down-regulated. Of those up-regulated, several belong to the TGF superfamily, including BMP4, BMP7, TGFA and TGFB1. Angiogenesis is an essential biological process, not only in embryogenesis but also in the progression of diabetic vascular complications. One member of the TGF superfamily, TGF-β, plays a critical role in angiogenesis [2], and also mediates expression of angiogenic factors such as VEGF [3], which we also found to be up-regulated in livers of diabetic patients. Another member, BMP-4, was recently found to stimulate VEGF synthesis [17, 18]. In development, VEGF acts upstream of the Notch pathway to determine arterial cell fate [19]. Notch signalling plays a critical role in vasculogenesis and angiogenesis [20]. In the absence of VEGF signalling, activation of the Notch pathway can rescue gene expression of arterial markers such as ephrin [20].
We also found that other cytokines involved in angiogenesis and the development of diabetic angiopathy, including endothelin [21] and PDGF [22], were up-regulated in diabetic livers. High concentrations of glucose enhance endothelin-1 expression, partly due to activation of protein kinase C, and may act to reduce retinal blood flow during the development of diabetic retinopathy [23]. In addition, receptors for PDGF and endothelin are up-regulated in the skeletal muscle of insulin-resistant Pima Indians [7]. Together, these alterations in gene expression in the diabetic liver could increase the risk for the systemic vascular complications associated with Type 2 diabetes, including diabetic retinopathy and ischaemic heart disease.
Plasmin is responsible for the proteolysis of extracellular matrix components, which prevent fibrosis and angiogenesis [24]. The down-regulation of plasminogen observed by us in the diabetic liver could thus result in reduced fibrinolysis, leading to thrombosis and atherosclerosis. Down-regulation of plasminogen has also been reported in the livers of ob/ob mice [11].
Recently, thrombospondin 2 gene polymorphism [25] and serum paraoxonase activity [26] were reported to predict coronary events. Mice deficient in thrombospondin 2 have a phenotype that could reduce the risk of myocardial infarction [27]. Thus the up-regulation of thrombospondin 2 and down-regulation of paraoxonase in the diabetic liver, as observed in this study, might be associated with increased risk of coronary artery disease.
Hyperglycaemia increases the production of reactive oxygen species, which may activate protein kinase C, induces advanced glycation end-product formation and activates the pleiotropic transcription factor, nuclear factor-kappa B [28]. Increases in reactive oxygen species are prevented by SOD2 [28]. Impaired expression of SOD2 or other genes associated with a stress defence system, including FMO5, as observed here and in ob/ob mice [11], could increase oxidative stress in the diabetic state. Similar alterations in gene expression in blood vessels may increase the risk of atherosclerosis.
Several of the genes we found aberrantly expressed in livers of patients with Type 2 diabetes can also be altered in other organs of diabetic patients. For example, up-regulation of Rad (a Ras-oncogene associated with diabetes), heat shock 70kD protein, and receptors for PDGF and endothelin, and down-regulation of general transcription and translation factors, and of cadherin and MHC have been observed in the skeletal muscle of insulin-resistant Pima Indians [7] and in Type 2 diabetic patients [8]. In addition, up-regulation of epidermal growth factor receptor and down-regulation of fatty acid-associated enzymes, fibronectin, VCAM1, MHC, plasminogen, SOD2, FMO5 and complement C3 have been observed in the liver of ob/ob mice [10, 11, 12].
We also observed, in the livers of Type 2 diabetic patients, reduced expression of the genes encoding some enzymes associated both with glucose metabolism (glucose phosphate isomerase, aspartylglucosaminidase and cytosolic acetoacetyl-coenzyme A thiolase) and with fatty acid metabolism (fatty acid desaturase and fatty acid translocase). Whilst this paper was being prepared, two other groups independently found a coordinated reduction of PGC1-responsive genes involved in oxidative metabolism in the skeletal muscle of Type 2 diabetic patients [29, 30]. A large-scale cDNA microarray containing comprehensive genes associated with metabolism could be used to find out whether these alterations also exist in the Type 2 diabetic liver.
In summary, the gene expression profile of Type 2 diabetic livers is different to that of the normal liver. Caution is necessary when generalising on the relevance of this observation for global cardiovascular complications. And while the direct contribution of these differential genes to systemic complications will, no doubt, be explained in the future, we suggest from our findings that clustering analyses and the identification of differentially expressed genes in the liver could be useful for clarifying the pathogenesis of Type 2 diabetes and its related complications.
Abbreviations
- aRNA:
-
antisense RNA
- BMP:
-
bone morphogenetic protein
- cDNA:
-
complementary DNA
- Cy:
-
cyanine
- FMO:
-
flavin-containing monooxygenase
- NASH:
-
non-alcoholic steatohepatitis
- PDGF:
-
platelet-derived growth factor
- SSC:
-
standard saline citrate
- SOD:
-
superoxide dismutase
- VCAM:
-
vascular cell adhesion molecule
- VEGF:
-
vascular endothelial growth factor
References
Expert committee on the diagnosis and classification of diabetes mellitus (2003) Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 25:S5–S20
Larsson J, Goumans MJ, Sjostrand LJ et al. (2001) Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. Embo J 20:1663–1673
Pertovaara L, Kaipainen A, Mustonen T et al. (1994) Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem 269:6271–6274
Schena M, Shalon D, Davis R, Brown P (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470
Honda M, Kaneko S, Kawai H, Shirota Y, Kobayashi K (2001) Differential gene expression between chronic hepatitis B and C hepatic lesion. Gastroenterology 120:955–966
Shirota Y, Kaneko S, Honda M, Kawai HF, Kobayashi K (2001) Identification of differentially expressed genes in hepatocellular carcinoma with cDNA microarrays. Hepatology 33:832–840
Yang X, Pratley RE, Tokraks S, Bogardus C, Permana PA (2002) Microarray profiling of skeletal muscle tissues from equally obese, non-diabetic insulin-sensitive and insulin-resistant Pima Indians. Diabetologia 45:1584–1593
Sreekumar R, Halvatsiotis P, Schimke JC, Nair KS (2002) Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 51:1913–1920
Rome S, Clement K, Rabasa-Lhoret R et al. (2003) Microarray profiling of human skeletal muscle reveals that insulin regulates approximately 800 genes during a hyperinsulinemic clamp. J Biol Chem 278:18063–18068
Ferrante AW Jr, Thearle M, Liao T, Leibel RL (2001) Effects of leptin deficiency and short-term repletion on hepatic gene expression in genetically obese mice. Diabetes 50:2268–2278
Liang CP, Tall AR (2001) Transcriptional profiling reveals global defects in energy metabolism, lipoprotein, and bile acid synthesis and transport with reversal by leptin treatment in ob/ob mouse liver. J Biol Chem 276:49066–49076
Lan H, Rabaglia ME, Stoehr JP et al. (2003) Gene expression profiles of nondiabetic and diabetic obese mice suggest a role of hepatic lipogenic capacity in diabetes susceptibility. Diabetes 52:688–700
Reid AE (2001) Nonalcoholic steatohepatitis. Gastroenterology 121:710–723
Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR (1999) Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 94:2467–2474
Lee RG (1998) Alcoholic and nonalcoholic steatohepatitis pathology. In: Bloomer J, Goodman ZD, Ishak KG (eds) Clinical and pathological correlations in liver disease: approaching the next millennium. The Armed Forces Institute of Pathology, the American Registry of Pathology, the American Association for the Study of Liver Disease, Washington DC, pp 274–283
Ye QH, Qin LX, Forgues M et al. (2003) Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med 9:416–423
Deckers MM, Bezooijen RL van, Horst G van der et al. (2002) Bone morphogenetic proteins stimulate angiogenesis through osteoblast-derived vascular endothelial growth factor A. Endocrinology 143:1545–1553
Tokuda H, Hatakeyama D, Shibata T, Akamatsu S, Oiso Y, Kozawa O (2003) p38 MAP kinase regulates BMP-4-stimulated VEGF synthesis via p70 S6 kinase in osteoblasts. Am J Physiol Endocrinol Metab 284:E1202–E1209
Lawson ND, Vogel AM, Weinstein BM (2002) Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell 3:127–136
Liu ZJ, Shirakawa T, Li Y et al. (2003) Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol Cell Biol 23:14–25
Sarman B, Toth M, Somogyi A (1998) Role of endothelin-1 in diabetes mellitus. Diabetes Metab Rev 14:171–175
Inaba T, Ishibashi S, Gotoda T et al. (1996) Enhanced expression of platelet-derived growth factor-beta receptor by high glucose. Involvement of platelet-derived growth factor in diabetic angiopathy. Diabetes 45:507–512
Park JY, Takahara N, Gabriele A et al. (2000) Induction of endothelin-1 expression by glucose: an effect of protein kinase C activation. Diabetes 49:1239–1248
Blakesley VA (2000) The role of growth factors in the pathogenesis of diabetic microvascular complications. In: LeRoith D, Taylor SI, Olefsky JM (eds) Diabetes mellitus. A fundamental and clinical text. Lippincott Williams & Wilkins, Philadelphia, pp 1000–1008
Boekholdt SM, Trip MD, Peters RJ et al. (2002) Thrombospondin-2 polymorphism is associated with a reduced risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol 22:e24–e27
Mackness B, Durrington P, McElduff P et al. (2003) Low paraoxonase activity predicts coronary events in the Caerphilly Prospective Study. Circulation 107:2775–2779
Kyriakides TR, Zhu YH, Smith LT et al. (1998) Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol 140:419–430
Nishikawa T, Edelstein D, Du XL et al. (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Patti ME, Butte AJ, Crunkhorn S et al. (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100:8466–8471
Mootha VK, Lindgren CM, Eriksson KF et al. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273
Acknowledgements
We thank Dr Gen-ichi Nishimura and Professor Koichi Miwa from the Department of Surgery II, Kanazawa University for generously providing the liver tissue samples.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Takamura, T., Sakurai, M., Ota, T. et al. Genes for systemic vascular complications are differentially expressed in the livers of Type 2 diabetic patients. Diabetologia 47, 638–647 (2004). https://doi.org/10.1007/s00125-004-1366-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1366-y