
Abstract
Key message
Modifying morphometric inflorescence traits is important for increasing grain yield in wheat. Mapping revealed nine QTL, including new QTL and a new allele for the q locus, controlling wheat spike morphometric traits.
Abstract
To identify loci controlling spike morphometric traits, namely spike length (SL), internode length (IL), node number per spike (NPS), and node density (ND), we studied 146 Recombinant Inbred Lines of tetraploid wheat (Triticum turgidum L.) derived from standard spike and spike-branching mutant parents. Phenotypic analyses of spike morphometric traits showed low genetic coefficients of variation, resulting in high heritabilities. The phenotypic correlation between NPS with growing degree days (GDD) suggested the importance of GDD in the determination of node number in wheat. The major effect QTL for GDD or heading date was mapped to chromosome 7BS carrying the flowering time gene, Vrn3-B1. Mapping also identified nine QTL controlling spike morphometric traits. Most of these loci controlled more than a single trait, suggesting a close genetic interrelationship among spike morphometric traits. For example, this study identified a new QTL, QND.ipk-4AL, controlling ND (up to 17.6% of the phenotypic variance), IL (up to 11% of the phenotypic variance), and SL (up to 20.8% of the phenotypic variance). Similarly, the major effect QTL for IL was mapped to the q locus. Sequencing of the Q/q gene further revealed a new q allele, qdel-5A, in spike-branching accessions possessing a six base pair deletion close to the miR172 target site. The identification of qdel-5A suggested that the spike-branching tetraploid wheats are double mutants for the spikelet meristem (SM) identity gene, i.e., branched headt (TtBHt), and the q gene, which is believed to be involved in the SM indeterminacy complex in wheat.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Wheat (Triticum sp.) domestication took place in the Fertile Crescent some 10,000 years ago (Heun et al. 1997; Faris 2014). Since then, it has become one of the largest and most important food crops grown across the world (Shiferaw et al. 2013). Like in other cereal crops, the domestication syndrome: a suite of phenotypic traits differentiating domesticated crops from their wild ancestors (Doebley et al. 2006; Meyer et al. 2012), is also evident in wheat. These include non-brittle rachis, grain threshability, spike length, tillering, photoperiod, and vernalization requirements (Salamini et al. 2002; Kilian et al. 2009; Faris et al. 2014).
Among several factors that have contributed to the broad adaptability of wheat in different parts of the world is the variation in flowering time (Beales et al. 2007; Cockram et al. 2007; Campoli and von Korff 2014). The time to flowering in wheat is predominantly influenced by developmental and environmental factors that are generally controlled by three sets of genes: the vernalization (Vrn), photoperiod (Ppd), and earliness per se genes (Yan et al. 2003, 2004, 2006; Beales et al. 2007; Cockram et al. 2007; Kippes et al. 2015).
Accelerated flowering in wheat resulted in reduced plant height, tillering, and spikelet number (Worland et al. 1998; Shaw et al. 2013). Furthermore, spikelet and floret primordia initiation in wheat are sensitive to temperature and photoperiod (Rawson 1970; Rahman and Wilson 1977, 1978; Cottrell et al. 1982; Baker and Gallagher 1983a, b; Worland et al. 1998; Boden et al. 2015; Steinfort et al. 2017). The sub-phase duration during spike development also affects spikelet number, thereby affecting grain yield (Rawson 1970; Baker and Gallagher 1983b; Guo et al. 2018). Therefore, temperature, light conditions, and phase duration are key factors for the variation in the spike morphometric traits, i.e., spike length, internode length, node density, and spikelet number (Friend 1965; Rawson 1970, 1971; Pinthus and Millet 1978; Fischer 1985; Rawson and Richards 1993; Shaw et al. 2013; Steinfort et al. 2017). As wheat spikelets bear the grains and are the building blocks of the wheat spike, spike morphometric traits are essential agronomic traits in wheat (Kumar et al. 2007).
At least three loci were known to control spike length in hexaploid wheat. These are the q/Q, C or compactum (i.e., club wheat) and the s/S (i.e., the shot wheat characterized by short dense spike) (Salina et al. 2000; Johnson et al. 2008; Faris et al. 2014). Therefore, common hexaploid wheat is suggested to have the genotype QcS (Faris et al. 2014). Among these loci, only the gene underlying the q locus has been identified as one of the domestication genes controlling spike length and other spike-related traits (Simons et al. 2006; Debernardi et al. 2017; Greenwood et al. 2017). Since the C and s loci were reported to reside on chromosome 2D and 3D, respectively, it is not yet clear whether the homoeo-loci also exist in tetraploid wheat.
As it has been suggested, if wheat yield is genuinely sink-limited (Slafer and Savin 1994; Borras et al. 2004; Shearman et al. 2005; Miralles and Slafer 2007), then increasing the sink (spike) size and improving spikelet fertility are key for increasing the grain yield (Donald 1968; Foulkes et al. 2011). In this regard, three approaches can be suggested for increasing the spike size in wheat. The first approach is increasing spikelet number per spike (Boden et al. 2015; Dobrovolskaya et al. 2015; Poursarebani et al. 2015; Dixon et al. 2018b). The second approach is increasing the number of florets and/or grains and grain size per spikelet (Debernardi et al. 2017; Greenwood et al. 2017; Guo et al. 2017; Sakuma et al. 2019). The third approach is increasing spikelet as well as floret/grain number and grain size simultaneously. The key challenge is, however, whether spike and/or spikelet fertility are proportionately improved with the increased sink size without trade-offs among these traits. Therefore, understanding the genetic, developmental and physiological basis of wheat spike morphology is key not only for increasing spikelet number and arrangements but also for maximizing the fruiting or grain setting efficiency of the spikelets (Slafer et al. 2015).
Despite the importance of wheat spike morphology, a relatively small number of genes have been identified (Simons et al. 2006; Dobrovolskaya et al. 2015; Poursarebani et al. 2015; Avni et al. 2017; Debernardi et al. 2017; Greenwood et al. 2017; Dixon et al. 2018b).
In this study, we used 146 F7- and F8-derived Recombinant Inbred Lines (RILs) derived from a cross between Bellaroi (an elite durum wheat cultivar with spring growth habit) and TRI 19165 (a traditional tetraploid landrace with winter growth habit and spike-branching commonly known as ‘Miracle Wheat’) for mapping the spike morphometric traits, namely spike length (SL), internode length (IL), node number per spike (NPS), node density (ND). The phenotypic and genetic analysis of spike morphometric traits generally shows high genetic correlation and heritability. Mapping also identified several genomic regions, including a new q allele (henceforth, qdel-5A) controlling more than one trait, suggesting strong genetic interrelationship of these traits contributing to unique developmental outcomes affecting the wheat spike morphometric traits. Moreover, this study also identified a previously unidentified locus on chromosome 4AL, QND.ipk-4AL, controlling SL, ND, and IL. The QTL controlling ND and SL on chromosome 2AL (QND.ipk-2AL) was mapped to the region harboring the C (Compactum) locus of hexaploid wheat, suggesting that QND.ipk-2AL is likely to be the homoeolog in tetraploid wheat.
Materials and methods
Development of the RILs
One hundred forty-six F7-derived RILs were developed through single-seed descent (SSD) from an F2 population derived from a cross between Bellaroi (an elite durum wheat variety with spring growth habit) and TRI 19165 (a ‘Miracle Wheat’ and winter type). Since the mapping population segregated for winter/spring growth habit, all RILs with winter growth habit were excluded from the mapping population by growing all the F2 plants without vernalization in the greenhouse. Those that were able to complete their life cycle and give grains without vernalization were all spring types (n = 146) and were used for this study. The RILs were evaluated under field conditions for two consecutive years in 2014 and 2015 growing seasons in three different environments (IPK14, IPK15, and HAL15) in Germany. In 2014, the F7-derived RILs were evaluated in Gatersleben (IPK14), 51.49° N and 11.16° E, Germany. In the following season, i.e., 2015, the F8-derived RILs were evaluated in two environments: Gatersleben (IPK15) and Halle/Saale (HAL15), Germany. All field evaluations were conducted on 3.75 m2 plots in a Randomized Complete Block Design (RCBD) in two replications. Three hundred grains per m2 were sown in rows spaced at 20 cm. Plants in all locations were fertilized according to the soil conditions in each environment. Similarly, herbicides and fungicides were also applied in order to control weeds and fungal infestations.
Phenotyping
Heading date was recorded when 50% of the spike appears from the flag leaf sheath in 50% of the plants following Zadoks scale (Zadoks 1985). Heading date was converted to growing degree days (GDD) (Miller et al. 2001) by summing the average daily maximum and minimum temperatures. Spike length, excluding the awns, was recorded from five to ten randomly sampled plants from the middle of each plot. Node number per spike was recorded from 10 to 15 spikes sampled from the middle row of each plot from all environments. Average spike internode length was calculated from spike length and node number per spike. Node density (average node number per cm of the spike) was computed from node number and spike length of the main culm spike by dividing node number by spike length. Basic statistical analysis, such as analysis of variance (ANOVA) and correlation analysis, was performed using Genstat 17 and SPSS 20 (IBM 2011; Payne et al. 2014). Narrow sense heritability was determined using the general formula: h2 = σ2G/(σ2G + σ2GEI + σ2e) (Nyquist 1991; Singh et al. 1993), where σ2G is genotypic variance due to additive effects, σ2GEI is variance component due to genotype-by-environment interaction, and σ2e is an error variance.
DNA extraction and genotyping
Genomic DNA from all F7-derived RILs was isolated following the modified CTAB method as described by Doyle (1990) and used for mapping. The final concentration was measured and samples were used for CAPS marker analysis as well as for genotyping-by-sequencing (GBS) library preparation following two-enzyme genotyping-by-sequencing approach (Poland et al. 2012; Wendler et al. 2014).
Marker generation from F7-RILs
DNA markers from RILs were generated through genotyping-by-sequencing (GBS) following the two-enzyme approach (Poland et al. 2012). Adapters were trimmed from reads with cutadapt version 1.8.dev0 (Martin 2011). Trimmed reads were mapped to the chromosome-shotgun assemblies of bread wheat cultivar Chinese Spring (The International Wheat Genome Sequencing Consortium (IWGSC) 2014) with BWA mem version 0.7.12 (Li 2013), converted to BAM format with SAMtools (Li et al. 2009) and sorted with Novosort (Novocraft Technologies Sdn Bhd, Malaysia, http://www.novocraft.com/). Multi-sample variant calling was performed with SAMtools version 0.1.19 (Li 2011). The command “mpileup” was used with the parameters “-C50 –DV”. The resultant VCF file was filtered with an AWK script provided as Text S3 by Mascher et al. (Mascher et al. 2013). Only bi-allelic SNPs were used. Homozygous genotype calls were set to missing if their coverage was below 1 or their genotype quality was below 3. Heterozygous genotype calls were set to missing if their coverage below 4 or their genotype quality was below 10. An SNP was discarded (i) if its quality score was below 40, (ii) its heterozygosity was above 20%, (iii) its minor allele frequency was below 10%, or (iv) had more than 66% missing data. Genotype calls were filtered and converted into genotype matrix with an AWK script available as Text S3 of Mascher et al. (2013) (Mascher et al. 2013). Chromosomal locations and genetic positions were taken from population sequence (POPSEQ) data (Chapman et al. 2015).
Construction of the linkage map
The genetic linkage map was constructed using Joinmap 4.0 (Stam 1993). All distorted markers were removed based on goodness-of-fit using a Chi-squared test. Regression and maximum likelihood mapping algorithms were used for the linkage construction. Linkage groups were determined using Haldane’s mapping function. The maximum distance of 50 cM was used to determine linkage between two markers. The maps were drawn using Map chart version 2.3 (Voorrips 2002).
QTL mapping
Phenotypic data from all 146 RILs and all environments (IPK14, IPK15, and HAL15) were used for QTL mapping using Genstat 17. A step size of 10 cM, minimum cofactor proximity of 50 cM, a minimum separation of selected QTL of 30 cM, and genome-wide significance level (alpha) of 0.05 were used for QTL analysis. Based on the mixed-model approach, the whole genome was scanned first using simple interval mapping (SIM) approach. Then, based on the SIM result, cofactors were selected for composite interval mapping (CIM). The final QTL model was selected using the backward selection on the selected cofactors, where QTL boundaries (lower and upper), QTL effect, and phenotypic variance explained (PVE) by QTL were determined.
Development of CAPS marker
The diagnostic CAPS marker for the Q/q gene was developed based on SNP T3142C, located in the miR172 target site (Debernardi et al. 2017). The T3142C substitution differentiates the ancestral q allele from the derived, domesticated Q allele. Genome-specific primers PJG14 and PJG18 were used to amplify 230 bP fragments spanning T3142C (Greenwood et al. 2017). Hinf I, New England Biolabs®, USA, was used for restriction digestion analysis following manufacturer’s protocol. The Hinf I restriction site has been eliminated from the Q-5A allele due to the T3142C substitution. Thus, Hinf I digests only the amplicon from the ancestral allele, i.e., q-5A into two fragments, i.e., 158 and 72 bp (see Supplementary Figure. 1).
Sequencing and sequence analysis
The complete Q/q gene (3229 bp) was amplified from genomic DNA of 44 spike-branching tetraploid wheat mutants (see Supplementary Table 2) using genome-specific primers (see Supplementary Table 1) and highly specific polymerase for amplification (HotStarTaq DNA Polymerase from QIAGEN). The amplicons were Sanger-sequenced from both sides using ten overlapping primer pairs of the Q/q gene. Sequences were assembled using Geneious version 11 (Kearse et al. 2012).
Results
Phenotypic correlation and heritability of spike morphometric traits
The mapping population used in this study was developed from parents with contrasting spike morphology (Fig. 1). Pairwise correlations among spike morphometric traits: SL, NPS, IL, ND, and HD (GDD), are shown in Table 1. SL was positively correlated with NPS, HD, and IL but negatively correlated with the ND. ND showed a negative correlation with SL and IL, suggesting a trade-off between SL or IL with the spikelet density. NPS, which is the total number of nodes per spike, was also positively correlated with ND and HD but showed a negative correlation with IL. The effect of HD on SL and NPS was likely to be a pleiotropic effect. Previous studies have also demonstrated that accelerated flowering in wheat shortens the duration of the vegetative phase, including inflorescence primordia development (Worland et al. 1998; Miralles and Richards 2000; Shaw et al. 2013), thereby affecting the size and number of organs to be produced. Estimates of the phenotypic (σ2P), genotypic (σ2G), genotype-by-environment interaction (σ2GEI), and error variances (σ2e) for SL, NPS, ND, HD, and IL are also shown in Table 1. The σ2G for all the traits were higher than that of the σ2GEI, suggesting that the measured phenotypic variance was indeed controlled by the genetic factors underlying each trait. This conclusion was further supported by the low genetic coefficients of variation (GCV) and high heritabilities (h2) of the traits (Table 1).

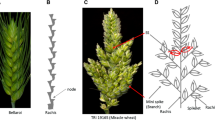
Spike morphological differences between parent Bellaroi (A) and TRI 19165 (C). For clarity, awns were removed from TRI 19165. Spike from Bellaroi shows a canonical or unbranched spike, where each node or spikelet is arranged in a distichous order along the axis (B). Red arrows in C show the mini-spike-like structures arising from the nodes of the bottom half of the rachis of TRI 19165, whereas the blue arrows show supernumerary spikelets (SS) sharing the same rachis node. (D) Schematic sketch of spike-branching wheat shown in C (color figure online)
Identification of loci controlling spike morphometric traits
Mapping identified several loci controlling spike morphometric traits. All the QTL, their chromosomal locations, QTL-by-environment additive effects (QTLxE AEs), and the phenotypic variance explained (PVE) by each of these QTL are shown in Table 2. Three QTL, i.e., QHD.ipk-7BS, QHD.ipk-2AS, and QHD.ipk-3AL, were detected for HD or GDD (Table 2, and Fig. 2A, B, F). The major effect QTL was located on chromosome 7BS (i.e., QHD.ipk-7BS). Interestingly, wheat chromosome 7BS carries the wheat flowering time gene VRN3-B1 or FT-B1 (Yan et al. 2006), suggesting that the underlying gene for QHD-ipk-7BS may indeed be FT-B1. QHD.ipk-7BS consistently appeared in all environments as a major effect QTL controlling about 59% of phenotypic variation for heading date. Similarly, wheat chromosome 2AS is known to carry the PHOTOPERIOD RESPONSE locus Ppd-A1 (Beales et al. 2007), suggesting that the underlying locus for QHD.ipk-2AS is most likely Ppd-A1. The winter wheat accession TRI 19165 contributed the high-value alleles (HVAs; i.e., late flowering) for all the three-heading time QTL. Besides controlling heading date, QHD.ipk-7BS was also colocated with QTL for SL and NPS in all environments (Table 2, and Fig. 2A). The effect is likely to be pleiotropic due to delayed flowering (Shaw et al. 2013; Dixon et al. 2018a). This result was also supported by significant positive phenotypic correlations of HD with SL (0.26; p = 0.01) and NPS (0.58; p = 0.01; Table 1).



Genetic maps of chromosomes showing QTL for spike length (SL), node number per spike (NPS), node density (ND), heading date (HD), and internode length (IL). Effect of the QTL is shown as box plots computed after grouping the RILs into two sets based on the closest marker flanking each QTL. Grouping was made based on the closest marker for each QTL (shown below the box plot). (+) shows the phenotype of the RILs carrying the QTL, and (−) shows the phenotype of the RILs without the QTL. B, allele from Bellaroi; T, allele from TRI 19165. ‘Miracle Wheat’ allele or the bht-A1 allele on chromosome 2A (39.1 cM) is underlined. Pink dash shows the putative centromeric position per chromosome
The other spike morphometric trait studied in the current mapping population is spike internode length (IL). IL, also known as rachis IL, was defined as the length between two successive nodes on the rachis, which is the central axis of the spike (Fig. 1B). In total, four QTL were identified controlling IL (Table 2, Fig. 2C, E, F). By controlling up to 50% of the phenotypic variance, the QTL on chromosome 5AL, QIL.ipk-5AL, was the major effect QTL. QIL.ipk-5AL also affected SL and spikelet density (ND). Interestingly, chromosome 5AL carries the q locus, which is one of the domestication-related wheat genes controlling the square-head spike shape and other several spike-related traits (Simons et al. 2006; Debernardi et al. 2017; Greenwood et al. 2017). This result suggested that the underlying phenotypic variation for IL, SL, and ND at QIL.ipk-5AL was likely to be controlled by allelic variation at the q locus. A Q/q gene derived CAPS marker analysis further revealed allelic variation at the q locus between Bellaroi and TRI 19165. Expectedly, the commercial variety Bellaroi carried the modern Q allele (Supplementary Figure 1). To our surprise, TRI 19165 carried the ancestral q allele, which lacks the mutation within the miRNA172 binding site (Debernardi et al. 2017). Linkage analysis also placed the q-derived CAPS marker at cM position 147.5 on chr 5AL (Fig. 2C). Re-mapping of IL, ND, and SL after including the q-derived CAPS marker further suggested linkage of q with the phenotypes. Therefore, it is highly likely that allelic variation at the q locus, QIL.ipk-5AL, is responsible for the phenotypic variation in IL, ND, and SL.
Apart from QIL.ipk-5AL, two other loci from chromosome 4AL (QND.ipk-4AL) and 2AL (QND.ipk-2AL) also controlled ND, IL, and SL. By controlling ND as a primary effect, QND.ipk-4AL also controlled IL, thereby affecting SL as well. As far as we know, QND.ipk-4AL is a new QTL having strong effects on spike morphometric traits in tetraploid wheat. Given the strong negative correlation between ND and IL (− 0.98, p = 0.01; Table 1), TRI 19165 contributed the HVA for increased IL, thereby increasing SL. The corresponding allele from Bellaroi increased ND, maximizing spikelet number per unit length of the rachis. Similarly, QND.ipk-2AL was mapped to chromosome 2AL (Fig. 2F). TRI 19165 contributed the HVA for ND while the corresponding allele from Bellaroi increased the IL. Interestingly, Johnson et al. (2008) also mapped the C (Compactum) locus in hexaploid wheat on the long arm of chromosome 2D close to the centromeric region (Johnson et al. 2008), indicating that the QND.ipk-2AL is likely to be the homoeo-locus of Compactum in the tetraploid wheat, which was not reported before. Similarly, we cannot completely rule out the possibility that the underlying gene for QND.ipk-2AL could also be the wheat ortholog of barley APELATA2 (HvAP2) gene which has been known to affect the density of grains along the rachis (Houston et al. 2013).
QIL.ipk-2AS controlling IL resides in a 4.6-cM interval distal to the bht-A1 locus (Poursarebani et al. 2015). Apart from controlling IL, QIL.ipk-2AS also had an effect on NPS (Table 2, Fig. 2F). The allele from Bellaroi increased IL, while the corresponding allele from TRI 19165 increased NPS, suggesting that QIL.ipk-2AS is either pleiotropic or closely linked to the ‘Miracle Wheat’ or the bht-A1 allele. Consistent with this, earlier studies have also mapped ND in the region harboring the wheat frizzy panicle (WFZP) gene which is the ortholog of TtBHt1 in hexaploid wheat (Sourdille et al. 2000; Ma et al. 2007; Cui et al. 2012; Echeverry-Solarte et al. 2014; Dobrovolskaya et al. 2015; Echeverry-Solarte et al. 2015).
Genetic interrelationships of spike morphometric traits are revealed by shared QTL
Besides the phenotypic correlations among spike morphometric traits, shared QTL among traits suggested genetic interrelationships between spike morphometric traits (Fig. 3). Correlation analysis showed that SL and NPS were positively correlated with HD (Table 1). Although the effect of the heading date QTL on chr 7BS, QHD.ipk-7BS, on SL and NPS seemed to be a pleiotropic effect, it might be the cause for the genetic correlation between SL and NPS with HD. Similarly, IL was strongly positively correlated with SL and negatively correlated with ND. Likewise, QIL.ipk-5AL, which is a major effect QTL for IL, also colocated with SL as well as ND. QND.ipk-4AL, which is the major effect QTL for ND, also colocated with SL and IL (Table 2). The strong negative correlation between IL and ND was also clearly manifested by the reciprocal action of the parental alleles at QIL.ipk-5AL and QND.ipk-4AL, where alleles from Bellaroi increased ND, while the corresponding alleles from TRI 19165 increased the IL (Fig. 3). Taken together, these results suggest a tight interplay among common gene sets that control spike morphometric traits providing unique developmental outcomes during spike development.

Genetic interrelationship of the spike morphometric traits as revealed by shared QTL. Solid lines connect the major effect QTL with the trait. Broken lines connect the extended effect of the QTL on the correlated traits. T(+) or B (+) indicates the source of the high-value allele (HVA) where T indicates allele from TRI 19165 and B indicates the corresponding allele from Bellaroi. HD heading date, IL internode length, SL spike length, ND node density, NPS node number per spike
Sequence analysis from the spike-branching wheat mutants revealed a new q allele
After discovering allelic variation at the Q/q locus between Bellaroi and TRI 19165, we sequenced this locus in 44 different spike-branching wheat mutants (Supplementary Table 2.). Forty-two out of the 44 spike-branching mutants carried a six bp deletion in exon 10, designated as qdel-5A (Fig. 4 and Supplementary Figure 2). The six bp deletion resulted in the deletion of two concurrent amino acids (L410del and Y411del) close to the miR172 target site; however, deletion and miR172 binding site do not overlap. From the fifteen SNPs identified (Fig. 4), eight of them were identified in this study. From these eight SNPs, six SNPs, i.e., G100T, C1896T, A2318G, T3113C, T3114C, and T3124G, were identified in the protein coding sequences. Among these, four SNPs, namely G100T, T3113C, T3114C, and T3124G, were non-synonymous (Fig. 4). While the T3113C and T3114C occurred outside of the miR172 target site, T3124G was located within the 3′-end of the miR172 target site, which is generally considered to be a less conserved region for miRNAs binding as compared to the 5′-end, or the so-called seed region (Bartel 2009). From previous studies, it was known that the interaction between miR172 and modern Q allele was reduced due to the pivotal SNP (C3142T), resulting in high Q transcript levels and shorter IL in modern square-headed spikes (Debernardi et al. 2017). Carriers of the new qdel-5A allele, however, rather show relaxed spike architecture (larger IL), implying that either transcript levels (less likely by T3124G) and/or amino acid substitutions (L409P, L410del, Y411del, A412P) might reduce or impair qdel-5A protein function in ‘Miracle Wheats.’ Even if we have not yet established the exact effect of the mutation for the qdel-5A allele, the increased IL or SL was likely to be the effect of the qdel-5A allele (Fig. 2C). Taken together, these results unambiguously showed that most ‘Miracle Wheats’ carry two mutations in two independent genes: one for spike-branching (i.e., bht-A1 allele) and one for the speltoid-like or spear-shaped spike form (i.e., qdel-5A allele).

Structural variations at the q locus in the spike-branching ‘Miracle Wheat’ mutants. A The q gene model. The box indicates exons. The orange filled boxes indicate the coding regions for the two AP2 domains of the q protein. SNP positions are indicated at the top. The numbers indicate the position of each SNP from the start codon. Each SNP was designated by taking Zavitan as a reference sequence. SNPs in red fonts are identified in this study, whereas those with blue fonts are also previously reported including the T3142C substitution that differentiated the two alleles at the q locus (Simons et al. 2006; Debernardi et al. 2017). The red and blue boxes in exon 10 indicate the six bp deletion and miR172 binding site, respectively. B Haplotypes of the spike-branching mutants at the q locus. Amino acid changes due to the non-synonymous substitutions are indicated beneath the figure. L410del and Y411del are deleted amino acids due to the deleted nucleotides. NC, no change; HAP, haplotype. The sequences from each haplotype reported in this study are deposited in the GenBank database with accession numbers MK423900, MK423901, MK423902, and MK423903 (color figure online)
Discussion
Wheat spike morphometric traits are phenotypically and genetically correlated with high heritabilities
Since its domestication, wheat has undergone important morphological changes in the inflorescence (Faris 2014). Although only a few genes were identified in this regard (Simons et al. 2006; Avni et al. 2017), the molecular understanding of the morphological changes of the inflorescence is not only important for the understanding of crop evolution and domestication, but also important for increasing grain yield through genetic manipulation of inflorescence architecture. Due to the complex genome structure of wheat (Mayer et al. 2014), its inflorescence architecture is controlled in a complex way. This often creates difficulties of visualizing the morphological changes caused by any recessive mutations due to the buffering effect of other functional gene(s) from the sub-genomes (Krasileva et al. 2017).
Wheat spike morphometric traits related to the domestication of the spike include SL, ND, NPS, and IL. Although several genetic loci were detected for these traits (Echeverry-Solarte et al. 2015; Zhai et al. 2016; Zhou et al. 2017), the Q gene is the only one that has been identified (Simons et al. 2006). Similarly, in the current population QTL for SL, ND, and IL were also mapped to the region of the q locus on 5AL (Fig. 2C). Interestingly, another important genomic region on 4AL, QND.ipk-4AL, similarly controls SL, ND, and IL. Most of the previous studies mapped SL QTL on 4AL in a region spanning from 75 to 93 cM (Jantasuriyarat et al. 2004; Kumar et al. 2007; Chu et al. 2008; Echeverry-Solarte et al. 2015). By controlling NPS and SL, QNPS.ipk-4AL was mapped at cM position 112 (Table 2, and Fig. 2E) and therefore is likely to be identical with previous studies. However, QND.ipk-4AL was mapped at cM position 265 (Table 2, and Fig. 2E) and hence is likely to be a new QTL.
As the wheat chromosome 4A is well characterized by two reciprocal translocations and two inversions, a portion of 4AL corresponds to 4DS (Mickelson-Young et al. 1995; Miftahudin et al. 2004; Dvorak et al. 2018). Likewise, the QTL reported by Cui et al. (2012) on 4DS controlling SL, NPS, and ND might correspond to QND.ipk-4AL (Cui et al. 2012). Recently, Dixon et al. (2018a, b) also reported the interaction between TEOSINTE BRANCHED1(TB1) and FLOWERING LOCUS T1, (i.e., Vrn3-B1) regulating inflorescence architecture in bread wheat (T. aestivum L.). Since TB1 is located on the short arm of chromosome 4D (i.e., 4DS), we checked whether TB-A1 could be the underlying gene for QND.ipk-4AL. QND.ipk-4AL was approximately mapped between 629,347,058 and 634,848,225 bp, while TB-A1 was mapped between 582,841,008 and 582,839,894 bp, indicating that TB-A1 is most likely not the candidate gene for QND.ipk-4AL but likely for QNPS.ipk-4AL which was flanked by marker 4AL_7091960:3169, mapped at position 544,598,274 bp. Therefore, the identification of genes underlying loci controlling spike morphometric traits, especially QND.ipk-2AL, QND.ipk-4AL, and QNPS.ipk-4AL, is essential for a better genetic and molecular understanding of spike morphogenesis in wheat.
Rachis, spikelets, rachilla, and florets are the building blocks of wheat spike architecture
Inflorescence architecture is determined by the activity of the inflorescence meristem (Tanaka et al. 2013). In wheat, the inflorescence meristem (IM) directly produces the spikelet meristems (SMs) on its flanks. The SMs then differentiate into glume primordia (GP) and floral meristems (FMs) (Barnard 1955; Kirby and Appleyard 1984). Due to the determinate meristematic activity of the IM, the wheat spike initiates a terminal spikelet, thereby terminating the initiation of new SMs from the IM (Bonnet 1967; McMaster 1997). However, unlike other cereal crops, the wheat SMs differentiate indeterminately, thereby initiating several floral meristems (FMs) on the flanks of the central spikelet axis, known as the rachilla (Fig. 5). Each FM finally differentiates into floral organs: lemma, palea, stamen, lodicules, and pistil. In standard or canonical wheat spike architecture, branching from the rachis node or the development of supernumerary spikelets (SS) is highly suppressed, while primary sessile spikelets are distichously attached to the central axis of the inflorescence, known as rachis (Fig. 5A).

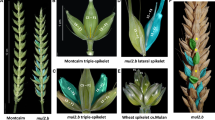
Wheat spike architecture. A The canonical or standard wheat spike where spikelets are arranged in a distichous order along the spike axis or the rachis. B The Q gene null mutants showing elongated rachilla (broken line) carrying more florets (not shown) as reported by Debernardi et al., (2017). C Spike-branching wheat mutant showing mini-spike arising from rachis node in place of the spikelets. SS supernumerary spikelet
Spike-branching and/or SS formation is prevented by TtBHt-A1/WFZP that provides SM identity to the axillary meristems (AxMs) of the IM (Dobrovolskaya et al. 2015; Poursarebani et al. 2015). Thus, spike-branching TtBHt-A1 mutants rather re-initiate inflorescence-like meristems instead of directly forming SMs, implicating that these mutants have partially lost their SM identity. Interestingly, the newly developed inflorescence-like meristems also initiate new AxMs, which will finally acquire the identity of SMs, thereby producing complete sessile spikelets arranged distichously on a central axis that resembles the main rachis (Fig. 5C).
Recent studies have shown that the wheat Q gene is a suppressor of floret number through the suppression of rachilla development (Debernardi et al. 2017; Greenwood et al. 2017). This has been confirmed by Q gene mutants of wheat which are typically characterised by rachis (Simons et al. 2006) and rachilla extension with extra florets (Debernardi et al. 2017; Greenwood et al. 2017). Therefore, the Q protein is also involved in the SM indeterminacy complex, i.e., by regulating rachilla development and the morphogenesis of the rachis. Based on this, it is very likely that lower abundance of the qdel-5A protein in ‘Miracle Wheats’ might also lead to a similar phenotype, i.e., extension of the rachilla with more florets. In fact, we found one such case in our RIL population, implying that ‘Miracle Wheats’ are indeed double mutants of SM identity and determinacy genes, i.e., bht-A1 and qdel-5A, respectively.
As SM identity and determinacy are two interdependent processes during grass inflorescence development (Chuck et al. 2002; Laudencia-Chingcuanco and Hake 2002; Whipple 2017; Bommert and Whipple 2018); and the fact that SM identity and regular spikelet patterning/determinacy are not affected by the loss-of-function of the Q protein (Debernardi et al. 2017; Greenwood et al. 2017), it is likely that the TtBHt/WFZP and q proteins have rather a non-redundant function in wheat.
Flowering time genes pleiotropically control spikelet number in wheat
Among the key developmental decisions plants have to make in their life cycle is whether and when to flower (Lin 2000). Such decisions, however, are dependent on the internal (genetic components) and external (environmental) factors including nutrients, temperature, day length, drought, salinity, exogenously applied hormones and chemicals (Cho et al. 2017). Furthermore, the time of flowering in plants is also accompanied by massive developmental reprogramming, resulting in the morphological, physiological, and biochemical changes (Poethig 1990). The most noticeable of all is the emergence of the inflorescence or reproductive structures. The fact that the current mapping population has been developed from contrasting parents, originating from distinct gene pools differing for flowering time (i.e., winter vs. spring), showed that early-heading RILs are characterized by a relatively short spike with less number of nodes (spikelets), while the late-heading RILs had relatively longer SL carrying more spikelets (Fig. 2A). This suggests the importance of phase duration, especially from spike initiation to the appearance of the terminal spikelet (Rawson 1970; Rahman and Wilson 1977; Guo et al. 2018) for increasing spikelet number in wheat. Likewise, accelerated flowering has been linked to reduced spikelet and tiller numbers in wheat (Worland et al. 1998; Shaw et al. 2013; Boden et al. 2015), suggesting that besides the benefits of flowering time genes for adjusting flowering time to certain climatic conditions and requirements, they are also important for the genetic modification of inflorescence architecture in wheat. Given the apparent effect of flowering time on inflorescence development and architecture (Guo et al. 2018), unraveling the molecular mechanisms of flowering may have a major impact for improving grain yield in wheat.
Author contribution statement
TS conceived and supervised the project. GMW conducted the experiments, collected, and analyzed the data. CT contributed in developing the RILs, isolation of DNA, and performed PCR and q gene sequencing. MM analyzed GBS data. GMW and TS wrote the manuscript. All authors read and approved the final version of the manuscript.
References
Avni R, Nave M, Barad O, Baruch K, Twardziok SO, Gundlach H, Hale I, Mascher M, Spannagl M, Wiebe K, Jordan KW, Golan G, Deek J, Ben-Zvi B, Ben-Zvi G, Himmelbach A, Maclachlan RP, Sharpe AG, Fritz A, Ben-David R, Budak H, Fahima T, Korol A, Faris JD, Hernandez A, Mikel MA, Levy AA, Steffenson B, Maccaferri M, Tuberosa R, Cattivelli L, Faccioli P, Ceriotti A, Kashkush K, Pourkheirandish M, Komatsuda T, Eilam T, Sela H, Sharon A, Ohad N, Chamovitz DA, Mayer KFX, Stein N, Ronen G, Peleg Z, Pozniak CJ, Akhunov ED, Distelfeld A (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357:93–97
Baker C, Gallagher J (1983a) The development of winter wheat in the field. 2. The control of primordium initiation rate by temperature and photoperiod. J Agric Sci 101:337–344
Baker CK, Gallagher JN (1983b) The development of winter wheat in the field. 1. Relation between apical development and plant morphology within and between seasons. J Agric Sci 101:327–335
Barnard C (1955) Histogenesis of the inflorescence and flower of Triticum aestivum L. Aust J Bot 3:1–20
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Beales J, Turner A, Griffiths S, Snape JW, Laurie DA (2007) A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor Appl Genet 115:721–733
Boden SA, Cavanagh C, Cullis BR, Ramm K, Greenwood J, Finnegan EJ, Trevaskis B, Swain SM (2015) Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat Plants 1:1–6
Bommert P, Whipple C (2018) Grass inflorescence architecture and meristem determinacy. Semin Cell Dev Biol 79:37–47
Bonnet OT (1967) Inflorescences of maize, wheat, rye, barley, and oats: their initiation and development. University of Illinois College of Agric. Exp. Stn. Bull. 721
Borras L, Slafer GA, Otegui ME (2004) Seed dry weight response to source-sink manipulations in wheat, maize and soybean: a quantitative reappraisal. Field Crops Res 86:131–146
Campoli C, Von Korff M (2014) Genetic control of reproductive development in temperate cereals. Mol Genet Flor Transit Flower Dev 72:131–158
Chapman JA, Mascher M, Buluc A, Barry K, Georganas E, Session A, Strnadova V, Jenkins J, Sehgal S, Oliker L, Schmutz J, Yelick KA, Scholz U, Waugh R, Poland JA, Muehlbauer GJ, Stein N, Rokhsar DS (2015) A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol 16:26
Cho LH, Yoon J, An G (2017) The control of flowering time by environmental factors. Plant J 90:708–719
Chu CG, Xu SS, Friesen TL, Faris JD (2008) Whole genome mapping in a wheat doubled haploid population using SSRs and TRAPs and the identification of QTL for agronomic traits. Mol Breed 22:251–266
Chuck G, Muszynski M, Kellogg E, Hake S, Schmidt RJ (2002) The control of spikelet meristem identity by the branched silkless1 gene in maize. Science 298:1238–1241
Cockram J, Jones H, Leigh FJ, O’sullivan D, Powell W, Laurie DA, Greenland AJ (2007) Control of flowering time in temperate cereals: genes, domestication, and sustainable productivity. J Exp Bot 58:1231–1244
Cottrell JE, Dale JE, Jeffcoat B (1982) The effects of daylength and treatment with gibberellic acid on spikelet initiation and development in Clipper Barley. Ann Bot 50:57–68
Cui F, Ding AM, Li J, Zhao CH, Wang L, Wang XQ, Qi XL, Li XF, Li GY, Gao JR, Wang HG (2012) QTL detection of seven spike-related traits and their genetic correlations in wheat using two related RIL populations. Euphytica 186:177–192
Debernardi JM, Lin H, Chuck G, Faris JD, Dubcovsky J (2017) microRNA172 plays a crucial role in wheat spike morphogenesis and grain threshability. Development 144:1966–1975
Dixon LE, Farré A, Finnegan EJ, Orford S, Griffiths S, Boden SA (2018a) Developmental responses of bread wheat to changes in ambient temperature following deletion of a locus that includes FLOWERING LOCUS T1. Plant Cell Environ 41:1715–1725
Dixon LE, Greenwood JR, Bencivenga S, Zhang P, Cockram J, Mellers G, Ramm K, Cavanagh C, Swain SM, Boden SA (2018b) TEOSINTE BRANCHED1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum). Plant Cell 30:563–581
Dobrovolskaya O, Pont C, Sibout R, Martinek P, Badaeva E, Murat F, Chosson A, Watanabe N, Prat E, Gautier N, Gautier V, Poncet C, Orlov YL, Krasnikov AA, Berges H, Salina E, Laikova L, Salse J (2015) FRIZZY PANICLE drives supernumerary spikelets in bread wheat. Plant Physiol 167:189–199
Doebley JF, Gaut BS, Smith BD (2006) The molecular genetics of crop domestication. Cell 127:1309–1321
Donald CM (1968) The breeding of crop ideotypes. Euphytica 17:385–403
Doyle JJ (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Dvorak J, Wang L, Zhu T, Jorgensen CM, Luo MC, Deal KR, Gu YQ, Gill BS, Distelfeld A, Devos KM, Qi P, Mcguire PE (2018) Reassessment of the evolution of wheat chromosomes 4A, 5A, and 7B. Theor Appl Genet 131:2451–2462
Echeverry-Solarte M, Kumar A, Kianian S, Mantovani EE, Simsek S, Alamri MS, Mergoum M (2014) Genome-wide genetic dissection of supernumerary spikelet and related traits in common wheat. Plant Genome. https://doi.org/10.3835/plantgenome2014.03.0013
Echeverry-Solarte M, Kumar A, Kianian S, Mantovani EE, Mcclean PE, Deckard EL, Elias E, Simsek S, Alamri MS, Hegstad J, Schatz B, Mergoum M (2015) Genome-wide mapping of spike-related and agronomic traits in a common wheat population derived from a supernumerary spikelet parent and an elite parent. Plant Genome. https://doi.org/10.3835/plantgenome2014.12.0089
Faris JD (2014) Wheat domestication: key to agricultural revolutions past and future. In: Tuberosa R, Graner A, Frison E (eds) Genomics of plant genetic resources: volume 1. Managing, sequencing and mining genetic resources. Springer, Dordrecht, pp 439–464
Faris JD, Zhang Z, Garvin DF, Xu SS (2014) Molecular and comparative mapping of genes governing spike compactness from wild emmer wheat. Mol Genet Genom 289:641–651
Fischer RA (1985) Number of kernels in wheat crops and the influence of solar-radiation and temperature. J Agric Sci 105:447–461
Foulkes MJ, Slafer GA, Davies WJ, Berry PM, Sylvester-Bradley R, Martre P, Calderini DF, Griffiths S, Reynolds MP (2011) Raising yield potential of wheat. III. Optimizing partitioning to grain while maintaining lodging resistance. J Exp Bot 62:469–486
Friend DJC (1965) Ear length and spikelet number of wheat grown at different temperatures and light intensities. Can J Bot 43:345–353
Greenwood JR, Finnegan EJ, Watanabe N, Trevaskis B, Swain SM (2017) New alleles of the wheat domestication gene Q reveal multiple roles in growth and reproductive development. Development 144:1959–1965
Guo Z, Chen D, Alqudah AM, Röder MS, Ganal MW, Schnurbusch T (2017) Genome-wide association analyses of 54 traits identified multiple loci for the determination of floret fertility in wheat. New Phytol 214:257–270
Guo Z, Chen D, Roder MS, Ganal MW, Schnurbusch T (2018) Genetic dissection of pre-anthesis sub-phase durations during the reproductive spike development of wheat. Plant J 95:909–918
Heun M, Schaferpregl R, Klawan D, Castagna R, Accerbi M, Borghi B, Salamini F (1997) Site of einkorn wheat domestication identified by DNA fingerprinting. Science 278:1312–1314
Houston K, Mckim SM, Comadran J, Bonar N, Druka I, Uzrek N, Cirillo E, Guzy-Wrobelska J, Collins NC, Halpin C, Hansson M, Dockter C, Druka A, Waugh R (2013) Variation in the interaction between alleles of HvAPETALA2 and microRNA172 determines the density of grains on the barley inflorescence. Proc Natl Acad Sci USA 110:16675–16680
IBM (2011) IBM SPSS statistics for windows, version 20.0. IBM Corp, Armonk, NY
Jantasuriyarat C, Vales MI, Watson CJ, Riera-Lizarazu O (2004) Identification and mapping of genetic loci affecting the free-threshing habit and spike compactness in wheat (Triticum aestivum L.). Theor Appl Genet 108:261–273
Johnson EB, Nalam VJ, Zemetra RS, Lizarazu OR (2008) Mapping the compactum locus in wheat (Triticum aestivum L.) and its relationship to other spike morphology genes of the Triticeae. Euphytica 163:193–201
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649
Kilian B, Özkan H, Pozzi C, Salamini F (2009) Domestication of the Triticeae in the Fertile Crescent. In: Muehlbauer GJ, Feuillet C (eds) Genetics and genomics of the Triticeae. Springer, New York, pp 81–119
Kippes N, Debernardi JM, Vasquez-Gross HA, Akpinar BA, Budak H, Kato K, Chao S, Akhunov E, Dubcovsky J (2015) Identification of the VERNALIZATION 4 gene reveals the origin of spring growth habit in ancient wheats from South Asia. Proc Natl Acad Sci USA 112:E5401–E5410
Kirby EM, Appleyard M (1984) Cereal development guide. NAC Cereal Unit, Stoneleigh, Kenilworth, UK
Krasileva KV, Vasquez-Gross HA, Howell T, Bailey P, Paraiso F, Clissold L, Simmonds J, Ramirez-Gonzalez RH, Wang X, Borrill P, Fosker C, Ayling S, Phillips AL, Uauy C, Dubcovsky J (2017) Uncovering hidden variation in polyploid wheat. Proc Natl Acad Sci USA 114:E913–e921
Kumar N, Kulwal PL, Balyan HS, Gupta PK (2007) QTL mapping for yield and yield contributing traits in two mapping populations of bread wheat. Mol Breed 19:163–177
Laudencia-Chingcuanco D, Hake S (2002) The indeterminate floral apex1 gene regulates meristem determinacy and identity in the maize inflorescence. Development 129:2629–2638
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.399700, 1–3
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Proc GPD (2009) The Sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Lin C (2000) Photoreceptors and regulation of flowering time. Plant Physiol 123:39–50
Ma Z, Zhao D, Zhang C, Zhang Z, Xue S, Lin F, Kong Z, Tian D, Luo Q (2007) Molecular genetic analysis of five spike-related traits in wheat using RIL and immortalized F2 populations. Mol Genet Genom 277:31–42
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12
Mascher M, Wu S, Amand PS, Stein N, Poland J (2013) Application of genotyping-by-sequencing on semiconductor sequencing platforms: a comparison of genetic and reference-based marker ordering in barley. PLoS ONE 8:e76925
Mayer KF, Rogers J, Doležel J, Pozniak C, Eversole K, Feuillet C (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788
Mcmaster GS (1997) Phenology, development, and growth of the wheat (Tritimm aestivum L.) shoot apex: a review. In: DL Sparks (ed) Advances in agronomy. Academic Press, Cambridge, pp 63–118. http://www.apnet.com
Meyer RS, Duval AE, Jensen HR (2012) Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol 196:29–48
Mickelson-Young L, Endo TR, Gill BS (1995) A cytogenetic ladder-map of the wheat homoeologous group-4 chromosomes. Theor Appl Genet 90:1007–1011
Miftahudin, Ross K, Ma XF, Mahmoud AA, Layton J, Milla MA, Chikmawati T, Ramalingam J, Feril O, Pathan MS, Momirovic GS, Kim S, Chema K, Fang P, Haule L, Struxness H, Birkes J, Yaghoubian C, Skinner R, Mcallister J, Nguyen V, Qi LL, Echalier B, Gill BS, Linkiewicz AM, Dubcovsky J, Akhunov ED, Dvorak J, Dilbirligi M, Gill KS, Peng JH, Lapitan NL, Bermudez-Kandianis CE, Sorrells ME, Hossain KG, Kalavacharla V, Kianian SF, Lazo GR, Chao S, Anderson OD, Gonzalez-Hernandez J, Conley EJ, Anderson JA, Choi DW, Fenton RD, Close TJ, Mcguire PE, Qualset CO, Nguyen HT, Gustafson JP (2004) Analysis of expressed sequence tag loci on wheat chromosome group 4. Genetics 168:651–663
Miller P, Lanier W, Brandt S (2001) Using growing degree days to predict plant stages. Ag/Extension Communications Coordinator, Communications Services, Montana State University-Bozeman, Bozeman, MO
Miralles DJ, Richards RA (2000) Responses of leaf and tiller emergence and primordium initiation in wheat and barley to interchanged photoperiod. Ann Bot 85:655–663
Miralles DJ, Slafer GA (2007) Sink limitations to yield in wheat: how could it be reduced? J Agric Sci 145:139–149
Nyquist WE (1991) Estimation of heritability and prediction of selection response in plant-populations. Crit Rev Plant Sci 10:235–322
Payne RW, Murray DA, Harding SA, Baird DB, Soutar DM (2014) An introduction to GenStat for windows, 17th edn. VSN International, Hemel Hempstead
Pinthus MJ, Millet E (1978) Interactions among number of spikelets, number of grains and grain weight in spikes of wheat (Triticum-Aestivum L.). Ann Bot 42:839–848
Poethig RS (1990) Phase change and the regulation of shoot morphogenesis in plants. Science 250:923–930
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253
Poursarebani N, Seidensticker T, Koppolu R, Trautewig C, Gawronski P, Bini F, Govind G, Rutten T, Sakuma S, Tagiri A, Wolde GM, Youssef HM, Battal A, Ciannamea S, Fusca T, Nussbaumer T, Pozzi C, Borner A, Lundqvist U, Komatsuda T, Salvi S, Tuberosa R, Uauy C, Sreenivasulu N, Rossini L, Schnurbusch T (2015) The genetic basis of composite spike form in barley and ‘miracle-wheat’. Genetics 201:155–165
Rahman M, Wilson J (1977) Determination of spikelet number in wheat. I. Effect of varying photoperiod on ear development. Aust J Agric Res 28:265–274
Rahman M, Wilson J (1978) Determination of spikelet number in wheat. III.* Effect of varying temperature on ear development. Aust J Agric Res 29:459–467
Rawson HM (1970) Spikelet number, its control and relation to yield per ear in wheat. Aust J Biol Sci 23:1–16
Rawson H (1971) An upper limit for spikelet number per ear in wheat as controlled by photoperiod. Crop Pasture Sci 22:537–546
Rawson HM, Richards RA (1993) Effects of high temperature and photoperiod on floral development in wheat isolines differing in vernalisation and photoperiod genes. Field Crops Res 32:181–192
Sakuma S, Golan G, Guo Z, Ogawa T, Tagiri A, Sugimoto K, Bernhardt N, Brassac J, Mascher M, Hensel G, Ohnishi S, Jinno H, Yamashita Y, Ayalon I, Peleg Z, Schnurbusch T, Komatsuda T (2019) Unleashing floret fertility in wheat through the mutation of a homeobox gene. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1815465116
Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W (2002) Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet 3:429–441
Salina E, Börner A, Leonova I, Korzun V, Laikova L, Maystrenko O, Röder MS (2000) Microsatellite mapping of the induced sphaerococcoid mutation genes in Triticum aestivum. Theor Appl Genet 100:686–689
Shaw LM, Turner AS, Herry L, Griffiths S, Laurie DA (2013) Mutant alleles of Photoperiod-1 in wheat (Triticum aestivum L.) that confer a late flowering phenotype in long days. PLoS ONE 8:e79459
Shearman VJ, Sylvester-Bradley R, Scott RK, Foulkes MJ (2005) Physiological processes associated with wheat yield progress in the UK. Crop Sci 45:175–185
Shiferaw B, Smale M, Braun H-J, Duveiller E, Reynolds M, Muricho G (2013) Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Secur 5:291–317
Simons KJ, Fellers JP, Trick HN, Zhang Z, Tai YS, Gill BS, Faris JD (2006) Molecular characterization of the major wheat domestication gene Q. Genetics 172:547–555
Singh M, Ceccarelli S, Hamblin J (1993) Estimation of heritability from varietal trials data. Theor Appl Genet 86:437–441
Slafer GA, Savin R (1994) Source-sink relationships and grain mass at different positions within the spike in wheat. Field Crops Res 37:39–49
Slafer GA, Elia M, Savin R, García GA, Terrile II, Ferrante A, Miralles DJ, González FG (2015) Fruiting efficiency: an alternative trait to further rise wheat yield. Food Energy Secur 4:92–109
Sourdille P, Tixier MH, Charmet G, Gay G, Cadalen T, Bernard S, Bernard M (2000) Location of genes involved in ear compactness in wheat (Triticum aestivum) by means of molecular markers. Mol Breed 6:247–255
Stam P (1993) Construction of integrated genetic linkage maps by means of a new computer package: Join Map. Plant J 3:739–744
Steinfort U, Fukai S, Trevaskis B, Glassop D, Chan A, Dreccer MF (2017) Vernalisation and photoperiod sensitivity in wheat: the response of floret fertility and grain number is affected by vernalisation status. Field Crops Res 203:243–255
Tanaka W, Pautler M, Jackson D, Hirano HY (2013) Grass meristems II: inflorescence architecture, flower development and meristem fate. Plant Cell Physiol 54:313–324
The International Wheat Genome Sequencing Consortium (IWGSC) (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wendler N, Mascher M, Noh C, Himmelbach A, Scholz U, Ruge-Wehling B, Stein N (2014) Unlocking the secondary gene-pool of barley with next-generation sequencing. Plant Biotechnol J 12:1122–1131
Whipple CJ (2017) Grass inflorescence architecture and evolution: the origin of novel signaling centers. New Phytol 216:367–372
Worland A, Börner A, Korzun V, Li W, Petrovic S, Sayers E (1998) The influence of photoperiod genes on the adaptability of European winter wheats. Euphytica 100:385–394
Yan L, Loukoianov A, Tranquilli G, Helguera M, Fahima T, Dubcovsky J (2003) Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci 100:6263–6268
Yan L, Loukoianov A, Blechl A, Tranquilli G, Ramakrishna W, Sanmiguel P, Bennetzen JL, Echenique V, Dubcovsky J (2004) The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303:1640–1644
Yan L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yasuda S, Dubcovsky J (2006) The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc Natl Acad Sci 103:19581–19586
Zadoks JC (1985) Decimal code for the growth-stages of cereals. Curr Contents Agric Biol Environ Sci 16:14-14
Zhai H, Feng Z, Li J, Liu X, Xiao S, Ni Z, Sun Q (2016) QTL analysis of spike morphological traits and plant height in winter wheat (Triticum aestivum L.) using a high-density SNP and SSR-based linkage map. Front Plant Sci 7:1617
Zhou Y, Conway B, Miller D, Marshall D, Cooper A, Murphy P, Chao S, Brown-Guedira G, Costa J (2017) Quantitative trait loci mapping for spike characteristics in hexaploid wheat. Plant Genome https://doi.org/10.3835/plantgenome2016.10.0101
Acknowledgements
We are thankful to Mechthild Pürschel for her excellent technical assistance. We are also grateful to IPK for providing ‘Miracle Wheat’ accessions and maintaining and conducting field experiments in Gatersleben. We thank Dr. Shun Sakuma for kindly providing some of his spike-branching wheat accessions. We are similarly thankful to Prof. Klaus Pillen, Martin-Luther-University Halle-Wittenberg, and his team (Vera Draba, and Markus Hinz) for providing, preparing, and maintaining the field experiment in Halle.
Funding
This research was supported Wissenschaftsgemeinschaft Gottfried Wilhelm Leibniz (WGL) and the Leibniz-Graduate School Gatersleben “Yield formation in cereals - overcoming yield-limiting factors” (FKZ 401410) for funding this study. During parts of this study, TS received financial support from the HEISENBERG Program of the German Research Foundation (DFG), grant no. SCHN 768/8-1, and IPK core budget.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Communicated by Takao Komatsuda.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Gizaw M. Wolde, Corinna Trautewig, Thorsten Schnurbusch: Independent HEISENBERG-Research Group Plant Architecture; Martin Mascher: Research Group Domestication Genomics.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wolde, G.M., Trautewig, C., Mascher, M. et al. Genetic insights into morphometric inflorescence traits of wheat. Theor Appl Genet 132, 1661–1676 (2019). https://doi.org/10.1007/s00122-019-03305-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03305-4