
Abstract
Aims/hypothesis
Type 2 diabetes is associated with impaired insulin action and secretion, including disturbed pulsatile release. Impaired pulsatility has been related to impaired insulin action, thus providing a possible link between release and action of insulin. Furthermore, progressive loss of beta-cell mass has been implicated in the pathogenesis of Type 2 diabetes. The aim of this study was to evaluate a possible link between loss of beta-cell mass and impaired pulsatile insulin secretion with special focus on glucose responsiveness of insulin secretion.
Methods
The kinetic and dynamic profiles of insulin in Göttingen minipigs are favourable for studies on pulsatility and a model of diabetes with reduced beta-cell mass has recently been established. Pigs were studied before (n=14) and after (n=10) reduction of beta-cell mass by nicotinamide (67 mg/kg) and streptozotocin (125 mg/kg) from 17.7±4.7 (normal animals,n=5) to 6.1±2.0 mg/kg. Pulsatile insulin secretion was examined during basal (n=8 normal, n=6 beta-cell reduced) and glucose entrained (n=6 normal, n=4 beta-cell reduced) conditions. Insulin concentration time series were analysed by autocorrelation and spectral analyses for periodicities and regularity, and by deconvolution for pulse frequency, mass and amplitude.
Results
Reduction of beta-cell mass and secondary hyperglycaemia resulted in correspondingly (r=0.7421, p=0.0275) reduced pulse mass (42% of normal during basal and 31% during entrained conditions) with normal periodicity (6.6±2.2 vs 5.8±2.4 min, p=0.50), regularity and entrainability of insulin secretion.
Conclusion/interpretation
Neither beta-cell loss, nor 2 weeks of slight hyperglycaemia, as seen in the beta-cell-reduced minipig, probably accounts for the disturbed insulin pulsatility observed in human Type 2 diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Type 2 diabetes is associated with defects in both beta-cell function and insulin action, but the sequence of events leading to overt Type 2 diabetes is still not clear. It has been speculated that long-term insulin resistance will cause secondary beta-cell failure, but the majority of insulin resistant (i.e. obese) subjects do not suffer from Type 2 diabetes [1, 2, 3] and, therefore, a beta-cell defect seems to be involved in the development of the disease.
The beta-cell dysfunction in Type 2 diabetes includes decreased overall insulin secretion (compared to ambient glucose concentrations) [4], reduced first phase insulin secretion [5], delayed and impaired post-prandial insulin secretion [4] and impaired pulsatile insulin secretion [6].
Evaluation of pulsatile insulin secretory defects seems to be a very sensitive method for predicting beta-cell failure, since irregular patterns of secretion have been shown in both glucose intolerant [7], glucose tolerant first-degree relatives of Type 2 diabetic patients [8] and islet cell antibody positive non-diabetic subjects [9]. The recently introduced glucose pulse induction technique seems to further improve the sensitivity in detecting early defects in pulsatility in humans [10].
Since pulsatile secretion of insulin is known to be important for insulin action in the liver [11], muscle [12, 13] and adipose tissues [14], impaired pulsatile secretion could provide a possible link between release and action of insulin.
Progressive loss of beta-cell mass, leading to mild hyperglycaemia, is believed to be important for the development of Type 2 diabetes [15, 16]. Thus, if loss of beta-cell mass is associated with impaired insulin pulsatility, this could link a primary beta-cell defect to impaired insulin action. A model linking a single inherited defect in beta-cell function with insulin resistance is, therefore, plausible, although it could equally be possible that the two distinct defects are present at the same time.
Pulsatile insulin secretion after a primary reduction of beta-cell mass has been investigated in baboons [17] and minipigs [18], and reveals a reduced amplitude but normal frequency of insulin pulses.
The Göttingen minipig offers several major advantages as an animal model for studies of insulin pulsatility because of the very rapid insulin kinetics in this species and a pulse pattern similar to that in humans. In addition, pulsatility is entrainable to achieve a very distinct on/off secretion pattern, strongly facilitating pulse detection [19]. The nicotinamide (NIA) and streptozotocin (STZ) treated Göttingen minipig [20] is a model of mild diabetes caused by a primary reduction of beta-cell mass. We assessed whether loss of beta-cell mass and the following moderate hyperglycaemia can cause secondary defects in pulsatile insulin secretion in this model. In addition to observations made during basal conditions, we evaluated pulsatile insulin secretion in response to glucose entrainment as an approach to improving detection of early beta-cell dysfunction and to determine whether glucose responsiveness is affected by a reduction in beta-cell mass.
Disturbed pulsatility in the beta-cell reduced minipig would suggest that the impaired pulsatile insulin secretion seen in human Type 2 diabetes could be caused by loss of beta-cell mass and/or slight hyperglycaemia whereas a preserved oscillatory pattern would indicate distinct and different mechanisms causing the disturbed pulsatility seen in human Type 2 diabetes. Such different mechanisms could be some of the characteristics of Type 2 diabetes not displayed by the animal model used in our study, such as insulin resistance.
Materials and methods
Animals
Adult male Göttingen minipigs 11 to 14 months of age were obtained from the barrier unit at Ellegaard Göttingen minipigs ApS, Dalmose, Denmark. Animals were housed in single pens under controlled conditions (temperature was kept between 18 and 22°C, relative air humidity was 30 to 70% with four air changes per hour) with a 12-h light to 12-h dark cycle and fed twice daily: 140 g of SDS minipig diet (SDS, Essex, England) and 240 g of a commercial swine fodder (“Svinefoder 22”, Slangerup, Denmark) and allowed free access to water. The pigs were studied at least 2 weeks after surgery and were trained carefully in all experimental procedures before the experiments began. In total 15 animals were used in the studies. Data from non-diabetic control animals (n=14) have been reported [19]. Non-diabetic control animals weighed 23±4 kg (range 19 to 31) and animals examined after NIA+STZ weighed 29±3 kg (range 24–33).
Principles of laboratory animal care were followed and the type of study was approved by the Animal Experiments Inspectorate, Ministry of Justice, Denmark.
Surgical implantation of central venous catheters
Two central venous catheters (Certo 455, B. Braun Melsungen AG, Melsungen, Germany) were inserted surgically in the cranial vena cava and exteriorised in the dorsal part of the neck under general anaesthesia induced with a combination of zolazepam 0.83 mg/kg, tiletamine 0.83 mg/kg (Zoletil 50 vet., Boehringer Ingelheim, Copenhagen, Denmark), xylazine 0.90 mg/kg (Rompun vet, 20 mg/ml; Bayer, Lyngby, Denmark), ketamine 0.83 mg/kg (Ketaminol vet, 100 mg/ml; Rosco, Taastrup, Denmark) and methadone 0.20 mg/kg (Metadon “DAK”, 10 mg/ml; Nycomed, Roskilde, Denmark) intra muscularly and maintained with isoflurane (1–3%) (Forene, Abbot, Gentofte, Denmark) in 100% oxygen. Post surgical analgesia was maintained by intra-muscular injection of buprenorphine 0.03 mg/kg (Anorfin, 0.3 mg/ml; GEA, Frederiksberg, Denmark) and carprofen 4 mg/kg (Rimadyl vet.,50 mg/ml; Pfizer, Ballerup, Denmark) before the end of anaesthesia and for 3 days after surgery by intra-muscular injecting carporfen 4 mg/kg once daily. Post-surgical infection was prevented by injection of dihydrostreptomycin sulfate (25 mg/kg) and benzylpenicillinprocain (20,000 IE/kg) (Streptocillin vet., 250 mg DHS+200 000 IE benzylpenicillinprocain/ml; Boehringer Ingelheim) immediately after surgery and once daily for the following 2 days. At the start of the study period, all animals had recovered fully from the surgical procedure as evaluated by normal behavioural and eating patterns.
Experimental groups: Protocol 1
A mixed meal OGTT was carried out in all animals (n=14 normal animals and n=10 after beta-cell reduction). In beta-cell reduced animals, the OGTT was done between 1 and 2 weeks after exposure to NIA and STZ. After an 18-h overnight fast, animals were given an OGTT of 25 g SDS minipigfodder and 2 g/kg glucose (500 g/l, SAD, Copenhagen, Denmark). The meal was eaten, rapidly and without stress, from a bowl under supervision. Blood samples were obtained from the jugular vein catheters at t=−15, −5, 0, 15, 30, 45, 60, 90, 120, 150, and 180 min relative to the fodder and glucose load.
Protocol 2
Basal pulsatile insulin secretion was evaluated in fasted (18 h) conscious animals [n=8 before NIA+STZ and n=6 after NIA+STZ (including one animal that was not investigated before NIA+STZ)]. Blood samples (0.8 ml) were taken from a jugular vein catheter every minute for 1 h. Before each blood sample was collected, 1.5 ml blood, corresponding to the catheter dead space was withdrawn and returned aseptically after each sample. Catheters were flushed with 0.8 ml of sterile saline (0.9% SAD, Copenhagen, Denmark) after each blood sample.
Protocol 3
Entrainment of pulsatile insulin secretion was evaluated in fasted (18 h) conscious animals (n=6 before NIA+STZ and n=4 after NIA+STZ). Blood samples (0.8 ml) were taken from a jugular vein catheter every minute for 1 h as described above. Every 10th min, starting at t=0 min, a bolus of glucose (4 mg kg−1 min−1 glucose 200 mg/ml, SAD) was infused over 1 min via the other jugular vein catheter.
Handling and analysis of blood samples
Blood samples were immediately transferred to vials containing EDTA (1.6 mg/ml final concentration) and aprotinin 500 KIU/ml full blood (Trasylol, 10,000 KIU/ml, Bayer, Lyngby, Denmark) and were kept on ice until centrifugation. Samples were centrifuged (4°C, 10 min, 3500 rpm) and plasma separated and stored at −20°C until analysis. Plasma glucose was analysed using the immobilized glucose oxidase method, 10 μl of plasma in 0.5 ml buffer (EBIO plus autoanalyser and solution, Eppendorf, Hamburg, Germany). Plasma insulin was analysed in a two-site immunometric assay with monoclonal antibodies as catching and detecting antibodies (Catching antibody HUI-018 raised against the A-chain of human insulin. Detecting antibody OXI-005 raised against the B-chain of bovine insulin) [21] and using purified porcine insulin to calibrate the assay. The minimal detectable concentration was 3.2 pmol/l, the upper limit was 1200 pmol/l (no sample dilution) and the inter-assay and intra-assay variations at three concentration were 15.3% and 3.2% (at 342 pM), 9.9% and 7.6% (at 235 pM) and 14.6% and 4.4% (at 87 pM). Recovery at high, medium and low concentrations was 97.1%, 97.9% and 101% respectively. Cross reactivity against a number of peptides has been tested previously: IGF-1 (human)=0.03%, growth hormone (porcine)=0.001%, glucagon (porcine)=0.4%, insulin (rat)=0.03%, somatostatin=0.2%, pancreatic polypeptide (porcine)=0.2%, C-peptide (porcine)=0.01%, intact proinsulin (human)=0.3%, 32–33 split proinsulin (human)=0.3%, des 31–32 split proinsulin (human)=0.5%, 65–66 split proinsulin (human)=30% and des 64–65 split proinsulin (human)=63% [21].
Reduction of beta-cell mass
Beta-cell mass was reduced by intravenous administration of NIA (Sigma N-3376) 67 mg/kg to accomplish a partial protection of the beta-cells 15 min prior to STZ, (Sigma S-0130) 125 mg/kg. NIA and STZ was injected after an 18 h overnight fast in conscious animals as described [20]. Animals were given SDS fodder 2 h after treatment and were observed frequently during the first 48 h after administration of NIA and STZ where blood glucose was monitored regularly to avoid episodes of hypoglycaemia due to sudden hyperinsulinaemia caused by necrosis of beta-cells.
Formulation of compounds
NIA was weighed out in individual portions and protected from light, and was dissolved in sterile saline (0.9%, SAD, Copenhagen, Denmark) to a concentration of 300 mg/ml immediately before injection. STZ was weighed out in individual portions and dissolved in sodium citrate buffer (no. 929546, Bie and Berntsen, Roedovre, Denmark, pH=4.7) to a concentration of 62.5 mg/ml immediately before injection.
Histological examination of pancreas
Histological examination was done 1 month (27±8 days) (n=7) or 2 months (60±0 days) (n=4) after dosing of NIA (67 mg/kg) and STZ and compared with data from normal animals (n=5). Furthermore, an animal that received STZ alone was included for comparison. After euthanasia with pentobarbitone (20 ml per animal) (200 mg/ml, Pharmacy of the Royal Veterinary and Agricultural University, Copenhagen, Denmark) at the end of the study, the pancreas was isolated in toto for histological examination [20]. In short, pancreases were fixed in paraformaldehyde (Bie and Berntsen, Copenhagen, Denmark), embedded in 3% agar solution (Meco-Benzon Cat. 303289, Copenhagen, Denmark) and sectioned as practiced in the smooth fractionator method [22, 23]. The deparaffinised sections were stained for insulin and a mixture of antibodies to glucagon, somatostatin and pancreatic polypeptide to visualise β and non-β endocrine cells. Furthermore, sections were counterstained with Mayer’s hematoxyline. Beta and non-beta endocrine cell mass was evaluated stereologically in two to three sections with the origin of the sections blinded to the observer. Mass of endocrine cells is expressed as mg/kg body weight.
Evaluation of results
The plasma insulin concentration time series were analysed by deconvolution to detect and quantify insulin secretory bursts. Deconvolution of venous insulin concentration data was carried out with a multi-parameter technique [24], which requires the following assumptions. The venous plasma insulin concentrations measured in samples collected at 1-min intervals were assumed to have resulted from five determinable and correlated parameters; (i) A finite number of discrete insulin secretory bursts occurring at specific times and having; (ii) individual amplitudes (maximal rate of secretion attained within a burst); (iii) a common half-duration (duration of an algebraically Gaussian secretory pulse at half-maximal amplitude), which are superimposed upon a (iv) basal time-invariant insulin secretory rate; and (v) a bi-exponential insulin disappearance model in the systemic circulation. The insulin disappearance kinetics were estimated based upon model fitting to the observed insulin concentration profiles. Assuming slower or faster kinetics for both first and second half-life (20% changes) resulted in markedly impaired fit as evaluated by sum of squared residuals. In some data sets without entrainment (four out of eight), different values could arise. In these cases, data was fitted to the observed concentrations, but only allowing kinetics to be within the range of values observed in other data sets. Subsequently kinetics could still vary, all yielding comparable fits, and therefore values were fixed in agreement with values in other data sets (mean). Assuming the fitted insulin disappearance values, we estimated the number, locations, amplitudes and half-duration of insulin secretory bursts, as well as a non-negative basal insulin secretory rate, for each data set by nonlinear least-squares fitting of the multi-parameter convolution integral for each insulin time series. A modified Gauss-Newton quadratically convergent iterative technique was used with an inverse (sample variance) weighting function. Parameters were estimated until their values and the total fitted variance both varied by less than 1 part in 100000. Asymmetric highly correlated variance spaces were calculated for each parameter by the Monte Carlo support-plane procedure. Secretory rates were expressed as mass units of insulin (pmol) released per unit distribution volume (litre) per unit time (min). The mass of hormone secreted per burst (time-integral of the calculated secretory burst) was thus computed as pmol insulin released per litre of systemic distribution volume. In the deconvolution analysis, basal secretion was adjusted to allow accommodation of most troughs. Likewise, the secretory burst half duration was adjusted to fit individual obvious secretory bursts, consisting of a series of data points building up to a peak and down to a trough. All data was analysed in a blinded manner.
Auto-correlation analysis
The periodic nature of individual insulin profiles was assessed by autocorrelation analysis. Because trends can distort the subsequent correlation analyses, they were removed by subtracting from each profile its best-fit line calculated by linear regression analysis. All auto-correlation analyses were done on series from which this procedure was first carried out. In the autocorrelation analyses, the correlation coefficients between the time series and a copy of itself at lags of 0, 1, 2, 3, etc. up to 25 min were calculated. For auto-correlation analysis, group statistical analysis of the correlation coefficients was done after using Fisher’s z transformation [25].
Spectral analysis
As an alternative way of quantifying the degree of periodicity in the series, a spectral analysis was carried out. Each time series was detrended by using the first-difference filter. The spectral estimates were then calculated with the use of a Tukey window as described [26]. The width of the window was chosen to be 35 min, yielding a good compromise between stability and fidelity.
Quantification of irregularity
The regularity of serum insulin concentration time series was assessed by application of Approximate Entropy (ApEn), which is a model independent statistic [27]. ApEn has been introduced to discriminate between insulin secretory profiles in glucose tolerant first degree relatives of Type 2 diabetic patients compared with healthy people [8]. In our study, there were a large number of consecutive insulin concentration measurements below the detection limit. Data was then assumed to be at the detection limit and ApEn was decreased in these data sets due to this artefact. ApEn is therefore not included in data analysis.
Statistics
Calculations of fasting values and AUC (baseline=0) for glucose and insulin during OGTT and statistical evaluation of results was done by using non-paired and paired two tailed Students t test and linear regression using Excel (2000) and GraphPad Prism version 3.00 for Windows (GraphPad Software, San Diego, Calif., USA). Data are presented as means ± standard deviation.
Results
Characteristics of beta-cell reduced animals
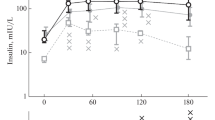
After administration of NIA+STZ (n=10), fasting plasma glucose (mmol/l) was increased slightly to 4.0±0.4 vs 3.5±0.5 in normal animals (n=14) (unpaired two-tailed t test, p<0.05). Due to different standard deviations before and after NIA+STZ, plasma concentrations of insulin were log-transformed for statistical analysis, showing that plasma insulin (pmol/l) was reduced to 12±10 (log-transformed: 0.93±0.37) from 32±26 (log-transformed: 1.38±0.32) in normal animals (unpaired two tailed t test, p<0.01). The AUC for glucose (mmol∙l−1∙min) during an OGTT was increased to 1257±201 vs 874±117 in normal animals (p<0.001) whereas the insulin response to the OGTT (pmol∙l−1∙min) was unchanged (20640±14760 vs 27960±11300 in normal animals (p=0.21)) (Fig. 1). During OGTT, relative hypoinsulinaemia [AUCinsulin/AUCglucose: 17.6±13 vs. 31.8±11.9 pmol l−1∙(mmol∙l−1)−1, p<0.05] was observed in animals dosed with NIA and STZ.

Plasma glucose (A) and insulin (B) during an oral glucose tolerance test in normal animals (1) and in animals dosed with NIA (67 mg/kg) and STZ (125 mg/kg) (2). Data are means ± SD. A n=14 normal and 10 NIA+STZ dosed, B n=12 normal and 10 NIA+STZ dosed
Beta-cell mass was reduced after dosing of NIA+STZ (6.1±2.0 mg/kg vs 17.7±4.7 mg/kg in five normal control animals, p<0.0001). The pancreases of non-diabetic minipigs had islets of quite irregular shape and the islets were not separated from the surrounding exocrine tissue by a complete collagen capsule. Furthermore, the pancreas contained many small clusters of beta and non-beta cells and numerous isolated single endocrine cells, especially alfa cells. In the pancreas of minipigs dosed with NIA+STZ, the disappearance of the majority of beta cells led to highly irregularly shaped islets and to an apparent increase in small clusters of islet cells and of single beta and non-beta cells (Fig. 2).

Pancreatic sections immunohistochemically stained for beta cells (insulin, brown cytoplasm) and non-beta endocrine cells (combination of glucagon, somatostatin, and pancreatic polypeptide, black cytoplasm). Upper row shows overviews at low magnification (4× objective); black bar=500 μm. Lower row shows details at higher magnification (40× objective); black bar=50 μm. Left column, pancreas from non-diabetic minipig (beta-cell mass=17.6 mg/kg). Right column, pancreas from diabetic minipig (beta-cell mass=3.5 mg/kg)
Insulin data
Table 1 summarises regularity and deconvolution statistics in individual animals after NIA+STZ, for comparison, mean values from normal animals [19] are included.
The insulin concentration time series showed obvious oscillations in each case studied. Representative data from animals before and after dosing of NIA+STZ are shown (Fig. 3). The induction of pulsatile insulin secretion resulted in large amplitude insulin oscillations shortly after glucose infusion both before and after induction of diabetes (Fig. 3). Note that the insulin concentration profiles showed sharp peaks and little or no insulin between pulse induction. The mean peak concentration (pmol/l) was decreased by NIA+STZ both during basal conditions (44±17 before vs 19±10 after NIA+STZ, p<0.01) and during pulse induction (82±21 before vs 42±23 after NIA+STZ, p<0.05), showing a reduced pulse amplitude after reduction of beta-cell mass (Fig. 3, Table 1).

Jugular vein plasma insulin concentration in conscious animals during basal conditions (A +C) or during entrainment of insulin pulses with glucose infusion (B+D) before (1) and after (2) dosing of with NIA (67 mg/kg) and STZ (125 mg/kg)
Regularity statistics
Spectral analysis (15.2±7.0 before vs 12.8±5.8 after NIA+STZ, p=0.51) and autocorrelation analysis (0.350±0.197 before vs 0.321±0.211 after NIA+STZ, p=0.80) showed the presence of oscillatory insulin patterns even after reduction of beta-cell mass in the basal state. Furthermore, pulse induction studies showed preserved ability to entrain pulsatile insulin secretion (spectral power: 19.7±2.5 before vs 17.0±4.5 after NIA+STZ, p=0.25; auto-correlation function: 0.555±0.148 before vs 0.459±0.173 after NIA+STZ, p=0.37). Data on individual animals for regularity statistics are given in Table 1. An unpaired t test was used for all comparisons. However, paired tests for all regularity statistics resulted in p values above 0.50.
Deconvolution
As expected from inspecting the concentration time series, deconvolution showed pulsatile insulin secretion in all animals both before and after reduction of beta-cell mass. During basal conditions, the frequency (6.6±2.2 vs 5.8±2.4 min, p=0.50) pulse amplitude (24.3±14.3 vs 13.7±14.0 pmol∙l−1∙min−1, p=0.19) as well as relative pulsatile insulin secretion (68.6±20.1 vs 73.0±33.5%, p=0.76) was unchanged before compared to after reduction of beta-cell mass, whereas pulse mass was decreased by NIA+STZ (41.2±18.6 vs 17.6±6.7 pmol∙l−1∙pulse−1, p<0.05). Furthermore, there was a correlation between pulse mass and beta-cell mass (r 2=0.7421, p=0.0275 (n=6)) (Fig. 4). During glucose pulse induction, pulse mass was reduced (228±117 vs 70.7±60.2 pmol∙l−1∙pulse−1, p<0.05) (Fig. 5) whereas both pulse amplitude (290±267 vs 46.9±41.1 pmol∙l−1∙min−1, p=0.115) and relative pulsatile insulin secretion (99.5±1.2 vs 71.5±33%, p=0.06) only tended to decrease (due to small sample size) before compared to after reduction of beta-cell mass. Pulse amplitude was reduced during pulse induction after NIA+STZ when using paired Student’s t test (p<0.01). No changes in kinetics of endogenous insulin were detected in the basal state or after glucose pulse induction.

Relation between beta-cell mass and insulin pulse mass during basal conditions in Göttingen minipigs dosed with NIA (67 mg/kg) and STZ (125 mg/kg). Note that regression line intercepts axes very close to the origin

–H. Changes in auto correlation (A+E), spectral power (B+F), pulse mass (C+G) and amplitude (D+H) in animals before (pre STZ) and after (post STZ) dosing of NIA+STZ. The upper row represents data obtained under basal conditions whereas the lower row represents data obtained with glucose entrainment
Discussion
Our studies were undertaken to evaluate whether a reduction of beta-cell mass (to approximately 35% of normal), would cause secondary defects in the pulsatile pattern of insulin secretion. STZ+NIA induced mild hyperglycaemia, mostly pronounced in the postprandial state. The decreased insulin secretion, failing to maintain euglycaemia showed a marked beta-cell defect. The results indicate that a reduction in beta-cell mass caused a correspondingly reduced secretory pulse mass. Together with normal regularity and normal response to entrainment of pulsatile insulin secretion, the data suggest a normal function of the remaining beta-cells, both individually and as a coordinated population. Thus, no signs of any secondary beta-cell dysfunction could be shown from the present study. Reduction of beta-cell mass in minipigs has been reported to reduce insulin decay rates in the fasted and the fed state [18]. We did not find changes in decay rates determined by deconvolution of endougenous insulin concentrations. The decreased secretory burst mass after a selective loss of beta cells is in accordance with previously published data in pigs [18] and baboons [17], as is the normal regularity of basal pulsatile insulin secretion in the fasted state [18]. Loss of beta-cell mass in minipigs has been reported to result in a lack of regular oscillations in insulin concentrations in response to orally and intravenously administered glucose when evaluated by auto-correlation, whereas devoncolution of the same data showed no change in pulse frequency after a reduction of beta-cell mass [18]. In our study, it seemed that the responsiveness of pulsatile insulin secretion to small changes in glucose concentration was maintained after a reduction in beta-cell mass. The lack of regularity reported in another study [18] could to some extent be attributable to the nonstationarity of insulin concentration profiles during glucose stimulation. Another important factor could be the reduced insulin secretory capacity due to a reduced beta-cell mass. Since most insulin secretion in the minipig seems to be pulsatile, reduction in insulin secretory capacity leads to a decrease in secretory burst mass, thereby making detection of pulses more difficult. We have shown that insulin secretory response to small changes in glucose concentration is preserved with respect to regularity after reduction of beta-cell mass. Defects in regularity of basal pulsatile insulin secretion has been reported at very early stages of Type 2 diabetes i.e. glucose intolerance [7] and in glucose tolerant first-degree relatives of Type 2 diabetic patients [8]. Furthermore, glucose entrainment has been reported to improve the ability to detect abnormal insulin secretion in Type 2 diabetes [10]. The failure to show impairment of regularity suggests that loss of beta-cell mass might not be the cause of impaired pulsatility in Type 2 diabetes. Furthermore, 2 weeks of slight fasting hyperglycaemia and pronounced postprandial hyperglycaemia did not cause perturbation of pulsatility nor entrainability, although pulse amplitude and mass were reduced. Although the duration of hyperglycaemia was only 2 weeks in our study, this time span exceeds what has been achieved through glucose infusions [28, 29]. The proportionally normal pulsatile insulin secretion, therefore, fails to support glucotoxicity as a primary cause of impaired pulsatile insulin secretion in Type 2 diabetes. Thus, the data on reduced beta-cell mass in the minipig suggest an expected (and correlated) loss of pulse mass but preserved pulsatile secretion of insulin in the remaining beta-cell population. This is supported by the striking correlation between pulse mass and beta-cell mass, as has also been reported in a similar group of animals [18].
The use of NIA+STZ, therefore, results in disease with loss of beta-cell mass but does not appear to represent Type 2 diabetes. The distinction between autoimmune and non-autoimmune diabetes has drawn attention to the slow immune mediated loss of beta-cell mass in approximately 10 to 15% of presumed Type 2 diabetic patients (LADA) [30]. In this large group of patients (in size similar to Type 1 diabetes), there seems to be a distinct and different aetiology compared to Type 2 diabetes. Combined with dietary-induced insulin resistance, the present model could display insulin resistance combined with blunted insulin secretion, thus carrying the two major characteristics of Type 2 diabetes, but in our study, defects more likely reflect LADA. To further support that primary beta-cell loss fails to result in Type 2 diabetes, data in other animals show normal insulin sensitivity after NIA+STZ, as is seen in honeymoon diabetic patients [31, 32]. The present model, however, might be very suitable for studies on pharmacological intervention to improve residual beta-cell mass. Little research in the field of pulsatile insulin secretion in humans has focused specifically on subgroups of patients with LADA. In evaluating beta-cell function in this group, pulse mass might be a parameter reflecting the progressive loss of beta-cell mass.
In conclusion, loss of beta-cell mass results in a correlated loss of pulse mass with no changes in regularity or entrainability of insulin secretion. Therefore, the remaining beta-cell population has a preserved insulin release pattern and responsiveness to glucose despite 2 weeks of slight hyperglycaemia. This indicates that the impaired pulsatility in Type 2 diabetes could have a different cause than loss of beta-cell mass.
Abbreviations
- ApEn:
-
Approximate entropy
- NIA:
-
nicotinamide
- LADA:
-
late onset autoimmune diabetes of the adult
- STZ:
-
streptozotocin
References
Hartz AJ, Rupley DC, Kalkhoff RD, Rimm AA (1983) Relationship of obesity to diabetes: influence of obesity level and body fat distribution. Prev Med 12:351–357
Ohlson LO, Larsson B, Bjørntorp P et al. (1988) Risk factors for type 2 (non-insulin-dependent) diabetes mellitus. Thirteen and one-half years of follow-up of the participants in a study of Swedish men born in 1913. Diabetologia 31:798–805
Chan JM, Rimm EB, Colditz GA, Stampfer MJ, Willett WC (1994) Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men. Diabetes Care 17:961–969
Polonsky KS, Given BD, Hirsch LJ et al. (1988) Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med 318:1231–1239
Brunzell JD, Robertson RP, Lerner RL et al. (1976) Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab 42:222–229
Lang DA, Matthews DR, Burnett M, Turner RC (1981) Brief, irregular oscillations of basal plasma insulin and glucose concentrations in diabetic man. Diabetes 30:435–439
O’Rahilly S, Turner RC, Matthews DR (1988) Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med 318:1225–1230
Schmitz O, Porksen N, Nyholm B et al. (1997) Disorderly and nonstationary insulin secretion in relatives of patients with NIDDM. Am J Physiol 272:E218–226
Bingley PJ, Matthews DR, Williams AJ, Bottazzo GF, Gale EA (1992) Loss of regular oscillatory insulin secretion in islet cell antibody positive non-diabetic subjects. Diabetologia 35:32–38
Hollingdal M, Juhl CB, Pincus SM et al. (2000) Failure of physiological plasma glucose excursions to entrain high-frequency pulsatile insulin secretion in type 2 diabetes. Diabetes 49:1334–1340
Komjati M, Bratusch Marrain P, Waldhäusl W (1986) Superior efficacy of pulsatile versus continuous hormone exposure on hepatic glucose production in vitro. Endocrinology 118:312–319
Matthews DR, Naylor BA, Jones RG, Ward GM, Turner RC (1983) Pulsatile insulin has greater hypoglycemic effect than continuous delivery. Diabetes 32:617–621
Schmitz O, Arnfred J, Nielsen OH, Beck Nielsen H, Orskov H (1986) Glucose uptake and pulsatile insulin infusion: euglycaemic clamp and [3-3H]glucose studies in healthy subjects. Acta Endocrinol Copenh 113:559–563
Schmitz O, Pedersen SB, Mengel A et al. (1994) Augmented effect of short-term pulsatile versus continuous insulin delivery on lipid metabolism but similar effect on whole- body glucose metabolism in obese subjects. Metabolism 43:842–846
Weir GC, Leahy JL, Bonner-Weir S (1986) Experimental reduction of B-cell mass: implications for the pathogenesis of diabetes. Diabetes Metab Rev 2:125–161
Leahy JL, Bonner-Weir S, Weir GC (1988) Minimal chronic hyperglycemia is a critical determinant of impaired insulin secretion after an incomplete pancreatectomy. J Clin Invest 81:1407–1414
Goodner CJ, Koerker DJ, Weigle DS, McCulloch DK (1989) Decreased insulin- and glucagon-pulse amplitude accompanying beta-cell deficiency induced by streptozocin in baboons. Diabetes 38:925–931
Kjems LL, Kirby BM, Welsh EM et al. (2001) Decrease in beta-cell mass leads to impaired pulsatile insulin secretion, reduced postprandial hepatic insulin clearance, and relative hyperglucagonemia in the minipig. Diabetes 50:2001–2012
Larsen MO, Elander M, Sturis J et al. (2002) The conscious Göttingen minipig as a model for studying rapid pulsatile insulin secretion in vivo. Diabetologia 45:1389–1396
Larsen MO, Wilken M, Gotfredsen CF, Carr RD, Svendsen O, Rolin B (2002) Mild streptozotocin diabetes in the Göttingen minipig. A novel model of moderate insulin deficiency and diabetes. Am J Physiol Endocrinol Metab 282:E1342–E1351
Andersen L, Dinesen B, Jørgensen PN, Poulsen F, Røder ME (1993) Enzyme immunoassay for intact human insulin in serum or plasma. Clinical Chemistry 39:578–582
Mayhew TM, Gundersen HJ (1996) ‘If you assume, you can make an ass out of u and me’: a decade of the disector for stereological counting of particles in 3D space. J Anat 188:1–15
Bock T, Svenstrup K, Pakkenberg B, Buschard K (1999) Unbiased estimation of total beta-cell number and mean beta-cell volume in rodent pancreas. APMIS 107:791–799
Veldhuis JD, Carlson ML, Johnson ML (1987) The pituitary gland secretes in bursts: appraising the nature of glandular secretory impulses by simultaneous multiple-parameter deconvolution of plasma hormone concentrations. Proc Natl Acad Sci USA 84:7686–7690
Fisher RA (1958) Statistical methods for research workers. Churchill Livingstone, Edinburgh
Jenkins GM, Watts DG (1968) Spectral analysis and its applications. Holden Day, San Fransisco
Pincus SM (1991) Approximate entropy as a measure of system complexicity. Proc Natl Acad Sci USA 88:2297–2301
Leahy JL, Weir GC (1991) Beta-cell dysfunction in hyperglycaemic rat models: recovery of glucose- induced insulin secretion with lowering of the ambient glucose level. Diabetologia 34:640–647
Leahy JL, Cooper HE, Weir GC (1987) Impaired insulin secretion associated with near normoglycemia. Study in normal rats with 96-h in vivo glucose infusions. Diabetes 36:459–464
Zimmet PZ (1995) The pathogenesis and prevention of diabetes in adults. Genes, autoimmunity, and demography. Diabetes Care 18:1050–1064
Gray RS, Cowan P, Duncan LJP, Clarke BF (1986) Reversal of insulin resistance in type 1 diabetes following initiation of insulin treatment. Diabet Med 3:18–23
Yki-Jarvinen H, Koivisto VA (1986) Natural course of insulin resistance in type 1 diabetes. N Engl J Med 315:224–230
Acknowledgements
We thank H. Nygaard, L. Sejersen Winther, M. Jeppesen, A.-G. Juul, L. Gottlieb Sørensen, N.K. Nowa Hansen, A. Petersen, K. Larsen and H. Rasmussen for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00125-003-1281-7
Rights and permissions
About this article
Cite this article
Larsen, M.O., Gotfredsen, C.F., Wilken, M. et al. Loss of beta-cell mass leads to a reduction of pulse mass with normal periodicity, regularity and entrainment of pulsatile insulin secretion in Göttingen minipigs. Diabetologia 46, 195–202 (2003). https://doi.org/10.1007/s00125-002-1011-6
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-002-1011-6