
Abstract
Aims/hypothesis
The ATP-sensitive potassium (KATP) channel couples beta cell electrical activity to glucose-stimulated insulin secretion. Loss-of-function mutations in either the pore-forming (inwardly rectifying potassium channel 6.2 [Kir6.2], encoded by KCNJ11) or regulatory (sulfonylurea receptor 1, encoded by ABCC8) subunits result in congenital hyperinsulinism, whereas gain-of-function mutations cause neonatal diabetes. Here, we report a novel loss-of-function mutation (Ser118Leu) in the pore helix of Kir6.2 paradoxically associated with sulfonylurea-sensitive diabetes that presents in early adult life.
Methods
A 31-year-old woman was diagnosed with mild hyperglycaemia during an employee screen. After three pregnancies, during which she was diagnosed with gestational diabetes, the patient continued to show elevated blood glucose and was treated with glibenclamide (known as glyburide in the USA and Canada) and metformin. Genetic testing identified a heterozygous mutation (S118L) in the KCNJ11 gene. Neither parent was known to have diabetes. We investigated the functional properties and membrane trafficking of mutant and wild-type KATP channels in Xenopus oocytes and in HEK-293T cells, using patch-clamp, two-electrode voltage-clamp and surface expression assays.
Results
Functional analysis showed no changes in the ATP sensitivity or metabolic regulation of the mutant channel. However, the Kir6.2-S118L mutation impaired surface expression of the KATP channel by 40%, categorising this as a loss-of-function mutation.
Conclusions/interpretation
Our data support the increasing evidence that individuals with mild loss-of-function KATP channel mutations may develop insulin deficiency in early adulthood and even frank diabetes in middle age. In this case, the patient may have had hyperinsulinism that escaped detection in early life. Our results support the importance of functional analysis of KATP channel mutations in cases of atypical diabetes.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
The central role of the ATP-sensitive potassium (KATP) channel in glucose-stimulated insulin secretion from pancreatic beta cells is well understood [1]. Glucose uptake and metabolism leads to an increase in the ATP/ADP concentration that causes a reduction in KATP channel activity. This triggers electrical activity and elevation of the cytosolic calcium concentration that initiate insulin granule exocytosis, thereby linking glucose metabolism to electrical activity. Mutations in either the pore-forming subunit (the inwardly rectifying potassium channel 6.2 [Kir6.2], encoded by KCNJ11) or the regulatory subunit (the sulfonylurea receptor 1 [SUR1], encoded by ABCC8) of the KATP channel hetero-octameric complex cause human disease. Loss-of-function mutations cause persistent and unregulated insulin secretion and congenital hyperinsulinism [2,3,4], whereas gain-of-function mutations prevent KATP channel closure in response to glucose metabolism, resulting in a failure of insulin secretion and neonatal diabetes [5, 6]. Sulfonylurea drugs, widely used to treat type 2 diabetes, bypass metabolism and close the KATP channel directly, thus stimulating insulin release in both neonatal and type 2 diabetes [7].
Recessively inherited loss-of-function mutations in either ABCC8 or KCNJ11 are the most common cause of congenital hyperinsulinism [8, 9]. They principally result in a failure of KATP channel assembly or trafficking to the cell surface. Diazoxide therapy is often ineffective because of the absence of KATP channels, which may necessitate partial pancreas resection to control the hypoglycaemia. This inevitably leads to diabetes in later life and has prompted a more conservative approach to disease management in recent years. Several small molecules have been shown to correct channel biogenesis and trafficking defects but none are yet used clinically [10,11,12]. KATP mutations causing dominantly inherited hyperinsulinism are less common than recessive mutations. They generally produce channels that traffic normally to the cell surface but have altered gating or impaired opening in response to MgADP [2, 3]. This results in a milder hypoglycaemia phenotype that is usually responsive to diazoxide or can be managed by diet, and may even escape detection. Adult carriers are frequently asymptomatic unless challenged with fasting or protein tolerance tests [13, 14].
It is becoming increasingly evident that individuals who have hyperinsulinism caused by dominant KATP channel mutations may show lessening of symptoms with age and progress to spontaneous remission, glucose intolerance or even diabetes [13,14,15,16,17,18,19,20,21,22,23]. It has been argued that the incidence of diabetes in individuals with hyperinsulinism is no greater than that in the general population [13]. However, this has been contested [14]. It is noteworthy that in one Finnish family, of the 11 family members who were heterozygous carriers of a loss-of-function KATP channel mutation (ABCC8-E1506K), all reported symptoms of hypoglycaemia in childhood, four had overt diabetes and five had glucose intolerance with severely blunted first-phase glucose-stimulated insulin secretion in adult life [15], an incidence substantially higher than in the general population. In contrast, the frequency of diabetes in a second family with this mutation was comparable to that of the general population [13]. However, subsequent studies have provided additional support for an association between hyperinsulinism in childhood and glucose intolerance, gestational diabetes and diabetes in later life [14, 16,17,18,19,20,21,22,23]. Clearly, this possibility requires further investigation, both because it may shed light on the aetiology of diabetes and because of its implications for therapy.
In this paper, we therefore study the functional effects of a dominant loss-of function KCNJ11 mutation that did not cause detectable hyperinsulinism in infancy but was associated with sulfonylurea-sensitive diabetes in the absence of an autoimmune or other monogenic cause, in early adult life.
Methods
Ethics statement
Our research studies followed the University of Chicago Institutional Review Board protocols (http://monogenicdiabetes.uchicago.edu). Written informed consent was obtained from the patient for publication of the submitted article.
Molecular genetics
Next-generation sequencing (NGS) panel for monogenic diabetes, including 14 monogenic diabetes-related genes (ABCC8, APPL1, BLK, CEL, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, KLF11, NEUROD1, PAX4, PDX1) and deletion/duplication analysis of the coding region of the same panel of genes, was performed by array-CGH at the University of Chicago Services laboratory.
Chemicals and antibodies
Table 1 gives the source of chemicals and antibodies used. Antibodies have been extensively validated by the companies and in numerous publications.
Molecular biology and cell preparation
pcDNA4/TO or pBF expression vectors were used to express the human Kir6.2 or SUR1 genes (KCNJ11 and ABCC8, respectively). The QuikChange XL system (Stratagene, UK) was used for site-directed mutagenesis, and mutant clones were verified by sequencing (SourceBioScience, UK). SP6 mMESSAGE MACHINE transcription kit (catalogue no. AM1340, ThermoFisher, UK) was used for mRNA synthesis [24].
HEK-293T cells were obtained from LGC Standards, UK (ATTC CRL-3216). Our working stock tested negative for mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza Bioscience, UK). Cells were maintained at 50% to 90% confluency at 37°C, and 5% CO2/95% air, in DMEM (4.5 g/l glucose; Sigma, UK), supplemented with 10% (vol./vol.) FBS (Life Technologies, UK), 100U/ml penicillin and 100µg/ml streptomycin (ThermoFisher Scientific, UK). When close to confluency, cells were gently dissociated with TryPLE (ThermoFisher Scientific) and seeded in 25 cm2 flasks. After 3 h, cells were transfected with 4.5 µg of wild-type SUR1 cDNA and 1.5 µg of either wild-type KCNJ11 cDNA, mutant KCNJ11 cDNA (homozygous [hom]), or a 50:50 mixture of wild-type and mutant KCNJ11 cDNAs (to simulate the heterozygous [het] state of the patient), using TransIT-LT1 (Mirus Bio, USA). Where indicated, 5 µmol/l glibenclamide was added to the media just before transfection. After 48 h, transfected cells were plated onto 35 mm petri dishes coated with poly-l-lysine (Corning, UK) and currents were recorded 24 h later.
Xenopus oocytes were prepared as previously described [25], injected with 0.8 ng wild-type or mutant KCNJ11 mRNA and ∼4 ng SUR1 mRNA, and maintained in Barth’s solution (in mmol/l: 88 NaCl, 1 KCl, 1.68 MgSO4, 0.41 CaCl2, 0.47 Ca[NO3]2, 2.4 NaHCO3, 10 HEPES, adjusted to pH 7.4 with NaOH) at 18°C. Currents were recorded 2–4 days after injection.
Electrophysiology
Giant inside-out patches were excised from HEK-293T cells expressing wild-type or mutant KATP channels. Macroscopic currents were recorded at −60 mV with an Axopatch 200B amplifier (Molecular Devices, UK), filtered at 1 kHz, and digitised at 10 kHz with a Digidata 1322A A/D interface driven by pClamp9 software (Molecular Devices). The extracellular (pipette) solution contained (in mmol/l) 140 KCl, 1.2 MgCl2, 2.6 CaCl2 and 10 HEPES, pH 7.4 with KOH. The intracellular (bath) solution contained (in mmol/l) 107 KCl, 2 MgCl2, 1 CaCl2, 10 EGTA and 10 HEPES (pH 7.3 with KOH), plus MgATP and/or MgADP as indicated.
For each MgATP concentration tested, current was expressed as a fraction of its mean value in the control intracellular solution before and after each ATP application [26]. The MgATP concentration producing half-maximal block of the KATP current (IC50) and the Hill coefficient were calculated from the Hill fit to each individual current–inhibition curve. For each construct, currents were recorded from at least four or five separate cells, and at least two independent transfections. However, the heterozygous channel recordings were from two cells. To test channel activation, 100 μmol/l MgADP was added in the presence of 100 μmol/l MgATP. Current was expressed as a fraction of the mean values in control solution (no nucleotides) before and after each application.
Whole-oocyte KATP currents were recorded at room temperature in response to ±20 mV steps from a holding potential of −10 mV using a two-electrode voltage-clamp (GeneClamp 500B amplifier; Molecular Devices). Data were filtered online at 500 Hz, digitised at 4 kHz with a Digidata 1440A acquisition system (Molecular Devices) and acquired with pCLAMP10 software (Molecular Devices). Oocytes were perfused with control solution containing (in mmol/l) 90 KCl, 1 MgCl2, 1.8 CaCl2 and 5 HEPES (adjusted to pH 7.4 with KOH). Na-azide (3 mmol/l), diazoxide (340µmol/l) or tolbutamide (500µmol/l) were added as indicated. Data were recorded from ‘n’ oocytes, from at least two separate frogs, and analysed with ClampFit (pClamp 11.2.0.59; Molecular Devices) and Prism 10.1.2 (GraphPad, USA).
Surface expression
Surface expression of wild-type or mutant Kir6.2 was measured based on their ability to chaperone a site-specific tagged SUR1 to the plasma membrane [27]. Briefly, a haemagglutinin (HA) tag (YAYMEKGITDLAYPYDVPDYA) was inserted into the last extracellular loop of SUR1 (TM16-TM17). Transfections were performed as described above and 72 h post-transfection cells were rinsed with PBS, fixed for 30 min in 10% neutral buffered formalin, rinsed twice with PBS and blocked with PBS+1% BSA for 30 min at 4°C. Anti-HA monoclonal antibody was added for 1 h at 4°C at a 1:1000 dilution, followed by multiple washes on ice with PBS+1% BSA. Cells were then incubated with horseradish peroxidase-conjugated goat anti-rat polyclonal antibody (diluted 1:1000) for 30 min at 4°C. After multiple washes with PBS+1% BSA and PBS, 500 μl of SuperSignal ELISA Femto Maximum Sensitivity Substrate (ThermoFisher Scientific) was added to each 35 mm dish and the chemiluminescence signal was measured using a Glomax 20/20 Luminometer (Promega, UK) after a 10 s incubation. Total protein in each dish was determined by BCA (ThermoFisher Scientific) and used to normalise the chemiluminescent signal. Each experimental point is the mean of three dishes from one transfection. Data were collected from three to five independent transfections and plotted as a percentage of the expression of the wild-type channel, for each individual experiment.
Western blotting
Western blotting was performed on total protein from HEK-293T cells transfected as described above (from three independent transfections), using an HA tag inserted into SUR1 to detect SUR1 abundance. Cells were harvested and solubilised in lysis buffer (0.5% Triton X-100, 100 mmol/l potassium acetate, pH 7.4), supplemented with cOmplete protease inhibitor cocktail (Roche Products, UK). Benzonase (Sigma) was added to each sample (250 U per 100 μl of solubilised cells) and incubated for 30 min at room temperature. Protein concentrations were measured by BCA and 20 µg of protein lysate was mixed with a reducing agent and loading buffer (NuPAGE; Invitrogen, UK). α-Tubulin was used as a control for equal loading. After running a precast NuPAGE 7% Tris-acetate poly-acrylamide gels at 150 V for 60 min, proteins were wet transferred overnight onto polyvinylidene difluoride membranes (Immobilon P; Merck Millipore, Germany) at 10 V in Tris-glycine buffer (25 mmol/l Tris, 192 mmol/l glycine, 10% methanol). Non-specific protein signal was blocked by incubating the membranes with 5% milk in TBS-T (150 mmol/l NaCl, 1% Tween 20, 25 mmol/l Tris, pH 7.2). Primary (1:1000 dilution) and secondary (1:20,000) antibodies were incubated for 30 min at room temperature, each time followed by multiple washes with TBS-T. Immunoreactive proteins were detected by chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Thermo Fisher). Analysis was performed using ImageJ 1.53K (NIH, USA).
Statistics
Results are reported as individual data points and mean±SEM. Statistical significance was determined using Student’s t test or one-way ANOVA followed by the post hoc Dunnett’s test for multiple comparisons.
Results
Clinical history
A previously healthy, non-obese (BMI 20–21 kg/m2) 31-year-old woman of white/European ancestry was diagnosed with mild hyperglycaemia and an HbA1c of 38 and 39 mmol/mol (5.6 and 5.7%) during successive employee screens at her place of work. She was not aware of any symptoms of either hypoglycaemia or hyperglycaemia in early life. There was no history of diabetes in her parents or siblings but her father and two siblings had a history of obesity. Two years later, during her first pregnancy, she had persistently elevated blood sugar levels: 1 h OGTT of 12.8 mmol/l; HbA1c of 42 mmol/mol (6.0%); and fasting blood glucose of 8.6 mmol/l at 27 weeks and 6 days of gestation. She was initially started on sulfonylurea therapy (glibenclamide 2.5 mg/day), which helped her glycaemic control, but her medication was subsequently switched to insulin.
The patient’s glycaemic control improved after her first pregnancy but did not recover completely, and she required insulin during her next two pregnancies (three pregnancies in total). Between her first and second pregnancies, and while off medication, her fasting insulin was 25 pmol/l and her C-peptide was 0.51 nmol/l (with fasting glucose of 6.7 mmol/l). Insulin was discontinued 6 months after the birth of her third child but she continued to experience increases in blood glucose (fasting glucose 7.3 mmol/l, HbA1c 48 mmol/mol [6.5%]). She was therefore treated with glibenclamide (7.5 mg daily) plus metformin (1000 mg twice daily). At follow-up, her HbA1c was 43 mmol/mol (6.1%), 88% of time within the target range, with a continuous glucose monitor report of modest hypoglycaemia (2% of the time) and hyperglycaemia (9% of the time). The following year, when aged 38 years, her HbA1c was 56 mmol/mol (7.3%) while taking metformin (1000 mg at night) and glibenclamide (2.5–5 mg in the morning and 5 mg before the evening meal). Metformin was stopped due to gastrointestinal symptoms; oral semaglutide (Rybelsus, 3 mg daily) was started and glibenclamide was increased to 5 mg in the morning and 5 mg before dinner. At follow-up, the patient reported decreased appetite and mild weight loss (from 56 to 53 kg). Her HbA1c was 48 mmol/mol (6.5%; 92% in range as measured by continuous glucose monitoring) without hypoglycaemia (<1%) and modest hyperglycaemia (7%), while taking glibenclamide (5 mg twice daily) and oral semaglutide (7 mg daily). All three of the patient’s children were delivered by C-section. Their gestational age and birth weights were as follows: 36 weeks 6 days, 3.43 kg; 38 weeks 1 day, 4.43 kg; and 38 weeks 2days, 3.98 kg.
Molecular genetics
Given the young age of the patient (31 years at diagnosis), lack of islet cell antibodies and lack of obesity, a genetic origin for her insulin-independent diabetes was implicated. Genetic testing was undertaken at the age of 35 years using a panel of genes known to cause MODY. No mutations were found in ABCC8, APPL1, BLK, CEL, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, KLF11, NEUROD1, PAX4 and PDX1 but sequence analysis identified a novel heterozygous missense mutation, p.Ser118Leu (c.353C>T ), in exon 1 of the KCNJ11 gene. This variant has been previously reported as a ‘variant of uncertain significance’ [4], although no functional studies were carried out. We were unable to obtain details of the clinical history of the individual studied in this earlier report but note that after starting sulfonylureas their HbA1c dropped from 124 mmol/mol (13.5%) to 52 mmol/mol (6.9%). To evaluate whether the KCNJ11 variant could explain our patient’s diabetes, her response to sulfonylureas and her lack of insulin resistance, we undertook functional analysis of the S118L mutation in KCNJ11. Unfortunately, none of her children have been tested for this variant but their blood glucose levels have remained within normal limits.
The S118L mutation does not affect KATP channel ATP sensitivity
We examined whether the Kir6.2-S118L mutation affects the sensitivity of the KATP channel to MgATP inhibition by recording KATP currents in inside-out patches (Fig. 1a,b). HEK293T cells were co-transfected with wild-type SUR1 and either Kir6.2 (wild-type), Kir6.2-S118L (homS118L) or a 1:1 mixture of wild-type Kir6.2 and Kir6.2-S118L (hetS118L). The latter aims to simulate the heterozygous state of the patient. There was no significant difference in the dose–response curves for ATP inhibition or the corresponding half-maximal block (IC50) of wild-type (14±2 µmol/l, n=4), hetS118L (11.8–12.7 µmol/l, n=2) or homS118L (13.5±0.5 µmol/l, n=5) channels (Fig. 1c,d). However, the current immediately after patch excision was less for homS118L channels (738±279 pA, median 362 pA, n=8) than for wild-type channels (2639±911 pA, median 2238 pA, n=4).

Inhibition of KATP currents by MgATP. (a, b) Representative current traces for wild-type (a) and hetS118L (b) channels recorded at −60 mV from inside-out patches excised from HEK293T cells and exposed to different MgATP concentrations (as indicated by the bars; values are in μmol/l). The zero-current level (I=0) is shown by the dashed lines. (c, d) MgATP concentration–response curves (c) and corresponding IC50 values (d) for wild-type (black/grey, n=4), hetKir6.2-S118L/SUR1 (hetS118L; dark green, n=2) and homKir6.2-S118L/SUR1 (homS118L; light green, n=5) channels. In (c), current amplitude (I) is expressed as a fraction of the maximum KATP current measured in control solution in the same patch (IC). The lines are the best fit of the Hill equation to the mean data. In (d), IC50 values were obtained from individual MgATP concentration–response curves. Box plots show individual data points and mean ± SEM
The S118L mutation does not affect KATP channel stimulation by MgADP
We next examined the ability of MgADP to stimulate channel activity. Figure 2a,b shows that 100 μmol/l MgADP increased both wild-type and mutant KATP current amplitudes when added in the presence of inhibitory ATP. The mean increases in the hetS118L and homS118L KATP currents were not significantly different from that of the wild-type current (Fig. 2c).

Activation of KATP currents by MgADP. (a, b) Representative current traces for wild-type (a) and hetKir6.2-S118L/SUR1 (b, hetS118L) channels recorded at −60 mV from inside-out patches excised from HEK293T cells and exposed to 100 μmol/l MgATP with or without 100 μmol/l MgADP (as indicated by the bars, values are in μmol/l). The zero-current level (I=0) is shown by the dashed lines. (c) Current amplitude (I) expressed as a fraction of the maximum KATP current measured in control solution in the same patch (IC) for wild-type (grey), hetS118L (dark green) and homS118L (light green) channels. Individual data points and mean ± SEM are shown
Metabolic inhibition of hetS118L and homS118L channels was unaffected
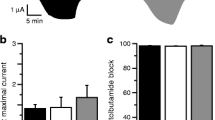
To investigate the effects of the S118L mutation on the metabolic regulation of the KATP channel, we recorded whole-cell currents from Xenopus oocytes. As previously reported [24, 25], wild-type KATP channels were closed in control solution, due to the high intracellular ATP/ADP concentration, but were opened by lowering ATP/ADP using the metabolic inhibitor sodium azide (Fig. 3a,b). The current was further increased by the potassium-channel opener diazoxide (340 µmol/l) and almost fully inhibited by the sulfonylurea tolbutamide (500 µmol/l).

Effects of metabolic inhibition and diazoxide on KATP currents. (a, b) Representative whole-cell currents recorded from Xenopus oocytes expressing wild-type (a) or hetS118L (b) KATP channels in response to ±20 mV steps from a holding potential of −10 mV every ~2 s. The horizontal bars indicate when 3 mmol/l Na-azide, 340 µmol/l diazoxide or 0.5 mmol/l tolbutamide (Tolb) were added to the external solution. (c–g) Mean current amplitudes for wild-type (grey bars), hetS118L (dark green bars) and homS118L (light green bars) recorded in control solution (Ictrl, black circles) (c), and in the presence of Na-azide (Iazide, black triangles) (d), Na-azide+diazoxide (Idiaz, black squares) (f) or Na-azide+tolbutamide (Itolb, black diamonds) (g). (e) Time constants of KATP current activation by Na-azide. (h–j) Current amplitudes in control solution (h), and in the presence of Na-azide (i) or Na-azide+tolbutamide (j), expressed as a fraction of the diazoxide-activated KATP current. This controls for variability in KATP channel expression, assuming diazoxide causes maximal KATP channel opening in all cases. Box plots show individual data points and mean ± SEM (n=6 or 7). There was no significant difference between the data
Mutations that reduce the channel ATP sensitivity normally increase the whole-cell current in both control solution and in the presence of azide, reflecting the fact that they are blocked to a lesser extent by the resting intracellular ATP concentration. Neither the hetS118L nor homS118L current amplitude was appreciably different from that of the wild-type current, either in control solution or in the presence of azide (Fig. 3a–d). This is consistent with the lack of a difference in ATP sensitivity (Fig. 1). The rate of current activation in response to azide was also similar (Fig. 3e). Diazoxide-activated wild-type, hetS118L and homS118L currents to similar extents (Fig. 3f), and tolbutamide blocked all three types of channel by ∼96% (Fig. 3g).
To control for variability in KATP channel expression between oocytes (and different batches of oocytes), we expressed currents in control solution (Fig. 3h), in azide solution (Fig. 3i) and in azide plus tolbutamide (Fig. 3j) as a percentage of that in the presence of diazoxide. Again, there was no difference between wild-type, hetS118L and homS118L currents.
Membrane trafficking of S118L channels is impaired
Because the ATP sensitivity and metabolic regulation of the mutant channels were unaltered, we explored if the mutation decreased the channel density in the plasma membrane. We used an HA tag, inserted into an extracellular epitope of SUR1, to detect KATP channels at the plasma membrane of transfected HEK293T cells. Surface expression was normalised first to protein content and then plotted as a percentage of the surface expression of the wild-type channel. No surface expression was detected in the absence of the HA tag. (Fig. 4a) There was an ~40% reduction in the surface expression of hetS118L and homS118L HA-tagged channels compared with wild-type channels (Fig. 4a), suggesting that the mutation disrupts surface trafficking and/or assembly of the KATP channel complex. However, membrane trafficking of hetS118L channels was partially rescued when cells were cultured in the presence of the sulfonylurea glibenclamide (5 µmol/l; olive bar, n=4, Fig. 4b) or at a lower temperature (28°C; red bar, n=3, Fig. 4b).

Glibenclamide rescues surface expression of hetKir6.2-S118L KATP channels. (a) Surface expression of HA-tagged or untagged SUR1 co-transfected with Kir6.2 (wild-type, grey bar, n=5), hetKir6.2-S118L (hetS118L; dark green bar, n=3) or homKir6.2-S118L (homS118L; light green bar, n=3). (b) Surface expression of HA-tagged or untagged SUR1 co-transfected with Kir6.2 (wild-type, dark grey bar, n=7), or hetKir6.2-S118L (dark green bar, n=7) cultured at 37°C in the absence of drug. Glibenclamide (5 µmol/l) was added to the media to promote surface membrane trafficking of wild-type and mutant constructs (wild-type, HA+Glib, light grey, n=4; hetKir6.2-S118L,_HA+Glib, olive bar, n=4). HetKir6.2-S118L was cultured at 28°C in the absence of drug (red bar, n=3). (c) Representative western blot for total SUR1 (~180 kDa) and α-tubulin (~50 kDa) proteins, in total protein lysates from HEK cells expressing wild-type Kir6.2 (ctrl), hetKir6.2-S118L (het) co-expressed with SUR1-HA, or not transfected (nt). Molecular mass markers shown on the right side of the blots are in kDa. (d) Quantification of the SUR1 bands, shown as a relative fold change against wild-type. Box plots show individual data points and mean ± SEM from three to seven independent transfections. **p<0.05, **p<0.01, ***p<0.001 (one-way ANOVA followed by the post hoc Dunnett’s test for multiple comparisons)
We also performed western blotting for SUR1 in total protein lysates from HEK cells expressing wild-type Kir6.2 or hetKir6.2-S118L co-expressed with SUR1-HA (Fig. 4c,d). Total SUR1 protein in hetKir6.2-S118L/SUR1-HA cells was less than in wild-type cells, consistent with the decrease in surface expression of the mutant channel. Glibenclamide co-culture corrected this effect of the mutation and increased expression of mutant channels.
Discussion
We describe a female patient who presented with glucose intolerance at the age of 31 years and who subsequently progressed to gestational and early-onset insulin-independent, anti-islet-antibody-negative non-obese diabetes. Her presentation suggested a diagnosis of MODY, and genetic screening identified a dominantly inherited mutation in KCNJ11, S118L. Surprisingly, functional analysis of this mutation indicated that it is not a gain-of-function mutation but a loss-of-function mutation that impairs surface expression of the KATP channel by ~40%. Thus, our data provide support for the idea [14,15,16, 20,21,22,23] that dominantly inherited loss-of-function KATP channel mutations may lead to glucose intolerance and diabetes.
Loss-of-function mutations that cause a similar reduction in KATP channel density are usually associated with congenital hyperinsulinism not diabetes. However, the penetrance of dominantly inherited loss-of-function KATP channel mutations is often weak and some carriers may have no symptoms or undiagnosed hyperinsulinism in childhood [14]. Furthermore, many show spontaneous remission of symptoms [14,15,16,17,18,19,20,21,22,23]. This may explain why our patient was not diagnosed with hyperinsulinism.
Several possibilities might account for our patient’s diabetes. First, her diabetes may be caused by an unidentified (novel) MODY gene. Second, she may carry a combination of gene variants that predispose to non-obese type 2 diabetes and the additional stress of pregnancy precipitated her diabetes. Both explanations assume her KCNJ11 mutation is an independent phenomenon without functional or clinical significance. Given the marked reduction in KATP channel density this seems unlikely. A third possibility is that her Kir6.2-S118L mutation did not cause symptomatic hyperinsulinism/hypoglycaemia in childhood but predisposed to glucose intolerance and diabetes in early adult life. We consider this explanation to best account for all the data. There is accumulating evidence in favour of the idea.
A progressive reduction in hypoglycaemia in patients with recessive inactivating KATP channel mutations is relatively frequent [14, 17,18,19] and in some individuals may evolve to glucose intolerance or, more rarely, diabetes [15, 16, 20,21,22,23]. The clinical history of our patient resembles that of individuals with the ABCC8-E1506K mutation, several of whom had gestational diabetes that remitted after pregnancy but subsequently returned [15, 16]. All ABCC8-E1506K carriers had low insulin values during a hyperglycaemic glucose clamp, indicating they had impaired insulin secretion. Likewise, individuals with another partial loss-of-function ABCC8 mutation (R1353H) have been diagnosed with hyperinsulinism [28, 29], gestational diabetes [29, 30] and non-obese antibody-negative early-onset diabetes [30]. However, whether the progression of hyperinsulinism to glucose intolerance is a general feature of the disease is a controversial area [13] and one that warrants further investigation.
Interestingly, the phenotype is shared with certain HNF4A (MODY1) and HNF1A (MODY4) mutations that cause hyperinsulinaemia at birth before evolving to decreased insulin secretion and diabetes in later life [31,32,33]. It has been proposed that the hyperinsulinaemia is due to downregulation of KATP channel genes [33], suggesting a common mechanism may be involved. Further evidence is needed to substantiate this hypothesis. However, if it is correct, it becomes somewhat semantic as to whether patients like ours, who have reduced KATP channel expression associated with diabetes in early adult life, should be classified as having a novel type of MODY (caused by her loss-of-function KATP channel mutation), or congenital hyperinsulinism.
Why a partial reduction in KATP conductance should lead to glucose intolerance or diabetes is still not understood. However, it is consistent with mouse models of hyperinsulinism. Partial genetic deletion of either Kir6.2 or SUR1 causes increased excitability and insulin secretion [34, 35]. However, total knockout does not result in neonatal hypoglycaemia but instead dramatically reduces insulin secretion and causes impaired glucose tolerance in adult mice that progresses to overt diabetes when fed a high-fat diet [36,37,38]. A similar phenotype is seen when human loss-of-function mutations are expressed in mice [39, 40]. Various hypotheses have been put forward to account for these paradoxical findings but none has yet been substantiated [41].
Mechanism of action of the S118L mutation
Both hetS118L and homS118L channels show normal ATP sensitivity and metabolic regulation, indicating that the mutation does not alter channel function. Instead, they reduce plasmalemmal KATP channel density. Multiple steps are involved in the surface expression of the KATP channel. Kir6.2 and SUR1 must be correctly translated, folded and co-assembled to form a hetero-octameric complex, which must be correctly trafficked to, and inserted into, the plasma membrane. A mutation may affect any of these processes, as well as the rate at which the channel is removed from the membrane. Neither Kir6.2 nor SUR1 traffic to the plasmalemma in the absence of the partner subunit, as they fail to exit the endoplasmic reticulum [27] and are then degraded [12]. The reduction in total SUR1 protein we observed favours the idea that channel assembly is affected, preventing surface trafficking. That surface expression was reduced to the same extent for hetS118L and homS118L channels is not unexpected. Given the tetrameric nature of the pore, the location of S118 at the interface between two pore subunits, and the known cooperativity between pore subunits, assembly of the whole tetramer is likely to be impaired by mutation of a single Kir6.2 subunit. Without correct assembly, SUR1 will be removed by the degradative machinery.
Structural studies of KATP channel open and closed states [42,43,44,45] reveal that S118 sits at the start of the pore helix, 3–4 Å from the adjacent Kir6.2 subunit (Fig. 5). However, it lies a significant distance from SUR1 (>14 Å), the closest approach being with the first set of transmembrane domains (TMD0) of SUR1. Thus, it is possible that the mutation affects channel assembly by disrupting interactions between Kir6.2 subunits and, thereby, indirectly influences interactions between Kir6.2 and SUR1. Interestingly, a heterozygous in-frame deletion of two adjacent residues, S116 and F117, led to a phenotype similar to that we report [23].

Location of S118L in Kir6.2. (a) Structural model of one pore-forming (Kir6.2, green) and one regulatory (SUR1, orange/yellow) subunit of KATP in an inhibited state (from Protein Data Bank [PDB] accession no. 6baa), with ATP bound to Kir6.2 and glibenclamide (not visible) bound to SUR1. (b) Side view, showing the position of S118 (cyan) at the start of the pore helix and E140 (green) at the top of transmembrane domain 2 (TM2). (c) Detail of two adjacent Kir6.2 subunits (one green, one blue) showing the close proximity (<4 Å) between S118 from one subunit with E140 from the neighbouring subunit
Glibenclamide correction of surface expression
Our data indicate that glibenclamide acts as a pharmacological chaperone, enhancing surface expression of mutant channels. Likewise, reduced temperature enhanced surface expression. Both have previously been reported for hyperinsulinism-inducing mutations in ABCC8 [10,11,12, 43]. Structural studies show the TMD0 of SUR1 (SUR1-TMD0) and the first transmembrane domain (TM1) of Kir6.2 make direct physical contact, explaining why SUR1-TMD0 mutations disrupt assembly and trafficking [46]. It has been proposed that this interaction is stabilised by pharmacological chaperones like glibenclamide, thus providing more time for SUR1-TMD0 and Kir6.2 to interact during channel assembly [47]. This may be how glibenclamide corrects assembly and trafficking of Kir6.2-S118L mutant channels.
Conclusions
We report a novel loss-of-function mutation in KCNJ11 (encoding Kir6.2) that results in a clinical phenotype largely indistinguishable from common gestational and type 2 diabetes, and was only identified by genetic screening. Because of the nature of the mutation our patient carried it was possible to manage her diabetes with glibenclamide and oral semaglutide rather than insulin injection, a significant advantage to the patient. Similarly, the diabetes of another patient, with a heterozygous loss-of-function mutation in SUR1, who progressed from hyperinsulinism to diabetes, was treatable with glibenclamide [16].
Determination of the cause of atypical diabetes is essential for proper management and understanding of possible syndromic effects. Cascade testing of family members, when there is a family history of diabetes, is essential and children at risk should be offered testing or monitoring following discussions with the family. Understanding the impact of variants of uncertain significance may require both functional and clinical studies, in addition to genetic studies. Individuals who present with diabetes in early adult life might have had undetected hyperinsulinaemia or diabetes in the neonatal period, leading to a misdiagnosis and potentially the wrong choice of therapy.
Abbreviations
- HA:
-
Haemagglutinin
- het:
-
Heterozygous
- HI:
-
Hyperinsulinism
- hom:
-
Homozygous
- IC50 :
-
Concentration producing half-maximal block of the KATP current
- KATP channel:
-
ATP-sensitive potassium channel
- Kir6.2:
-
Inwardly rectifying potassium channel 6.2
- SUR1:
-
Sulfonylurea receptor 1
- TMD0:
-
First set of transmembrane domains of SUR1
References
Rorsman P, Ashcroft FM (2018) Pancreatic β-cell electrical activity and insulin secretion: of mice and men. Physiol Rev 98(1):117–214. https://doi.org/10.1152/physrev.00008.2017
Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ (2004) Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev 84(1):239–275. https://doi.org/10.1152/physrev.00022.2003
Cosgrove KE, Shepherd RM, Fernandez EM et al (2004) Genetics and pathophysiology of hyperinsulinism in infancy. Horm Res 61:270–288. https://doi.org/10.1159/000076933
De Franco E, Saint-Martin C, Brusgaard K et al (2020) Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat 41(5):884–905. https://doi.org/10.1002/humu.23995
Pipatpolkai T, Usher S, Stansfeld PJ, Ashcroft FM (2020) New insights into KATP channel gene mutations and neonatal diabetes. Nat Rev Endocrinol 16:378–393. https://doi.org/10.1038/s41574-020-0351-y
Hattersley AT, Ashcroft FM (2005) Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights and new therapy. Diabetes 54(9):2503–2513. https://doi.org/10.2337/diabetes.54.9.2503
Pearson ER, Flechtner I, Njølståd PR et al (2006) Neonatal Diabetes International Collaborative Group (2006) Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. New Engl J Med 355(5):467–477. https://doi.org/10.1056/NEJMoa061759
Snider KE, Becker S, Boyajian L et al (2013) Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab 98(2):E355–E363. https://doi.org/10.1210/jc.2012-2169
Henwood MJ, Kelly A, Macmullen C et al (2005) Genotype-phenotype correlations in children with congenital hyperinsulinism due to recessive mutations of the adenosine triphosphate-sensitive potassium channel genes. J Clin Endocrinol Metab 90:789–794. https://doi.org/10.1210/jc.2004-1604
Yan F, Lin CW, Weisiger E, Cartier EA, Taschenberger G, Shyng SL (2004) Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J Biol Chem 279(12):11096–11105. https://doi.org/10.1074/jbc.M312810200
Yan FF, Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL (2007) Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes 56(9):2339–2348. https://doi.org/10.2337/db07-0150
Martin GM, Chen PC, Devaraneni P, Shyng SL (2013) Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Front Physiol 4:386. https://doi.org/10.3389/fphys.2013.00386
Pinney SE, MacMullen C, Becker S et al (2008) Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest 118(8):2877–2886. https://doi.org/10.1172/JCI35414
Kapoor RR, Flanagan SE, James CT et al (2011) Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia 54(10):2575–2583. https://doi.org/10.1007/s00125-011-2207-4
Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M (2003) A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet 361(9354):301–307. https://doi.org/10.1016/S0140-6736(03)12325-2
Abdulhadi-Atwan M, Bushman J, Tornovsky-Babaey S et al (2008) Novel de novo mutation in sulfonylurea receptor 1 presenting as hyperinsulinism in infancy followed by overt diabetes in early adolescence. Diabetes 57(7):1935–1940. https://doi.org/10.2337/db08-0159
Xu ZD, Hui PP, Zhang W et al (2021) Analysis of clinical and genetic characteristics of Chinese children with congenital hyperinsulinemia that is spontaneously relieved. Endocrine 72(1):116–123. https://doi.org/10.1007/s12020-020-02585-x
Kumaran A, Kapoor RR, Flanagan SE, Ellard S, Hussain K (2010) Congenital hyperinsulinism due to a compound heterozygous ABCC8 mutation with spontaneous resolution at eight weeks. Horm Res Paediatr 73(4):287–292. https://doi.org/10.1159/000284394
Salomon-Estebanez M, Flanagan SE, Ellard S et al (2016) Conservatively treated congenital hyperinsulinism (CHI) due to K-ATP channel gene mutations: reducing severity over time. Orphanet J Rare Dis 11(1):163. https://doi.org/10.1186/s13023-016-0547-3
Vieira TC, Bergamin CS, Gurgel LC, Moisés RS (2010) Hyperinsulinemic hypoglycemia evolving to gestational diabetes and diabetes mellitus in a family carrying the inactivating ABCC8 E1506K mutation. Pediatr Diabetes 11:505–508. https://doi.org/10.1111/j.1399-5448.2009.00626.x
Gussinyer M, Clemente M, Cebrián R, Yeste D, Albisu M, Carrascosa A (2008) Glucose intolerance and diabetes are observed in the long-term follow-up of nonpancreatectomized patients with persistent hyperinsulinemic hypoglycemia of infancy due to mutations in the ABCC8 gene. Diabetes Care 31(6):1257–1259. https://doi.org/10.2337/dc07-2059
Leibowitz G, Glaser B, Higazi AA, Salameh M, Cerasi E, Landau H (1995) Hyperinsulinemic hypoglycemia of infancy (nesidioblastosis) in clinical remission: high incidence of diabetes mellitus and persistent beta-cell dysfunction at long-term follow-up. J Clin Endocrinol Metab 80:386–392. https://doi.org/10.1210/jcem.80.2.7852494
Liu L, Nagashima K, Yasuda T et al (2013) Mutations in KCNJ11 are associated with the development of autosomal dominant, early-onset type 2 diabetes. Diabetologia 56(12):2609–18. https://doi.org/10.1007/s00125-013-3031-9
Proks P, Girard C, Ashcroft FM (2005) Functional effects of KCNJ11 mutations causing neonatal diabetes: enhanced activation by MgATP. Hum Mol Genet 14(18):2717–2726. https://doi.org/10.1093/hmg/ddi305
Gribble FM, Ashfield R, Ämmälä C, Ashcroft FM (1997) Properties of cloned ATP-sensitive K-currents expressed in Xenopus oocytes. J Physiol 498(1):87–98. https://doi.org/10.1113/jphysiol.1997.sp021843
Proks P, de Wet H, Ashcroft FM (2010) Activation of the KATP channel by Mg-nucleotide interaction with SUR1. J Gen Physiol 136(4):389–405. https://doi.org/10.1085/jgp.201010475
Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22(3):537–548. https://doi.org/10.1016/s0896-6273(00)80708-4
Khoriati D, Arya VB, Flanagan SE, Ellard S, Hussain K (2013) Prematurity, macrosomia, hyperinsulinaemic hypoglycaemia and a dominant ABCC8 gene mutation. BMJ Case Rep 2013:bcr2013008767. https://doi.org/10.1136/bcr-2013-008767
Magge SN, Shyng SL, MacMullen C, Steinkrauss L, Ganguly A, Katz LE, Stanley CA (2004) Familial leucine-sensitive hypoglycemia of infancy due to a dominant mutation of the beta-cell sulfonylurea receptor. J Clin Endocrinol Metab 89:4450–6. https://doi.org/10.1210/jc.2004-0441
Koufakis T et al (2019) First report of diabetes phenotype due to a loss-of-function ABCC8 mutation previously known to cause congenital hyperinsulinism. Case Rep Genet 2019:3654618. https://doi.org/10.1155/2019/3654618
Pearson ER, Boj SF, Steele AM et al (2007) Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med 4:e118. https://doi.org/10.1371/journal.pmed.0040118
Stanescu DE, Hughes N, Kaplan B, Stanley CA, De León DD (2012) Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab 97(10):E2026-30. https://doi.org/10.1210/jc.2012-1356
Hermann FM, Kjærgaard MF, Tian C et al (2022) An insulin hypersecretion phenotype precedes pancreatic b-cell failure in MODY3 patient-specific cells. Cell Stem Cell 30(1):38-51.e8. https://doi.org/10.1016/j.stem.2022.12.001
Koster JC, Remedi MS, Flagg TP et al (2002) Hyperinsulinism induced by targeted suppression of beta cell KATP channels. Proc Natl Acad Sci 99(26):16992–16997. https://doi.org/10.1073/pnas.012479199
Remedi MS, Rocheleau JV, Tong A et al (2006) Hyperinsulinism in mice with heterozygous loss of K(ATP) channels. Diabetologia 49(10):2368–2378. https://doi.org/10.1007/s00125-006-0367-4
Miki T, Nagashima K, Tashiro F et al (1998) Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci 95(18):10402–10406. https://doi.org/10.1073/pnas.95.18.10402
Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J (2000) Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem 275(13):9270–9277. https://doi.org/10.1074/jbc.275.13.9270
Remedi MS, Koster JC, Markova K et al (2004) Diet-induced glucose intolerance in mice with decreased beta-cell ATP-sensitive K channels. Diabetes 53(12):3159–3167. https://doi.org/10.2337/diabetes.53.12.3159
Shimomura K, Tusa M, Iberl M et al (2013) A mouse model of human hyperinsulinism produced by the E1506K mutation in the sulphonylurea receptor SUR1. Diabetes 62(11):3797–3806. https://doi.org/10.2337/db12-1611
Hugill A, Shimomura K, Ashcroft FM, Cox RD (2010) A mutation in KCNJ11 causing human hyperinsulinism (Y12X) results in a glucose-intolerant phenotype in the mouse. Diabetologia 53(11):2352–2356. https://doi.org/10.1007/s00125-010-1866-x
Nichols CG, York NW, Remedi MS (2022) ATP-sensitive potassium channels in hyperinsulinism and type 2 diabetes: inconvenient paradox or new paradigm? Diabetes 71(3):367–375. https://doi.org/10.2337/db21-0755
Lee KPK, Chen J, MacKinnon R (2017) Molecular structure of human KATP in complex with ATP and ADP. eLife 6:e32481. https://doi.org/10.7554/eLife.32481
Zhao C, MacKinnon R (2021) Molecular structure of an open human K ATP channel. Proc Natl Acad Sci 118(48):e2112267118. https://doi.org/10.1073/pnas.2112267118
Driggers CM, Kuo YY, Zhu P, ElSheikh A, Shyng S-L (2023) Structure of an open KATP channel reveals tandem PIP2 binding sites mediating the Kir6.2 and SUR1 regulatory interface. bioRxiv preprint. https://doi.org/10.1101/2023.08.01.551546
Martin GM, Kandasamy B, DiMaio F, Yoshioka C, Shyng SL (2017) Anti-diabetic drug binding site in a mammalian KATP channel revealed by Cryo-EM. eLife 6:e31054. https://doi.org/10.7554/eLife.3105410.7554/eLife.31054
Yang K, Fang K, Fromondi L, Chan KW (2005) Low temperature completely rescues the function of two misfolded KATP channel disease-mutants. FEBS Lett 579:4113–4118. https://doi.org/10.1016/j.febslet.2005.06.039
Martin GM, Sung MW, Yang Z et al (2019) Mechanism of pharmacochaperoning in a mammalian KATP channel revealed by cryo-EM. Elife 8:e46417. https://doi.org/10.7554/eLife.46417
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Acknowledgements
We thank the patient for her participation.
Data availability
The datasets generated during the current study are available from the corresponding author.
Funding
NV and FMA are supported by UKRI | Biotechnology and Biological Sciences Research Council (BBSRC) (BB/R002517/1 and BB/R017220/1). FMA is also supported by the Medical Research Council (MRC) (MR/T002107/1). This work was also supported by the US National Institutes of Health R01DK104942 (LHP/SAWG), NIH DRTC P30DK020595, NIH K23DK094866 (SAWG), NIH CTSA UL1 TR000430 (REDCap support), and gifts from the Kovler Family Foundation. MVS is supported by Clinical Therapeutics Training Grant (T32GM007019).
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
NV, FMA and LHP designed the study. LHP, CHY, SAWG and MVS identified the mutation and collected and analysed the genetic and clinical data. NV collected and analysed the patch-clamp, two-electrode voltage-clamp and western blot data. FMA, NV, LHP, SAWG and MVS wrote the manuscript. All authors critically revised the paper and approved the final version. FMA is the guarantor of the electrophysiological studies, and LHP is the guarantor of the genetic studies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vedovato, N., Salguero, M.V., Greeley, S.A.W. et al. A loss-of-function mutation in KCNJ11 causing sulfonylurea-sensitive diabetes in early adult life. Diabetologia 67, 940–951 (2024). https://doi.org/10.1007/s00125-024-06103-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-024-06103-w