
Abstract
Aims/hypothesis
ATP-sensitive potassium (KATP) channels are crucial for the regulation of insulin secretion from pancreatic beta cells and mutations in either the Kir6.2 or SUR1 subunit of this channel can cause congenital hyperinsulinism (CHI). The aim of this study was to analyse the functional consequences of four CHI mutations (A1457T, V1550D and L1551V in SUR1, and K67N in Kir6.2) recently identified in the Finnish population.
Methods
Wild type or mutant Kir6.2 and SUR1 subunits were coexpressed in Xenopus oocytes. The functional properties of the channels were examined by measuring currents in intact oocytes or giant inside-out membrane patches. Surface expression was measured by enzyme-linked immunosorbance assay, using HA-epitope-tagged subunits.
Results
Two mutations (A1457T and V1550D) prevented trafficking of the channel to the plasma membrane. The L1551V mutation reduced surface expression 40-fold, and caused loss of MgADP and diazoxide activation. Both these factors will contribute to the lack of KATP current activation observed in response to metabolic inhibition in intact oocytes. The L1551V mutation also increased the channel open probability, thereby producing a reduction in ATP-sensitivity (from 10 µmol/l to 120 µmol/l). The fourth mutation (K67N mutation in Kir6.2) did not affect surface expression nor alter the properties of KATP channels in excised patches, but resulted in a reduced KATP current amplitude in intact cells on metabolic inhibition, through an unidentified mechanism.
Conclusion/interpretation
The four CHI mutations disrupted KATP channel activity by different mechanisms. Our results are discussed in relation to the CHI phenotype observed in patients with these mutations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Congenital hyperinsulinaemia (CHI) is characterised by unregulated insulin secretion that results in persistent and recurrent hypoglycaemia in infancy [1]. As in most populations, the incidence of CHI in Finland is only about 1:50,000 [2, 3], but it is a major cause of neurological damage and life-long handicap if not treated promptly [4]. In most families, CHI is inherited in an autosomal recessive manner, but dominant forms of the disease have also been described [5, 6].
Mutations in four genes are known to cause CHI: the pancreatic beta-cell ATP-sensitive potassium (KATP) channel subunits Kir6.2 and SUR1 [7, 8, 9], glucokinase [10] and glutamate dehydrogenase [6, 11]. Mutations in KATP channel subunits account for about 50% of cases. In up to 30% of these subjects, the condition is caused by focal adenomatous hyperplasia (focal CHI) due to loss of maternal imprinted alleles, allowing the penetrance of a paternal Kir6.2/SUR1 mutation [12, 13]. The incidence of CHI due to mutations in Kir6.2, glucokinase and glutamate dehydrogenase is low, and the molecular basis of CHI in about 50% of subjects has not been established [7].
KATP channels regulate the membrane potential of pancreatic beta cells [14, 15]. These channels are open at low glucose concentrations, allowing K+ efflux to maintain the plasma membrane at a hyperpolarised potential. Closure of KATP channels in response to rising glucose concentrations, or sulphonylureas, leads to beta-cell depolarization that triggers the opening of voltage-gated Ca2+ channels, Ca2+ influx and insulin secretion. The most important mediators of glucose-stimulated KATP channel closure are believed to be the rising intracellular concentration of ATP and the corresponding decrease in MgADP, because ATP is an inhibitor and MgADP an activator of the KATP channel [14, 15]. All CHI mutations are thought to lead to loss of beta-cell KATP channel function, either as a consequence of defects in the channel itself (SUR1, Kir6.2) or in its metabolic regulation (glucokinase, glutamate dehydrogenase). This produces a persistent depolarization that leads to Ca2+ influx and continuous insulin secretion that is independent of the blood glucose concentration [8, 15, 16].
The beta-cell KATP channel is a hetero-octameric complex of Kir6.2 and SUR1 subunits, members of the inwardly rectifying K+ channel and ABC transporter gene families, respectively (Fig. 1) [15]. Four Kir6.2 subunits assemble to form the channel pore, through which K+ flux occurs. This subunit also contains the site at which ATP binds to cause channel inhibition. Each Kir6.2 subunit associates with a regulatory SUR1 subunit that confers sensitivity to inhibition by sulphonylureas, and activation by diazoxide and MgADP [17, 18]. To date, about 50 mutations in SUR1 and three in Kir6.2 have been reported [7, 8]. Most, but not all, of these mutations show a recessive pattern of inheritance. The Finnish SUR1 mutation E1506K is one exception, showing a dominant mode of inheritance [5]. Mutations in KATP channel subunits have been shown to give rise to CHI by several mechanisms. These include the production of prematurely truncated, non-functional SUR subunits that are rapidly degraded, defective trafficking of the KATP channel to the surface membrane [19, 20], and loss of metabolic sensitivity due to the absence of MgADP activation [5, 21, 22].

Localisation of mutations in KATP-channel subunits associated with CHI. Membrane topology of the beta-cell KATP-channel subunits, Kir6.2 and SUR1, indicating the locations of the CHI mutations found in the Finnish population [23]. The mutations V187D and E1506K have been described previously [3, 5]. WA and WB indicate the position of the Walker A and B motifs of the nucleotide binding folds (NBDs) respectively
We have characterised the properties of four new KATP channel mutations (Fig. 1), recently identified in the Finnish population [23]. Three of these mutations are found in the C-terminal domain of SUR1 (L1551V, A1457T and V1550D), and one is found in the N-terminus of Kir6.2 (K67N). Using surface membrane expression assays and functional studies of KATP channel activity in Xenopus oocytes, we show that these mutations give rise to the CHI phenotype by different mechanisms.
Methods
Molecular biology
Mouse Kir6.2 (GenBank D50581) and rat SUR1 (GenBank L40624) were used in this study. Mutations were made in Kir6.2 or SUR1 as described [18]. cDNAs were cloned into the pBF vector and synthesis of capped mRNA was carried out using the mMessage mMachine large-scale in vitro transcription kit (Ambion, Austin, Tex., USA).
Electrophysiology
Female Xenopus laevis were anaesthetised with MS222 (2 g l−1 added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Once the wound had completely healed, the second ovary was removed in a similar operation and the animal was then killed by decapitation whilst under anaesthesia. Immature stage V-VI Xenopus oocytes were incubated for 60 min with 1.0 mg ml−1 collagenase A (Roche Molecular Biochemicals, Mannheim, Germany) and manually defolliculated. Oocytes were coinjected with 0.1 ng full-length wild-type or mutant Kir6.2 and 2 ng of wild-type or mutant SUR1 (giving a 1:20 ratio). The final injection volume was about 50 nl per oocyte. Isolated oocytes were maintained in tissue culture and studied 1 to 4 days after injection.
Whole-cell currents were recorded from intact oocytes at 20 to 24°C using a two-electrode voltage clamp (Geneclamp 500; Axon Instruments, Foster City, Calif., USA), filtered at 1 kHz and digitized at 4 kHz [24]. Whole-cell currents were measured 280 to 295 ms after the start of the voltage pulse. Oocytes were constantly perfused with a solution containing (in mmol/l): 90 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES (pH7.4 with KOH). Metabolic inhibition was produced by perfusion with 3 mmol/l Na-azide.
Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of 0 mV and at 20 to 24°C [24]. Currents were evoked by repetitive 3 s voltage ramps from −110 mV to +100 mV and recorded using an EPC7 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany). They were filtered at 0.2 kHz, digitised at 0.5 kHz using a Digidata 1200 Interface and analysed using pClamp software (Axon Instruments, Foster City, Calif., USA). For macropatch recordings, the pipette solution contained (mmol/l): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 HEPES (pH 7.4 with KOH) and the internal (bath) solution contained (mmol/l): 107 KCl, 2 MgCl2, 1 CaCl2, 30 KOH, 10 EGTA, 10 HEPES (pH 7.2 with KOH) plus nucleotides or drugs as indicated. In some experiments, a Mg-free internal solution was used (mmol/l): 107 KCl, 2.6 CaCl2, 30 KOH, 10 EDTA, 10 HEPES (pH 7.2 with KOH). Solutions containing nucleotides were made up fresh each day, and the pH subsequently readjusted, if required. Rapid exchange of internal solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath.
Whole-cell currents were monitored in response to voltage steps of ±20 mV from a holding potential of −10 mV. Currents evoked by the hyperpolarizing step were measured at steady state for each condition. For macropatch recordings, we measured the slope conductance by fitting a straight line to the current-voltage relation between −20 and −100 mV: the average of five consecutive ramps was calculated in each solution. To control for possible rundown, the control conductance was taken as the mean of that obtained in control solution before and after application of test compounds. ATP concentration-response curves were fit by the Hill equation: G/Gc=1/(1+([ATP]/IC50)h), where [ATP] is the ATP concentration, IC50 is the ATP concentration at which inhibition is half maximal and h is the slope factor (Hill coefficient).
Single-channel currents were recorded using the same solutions as for the macropatch experiments from small inside-out membrane patches at −60 mV. They were filtered at 5 kHz, sampled at 20 kHz, and analysed using a combination of pClamp and in-house software written by Dr. P. Smith (Oxford University, Oxford, UK) as described previously [25]. Open probability and lifetime distributions were determined from current records of about 1 min duration containing only one open level. Events were detected using a 50% threshold level method. Burst analysis was carried out as described [26].
Surface expression assay
Surface expression was assessed using an enzyme-linked immunosorbance assay (ELISA) [27]. An HA epitope (YPYDVPDYA) was introduced at residue 102 in the extracellular loop between TM1 and TM2 of Kir6.2, and at residue 1281 in the extracellular loop between TMs 16 and 17 of SUR1 [27]. Two days after mRNA injection, oocytes were incubated at 4°C for 30 min in ND96 solution (mmol/l: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4) plus 1% Bovine Serum Albumin (BSA) to block non-specific antibody binding. Subsequently, they were incubated at 4°C for 60 min with 1 mg/ml rat monoclonal anti-HA antibody (3F10, Boehringer, Lewes, UK, in 1% BSA/ND96), washed six times at 4°C with ND96 plus 1% BSA, and incubated for 40 min with 2 mg/ml horseradish peroxidase-coupled secondary antibody (goat anti-rat fab fragments, Jackson Immunoresearch, West Grove, Pa., USA). Oocytes were then thoroughly washed in ND96 plus 1% BSA (4°C, 60 min), followed by ND96 alone (4°C, 60 min). Individual oocytes were then added to 50 µl Power Signal Elisa solution (Pierce, Chester, UK) and, after an equilibration period of 10 s, chemiluminescence was quantified in a Turner TD-20/20 luminometer (Sunnyvale, Calif., USA) by integrating the signal over a period of 15 s. Results are given in relative light units (RLU). For calculation of current amplitude as a fraction of surface expression (nA/RLU), both surface expression and azide-induced whole-oocyte currents were measured in the same batch of injected oocytes, on the same day.
Data analysis
All data are given as means ± SEM. The symbols in the figures indicate the mean and the vertical bars one SEM. (where this is larger than the symbol). Statistical significance was tested by Student’s t test or ANOVA, as appropriate.
Results
Effects of metabolism on wild-type or mutant KATP channels
We first compared the effects of metabolic inhibition on oocytes expressing either wild-type or mutant KATP-channels. In control solution, only very small current amplitudes were detected in both wild-type and mutant channels (Fig. 2). Metabolic poisoning with 3 mmol/l azide induced a large increase in Kir6.2/SUR1 currents, a smaller increase in Kir6.2-K67N/SUR1 and Kir6.2/SUR1-L1551V currents, and no significant change in Kir6.2/SUR1-A1457T or Kir6.2/SUR1-V1550D currents. The increase in Kir6.2/SUR1-L1551V current (3.1±0.7 fold, n=8) was comparable to that observed for Kir6.2ΔC36 expressed in the absence of SUR (5.9±1.1 fold, n=5), but much smaller than that found for Kir6.2/SUR1 currents (89±15 fold, n=19). The increase in Kir6.2-K67N/SUR1 currents on metabolic inhibition (34±6fold, n=11) was also smaller than that found for the wild-type channel.

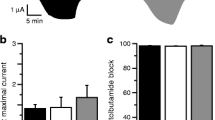
Effects of metabolic inhibition on wild-type or mutant Kir6.2/SUR1 currents. Mean whole-cell current evoked by a voltage step from −10 to −30 mV in control solution (black bars), then after reaching steady state in 3 mmol/l azide (hatched bars), then in the continued presence of azide plus 340 µmol/l diazoxide (white bars) and finally following the further addition of 500 µmol/l tolbutamide (shaded bars). Oocytes were co-injected (as indicated) with mRNAs encoding Kir6.2 plus either SUR1, SUR1-A1457T, SUR1-V1550D or SUR1-L1551V, or with Kir6.2-K67N plus SUR1, or with Kir6.2Δ36 alone. The number of oocytes is given above the bars. Statistical significance (paired t test) is indicated by: NS, non significant; *p<0.05, **p<0.01; ***p<0.001. Inset: mean whole-cell current amplitudes for Kir6.2-L1551V measured at −100 mV shown on an expanded scale
The latencies of current activation by azide and times to reach steady state activation are shown in Table 1. Kir6.2/SUR1 currents started to increase about 200 s after azide addition, and reached a steady state at about 700 s. Kir6.2/SUR1-L1551V currents showed a similar latency, but a very much slower rate of current increase. In contrast, Kir6.2-K67N/SUR1 currents showed delays in both latency and time to steady state amplitude. Kir6.2ΔC36 currents also increased only slowly, but were activated almost immediately after azide was added.
Addition of 340 μmol/l diazoxide, in the continued presence of azide, increased Kir6.2/SUR1 and Kir6.2-K67N/SUR1 currents but did not activate Kir6.2/SUR1-L1551V, Kir6.2/SUR1-A1457T or Kir6.2/SUR1-V1550D currents. Both diazoxide and azide-activated currents were inhibited by the subsequent addition of 500 μmol/l tolbutamide, indicating that they flowed through KATP channels.
Some mutant subunits form functional channels
To determine if the lack of metabolic activation of mutant KATP channels was due to impaired metabolic regulation, or the absence of functional KATP channels in the surface membrane, we measured current amplitudes in giant inside-out membrane patches (Fig. 3A). Oocytes expressing wild-type KATP channels showed small currents in cell-attached patches, which increased dramatically upon excision into nucleotide-free solution. This results from the loss of inhibition by intracellular nucleotides on patch excision. The KATP current amplitude in the inside-out patch was not altered by the K67N mutation in Kir6.2. In contrast, no measurable currents were observed in patches excised from oocytes coexpressing Kir6.2 and either SUR1-A1457T or SUR1-V1550D. The L1551V mutation caused a tenfold reduction in current.

Effects of the CHI-mutations on surface expression. (A) Mean current amplitudes recorded at −100 mV in the cell-attached or inside-out patch configuration from oocytes injected with the indicated mRNAs. The number of oocytes is given above the bars. Statistical significance (t test) is indicated by: NS, non significant; *p<0.05, **p<0.01; ***p<0.001. (B) Surface expression of the indicated KATP channels. Expression is given in relative light units. The number of oocytes is given above the bars. Statistical significance (t test) was tested against uninjected oocytes for Kir6.2HA coinjected with SUR1, SUR1-A1457T, SUR1-V1550D or SUR1-L1551V. Statistical significance (t test) was tested for Kir6.2/SUR1-HA against Kir6.2-K67/SUR1-HA. Increase in RLU:NS, non significant; **p<0.01; ***p<0.001
Surface expression
The reduced (or absent) currents observed for mutant KATP channels in excised patches might result from the lack of protein in the surface membrane, or from channels that fail to open even in the absence of nucleotide. To distinguish between these possibilities, we monitored surface expression using HA-epitope-tagged forms of either Kir6.2 or SUR1. Kir6.2-HA was detectable in the plasma membrane when coexpressed with wild-type SUR1, but not when coexpressed with either SUR1-A1457T or SUR1-V1550D (Fig. 3B). Although coexpression of SUR1-L1551V resulted in detectable surface expression of Kir6.2-HA, the signal was about 40-fold lower than that found for wild-type SUR1. Thus, these mutations prevent (A1457T, V1550D) or reduce (L1551V) plasma membrane targeting of KATP channels expressed in oocytes.
In contrast to the SUR1 mutations, no significant difference was seen in the surface expression of SUR1-HA on co-expression with wild-type Kir6.2 or Kir6.2-K67N (Fig. 3B). However, when expressed as a fraction of surface expression, the azide-activated current was reduced from 3.5±0.8 nA/RLU for Kir6.2/SUR1-HA (n=10) to 0.84±0.14 nA/RLU for Kir6.2-K67N/SUR1-HA (n=11) (p<0.0001). This suggests that the mutant channel is not activated by metabolic poisoning to the same extent as the wild-type channel.
Pharmacological and nucleotide sensitivity of mutant channels
We next investigated the properties of Kir6.2-K67N/SUR1 and Kir6.2/SUR1-L1551V channels in detail. First, we describe the functional effects of the SUR1 mutation and then those of the Kir6.2 mutation.
Kir6.2/SUR1-L1551V currents were inhibited by ATP with an IC50 of 119±12 µmol/l, and a Hill coefficient of 0.99±0.10 (n=8; Fig. 4A). They are thus less ATP sensitive than wild-type Kir6.2/SUR1 channels (IC50=11.7±2.2 µmol/l, n=5), but similar to Kir6.2ΔC36 channels expressed in the absence of SUR (IC50=152±10 μmol/l, n=5). The mutant channels were also blocked by 100 µmol/l MgADP (Fig. 4B), unlike wild-type KATP channels, which are activated by the nucleotide. The block of Kir6.2/SUR1-L1551V produced by MgADP reflects the inhibitory action of ADP on Kir6.2, which becomes evident when the stimulatory action of the nucleotide (mediated via SUR1) is abolished. Kir6.2/SUR1-L1551V channels were also unaffected by diazoxide and inhibited by tolbutamide to a lesser extent than wild type Kir6.2/SUR1 currents (Fig. 4B).

(A) ATP-sensitivity of Kir6.2/SUR1-L1551V and Kir6.2-K67N/SUR1 currents. Mean ATP concentration-response relations for Kir6.2/SUR1 (n=5), Kir6.2/SUR1-L1551V (n=8), Kir6.2-K67N/SUR1 (n=4) and Kir6.2ΔC36 (n=5) currents. The slope conductance (G) is expressed as a fraction of the mean (Gc) of that obtained in control solution before and after the exposure to ATP. The line is the best fit of the data to the Hill equation for Kir6.2/SUR1-L1551V (IC50=119±12 µmol/l, h=1.0±0.1) and Kir6.2-K67N/SUR1 (IC50=6.9±0.8 µmol/l, h=0.9±0.1). The dotted and dashed lines indicate the ATP concentration-response relation obtained for Kir6.2Δ36 (IC50=152±10 µmol/l, h=1.05±0.05) and Kir6.2/SUR1 (IC50=11.7±2.2 µmol/l, h=1.02±0.02). (B) Effects of MgADP, tolbutamide and diazoxide on Kir6.2/SUR1, Kir6.2/SUR1-L1551V and Kir6.2-K67N/SUR1 currents. Mean macroscopic slope conductance recorded in the presence of ADP (100 µmol/l), ATP (100 µmol/l), ATP plus diazoxide (100 and 340 µmol/l, respectively), or ATP plus ADP (100 µmol/l each), or tolbutamide (100 µmol/l), expressed as percentage of the slope conductance in control solution, for Kir6.2/SUR1, Kir6.2/ SUR1-L1551V and Kir6.2-K67N/SUR1 currents. The number of oocytes is given above the bars. Statistical significance (t test, comparing the mutant with the corresponding wild type data) is indicated by: NS, non significant; **p<0.01; ***p<0.001. (C) Spermine sensitivity of Kir6.2-K67N/SUR1 and Kir6.2/SUR1 currents. (i) Macroscopic Kir6.2-K67N/SUR1 and Kir6.2/SUR1 currents recorded from inside-out patches in response to a series of voltage ramps from −110 mV to +100 mV in Mg2+ free solution. Spermine (100 μmol/l and 1 mmol/l) was added to the internal solution as indicated by the bars. (ii) Voltage protocol (top) and mean current responses (bottom) of Kir6.2-K67N/SUR1 and Kir6.2/SUR1 in the presence of 100 μmol/l spermine. Currents from four consecutive ramps, shown in (i), were averaged and scaled to similar size
The altered properties of Kir6.2/SUR1-L1551V channels might be attributable either to an increase in the channel open probability [25], or to a functional uncoupling of Kir6.2 from mutant SUR1. To distinguish between these possibilities, we measured the single-channel properties of Kir6.2/SUR1-L1551V expressed in oocytes. The L1551V mutation did not significantly affect the single-channel conductance, but increased the channel open probability (Po) from 0.22 [25] to 0.65. The increase in Po was due to an increase in the mean open time (τo), an increase in the mean burst duration and a reduction in the percentage of long closed times (τs) (Table 2). As discussed below, these effects probably explain the observed reduction in the inhibitory effects of both ATP and tolbutamide on Kir6.2/SUR1-L1551V currents.
The properties of Kir6.2-K67N/SUR1 channels in excised patches were indistinguishable from those of wild-type channels (Fig. 4B). Thus, Kir6.2-K67N/SUR1 currents were half-maximally blocked by 6.9±0.8 μmol/l ATP (n=4), were activated by MgADP and diazoxide, and were inhibited by tolbutamide. The lysine affected by the K67N mutation is highly conserved throughout the Kir family and this mutation is predicted to remove a positive charge from the cytoplasmic mouth of the pore. We speculated that this mutation might result in an increased sensitivity to polyamines, which bind within the channel pore and contribute to inward rectification [28]. This would reduce the outward current and could therefore produce the CHI phenotype. However, no obvious difference in spermine sensitivity was detected between Kir6.2-K67N/SUR1 and wild-type currents (Fig. 4C). Similar results were observed with putrescine and spermidine (data not shown).
Discussion
Mutations in the genes encoding the beta-cell KATP channel subunits are the most common cause of CHI. Non-functional KATP channels result in continuous depolarization of the beta cell and persistent insulin secretion, which is not linked to the plasma glucose concentration [16]. We investigated the functional effects of four new CHI mutations found in the Finnish population [23]. Three of these mutations are in the SUR1 subunit and the fourth is in Kir6.2.
The new mutations impaired KATP channel activity by different mechanisms. Two of the mutations in SUR1 (A1457T and V1550D) prevented trafficking of the channel to the plasma membrane. This explains the lack of KATP current activation on metabolic poisoning in intact oocytes. KATP channels containing the L1551V mutation in SUR1 had impaired surface expression and altered channel properties, including loss of MgADP activation. Both these factors are likely to reduce KATP channel activity in pancreatic beta cells. The K67N mutation in Kir6.2 did not affect surface expression nor alter the properties of channels in excised patches, but resulted in a reduced current amplitude in intact cells on metabolic inhibition, through an unidentified mechanism.
Functional effects of CHI mutations
Kir6.2/SUR1-L1551V currents showed reduced sensitivity to ATP-inhibition, loss of activation by MgADP and diazoxide and impaired block by tolbutamide. The reduced ATP sensitivity and tolbutamide block are consistent with the increased channel open probability, as similar effects have been observed when the open probability was increased by certain mutations [25], by N-terminal truncation of Kir6.2 [29, 30], or by increasing the concentration of PIP2 [31]. The inhibitory effect of MgADP on Kir6.2/SUR1-L1551V currents was less than that observed when MgADP activation was abolished by mutation of the Walker A lysines in SUR1 [32, 33]. Because both ATP and ADP block the channel by interaction with the same site on Kir6.2, the reduced ADP block might also be explained by the increased channel open probability. However, it is also possible that the mutation does not abolish MgADP activation completely. The lower ATP sensitivity, loss of MgADP activation, and reduced surface expression probably all contribute to the smaller magnitude and slower activation of Kir6.2/SUR1-L1551V channels on metabolic poisoning in intact oocytes.
Kir6.2-K67N/SUR1 currents were indistinguishable from Kir6.2/SUR1 currents in excised patches, but the metabolically sensitive current (corrected for surface expression) in oocytes was approximately half that of the wild-type channel. The time course of current activation was also slower. The reason for this is not clear, but it cannot be explained by an altered sensitivity to ATP, MgADP or polyamines. One possible explanation is that an additional cytoplasmic factor, whose effect is modified by the K67N substitution, is involved in the metabolic response of the KATP channel.
Genotype/phenotype correlations
Two mutations in SUR1 (A1457T and V1550D), that did not result in functional channels in Xenopus oocytes, were each found only once in the Finnish population [23]. In both cases they occurred as part of a complex heterozygous genotype, with the SUR1 mutation V187D on the second allele [3]. As SUR1-V187D also abolished KATP channel activity [3], these two subjects would be predicted to have no functional KATP channels, consistent with the observed severe, drug-resistant CHI phenotype [23].
The third mutation in SUR1 (L1551V) was found in two siblings, who had a milder form of CHI, which was responsive to diazoxide [23]. The mild phenotype and diazoxide-sensitivity of these subjects is consistent with the finding that both siblings were heterozygous for the L1551V mutation and had an apparently normal second SUR1 allele. Although CHI is usually a recessive condition, dominant SUR1 mutations (e.g. SUR1-E1506K) have been reported [5]. The subjects in this study inherited the L1551V mutation from their father, who is not known to have had CHI [23]. Paternally inherited mutant SUR1 subunits have been implicated in focal adenomatous hyperplasia resulting from loss of maternal imprinted genes [9, 12, 13]. However, if this were the case for the L1551V family, the hyperinsulinaemia should not have been sensitive to diazoxide, as the hyperplastic tissue would express only diazoxide-insensitive SUR1-L1551V subunits. One possibility is that the siblings are compound heterozygotes and carry another maternally derived SUR1 mutation that is diazoxide sensitive, but which has not been identified in the genetic screening. An alternative idea is that the phenotypic effect of the L1551V mutation depends on the genetic background of the individual, and that it produces CHI only in those people who carry polymorphisms in other genes that predispose towards enhanced insulin secretion. Indeed there are now a number of cases in the literature of severe CHI associated with diffuse hyperplasia, in which only a single affected Kir6.2 or SUR1 allele could be identified [9].
The K67N mutation in Kir6.2 was found in one subject, who also had a mutation in the 5’UTR of Kir6.2 on the second allele [23]. The 5′UTR mutation is predicted to result in a premature start codon and a scrambled sequence that will result in a total loss of protein. Thus all KATP channel subunits in this individual are likely to contain the K67N mutation. This subject did not respond to octreotide and underwent a subtotal pancreatectomy at the age of 11 days [23]. Despite the severe CHI phenotype, he subsequently showed good insulin responses to glucose and tolbutamide [23], suggesting that his beta cells possessed some functional KATP channels. The CHI phenotype in this individual is likely to be the result of both gene dosage effects and the reduced metabolic response of the mutant KATP channel observed in in vitro experiments.
Change history
26 November 2003
An Erratum to this paper has been published: https://doi.org/10.1007/s00125-003-1211-8
Abbreviations
- CHI:
-
Congenital hyperinsulinism
- HA:
-
haemagluttinin
- KATP channel:
-
ATP-sensitive potassium channel
- Po:
-
open probability
- PIP2 :
-
phosphatidyl inositol bis-phosphate
- RLU:
-
relative light units
- SUR:
-
sulphonylurea receptor
- UTR:
-
untranslated region
References
Aynsley-Green A, Polak JM, Bloom SR et al. (1981) Nesidioblastosis of the pancreas: definition of the syndrome and the management of the severe neonatal hyperinsulinaemic hypoglycaemia. Arch Dis Child 56:496–508
Bruining GJ (1990) Recent advances in hyperinsulinism and pathogenesis of diabetes mellitus. Curr Opin Pediatr 2:758–765
Otonkoski T, Ammala C, Huopio H et al. (1999) A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycemia of infancy in Finland. Diabetes 48:408–415
Aynsley-Green A, Hussain K, Hall J et al. (2000) Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed 82: F98–F107
Huopio H, Reimann F, Ashfield R et al. (2000) Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest 106:897–906
Stanley CA, Lieu YK, Hsu BY et al. (1998) Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 338:1352–1357
Glaser B, Thornton P, Otonkoski T, Junien C (2000) Genetics of neonatal hyperinsulinism. Arch Dis Child Fetal Neonatal Ed 82:F79–F86
Sharma N, Crane A, Gonzalez G, Bryan J, Aguilar-Bryan L (2000) Familial hyperinsulinism and pancreatic β-cell ATP-sensitive potassium channels. Kidney Int 57:803–808
Lonlay P de, Fournet JC, Touati G et al. (2002) Heterogeneity of persistent hyperinsulinaemic hypoglycaemia. A series of 175 cases. Eur J Pediatr 161:37–48
Glaser B, Kesavan P, Heyman M et al. (1998) Familial hyperinsulism caused by an activating glucokinase mutation. New Engl J Med 338:226–230
Tanizawa Y, Nakai K, Sasaki T et al. (2002) Unregulated elevation of glutamate dehydrogenase activity induces glutamine-stimulated insulin secretion: identification and characterization of a GLUD1 gene mutation and insulin secretion studies with MIN6 cells overexpressing the mutant glutamate dehydrogenase. Diabetes 51:712–717
Glaser B, Ryan F, Donath M et al. (1999) Hyperinsulism caused by paternal-specific inheritance of a recessive mutation in the sulfonylurea-receptor gene. Diabetes 48:1652–1657
Lonlay P de, Fournet JC, Rahier J et al. (1997) Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J Clin Invest 100:802–807
Ashcroft FM, Rorsman P (1989) Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 54:87–114
Ashcroft FM, Gribble FM (1999) ATP-sensitive K+-channels and insulin secretion: their role in health and disease. Diabetologia 42:903–919
Kane C, Shepherd RM, Squires PE et al. (1996) Loss of functional KATP channels in pancreatic β-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nat Med 2:1344–1347
Aguilar-Bryan L, Bryan J (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev 20:101–135
Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM (1997) Truncation of Kir6.2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature 387:179–183
Cartier EA, Conti LR, Vandenberg CA, Shyng SL (2001) Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci USA 98:2882–2887
Partridge CJ, Beech DJ, Sivaprasadarao A (2001) Identification and pharmacological correction of membrane trafficking defect associated with a mutation in the sulphonylurea receptor causing familial hyperinsulinism. J Biol Chem 276:35947–35952
Nichols CG, Shyng SL, Nestorowicz A et al. (1996) Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272:1785–1787
Shyng SL, Ferrigni T, Shepard JB et al. (1998) Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes 47:1145–1151
Huopio H, Jääskeläinen J, Komulainen J et al. (2002) Acute insulin response tests for differential daignosis of congenital insulinism. J Clin Endocrinol Metab 87:4502–4507
Gribble FM, Ashfield R, Ämmälä C, Ashcroft FM (1997a) Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. J Physiol (Lond) 498:87–98
Trapp S, Proks P, Tucker SJ, Ashcroft FM (1998) Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J Gen Physiol 112:333–349
Jackson MB, Wong BS, Morris CE, Lecar H, Christian CN (1983) Successive openings of the same acetylcholine receptor channel are correlated in open time. Biophys J 42:109–114
Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22:537–548
Shyng SL, Ferrigni T, Nichols CG (1997) Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J Gen Physiol 110:141–153
Reimann F, Tucker SJ, Proks P, Ashcroft FM (1999) Involvement of the N-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J Physiol (Lond) 518:325–336
Koster JC, Sha Q, Shyng SL, Nichols CG (1999) ATP inhibition of K-ATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J Physiol (Lond) 515:19–30
Baukrowitz T, Schulte U, Oliver D et al. (1998) PIP2 and PIP as determinants for ATP-inhibition of KATP channels. Science 282:1141–1144
Gribble FM, Tucker SJ, Ashcroft FM (1997) The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J 16:1145–1152
Shyng SL, Ferrigni T, Nichols CG (1997b) Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol 110:643–654
Acknowledgements
We thank Dr. B. Schwappach for supplying the HA-epitope-tagged constructs. This work was supported by grants from the Wellcome Trust. F. Reimann is a Diabetes UK RD Lawrence Fellow, M. Dabrowski is a Robert Turner Visiting Scholar, P. Proks is a Beit Memorial Trust Fellow. F.M. Gribble is a Wellcome Clinician Scientist Fellow. F.M. Ashcroft is the Royal Society GlaxoSmithKline Research Professor.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reimann, F., Huopio, H., Dabrowski, M. et al. Characterisation of new KATP-channel mutations associated with congenital hyperinsulinism in the Finnish population. Diabetologia 46, 241–249 (2003). https://doi.org/10.1007/s00125-002-1014-3
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-002-1014-3