
Abstract
Key message
Intracellular factors differentially affected enzyme activities of N assimilation in the roots of maize testcrosses where alanine aminotransferase and glutamate synthase were the main enzymes regulating the levels of glutamate.
Abstract
N is a key macronutrient for plant growth and development. Breeding maize with improved efficiency in N use could help reduce environmental contamination as well as increase profitability for the farmers. Quantitative trait loci (QTL) mapping of traits related to N metabolism in the root tissue was undertaken in a maize testcross mapping population grown in hydroponic cultures. N concentration was negatively correlated with root and total dry mass. Neither the enzyme activities nor metabolites were appreciably correlated between the root and leaf tissues. Repeatability measures for most of the enzymes were lower than for dry mass. Weak negative correlations between most of the enzymes and dry mass resulted likely from dilution and suggested the presence of excess of enzyme activities for maximal biomass production. Glutamate synthase and alanine aminotransferase each explained more variation in glutamate concentration than either aspartate aminotransferase or asparagine synthetase whereas glutamine synthetase was inconsequential. Twenty-six QTL were identified across all traits. QTL models explained 7–43% of the variance with no significant epistasis between the QTL. Thirteen candidate genes were identified underlying QTL within 1-LOD confidence intervals. All the candidate genes were located in trans configuration, unlinked or even on different chromosomes, relative to the known genomic positions of the corresponding structural genes. Our results have implications in improving NUE in maize and other crop plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
After hydrogen, carbon, and oxygen, nitrogen (N) is the most abundant element in plant tissues. Nearly all of it is derived from the synthetic fertilizers applied to the soil. Variable proportions of the applied soil N are lost to the environment by leaching and denitrification. Leached N flows into the streams and rivers, and eventually into the ocean, supporting algal growth. Excessive algal growth forms “dead zones”, for example, in the Gulf of Mexico, by depleting oxygen in the water, and thus asphyxiating life (Goolsby and Battaglin 2000). Annual delivery of nitrate from the Mississippi river to the Gulf has nearly tripled in the last half century. The size of the dead zone of the Mississippi delta varies depending upon the frequency and the intensity of precipitation in the catchment area of the Mississippi river.
One approach to reduce N loss from the soil is to improve N use efficiency (NUE) of maize. NUE, which in cereals has been defined as the ratio of grain produced per unit of soil N, can be subdivided into two main components: N acquisition efficiency (total plant N/soil N) and N utilization efficiency (total grain yield/total plant N) (Moll et al. 1982; Dhugga and Waines 1989). A comprehensive understanding of N metabolism at the genetic level could provide new avenues to improve NUE in maize (Trucillo Silva et al. 2017).
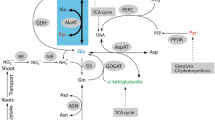
The model pathway for N reduction and incorporation of reduced N into organic molecules has been well described (Yemm and Folkes 1958; Lea et al. 1990; Lea and Miflin 2010; Plett et al. 2016; Trucillo Silva et al. 2017) (Fig. 1). Nitrate is reduced to nitrite by nitrate reductase (NR) in the cytoplasm, followed by reduction of nitrite in the plastids to ammonium by nitrite reductase (NiR). Ammonium thus generated is coupled to glutamate by glutamine synthetase (GS). Another enzyme, glutamine-2-oxoglutarate aminotransferase (GOGAT) or glutamate synthase, then converts glutamine back to glutamate, producing an additional glutamate from 2-oxoglutarate, thereby initiating the conversion of inorganic N into organic form. This pair of reactions is referred to as GS-GOGAT cycle. Asparagine synthase (ASN) produces asparagine and glutamate from glutamine and aspartate. Glutamate serves as an amino group donor for the formation of other amino acids, a reaction catalyzed by different amino transferases. For instance, alanine aminotransferase (AlaAT) catalyzes the amino group transfer to pyruvate to form 2-oxoglutarate and alanine, while aspartate aminotransferase (AspAT) forms 2-oxoglutarate and aspartate after transferring the amino group of glutamate to oxaloacetate. Following N assimilation, glutamate, asparagine, glutamine and other amino acids are transported via vasculature to the growing organs. Alternatively, they can be stored as vegetative storage proteins, which can aid plant growth during the periods of N deficiency (Dhugga et al. 2007).

(adapted from Trucillo Silva et al. 2017)
Enzymes and proteins involved in N-acquisition and assimilation in C4 plants (created with Adobe Illustrator CS2). AlaAT and GOGAT each explained more variation in glutamate than AspAT and ASN. AlaAT alanine aminotransferase, ASN asparagine synthase, AspAT aspartate aminotransferase, GDH glutamate dehydrogenase, GOGAT glutamate synthase, GS glutamine synthetase, NR nitrate reductase, NiR nitrite reductase, PEPC phosphoenolpyruvate carboxylase, PPDK pyruvate orthophosphate dikinase
Mapping quantitative trait loci (QTL) is a routine in plant genetic investigations and breeding programs. The procedure relies on differences among the trait means of genotypes at a marker locus (Bernardo 2010). The precision in the identification of a QTL is important for the success of further studies, for instance, identification of candidate genes and positional cloning (Remington et al. 2001). That precision in the estimation of the QTL position, referred to as resolution, varies depending on several factors, such as recombination frequency, marker density and population size (Yu et al. 2011).
Much of the current commercial maize germplasm originates from seven progenitor lines, including B73 and Mo17 (Mikel and Dudley 2006). Both inbred lines differ in their response to N fertilization (Balko and Russell 1980) and are parents of the IBM (intermated B73 × Mo17) mapping population (Lee et al. 2002). After ten rounds of random mating, 360 doubled haploid (DH) lines were generated from the IBMSyn10 population (Hussain et al. 2007), which had a higher-resolution for mapping that could be directly associated to the physical map established for the B73 inbred (http://www.maizesequence.org).
Several studies have shown association between QTL and N-metabolism related enzymes (Bertin and Gallais 2001; Hirel et al. 2001; Limami et al. 2002; Zhang et al. 2010, 2015; Liu et al. 2012; Trucillo Silva et al. 2017). We previously described the mapping of various enzymes of N metabolism in the leaf tissue of maize testcrosses (TCs) derived from the IBMSyn10-DH lines and an elite inbred, grown in hydroponics. In this study, we present the mapping of enzymes and metabolites related to N metabolism in the root tissue, and relate those observations to the preceding study of N metabolism in the shoot tissue. This is the first QTL analysis for N-metabolism related enzymes and metabolites in maize roots, particularly in a TC population.
Materials and methods
Plant material
From the cross between IBMSyn10-DH lines and an elite inbred (PEI), property of DuPont Pioneer, 176 TC genotypes were generated and used in this investigation. The IBMSyn10-DH population consists of a set of DH lines derived from a population after ten generations of random mating from the cross between B73 × Mo17 (Hussain et al. 2007).
Experimental design
The same experimental design as described previously was used (Trucillo Silva et al. 2017). Each TC genotype was germinated in autoclaved paper rolls and sterilized water, and subsequently grown under hydroponic conditions. Ten tanks (i.e., sets) containing appropriate growth media were planted with 264 seedlings per tank. In every set, 22 genotypes were grown, and each genotype was replicated 12 times. Two genotypes (B73 and Mo17 each crossed to the PEI) served as controls, and were included in every set and replication.
The growth media consisted of MgSO4·7H2O 0.5 mM, KH2PO4 0.5 mM, Fe-EDTA 0.1 mM, FeEDDHA 0.1 mM, Ca(NO3)2·4H2O 1.25 mM, KNO3 2.5 mM, Na(OH) 0.1 mM, and 0.4 L of trace elements (25 mM H3BO3, 2 mM MnSO4·H2O, 2 mM ZnSO4·7H2O, 0.5 mM CuSO4·5H2O, 0.5 mM Na2MoO4·2H2O and 50 mM KCl) in a total of 400 L solution per hydroponic tank. The pH was maintained between 5.9 and 6.1 as described in Garnett et al. (2013). A flux density at the canopy level of ~ 500 µmol m−2 s−1 was supplied at 14 h (25 °C) day: 10 h (20 °C) night cycle. The plants were randomized in the tank every 5 days to guard against the position effects. Two weeks after planting, the six most representative uniform plants of each genotype (based on root and shoot development), were selected and transplanted into another hydroponic tank with same media.
When plants reached V4 stage (Abendroth et al. 2011), usually 4–5 weeks from planting, 4–5 cm of the primary root from six plants were collected and stored at − 80 °C while the rest of the plant tissues were dried for 12 days at 48 °C.
The V4 stage of development was selected for assays because inter-plant shading became a factor after this. The potential border-row effect was mitigated by randomizing the plants more than once during their growth (Tuberosa et al. 2002).
Biochemical assays
All the biochemical assays were performed as previously described (Trucillo Silva et al. 2017). Activity of eight enzymes related with the N-metabolism pathway was determined in root samples of each genotype. The set of enzymes included NR, NiR, GS, GOGAT, AlaAT, ASN, AspAT and PEPC, and specific protocols were adapted by K. Dhugga, R. Abbaraju and L. Fallis and described in Plett et al. (2016). GS, GOGAT, Asp AT and PEPC assay protocols were adapted from Gibon et al. (2004), NR from Lea et al. (1990), NiR from Bourne and Miflin (1973), ASN from Joy and Ireland (1990), and AlaAT protocol was modified from Ashton et al. (1990). Metabolites nitrate and glutamate were measured as byproducts of enzyme reactions. All measurements were determined by the absorbance of each biochemical reaction compared to known standards using a spectrophotometer (Spectramax Plus 384 Microplate Reader, Molecular Devices).
Plant tissues were weighed and analyzed for N content by combustion analysis as described by DeBruin et al. (2013). Based on root biomass dry weight (RW) and percentage of N measurements (Nr), total amount of N present in root (TNr) tissues was calculated. In addition, Nratio was estimated as the ratio between total amount of N present in shoot tissues (TNs) and TNr. The analysis of shoot dry weight (SW), TNs and Ns is presented in Trucillo Silva et al. (2017).
Trait data analysis
Statistical analysis was implemented in R statistical program (RCoreTeam 2014) as described in Trucillo Silva et al. (2017). Ggplot2 (Wickham 2010) and GGally (Schloerke et al. 2014) R packages were used for initial analysis of the raw data. First, a univariate analysis, where a single variable is fitted in a model, followed by a multivariate approach, where multiple variables are analyzed simultaneously, was performed to comprehend the relationship among the variables. Then, based on a jackknife resampling strategy, outliers in the dataset were identified as described in Trucillo Silva et al. (2016). The main procedure consists on fitting a statistical model n times, systematically omitting one observation from the dataset, followed by the prediction of random effects for a subset of the most consistent genotypes each of the n times. The mixed model was fitted with ASReml R package (Butler et al. 2007) and correspondent mixed model equations were solved for the prediction of random effects and estimation of fixed effects.
The statistical model can be represented as follows:
where \(y\) denotes a n × 1 vector of observed response values, \(b\) is a p × 1 vector of fixed effects, \(X\) is a n × p design matrix, \(u\) is a q × 1 vector of random effects,\(Z\) is a n × q design matrix, and \(e\) being the error term.
The following assumptions were used: E (u) = 0, E (e) = 0, Cov (u, e) = 0, and Var (u) = G and, Var (e) = R. The G matrix had a compound symmetry structure on the genotype levels and R matrix is a diagonal matrix with different values for each set, allowing non-constant variance across sets. The response variable was the activity of the enzyme and the metabolite concentration, respectively. Set, the light replicate and plate were included as fixed effects in the model (where replicate and plate are nested in a set), and the check genotype effect was included as a continuous covariate. Finally, the genotype was included as a random effect in the linear model. Several genotypes were discarded depending on the trait (e.g., for both AlaAT and NR five genotypes were removed). Furthermore, one and four complete sets of data were removed for glutamate and nitrate, respectively, due to the contamination of samples and very low accuracy in the estimations.
Significance of genetic variance was calculated based on log-likelihood ratio test by comparing models with and without the TC random effect. Correlations were calculated among BLUP values for each pair of traits and significance was adjusted after the Bonferroni correction for multiple comparisons. Repeatability was derived from variance estimations from ASReml. The variance components were estimated for each different set. As a result, different values of repeatabilities were estimated and partial estimates were averaged. Path coefficient was performed as described by Wright (1921) and Trucillo Silva et al. (2017).
The studied traits followed Gaussian distribution as judged from the similarity of mean and median values along with skewness estimates (Supplementary Material 1).
Genotypic information and genetic maps
TC materials were genotyped with 5306 single nucleotide polymorphism (SNP) markers by the Beijing Genomics Institute. Physical and genetic positions of the markers were determined and genetic maps were created with R/qtl (Broman et al. 2003). Recombination fractions were estimated and the Kosambi mapping function was implemented to calculate genetic map distances (Kosambi 1944). In addition, mapping distances were adjusted to compare the results with previous investigations. The expansion factor was determined based on the following equation: \(\propto\, = \frac{j}{2} + (2{\text{i}} - 1)/2{\text{i,}}\) where j corresponds to the number of generations of intermating including the two generations for generating the F2, and i is the number of inbred generations after intermating (Teuscher et al. 2005).
The real map was 11,265.25 cM and map distances were reduced by a factor of 6.5 to estimate the adjusted F2 map. The final adjusted map was 1733.12 cM length with an average spacing between markers of 0.33 cM, while the maximal spacing between markers was nearly 7 cM, on chromosome 6. With regard to physical distance, the length of the total genome was 2051.75 Mb, with the biggest gap between markers of 69.80 Mb length (located on chromosome 2). On average there was a marker positioned every 400 Kb.
QTL mapping and identification of candidate genes
QTL Cartographer (Basten et al. 2002) was utilized to detect associations between phenotypes and genotypes. Single-marker analysis, linear regression analysis and composite interval mapping (CIM) was implemented. Zmap (model 6) was performed for CIM, using the ten most significant marker cofactors identified by forward and backward regression. QTL were scanned at intervals of 1 cM and at every marker while cofactors located within a window of 10 cM of the scanned position were excluded from the analysis. To determine LOD score thresholds of 5%, and significant QTL, 1000 permutations were performed for every trait. Two nearby QTL were considered as different when LOD peaks were localized 20 cM or greater apart. Effects of QTL are expressed relative to the B73 allele, where an effect with a positive sign represents an increasing allele from B73 and the one with a negative sign denotes an increasing allele from Mo17.
Multiple interval mapping (MIM) analysis was performed by fitting previously identified QTL from CIM analysis, and parameters were re-estimated and positions refined. All pairwise interactions between QTL in every model were examined for each trait. The significance was determined based on the information criterion: IC (k) = − 2 (log (L) − kc (n)/2), where the penalty function corresponds to: c (n) = log (n) and a threshold of 0.0 was used (Basten et al. 2002). The proportion of the total phenotypic variance associated with each model was estimated.
Candidate genes annotated on corresponding 1-LOD QTL confidence interval regions were examined from MaizeGDB (Lawrence et al. 2008) and Phytozome (Goodstein et al. 2012). Those candidate genes directly related to N-metabolism based on descriptions in model species, such as rice (Oryza sativa) and Arabidopsis (Arabidopsis thaliana), were proposed for further studies. Several other candidate genes may be promising candidates for further investigations, including transcription factors; however, they were not considered due to the difficulties to ascertain a direct relationship with N-metabolism in maize based on available descriptions.
Results
Plant dry mass and its relationship to biochemical traits
Shoot and root dry mass, respectively, explained 83 and 17% variation in total plant biomass as determined by path coefficient analysis, which mirrors the actual, average proportion of the two components of the plant across all the TC at 86 and 14% (data not shown). The coefficient of variation (CV) was, respectively, 26.2, 19.3, and 19.9% for the root, shoot, and total biomass. A relatively lower CV of 13.4% for the root/shoot ratio implies that partitioning of dry matter between these two plant parts was less variable than the total dry matter accumulation itself.
Root dry mass exhibited a negative, logarithmic relationship with N concentration, with an R2 of 0.51 (Fig. 2). Shoot N concentration did not correlate with shoot or total dry mass (data not shown). Root N concentration in fact was also negatively correlated with the shoot dry mass and total dry mass with R2 values of 0.35 and 0.39, respectively, which is not surprising because shoot/root ratio was less variable than either of these traits as discussed earlier. This implies that the roots of the rapidly growing plants retained less N, pointing to a limitation of sink in the shoot for the absorbed N. Apparently, N acquisition, unlike dry matter formation, was not a limiting factor in plant growth. A limitation in dry matter deposition seemingly limited dilution of N in the dry matter, which is manifested in a negative correlation between these two traits. These results are from the plants grown under non-limiting N levels, however, where the root surface was continuously bathed with nitrate. N uptake could become limiting, particularly under low soil N, where a depletion zone develops around the root, particularly during the peak period of transpiration.

Relationship of root dry mass to root N concentration (created with Adobe Illustrator CS2). The R2 values for root N versus shoot dry mass and root N versus total dry mass were 0.35 and 0.39, respectively; *** indicates statistically significant at P < 0.001
ASN and NR had higher activities on a protein basis in the leaves than in the roots, GOGAT was similar between these two tissues, but the activities of the remaining enzymes were higher in the roots at varying levels with NiR being approximately four-fold more active (Fig. 3). Nitrate concentration on dry mass basis was higher in the shoot, implying its efficient transport from the root to the shoot tissue. Glu was more abundant in the roots than in the leaves, suggesting significant nitrate reduction in the roots (Fig. 3).

Shoot to root ratios for various enzymes and metabolites in maize IBMsyn10-DH test crosses (created with Adobe Illustrator CS2). The ratio for shoot dry mass to root dry mass was 6.3
Enzyme activities and metabolite concentrations were poorly correlated between the root and the shoot tissues (Fig. 4). The highest correlation coefficient, 0.26, was observed for tissue N concentration. These results imply that the enzymes for N metabolism operate independently in the root and shoot tissues. It is possible that the leaf enzymes are primarily reflective of dry matter deposition, and root enzymes of maintaining N absorption and transport; the relative proportions of these two components in the plant are 84 and 16%, respectively. Furthermore, the reduction of nitrate in the roots may be a mechanism to maintain a favorable electrochemical potential gradient for its continued uptake.

Pearson correlation coefficients for enzyme activities and metabolite concentrations between root and shoot tissues in the IBMSyn10-DH testcross population (created with Adobe Illustrator CS2). *, **, *** indicate statistically significant at P < 0.05, 0.01, and 0.001, respectively
Regression of root and shoot dry matter on each of the enzyme activities revealed a weakly negative relationship for all but two of the enzymes (Table 1). GS explained the most variation, 9%, whereas AspAT and NR each explained approximately 4% of the variation in root dry mass. For the shoot dry mass, AspAT, ASN, GOGAT, NiR, GS, and AlaAT explained 9.5, 7.5, 6.6, 4.7, 4.6, and 3.3% variation, respectively. Of these, AspAT, NiR, and ASN subsumed the variation explained by GOGAT, GS, and AlaAT as indicated by the direct and indirect contributions of each of the enzymes (Trucillo Silva et al. 2017). These observations point to the dilution effect of cell expansion on the cellular contents and suggest that excess capacity for enzyme activities is reflective of metabolic homeostasis to maintain biomass productivity.
Repeatability of N-metabolism associated traits
Repeatability, also referred to as broad-sense heritability in forward selection, is a measure of consistency of a trait among the plants (replicates) of the same TC (Fig. 5). All traits were measured on the same plants grown in the same hydroponic culture system under controlled light and temperature (Table 2). Since the standard error of replicated assays from each plant was negligible, repeatability provides a measure of consistency of different traits among plants in each line. Nitrate concentration and ASN in the shoot tissue were the most and least consistent traits, respectively. Nitrite reductase (NiR) in the leaves was the most consistent enzyme although NR was a close second. PEPC was more uniform in the roots than in leaves, which could be because the light-inducible form in the leaves is the main enzyme for C4 photosynthesis and responds to small changes in light availability. The plants were in close proximity to each other and slight interference from variable shading was unavoidable. The root PEPC was perhaps more stable because its role is primarily anaplerotic, that is, to fix CO2 released by respiration into oxaloacetate using phosphoenolpyruvate as the other substrate to support amino acid formation (Fig. 1).

Repeatability values for traits measured on root and shoot tissues in the maize IBMSyn10-DH TC population (created with Adobe Illustrator CS2). AlaAT alanine aminotransferase, ASN asparagine synthase, AspAT aspartate synthase, GOGAT glutamine oxoglutarate aminotransferase or glutamate synthase, GS glutamine synthetase, NiR nitrite reductase, NR nitrate reductase, PEPC phosphoenol pyruvate carboxylase, Glu glutamate, DM dry matter. For the shoot tissue, enzyme activities were measured on the youngest, fully expanded leaf at V4 stage (Trucillo Silva et al. 2017)
Nitrate concentration was less variable among plants in the leaves than in the roots, which suggests that roots likely respond to feedback from the leaves for free N status, and thus adjust nitrate absorption to maintain homeostasis in the leaves, where most of it is reduced. This could particularly be the case because nitrate concentration was higher in the leaves than in the roots (Fig. 3).
Despite wide variation in repeatability among the enzymes of N assimilation, both root and shoot biomass were more uniform among the plants sampled for analysis (Fig. 5). Since the plants were selected for uniformity by rejecting the outliers (see Materials and Methods), the root and shoot dry masses are relatively uniform.
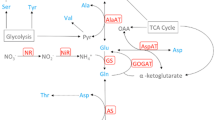
Enzymes for glutamate formation and utilization
Glutamate is a key amino acid in N assimilation as it constitutes the entry step of inorganic N into organic form (Trucillo Silva et al. 2017). A path coefficient diagram depicting the direct and indirect effects of various enzymes toward the cellular glutamate concentration is shown in Fig. 6. In the leaf tissue, AlaAT alone explained 58% of the variation in glutamate, suggesting a key role in maintaining the intracellular concentration of this amino acid (Trucillo Silva et al. 2017). No single enzyme was as strongly correlated with glutamate in the roots, however, perhaps because roots act more as conduits for nitrate uptake but not for primary N assimilation. AlaAT alone still explained 10% of the net variation in glutamate level, but GOGAT was just as important, explaining 11% of the variation (Fig. 6). AspAT and ASN each explained 4% variation in the glutamate level. GS was inconsequential in determining glutamate level, which implies that glutamine, a substrate for glutamate regeneration along with α-ketoglutarate (Fig. 1), was not a limiting factor in the root-cell glutamate level (Fig. 6). A limitation of carbon skeletons because of their utilization in dry matter deposition in expanding cells might favor N cycling between glutamate and alanine (C/N ratio 3) than between glutamate and aspartate (C/N ratio 4) (Trucillo Silva et al. 2017). This pattern appears less prominent in the roots, perhaps because they are not the primary sites of carbon fixation, N reduction and assimilation (Fig. 6). Pairwise correlation coefficients for all the traits are shown in Supplementary Material 2. Of all the pairwise correlations, 31% were highly significant (p value < 0.001). All significant correlations between enzyme activities, enzymes and metabolites, and between metabolites were positive.

Direct and indirect relationship between glutamate and enzymes directly involved in its formation and utilization (created with Adobe Illustrator CS2). See pathway in Fig. 1. On the right is the table showing partial R2 for each of the enzymes. Double arrows describe the correlation coefficients between various pairs of enzymes. Path coefficients from each of the enzymes are shown as lines with single arrows. Unexplained path coefficient, which is the square root of the unexplained variation (1 − R2), is shown separately. *, **, *** indicate statistically significant at P < 0.05, 0.01, and 0.001, respectively
Identification of quantitative trait loci
Composite interval mapping
Twenty-six QTL were identified across all the traits. Five QTL were detected on one chromosome, 7, whereas only one was identified on chromosome 8 (Fig. 7). AlaAT-3 and AspAT-2 were the only QTL that overlapped their respective 1-LOD CI on chromosome 10. The number of QTL varied for different traits, ranging from only one for some traits (GOGAT and NR) to four for NiR (Fig. 7).

Genetic map and distribution of QTL associated with N metabolism related enzymes and metabolites measured on root tissue in the maize IBMSyn10-DH TC population. Created with MapChart 2.2 (Voorrips 2002). QTL positions are shown at left of chromosomes (in cM) and the lengths of QTL bars are determined by 2-LOD confidence intervals. Only selected markers are displayed in the figure to the right of chromosomes. QTL associated with the enzyme activities are in blue, while QTL associated with metabolites are in red (color figure online)
A majority of the QTL, 19, explained less than 10% of the genetic variance. Six QTL explained 10–25%, while one explained > 25% of the variance. The QTL which accounted for the highest amount of variance (31.5%) and presented the highest LOD score (23.4) was for PEPC-1, located on chromosome 5. For that QTL, the B73 allele had a negative effect (− 23.78 µmole NADH/min/mg protein). Furthermore, for 70% of all QTL detected across traits, B73 alleles had negative additive effects. For certain traits, for example, AspAT, GOGAT, and GS, B73 alleles exhibited only a negative effect, however, for ASN QTL, B73 alleles had positive effects (Table 3).
Confidence intervals (CI 1-LOD) for QTL ranged from 1.04 to 24.46 cM (0.16–3.76 cM adjusted distance) length, with an average of 7.79 cM (1.2 cM adjusted distance). Those CI correspond to 0.2–21.1 Mb in physical distance, with a mean CI length of 2.46 Mb (Table 3).
Multiple interval mapping
First order epistatic interactions between QTL identified previously by CIM were not significant for any of the traits, thus epistatic digenic effects were excluded from genetic models. Even though 43% of the total variance was explained in PEPC by fitting two QTL in an MIM model, other genetic models captured less than 10% of the phenotypic variance, such as for ASN, GOGAT, GS and NR (Table 4). On average, multiple QTL models explained 15.1% of the variance when two QTL were included in each of the models.
Candidate genes
An average of 63 genes were annotated underlying QTL 1-LOD regions, with CI regions containing between six and 376 genes. Only a subset of the putative genes could be associated to N-metabolism pathway based on their descriptions in model species. The most promising genes were GRMZM2G028574, GRMZM2G111225, GRMZM2G136712, GRMZM2G155974, GRMZM2G166366, GRMZM2G374302, GRMZM2G409131, GRMZM2G466543, GRMZM2G473001, GRMZM2G481529, GRMZM5G817058, GRMZM2G575696 and GRMZM2G580894 (Table 5). Each of them was associated with a putative function relevant to NUE. Examples are PEPC, nitrilase, aspartate kinase, glutathione synthetase, aspartate kinase, arginine decarboxylase, phosphofructokinase, arogenate dehydratase, phosphopyruvate hydratase, phosphoribosyl transferase, and last two genes as S-adenosyl-methionine-dependent (SAM)-methyltransferase, respectively. In agreement with our earlier study on the leaves of this population (Trucillo Silva et al. 2017), all the QTL identified in this study are located on a different position to the known genomic location of each corresponding structural gene. For example, GS QTL were identified on chromosomes 7 and 9 at physical positions 158.15 and 23.85 Mb in this study, whereas GS1 and GS2 locus are located in chromosomes 1, 2, 4, 5, 9 (between 146.06 and 146.07 Mb), and 10, based on the following nearest loci on the IBM2 2008 Neighbors map, respectively. The candidate genes identified within the QTL regions might affect the enzyme activities in a trans-acting regulatory manner as previously described, most likely through metabolic pathways as all the genes are non-regulatory in nature (Zhang et al. 2010). The candidate genes we identified are located on chromosomes 1, 2, 3, 4, 6, and 7. No candidate genes related to N metabolism were identified underlying QTL for ASN, GS, NiR, PEPC, nitrate, and glutamate.
Discussion
Previously, we reported the genetic and biochemical analyses of shoot enzymes and metabolites in a TC mapping population (Trucillo Silva et al. 2017). In this report, we focus on the root tissue of the same population.
Variable repeatabilities for various enzymes and metabolites both for root and shoot tissues provide a window into the stability of each of these traits within a line (Fig. 5). Despite the fact that a controlled environment was used (light intensity, nutrients, and temperature) in which the testcrosses were grown to the maximum extent possible, repeatability for a majority of the biochemical traits was generally less than 50% both in the roots as well as in the leaves (Fig. 5). Higher repeatability for the root and shoot dry mass than most of the enzymes suggests that enzyme levels can fluctuate to maintain metabolic homeostasis such that dry matter deposition is maintained. A slight negative correlation between dry mass and enzyme activities suggests that the enzyme amounts are maintained above the threshold for optimal biomass production (Table 1). Nevertheless, variation for repeatability highlights how a trait can vary significantly among plants of the same genotype grown in a controlled environment. These observations are significant in that they imply that environmental variation may be difficult to control under the field conditions regardless of measures taken. Increasing the number of replications and years or locations for testing genetic variants could help reduce the chances of false positives.
A lack of correspondence between the enzyme activities of the root and shoot tissues suggests that metabolism in these tissues is optimized for different functions, apparently for dry matter accumulation in the shoot and nutrient absorption and transport in the root.
Even though numerous QTL associated with enzymes involved in N-metabolism were identified in previous studies (Agrama et al. 1999; Limami et al. 2002; Canas et al. 2012), only a few investigations were based on a representative and high-resolution mapping population, such as Zhang et al. (2010) and (2015). The performance of most traits in maize in the inbred lines is weakly, if at all, correlated with their hybrid (Hallauer et al. 2010). Yet, only a few studies have focused on mapping in testcross populations (Bertin and Gallais 2001; Gallais and Hirel 2004). Furthermore, we used TC derived from the IBMsyn10 population, which represent a higher recombination frequency, and thus narrower intervals of the identified QTL. To account for the higher recombination rate, we used a platform with 5303 SNP markers.
As reported in previous studies, the activity of enzymes investigated, constituents of the N-metabolism pathway (except PEPC, which is a member of the primary C-metabolism), seem to be co-regulated (Zhang et al. 2010; Trucillo Silva et al. 2017). Hence, a positive correlation between enzyme activities, as well as within metabolites concentration, was expected and our observations confirmed it. Significant correlations between enzyme activities and metabolites were also positive (Supplementary Material 2).
In comparison to previous studies (Zhang et al. 2010; Trucillo Silva et al. 2017), in which leaf tissue was investigated, determination of root enzyme activity was more prone to sample to sample variation, mainly because, unlike the leaf where the same position could be sampled from each plant, root lengths were more variable, as reflected in final samples. Performance of assays on six replications per genotype ensured that the repeatability measures are quite similar between the root and leaf enzymes (Fig. 5).
A few of the QTL identified in this study were found to be in analogous positions as in previous detected NUE-related QTL on leaf tissues (Trucillo Silva et al. 2017). For instance, a root QTL associated with ASN located on chromosome 5, corresponds to leaf QTL for PEPC, nitrate and GOGAT (LOD peak values identified 2, 4 and 7 adjusted cM apart, respectively). In agreement with Zhang et al. (2010), a QTL for AlaAT was detected on chromosomes 4, about 5 cM away from the detected position in this study. Nonetheless, most of the QTL reported in other maize studies (Agrama et al. 1999; Hirel et al. 2001; Canas et al. 2012), which failed to co-locate, were greater than 20 cM away or even on different chromosomes compared to the QTL identified in this investigation. For example, QTL for GS activity were determined on chromosomes 7 and 9 in this study, whereas on chromosomes 4 and 5 in a previous study (Canas et al. 2012).
A lower number of QTL was identified per trait compared to previous investigations on leaf tissues (Zhang et al. 2010; Trucillo Silva et al. 2017). This suggests that similar traits are differentially regulated in roots and leaf tissues. The power to identify a QTL depends on the magnitude of the QTL effect and the size of the segregating population (Beavis 1998). Because a large number of small-effect QTL segregating in the genome were expected, and due to the size of the segregating population (176 individuals), only a subset of the total number of QTL was expected to be identified. Moreover, in comparison to Zhang et al. (2010), the number of QTL detected were most likely affected by the six additional rounds of random mating before fixing the lines that constituted the IBMsyn10-DH population. It is possible that the QTL previously detected in large linkage blocks, might have been separated into several smaller-effect QTL after further recombination events occurred. Therefore, the power to detect a QTL, each with a very small effect, would be expected to be lower. Another difference between the two studies is the use of inbred versus hybrids for mapping QTL. Little evidence of common QTL detection between inbred per se and TC progeny has been found in previous investigations (Beavis et al. 1994; Schon et al. 1994). It is possible also that some of the QTL identified in the inbred lines could have been masked in heterozygous form.
As previously reported by Trucillo Silva et al. (2017), the MIM results across traits suggest that there might be several undetected small effect QTL responsible for the rest of the genetic variation, for example, for PEPC and ASN, two QTL explained 42.5 and 8.1% of the variance, respectively. The sum of the effect of numerous QTL, each with small marginal effect, plus any type of epistasis which they might be involved in, should account for all the unexplained genetic variance in the MIM QTL models. It has been established that epistasis can make a large contribution to the genetic regulation of complex traits (Carlborg and Haley 2004). However, statistically significant first order epistasis between identified QTL was not detected. Likewise, no significant epistasis between QTL was detected in a recent study based on the maize nested association mapping (NAM) population, which included the parents of this population (B73 and Mo73) (Zhang et al. 2015).
From a total of 60,000 annotated genes across the maize genome, a limited number was identified under 1-LOD QTL intervals. One of the genes, GRMZM2G368398, an oligopeptide transporter, was also identified in a previous meta-QTL investigation of candidate genes for NUE in maize (Liu et al. 2012). An additional gene (GRMZM2G053958), which codes for NAD(P)-binding Rossmann-fold superfamily protein was proposed as a candidate gene in a recent investigation based on C and N metabolism in the NAM population (Zhang et al. 2015). In this study, 13 candidate genes associated with N-metabolism are suggested for further studies. GRMZM2G028574 and GRMZM2G473001 are annotated as PEPC genes. GRMZM2G111225 is annotated as a nitrilase enzyme, which catalyzes the hydrolysis of nitriles to carboxylic acids and ammonia, and is implicated in auxin biosynthesis in maize (Park et al. 2003). GRMZM2G166366 and GRMZM2G136712, code for aspartate kinases, which catalyze the phosphorylation of aspartate to for β-aspartyl phosphate, and is responsible for the first step in the biosynthesis of the amino acids lysine, methionine, and threonine (Azevedo et al. 1992). GRMZM2G155974 catalyzes the addition of glycine to γ-glutamyl-cysteine, generating glutathione. Glutathione is a key water-soluble antioxidant, which represents the storage form and long-distance transport form of reduced sulfur (Zagorchev et al. 2013). GRMZM2G374302 codes for arginine decarboxylase, a key enzyme involved in polyamine biosynthesis that decreases in concentration under N-deficiency conditions (Amiour et al. 2012). In addition, GRMZM2G409131 catalyzes the phosphorylation of d-fructose 6-phosphate to fructose 1,6-biphosphate, the entry point into glycolysis (Plaxton and Podesta 2006). GRMZM2G466543 codes for arogenate dehydratase, a gene that functions in the final steps of the aromatic amino acid pathway that produces two essential amino acids, tyrosine and phenylalanine, which initiate lignin formation, releasing ammonium as a byproduct that is again absorbed by the GS/GOGAT cycle (Holding et al. 2010). GRMZM2G481529 is a cytosolic enolase or phosphopyruvate hydratase and is described as a metalloenzyme responsible for the conversion of 2-phosphoglycerate to PEP, necessary for sucrose synthesis from pyruvate in C4 plants (Karpilov et al. 1978). GRMZM5G817058 is a phosphoribosyltransferase and acts in amino acid metabolism by catalyzing the first step in the biosynthesis of histidine (Morot-Gaudry et al. 2001). Finally, GRMZM2G575696 and GRMZM2G580894, both S-adenosyl-l-methionine (SAM)-dependent methyltransferases, are responsible for transferring methyl groups from the methyl donor SAM to N, oxygen, sulfur, and C atoms of several biomolecules, such as DNA, RNA, histones, and other proteins. These modifications may affect the expression of a wide variety of genes involved in signaling, nuclear division, and metabolism (Bobenchik et al. 2011).
Conclusions
Enzymes for N metabolism exhibit relatively low repeatabilities as compared to dry mass, suggesting they might be overexpressed in the cells under normal N possibly to maintain biomass accumulation through metabolic homeostasis. Poor or no correlation between the root and leaf enzymes and metabolites signifies the importance of studying these two tissues separately. Mildly negative correlations between dry mass and tissue N concentration as well as between the enzymes and dry mass appear to arise from a lack of dilution of cellular contents because of constraints on cell expansion, which, in turn, might arise from factors other than N that limit dry matter formation. Glutamate synthase and alanine aminotransferase were the key enzymes in regulating the cellular levels of glutamate. Our high-throughput assays pave the way to study the enzymes and metabolites of N utilization at field scale.
Author contribution statement
KSD, ML, and ITS conceived and designed the experiments, carried out data analyses, and wrote the manuscript. ITS, HKRA, and LPF performed the laboratory analyses, HL provided molecular marker information. All authors revised and approved the final manuscript.
References
Abendroth LJ, Elmore RW, Boyer MJ, Marlay SK (2011) Corn growth and development. Extension Publication #PMR-1009, Iowa State University
Agrama HAS, Zakaria AG, Said FB, Tuinstra M (1999) Identification of quantitative trait loci for nitrogen use efficiency in maize. Mol Breed 5:187–195
Amiour N, Imbaud S, Clement G, Agier N, Zivy M, Valot B, Balliau T, Armengaud P, Quillere I, Canas R, Tercet-Laforgue T, Hirel B (2012) The use of metabolomics integrated with transcriptomic and proteomic studies for identifying key steps involved in the control of nitrogen metabolism in crops such as maize. J Exp Bot 63:5017–5033
Ashton AR, Burnell JN, Furbank RT, Jenkins CLD, Hatch MD (1990) Enzymes of C4 photosynthesis. Academic Press, London
Azevedo RA, Blackwell RD, Smith RJ, Lea PJ (1992) Three aspartate kinase isoenzymes from maize. Phytochemistry 31:3725–3730
Balko LG, Russell WA (1980) Response of maize inbred lines to N-fertilizer. Agron J 72:723–728
Basten CJ, Weir BS, Zeng ZB (2002) QTL Cartographer, Version 1.16. In: Department of Statistics, North Carolina State University, Raleigh, NC
Beavis WD (1998) QTL analyses: power, precision, and accuracy. In: Paterson AH (ed) Molecular dissection of complex traits. CRC Press, Boca Raton, FL, pp 145–162
Beavis WD, Smith OS, Grant D, Fincher R (1994) Identification of quantitative trait loci using a small sample of topcrossed and F4 progeny from maize. Crop Sci 34:882–896
Bernardo R (2010) Breeding for quantitative traits in plants, 2nd edn. Stemma Press, Woodbury
Bertin P, Gallais A (2001) Genetic variation for nitrogen use efficiency in a set of recombinant inbred lines II—QTL detection and coincidences. Maydica 46:53–68
Bobenchik AM, Augagneur Y, Hao B, Hoch JC, Ben Mamoun C (2011) Phosphoethanolamine methyltransferases in phosphocholine biosynthesis: functions and potential for antiparasite therapy. FEMS Microbiol Rev 35:609–619
Bourne WF, Miflin BJ (1973) Studies on nitrite reductase in barley. Planta 111:47–56
Broman KW, Wu H, Sen Ś, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:2990–2992
Butler D, Cullis BR, Gilmour AR, Gogel BJ (2007) Analysis of mixed models for S-language environments: ASReml-R reference manual. Queensland DPI, Brisbane, Australia
Canas RA, Quillere I, Gallais A, Hirel B (2012) Can genetic variability for nitrogen metabolism in the developing ear of maize be exploited to improve yield? New Phytol 194:440–452
Carlborg O, Haley CS (2004) Epistasis: too often neglected in complex trait studies? Nat Rev Genet 5:618–625
DeBruin J, Messina CD, Munaro E, Thompson K, Conlon-Beckner C, Fallis L, Sevenich DM, Gupta R, Dhugga KS (2013) N distribution in maize plant as a marker for grain yield and limits on its remobilization after flowering. Plant Breed 132:500–505
Dhugga KS, Waines JG (1989) Analysis of nitrogen accumulation and use in bread and durum-wheat. Crop Sci 29:1232–1239
Dhugga KS, Appenzeller LM, Gupta R, Abbaraju HKR (2007) Methods and compositions for increasing the nitrogen storage capacity of a plant. US Patent, CA2634544 A1
Gallais A, Hirel B (2004) An approach to the genetics of nitrogen use efficiency in maize. J Exp Bot 55:295–306
Garnett T, Conn V, Plett D, Conn S, Zanghellini J, Mackenzie N, Enju A, Francis K, Holtham L, Roessner U, Boughton B, Bacic A, Shirley N, Rafalski A, Dhugga K, Tester M, Kaiser BN (2013) The response of the maize nitrate transport system to nitrogen demand and supply across the lifecycle. New Phytol 198:82–94
Gibon Y, Blaesing OE, Hannemann J, Carillo P, Hohne M, Hendriks JHM, Palacios N, Cross J, Selbig J, Stitt M (2004) A robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell 16:3304–3325
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar DS (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40:D1178–D1186
Goolsby DA, Battaglin WA (2000) Nitrogen in the Mississippi basin: estimating sources and predicting flux to the Gulf of Mexico. In: U.S. Geological Survey Fact Sheet, FS-135-00. U.S. Geological Survey, Reston, Virginia
Hallauer AR, Carena MJ, Filho JBM (2010) Quantitative genetics in maize breeding, vol 6. Iowa State University Press, Ames
Hirel B, Bertin P, Quillere I, Bourdoncle W, Attagnant C, Dellay C, Gouy A, Cadiou S, Retailliau C, Falque M, Gallais A (2001) Towards a better understanding of the genetic and physiological basis for nitrogen use efficiency in maize. Plant Physiol 125:1258–1270
Holding DR, Meeley RB, Hazebroek J, Selinger D, Gruis F, Jung R, Larkins BA (2010) Identification and characterization of the maize arogenate dehydrogenase gene family. J Exp Bot 61:3663–3673
Hussain T, Tausend P, Graham G, Ho J (2007) Registration of IBM2 SYN10 doubled haploid mapping population of maize. J Plant Regist 1:81
Joy KW, Ireland RJ (1990) Enzymes of asparagine metabolism. In: Lea PJ (ed) Methods in plant biochemistry. Academic Press Inc., San Diego, CA, pp 287–296
Karpilov LS, Novitskaia IL, Kuz’min AN, Maslov AI, Popova EI (1978) Reversibility of glycolysis in leaves of C4-plants. Biokhimiia 42:1810–1816
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lawrence CJ, Harper LC, Schaeffer ML, Sen TZ, Seigfried TE, Campbell DA (2008) MaizeGDB: the maize model organism database for basic, translational, and applied research. Int J Plant Genom 496957:1–10
Lea PJ, Miflin BJ (2010) Nitrogen assimilation and its relevance to crop improvement. In: Foyer CH, Zhang H (eds) Nitrogen metabolism in plants in the post-genomic era, vol 42. Wiley-Blackwell. Chichester, UK, pp 1–40
Lea PJ, Blackwell RD, Chen FL, Hecht U (1990) Enzymes of ammonia assimilation. In: Lea PJ (ed) Methods in plant biochemistry, vol 3. Academic Press Limited, London, pp 257–276
Lee M, Sharopova N, Beavis WD, Grant D, Katt M, Blair D, Hallauer A (2002) Expanding the genetic map of maize with the intermated B73 × Mo17 (IBM) population. Plant Mol Biol 48:453–461
Limami AM, Rouillon C, Glevarec G, Gallais A, Hirel B (2002) Genetic and physiological analysis of germination efficiency in maize in relation to nitrogen metabolism reveals the importance of cytosolic glutamine synthetase. Plant Physiol 130:1860–1870
Liu R, Zhang H, Zhao P, Zhang Z, Liang W, Tian Z, Zheng Y (2012) Mining of candidate maize genes for nitrogen use efficiency by integrating gene expression and QTL data. Plant Mol Biol Report 30:297–308
Mikel MA, Dudley JW (2006) Evolution of north American dent corn from public to proprietary germplasm. Crop Sci 46:1193–1205
Moll RH, Kamprath EJ, Jackson WA (1982) Analysis and interpretation of factors which contribute to efficiency of nitrogen-utilization. Agron J 74:562–564
Morot-Gaudry JF, Job D, Lea PJ (2001) Amino acid metabolism. In: Lea PJ, Morot-Gaundry JF (eds) Plant nitrogen. Springer, Berlin, pp 167–211
Park WJ, Kriechbaumer V, Muller A, Piotrowski M, Meeley RB, Gierl A, Glawischnig E (2003) The nitrilase ZmNIT2 converts indole-3-acetonitrile to indole-3-acetic acid. Plant Physiol 133:794–802
Plaxton WC, Podesta FE (2006) The functional organization and control of plant respiration. Crit Rev Plant Sci 25:159–198
Plett D, Holtham L, Baumann U, Kalashyan E, Francis K, Enju A, Toubia J, Roessner U, Bacic A, Rafalski A, Dhugga KS, Tester M, Garnett T (2016) Nitrogen assimilation system in maize is regulated by developmental and tissue-specific mechanism. Plant Mol Biol 92:293–312
RCoreTeam (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Remington DL, Ungerer MC, Purugganan MD (2001) Map-based cloning of quantitative trait loci: progress and prospects. Genet Res 78:213–218
Schloerke B, Crowley J, Cook D, Hofmann H, Wickham H, Briatte F, Marbach M, Thoen E (2014) GGally: extension to ggplot2. R package, Ed 0.4.8, Vienna, Austria
Schon CC, Melchinger AE, Boppenmaier J, Brunklausjung E, Herrmann RG, Seitzer JF (1994) RFLP mapping in maize—quantitative trait loci affecting testcross performance of elite European flint lines. Crop Sci 34:378–389
Teuscher F, Guiard V, Rudolph PE, Brockmann GA (2005) The map expansion obtained with recombinant inbred strains and intermated recombinant inbred populations for finite generation designs. Genetics 170:875–879
Trucillo Silva I, Lee M, Cook D (2016) Outlier detection in a complex biological dataset. Int J Agron Crop Sci 1:1–13
Trucillo Silva I, Abbaraju HKR, Fallis LP, Liu H, Lee M, Dhugga KS (2017) Mapping of QTL for N-metabolism related enzymes and metabolites in a maize testcross population grown in hydroponics: I. Leaves. Theor Appl Genet 130:1453–1466
Tuberosa R, Sanguineti MC, Landi P, Giuliani MM, Salvo S, Conti S (2002) Identification of QTLs for root characteristics in maize grown in hydroponics and analysis of their overlap with QTLs for grain yield in the field at two water regimes. Plant Mol Biol 48:697–712
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wickham H (2010) A layered grammar of graphics. J Comput Gr Stat 19:3–28
Wright S (1921) Correlation and causation. J Agric Res 20:557–558
Yemm W, Folkes BF (1958) The metabolism of aminoacids and proteins in plants. Annu Rev Plant Physiol 9:245–280
Yu H, Xie W, Wang J, Xing Y, Xu C, Li X, Xiao J, Zhang Q (2011) Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 6:e17595
Zagorchev L, Seal CE, Kranner I, Odjakova M (2013) A central role for thiols in plant tolerance to abiotic stress. Int J Mol Sci 14:7405–7432
Zhang N, Gibon Y, Gur A, Chen C, Lepak N, Hoehne M, Zhang Z, Kroon D, Tschoep H, Stitt M, Buckler E (2010) Fine quantitative trait loci mapping of carbon and nitrogen metabolism enzyme activities and seedling biomass in the maize IBM mapping population. Plant Physiol 154:1753–1765
Zhang N, Gibon Y, Wallace JG, Kruger Lepak N, Li P, Dedow L (2015) Genome-wide association of carbon and nitrogen metabolism in the maize nested association mapping population. Plant Physiol 168:575–583
Acknowledgements
We thank R.F. Baker Center for Plant Breeding–Department of Agronomy–Iowa State University and DuPont Pioneer for making this research possible. We would like to also thank Iowa State University undergraduate students (especially Guan Yi Lai) who assisted with planting and harvesting in the hydroponic experiments. Requests for testcross materials and IBMSyn10-DH lines may be directed to DuPont Pioneer and Dr. Michael Lee, respectively.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
No conflict of interest declared.
Additional information
Communicated by Michael Gore.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2018_3071_MOESM1_ESM.tiff
Mean, median and skewness of the N-metabolism related enzymes and metabolites measured on root tissues in the maize IBMSyn10-DH TC population (created with Adobe Illustrator CS2) (TIFF 104 kb)
122_2018_3071_MOESM2_ESM.tiff
Correlation matrix-heatmap of the N-metabolism related enzymes and metabolites measured on root tissues in the maize IBMSyn10-DH TC population (created with corrplot package, R). Significant correlation values (p value <0.05) depicted in blue (positive correlation) and red (negative correlation) (TIFF 1183 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Silva, I.T., Abbaraju, H.K.R., Fallis, L.P. et al. Biochemical and genetic analyses of N metabolism in maize testcross seedlings: 2. Roots. Theor Appl Genet 131, 1191–1205 (2018). https://doi.org/10.1007/s00122-018-3071-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-018-3071-0