
Abstract
In Asian cultivated rice (Oryza sativa L.), aroma is one of the most valuable traits in grain quality and 2-ACP is the main volatile compound contributing to the characteristic popcorn-like odour of aromatic rices. Although the major locus for grain fragrance (frg gene) has been described recently in Basmati rice, this gene has not been characterised in true japonica varieties and molecular information available on the genetic diversity and evolutionary origin of this gene among the different varieties is still limited. Here we report on characterisation of the frg gene in the Azucena variety, one of the few aromatic japonica cultivars. We used a RIL population from a cross between Azucena and IR64, a non-aromatic indica, the reference genomic sequence of Nipponbare (japonica) and 93–11 (indica) as well as an Azucena BAC library, to identify the major fragance gene in Azucena. We thus identified a betaine aldehyde dehydrogenase gene, badh2, as the candidate locus responsible for aroma, which presented exactly the same mutation as that identified in Basmati and Jasmine-like rices. Comparative genomic analyses showed very high sequence conservation between Azucena and Nipponbare BADH2, and a MITE was identified in the promotor region of the BADH2 allele in 93–11. The badh2 mutation and MITE were surveyed in a representative rice collection, including traditional aromatic and non-aromatic rice varieties, and strongly suggested a monophylogenetic origin of this badh2 mutation in Asian cultivated rices. Altogether these new data are discussed here in the light of current hypotheses on the origin of rice genetic diversity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Based on a number of morphological, physiological, biochemical and molecular traits, Asian cultivated rices are organised in two major subspecies, i.e. Oryza sativa japonica and Oryza sativa indica (Second 1982; Glaszmann 1987; Oka 1988). These two subspecies are commonly associated with differences in growth habitat (Khush 1997) and are the products of independent domestication events from ancestral Oryza rufipogon populations in different locations and at different times (Vitte et al. 2004; Ma and Bennetzen 2004; Sang and Ge 2007a). In addition to this major genetic organisation, several other minor groups of varieties, usually based on more limited geographical distribution or special adaptation and characteristics, have been identified with genetic markers (Second 1982; Glaszmann 1987) and confirmed more recently (Bautista et al. 2001; Garris et al. 2005; Londo et al. 2006). For instance, these minor groups include Aus cultivars of India and Bangladesh, Ashinas varieties of Bangladesh, and aromatic Basmati rice of India, among others. Nevertheless, the identification of these minor secondary groups does not contradict the fundamental genetic organisation of the Asian rice in japonica and indica subspecies and Basmati rices. This latter forms a small group of closely related varieties with strong affinity to the japonica group, but which is genetically distinct (Garris et al. 2005; McCouch et al. 2007), as confirmed by specific marker studies and rare allelic associations. Therefore, it has been proposed that Basmati rices, may have been independently domesticated (Garris et al. 2005; McCouch et al. 2007).
Interestingly aromatic rices varieties are also appreciated to an increasing extent in Western societies and thus the world market for these scented rices is in continuous full expansion. Unfortunately, aromatic rice cultivars, particularly Basmati rices from India, often produce poor yields because of their low resistance to rice diseases and limited adaptation outside their original geographical distribution. Basmati rice types also have a poor combining ability when crossed with other rice genotypes.
Among the more than 100 volatile flavour compounds which constitute rice aroma, 2-acetyl-1-pyrroline (2-ACP) has been identified as the main agent in Basmati and Jasmine-style fragrant rices (Buttery et al. 1983; Paule and Powers 1989; Petrov et al. 1996). 2-ACP is actually detected in all parts of the rice plant, except in the roots (Lorieux et al. 1996). The detailed biosynthesis pathway of this compound has not yet been completely elucidated (Lorieux et al. 1996; Bradbury et al. 2005a). However, it was demonstrated, using the aromatic variety Thai Hom Mali, that the osmoprotectant proline was its precursor and the nitrogen source of 2-ACP (Yoshihashi et al. 2002). In higher plants, proline is synthesized from glutamate or ornithine and highly accumulated under osmotic stress conditions (Verbruggen et al. 1996; Tuteja 2007).
Initial genetic studies, performed by Tanksley’s group (Ahn et al. 1992), localised a gene controlling aroma or fragrance (frg gene) in Della (Jasmine-derived aromatic variety) on the long arm of chromosome 8. Later, in our group Lorieux et al. (1996) tagged this gene as a major and recessive quantitative trait locus (QTL) in the same region, but limited to a 12 cm genetic interval, and in a (IR64 × Azucena) DH population where the traditional upland variety Azucena was the donor of aroma. For the first time, this study demonstrated that 2-ACP detected by sensitive methods and gas chromatography (GC) was perfectly correlated and that large quantitative variations were observed among aromatic DH lines. Moreover, in the same study, two other possible minor QTLs were identified on chromosome 4 and 12, which may affect the strength of aroma. Since then, several authors have reported mainly identifying markers (SSR, PCR-based markers) associated with the frg locus that could be used to help in distinguishing aromatic and non-aromatic rice varieties for marker-assisted selection (MAS) (Garland et al. 2000; Cordeiro et al. 2002; Jin et al. 2003). More recently, in traditional Basmati and Jasmine-like rices, Bradbury et al. (2005a, b) further restricted the aroma region and identified a single recessive gene responsible for aroma. This gene is a defective allele of a gene encoding betaine aldehyde dehydrogenase BADH2. The deletion observed in exon 7 of this (BADH2) gene generates a premature stop codon and presumably results in loss of activity. It was hypothesized that loss of BADH2 activity causes 2-ACP accumulation (Bradbury et al. 2005a).
Up to now, most studies on aroma gene in rice have mainly concerned traditional aromatic rices to assist breeders in the development of new cultivated fragrant rice varieties better adapted to particular environmental constraints (Bradbury et al. 2005b; Wanchana et al. 2005). Here we report the identification and characterisation of the fragrance aroma gene in the Azucena cultivar, one of the few japonica rices referred as aromatic. This allowed us to analyse the diversity of the region surrounding this major aroma gene.
By using a population of recombinant inbred lines (RILs), we physically mapped the Azucena aroma allele in a 160 kbp interval. A suitable Azucena BAC library was then developed and screened to isolate a BAC clone of about 50 kbp containing the aroma gene. This Azucena BAC clone was sequenced, annotated and compared with available indica and japonica genomic sequences. Screening of a representative rice germplasm collection, including aromatic and non-aromatic forms, allowed us to analyse the diversity of the gene and speculate on its origin in Asian rice varieties.
Results
Genetic mapping of the Azucena frg gene
The Azucena aroma region had been previously mapped in our group (Lorieux et al. 1996) between the RG1 and RG28 RFLP markers by using a (IR64 × Azucena) population of double haploid (DH) lines (Guiderdoni et al. 1992). In this study, fine mapping was carried out using a new RIL population derived from the same cross between Azucena and IR64 (Table 1).
An F7 population of about 300 RILs was initially mapped with SSR markers RM42 and RM284, located within the RG28 and RG1, and RM72 and RM44, intervals instead of RG28 (Temnykh et al. 2000). Among more than 300 lines, about 190 recombinant lines (F7–F9 generation) displaying good fertility, were selected to develop an SSR core map (Tranchant et al. 2005) and multiplied. SSR markers RM42 and RM284 spanning the aroma gene region were selected to identify 40 recombinant lines for adding markers and for rapid screening of aroma by the smelling method after soaking seeds in KOH solution. Additional mapping of RM515, RM8265, RM223 markers allowed us to reduce the number of informative recombinants to nine independent RILs (16C, 19A, 35B, 122A, 159B, 164A, 186A, 259B and 274A) (Table 2). As the data obtained by direct sniffing assays could not be considered as completely reliable because of the subjective nature of this method, the 2-ACP concentration in each of the nine recombinants was also measured by GC (Table 3). This method validated the aroma evaluation previously obtained by the chemical test and the six recombinant lines identified as aromatic by the smelling method presented a 2-ACP concentration range of 77–254 ppb. In some cases, this concentration was even higher than those detected in the Azucena parent (183 ppb). Reciprocally, no 2-ACP could be detected in the three non-aromatic RILs, as in IR64 parent.
Finally, the Azucena aroma region was delimited in an interval of less than two BACs (AP005537 and AP004463) by comparison to the Nipponbare physical map and representing approximately 200 kbp. To further reduce the Azucena aroma candidate region, eleven new additional markers covering this region were designed by comparing Nipponbare (japonica) and 93–11 (indica) genomic sequences available in databases since they were supposed to be polymorphic between IR64 and Azucena parents. Genotyping of the nine (F9)-RILs with these eleven markers allowed us to narrow down the aroma locus to a region of 167 kbp between markers 5537-8 and 4463-15 (Table 2).
The interval delineating the aroma locus encompassed 24 predicted genes in Nipponbare (TIGR Genome Annotation Release version V) out of a total of 30 gene models annotated on BAC AP005537 and AP004463. Putative functions were assigned to 12 of the 24 predicted genes (Table 4). Based on the availability of either a full length cDNA sequence or an EST, only 10 were expressed and 7 of them encoded known enzymes. The BADH2 gene (Os08g32870) was among them, which was also previously reported as being the frg locus of traditional Basmati and Jasmine-style aromatic rices (Bradbury et al. 2005a).
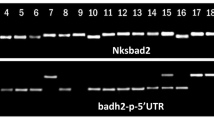
As we expected to find a modification in the nucleotide sequence in the Azucena orthologous badh2 allele, primers were designed in the Nipponbare sequence to amplify a 235 bp fragment spanning the mutation observed by Bradbury et al. (2005a). A Tilling experiment was performed on the 9 previous individual RILs. The results confirmed the presence of a mutation in the Azucena badh2 gene, in the 6 aromatic RILs and its absence in non-aromatic RILs, thus indicating that badh2 was the best aroma candidate locus in the Azucena japonica cultivar (Fig. 1).

Genotyping confirmation by Ecotilling and the presence of a deletion in the Azucena badh2 gene. Ecotilling was performed on genomic DNA of the nine individual RILs (16C, 19A, 35B, 122A, 159B, 166A, 186A, 259B and 274A) and the two parents Azucena and IR64. PCR amplification controls were performed on Nipponbare, Azucena and IR64. Control for heteroduplex formation and digestion was performed with Azucena and IR64 fragments
Sequence composition of the Azucena aroma region and comparison with reference regions in Nipponbare and 93–11
In order to analyse the exact composition of the Azucena aroma region with respect to gene content and other elements, we prepared a small Azucena BAC library of about 6,000 clones and isolated the orthologous genomic region corresponding to badh2. A BAC clone (05K17) of about 50 kbp was identified by PCR screening with primers designed from the Nipponbare BADH2 sequence and, following shotgun sequencing, a contig of 38,666 bp was finally assembled and compared with the Nipponbare and 93–11 sequences. As sequencing immediately revealed at the same position in Azucena the 8 bp deletion observed in the BADH2 gene of Basmati rices, we have compared the predicted (ORF) gene structure along the whole Azucena BAC clone sequence to Nipponbare and 93–11 sequences.
Sequence comparisons of the aroma orthologous region with Nipponbare and 93-11
Dot-plot alignment showed extensive nucleotide conservation, indicating an orthologous relationship between Nipponbare (BAC AP005537, TIGR pseudomolecule chromosome 8, position 20,230,406–20,268,978, i.e. a 38,572 bp interval) and Azucena sequences (Fig. 2a). The nucleotide alignment indicated 99% overall sequence conservation (in the ~38.5 kbp interval contig) and contained 26 InDels ranging from 1 to 173 bp and 85 SNP (Fig. 2a, b coloured boxes and vertical lines, respectively). The most important difference between the two sequences was a 173 bp deletion (position 7,326–7,498 on the Azucena contig of 38,666 bp) in a GC-rich region upstream of the Azucena badh2 allele in the Osjaz-05K17.1 gene (Fig. 2a, green arrow, and Fig. 2b). However, this 173 bp deletion was also observed in a Nipponbare cDNA (TIGR Genome browser), suggesting the presence of two potential start codons and possible alternative splicing for the Osjaz-05K17.1 gene.

a Dot-plot alignments of aroma orthologous regions from Nipponbare (Nipponbare badh2; 43 kbp, horizontal) and Azucena (Azucena badh2; 38.6 kbp, vertical) O. sativa ssp. japonica varieties. Nucleotide conservation between orthologs is indicated by diagonal lines. Horizontal annotations correspond to the aligned segments of Nipponbare and vertical annotations to Azucena. Black boxes represent predicted genes and coloured boxes represent the different types of TEs. Blue arrows indicate the location of the mutation in the Azucena badh2 allele in both the annotations and aligned sequences. Vertical bars represent SNPs. b Orthologous regions from Azucena. Comparisons of the SNP content in Azucena and Nipponbare and, Azucena and 93-11. Vertical bars represent SNPs
Since the 93-11 sequence is the result of shotgun sequencing, alignment was sometimes difficult. Hence, we observed, for example, a large (28,484 bp) insertion in the corresponding extracted region of 93-11 (pseudomolecule 08—BGI annotation), which was eliminated from the comparison. However, we were able to unambiguously aligne a 37,096 bp region spanning the badh2 region. In this interval, the overall sequence identity between Azucena and 93-11 was 96.1% and, in comparison with the previous analysed Azucena/Nipponbare region, 128 InDels and 243 SNP were observed (Fig. 2b).
Annotation of the Azucena aroma region
When annotating the 38,666 kbp Azucena sequence, we identified four predicted genes and nineteen transposable elements (Fig. 2a). The four genes correspond to those described in Nipponbare: The Osjaz-05K17.1 (Oryza sativa japonica Azucena), partially covered by the sequenced contig analysis, is similar to the Nipponbare propionyl-CoA gene (Os08g32850). The second gene, i.e. Osjaz-05K17.2, was identified as a badh2 allele, as described above. However, analysis of the Azucena badh2 (Osjaz-05K17.2) allele revealed the presence of a 12 bp sequence variation in exon 7, located 3,273 bp from the start codon (Fig. 2a, blue arrow; Fig. 3). In comparison to the Nipponbare BADH2 genes, the presence of this mutation introduced a premature stop codon, leading to a predicted truncated, and presumably inactive protein of 211 amino acids. This sequence variation in Azucena was exactly the same as that identified in the Basmati badh2 gene (Bradbury et al. 2005a). Indeed, it consisted of both an 8-bp deletion and a 4-bp insertion (Fig. 3). The last two genes (Osjaz-05K17.3 and Osjaz-05K17.4) showed similarities to putative disease resistance genes and notably an absence of introns, as observed in Nipponbare.

Identification of the Azucena aroma allele badh2 and sequence alignment of BADH2 alleles in different rice varieties harbouring aroma haplotypes. The 12 bp sequence variation identified in the badh2 Azucena aroma allele is identical to that identified in Basmati and Jasmine rice aroma gene candidates. Nipponbare and 93-11 displayed no sequence variation, showing that they only contain the no-aromatic allele badh2. Four varieties not classified as aromatic (Arias, JC120, Khao Kap Xang, JC157) show the 12 bp mutation in BAD (Table 5)
Analysis of the non-gene regions of the Azucena BAC clone and corresponding region of Nipponbare revealed a high density of transposable elements (TEs). Nineteen TEs were thus identified in the Azucena sequence, sixteen of which were inserted in the intergenic region between Osjaz-05K17.1 and Osjaz-05K17.2. Among these TEs, we found eight transposons, four MITE and four retroelements (Fig. 2, blue, yellow and red coloured boxes). This local accumulation of TEs contributed to 64.8% of the Osjaz-05K17.1 and Osjaz-05K17.2 Azucena intergenic sequence in a 10.7 kbp interval. These elements are highly similar to those present in Nipponbare, confirming the japonica nature of Azucena.
Sequence analysis of the Azucena badh2 allele and its promoter region with Nipponbare and 93-11
A comparative analysis of the upstream intergenic and coding regions of frg alleles of Azucena (badh2), Nipponbare (BADH2) and 93-11 (BADH2) was performed in an attempt to determine which variations could be related to fragrance and which were related to indica/japonica differentiation. First, a fragment of approximately 8.7 kbp containing the upstream intergenic and coding regions of the Azucena badh2 allele was extracted and compared by dot-plot to orthologous fragments in Nipponbare and 93-11. Dot-plot alignment showed almost perfect conservation between Azucena and Nipponbare, with nucleotide identity reaching 99.5% (Fig. 4a) and low InDel and SNP densities (five InDels ranging from 1 to 8 bp localised in an intergenic upstream region and a level of 1.5 SNP/kbp). Regarding the gene sequence, only one SNP was identified in intron 2 between Azucena and Nipponbare alleles. Second one was located downstream of the deletion in exon 7 (Fig. 4a).

Dot-plot alignments of badh2 orthologous alleles, from Nipponbare (Nipponbare BADH2, horizontal) and Azucena (Azucena badh2, vertical) O. sativa ssp. japonica varieties (a), from O. sativa ssp. indica 93-11 (93-11 BADH2, horizontal) and Azucena (Azucena badh2, vertical) (b). Nucleotide conservation between orthologous regions is indicated by diagonal lines. A break in the alignment between O. sativa 93-11 and Azucena shows the insertion of a TE in the promoter region of the 93-11 sequence (yellow arrow). Black boxes represent exons of badh2 alleles and coloured boxes represent different types of TEs. Blue arrows indicate the location of the mutation in the Azucena aroma badh2 allele in both the annotations and aligned sequences
In contrast, dot-plot alignment between Azucena and 93-11, pseudomolecule chromosome 8, position 21,698,225–21,707,164, indicated a conservation disruption in the region (Fig. 4b, yellow arrow). Sequence analysis revealed that this disruption was due to the insertion of a transposable element classified as a MITE (miniature interspersed transposable element) in the promotor region of the BADH2 allele in 93-11, compared to Azucena and Nipponbare, suggesting that this insertion could be a specific landmark of the indica sub-species lineage. In addition, sequence analysis revealed only 94.4% overall sequence identity (Fig. 4b) with high InDel and SNP densities, respectively (15 InDels ranged from 1 to 30 bp in non-genic regions and a level of 6.97 SNP/kbp). Fourteen SNP distinguished Azucena and 93-11 alleles and all of them were in introns. Thus, 7 were in intron 1 and 3 in intron 3, with the last 4 being in intron 6 and 11 (3 and 1, respectively).
In summary, altogether these analyses confirmed that Azucena and Nipponbare badh2 orthologous regions are much more closely related to each other than to the orthologous region between Azucena and 93-11 and demonstrated that the surrounding region of the Azucena badh2 allele was clearly in a japonica background (Fig. 2a, b). Regarding only badh2 alleles and their promotor regions, the absence of the MITE revealed an (even) higher percentage of overall sequence identity (99.5%), between Azucena and Nipponbare (compared to those observed between Azucena/93-11 and Nipponbare/93-11, 94.4 and 94.6%, respectively), thus confirming the higher identity between Azucena and Nipponbare (Fig. 4a, b) at this locus.
Genetic diversity of the frg gene region in traditional aromatic and non-aromatic cultivated Asian rice varieties
We extended this analysis to a survey of the diversity of the aroma locus among cultivated rice varieties, while taking the deletion in the BADH2 gene but also the presence or absence of the MITE insertion in the promoter into account. Primers were designed to specifically detect the 93-11 MITE insertion (primers MITE_5 and MITE_3) and/or the deletion in the Azucena badh2 allele (Primers AR_5 and AR_3), while using the Nipponbare BADH2 gene (Primers AR_5 and NAR_3) as control. DNA was amplified from 81 varieties extracted from a core collection representative of Asian rice isozyme diversity (Glaszmann 1987) (Table 5).
PCR amplifications showed that the presence of the MITE in the promoter region was more characteristic of indica varieties (Table 6) and rare in japonica varieties. In particular, it was totally absent in temperate varieties. Therefore, the variation in the frequency of this MITE in the rice collection reflects a pattern similar to that of other genetic markers derived from isozymes or RFLP, where one allele is specific to one group whereas balanced polymorphism is observed in the other group.
Screening the collection clearly showed that the presence of the 12 bp sequence variation was systematically associated with the absence of MITE in the three original aromatic rice varieties tested in this collection (Basmati 370, KDM105 and Azucena) (Table 4). This association was also found in several varieties belonging to different groups: JC120 in indica, Arias and Khao Kap Xang, two upland japonica varieties and JC157 in isozyme group V, which are not referred as aromatic varieties (Table 4).
Discussion
The aim of this study was to investigate, in Azucena—one of the few aromatic japonica upland varieties mainly cultivated in the Philippines—the nature of the aroma gene (frg) that has been up to now identified in Basmati and Jasmine-indica-related rices (Bradbury et al. 2005a) and to investigate its diversity amongst cultivated rice varieties. We thus used a combined approach of fine genetic mapping and comparative genomic analyses along with a survey of representative common cultivated Asian rice varieties. Our results showed that, in the Azucena cultivar, there is very high sequence conservation with the Nipponbare BADH2 gene and moreover all the attributes of a japonica variety are present, at least in the genomic region analysed here.
Interestingly, this work highlighted new evidence of the close relationship between the 12 bp mutation observed in traditional aromatic rices—leading a priori to a truncated predicted protein—and the 2-ACP synthesis and aroma expression. Finally, apart from a 173 bp Indel found in the Osjaz-05K17.1 gene, corresponding to a possible sequence variation or an unreliable annotation present at the beginning of the compared sequences, the only striking differences concerned this mutation in exon 7 of the Azucena badh2 allele and a MITE in the promoter region of indica 93-11. This transposable element clearly appears to be specific to the majority indica subspecies and its absence associated with the mutation for aroma expression.
Therefore, 2-ACP synthesis involves a biochemical pathway in which badh2 is supposed to play a critical role since only its predicted non-functionality is associated with aroma expression. Indeed, a BADH gene is a good candidate gene since the corresponding protein oxidises betaine aldehyde into glycine betaine, an osmolyte-like proline, one of the known potential precursors of 2ACP (Weretilnyk and Hanson 1990; Sakamoto and Murata 2002). The BADH enzyme also displays a wide range of substrates (Trossat et al. 1997; Livingstone et al. 2003), with a structure similar to 2-ACP and generally reported to be associated with the polyamine pathway. Bradbury et al. (2005a) proposed that 2-ACP or its probable precursor(s) are also likely substrates for BADH enzyme. The hypothesis put forward so far is that in the presence of the functional non-truncated BADH2 allele, betaine aldehyde or another related substrate is oxidised and 2-ACP is consumed and not accumulated. In contrast, when the aroma badh2 allele is not functional, aroma expression could be explained by the modification or interruption of a natural upstream pathway, leading to the accumulation of 2-ACP or its precursor(s). Lastly, a direct role of regulating genes on the badh2 aroma allele also cannot be definitively excluded. Ultimately, these different possible 2-ACP synthesis scenarios would necessarily require attention to select appropriate strategies for the functional validation of any candidate gene assumed to play a role in aroma biosynthesis.
If many convergent data are accumulating to give a major role to the badh2 locus in the presence/absence of 2-ACP, this trait remains at a phenotypic level a quantitative character that is largely dependent on environmental conditions and the genetic background, thus suggesting that 2-ACP synthesis has a polygenic aspect. The high variation noted in 2-ACP (ranging from 77 to 254 ppb) in the nine independent RILs has also already been observed in our previous studies (Lorieux et al. 1996), thus suggesting that additional minor QTLs on chromosome 4 and 12 have a marked influence on the aroma strength among recombinant lines that have the badh2 gene mutation. This observation is supported by the results of Amarawathi et al. (2007) who very recently identified a new QTL for aroma on chromosome 3 in a different Basmati cultivar. The second betaine aldehyde dehydrogenase gene, i.e. BADH1, observed in the rice genome (Os04g39020; 92% homology and 76% identity in Nipponbare protein) (IRGSP, 2005) is localised under the minor QTL observed on chromosome 4. Genomic analyses indicated that in Nipponbare BADH1 and BADH2 genes were located in a duplicated region and were likely paralogous genes issued from the extensive gene duplication that japonica and indica genomes have undergone, and which is shared by most cereals (Salse et al. 2008). The more complex genetic basis of aroma has also been confirmed by breeders, who noted that transfer of the aroma trait by MAS sometimes fails to give recipient varieties with high aroma expression. Finally, in these studies, the few lines that were found to harbour the badh2 mutation, but which are not recognised as true aromatic or economically important varieties, might correspond to lines with only faint or transient aromatic expression which is not reliably detectable by the smelling method.
Interestingly, the survey of genetic diversity in and around the badh2 gene revealed a much more frequent MITE insertion in the indica variety than in the japonica group in the broad sense (Tables 5, 6). Identification of such an association between the absence of a MITE and the presence of a mutated badh2 gene in unrelated aromatic varieties (aromatic, Basmati, indica KDM 105), combined with the nature of the 12 bp sequence variation (deletion and insertion), strongly suggest that this allele has a monophylogenetic origin rather than being the result of independent mutational and domestication events which would result in different polymorphisms. The data are also more consistent with respect to highlighting a single origin of the badh2 mutation in one or the other lineage, which would have occurred after domestication and followed by introgressions with rice varieties according to the dispersion of varieties by man. Recent results of varietal screening confirmed, for example, the observation of the mutation in badh2 exon 7 in 3 Chinese indica aromatic varieties (Shi et al. 2008). As aroma was not an essential trait, selection for aroma in this badh2-mutated context could have largely depended on environmental factors and the genetic background favourable for its expression, or special interest shown by local farmers, as was the case with Basmati rices (Tang et al. 2007). Indeed, it is well known that cold temperature at flowering time increases aroma expression in Basmati varieties grown in the foothills of Nepal. In the same way, the indica variety KDM105 exhibits stronger aroma when it is grown in drought-prone areas (Yoshihashi et al. 2002).
Interestingly, the badh2 mutation observed in this study has also occasionally been reported in wild Thai rice (Vanavichit et al. 2004). However, this more likely reflects natural introgressions that have given rise to weedy forms surrounding fields largely planted with aromatic varieties, and is in line with the dynamics of weedy O. rufipogon populations (Oka 1988). Genes corresponding to mutations selected during domestication have been very recently cloned. A single gene conferring red pericarp (rc gene) was thus cloned and gave a gene function pattern similar to that observed in badh2, since a 14 bp frameshift deletion truncates the Rc protein before a bHLH domain (Sweeney et al. 2006, 2007). In the same way, the pattern of variation of the frequency of the badh2 mutation and MITE shows also high similarity with the frequency pattern of very critical genes involved in domestication, e.g., genes affecting shattering (sh4, qSH1; Sweeney and McCouch 2007). Then, sh4 gene was previously mapped in O. nivara x indica cultivars, but the non-shattering allele was found to be present in all O. sativa varieties surveyed and along with a varied panel of indica and as well as tropical and temperate japonica cultivars (Li et al. 2006a, b). In this case, functional nucleotide polymorphism (FNP) was clearly identified and accompanied by five other SNPs within the gene shared by all O. sativa varieties. The corresponding “non-shattering haplotypes” was also found in some O. nivara populations, thus confirming introgressions within weedy forms.
The next step will be to further extend the characterisation of the region around the aroma badh2 allele in order to more precisely define the concept of an “aroma haplotype” in indica and japonica varieties. A larger sampling of cultivated varieties as well as wild and weedy O. rufipogon accessions should thus be screened for gene diversity. Recently a new badh2 null allele characterised by a different deletion in exon 2 has been evidenced in Chinese japonica varieties (Shi et al. 2008) Fine characterisation of aroma in these varieties presenting the badh2 mutation, but not necessarily with high aroma expression, would be required to identify other genes which might also be involved in aroma synthesis and expression. This could contribute to the selection of a better genetic background to promote high aroma expression in rice varieties.
Finally, as an increasing number of key rice genes will be cloned and analysed, this will help to gain greater insight into the domestication (Konishi et al. 2006; Kovach et al. 2007; Sang and Ge 2007b) process and the importance of introgressions in the origin of genetic diversity in Asian rice.
Material and methods
Genetic mapping
The population of recombinant inbred lines (RILs) used to analyse the aroma locus in this study is derived from a cross between IR64 × Azucena varieties. Azucena is a scented japonica landrace from the Philippines, and IR64 is a non-scented indica variety developed by IRRI and characterised by high productivity and high pest resistance. This population mapping involved 100 RILs constructed according to the single-seed descent method (SSD) with selection from F7–F9 generations.
The markers used were either published markers selected for their polymorphism between Azucena and IR64 (Temnykh et al. 2000; McCouch et al. 2002) or designed from the public rice genomic sequence of Nipponbare (http://www.gramene.org; http://www.TIGR.org). The database developed by Shen et al. (2004) was used to identify putative japonica-indica polymorphisms through a sequence comparison between Nipponbare (japonica) and 93-11 (indica). Out of all primers used, 5 were already designed and 11 were newly generated: seven SSR markers, five PCR-based markers (Azucena or IR64-specific) and five cleaved amplified polymorphism sequence (CAPS) markers (Table 1).
Phenotypic evaluation
Aroma expression was assessed in all of recombinant plant progenies by smelling KOH extracts of four seeds to score these plants as aromatic or non-aromatic. Seeds were ground mechanically with a TissueLyser (Qiagen; 2 × 30 s at 30 Hz) in a 1.5 ml tube with a 5 mm tungsten bead and then incubated in 1 ml KOH 1.7% for 30 min at 37°C (Sood and Siddiq 1978). Several evaluations (minimum of nine measurements) were carried out for each plant tested, both on dried and immature seeds, and by at least three persons. Aroma detection was always carried out in comparison with the two parents, i.e., Azucena and IR64. As the data obtained with this method are not considered as completely reliable because of the subjective nature of human sniffing, the 2-ACP concentration in the nine independent RILs was also measured by gas chromatography (GC). The 2-APC measurement protocol (Petrov et al. 1996) was slightly modified in order to work with 20 g of seeds instead of 100 g.
DNA extraction and molecular markers
The genomic DNA of RILs was isolated from flash-frozen leaves (~4 cm of the extremity of young leaves) in liquid nitrogen. Leaves were ground with a TissueLyser (Qiagen; 2 × 30 s at 30 Hz) in racks of 96-collection microtubes of 1.2 ml (Qiagen) with one 2 mm tungsten bead. DNA extractions were carried out according to the protocol described by Edwards et al. (1991) but adapted for a routine polymorphism detection and genetic mapping procedure. DNA was resuspended in 50 μl TE and 2–4 μl were used for PCR reactions (in a final volume of 15 μl). The CTAB method was used for larger amounts of genomic DNA (Dellaporta et al. 1983).
SSR markers were amplified in a final volume of 15 μl (0.67 mm dNTPs, 1X Taq buffer, 0.66 μm Primer-Forward and 0.66 μm Primer-Reverse, MgCl2 2 mm, 1 unit Taq DNA polymerase) in the following conditions: one cycle at 94°C for 3 min, 30 cycles (94°C for 30 s, 50°C for 30 s and 72°C for 45°C) and one cycle at 72°C for 10 min. PCR reactions were carried out on Perkin Elmer or Biometra thermocyclers. Large size polymorphisms were detected on agarose gel, whereas small size polymorphisms were detected on 6.5% acrylamide gel. In the latter case, one primer of each pair was tailed during synthesis with the M13 sequence as described in Albar et al. (2006) and runs were performed with a LICOR-Genotyper (LI-COR Inc., Lincoln, NE, USA) and image analysis with Adobe Photoshop software (Adobe Systems Incorporated).
PCR-based markers were designed on both sides of an intron on the Nipponbare sequence. PCR reactions were performed on the Azucena and IR64 in a final volume of 25 μl with 100 ng of genomic DNA, 0.4 μm each of forward and reverse primers, 0.66 mm each of dNTPs, and 1 unit of Taq DNA polymerase. The reaction mixture was incubated at 94°C for 2 min followed by 35 cycles at 94°C for 15 s, one cycle at 55°C for 15 s and finally an extension cycle at 72°C for 30 s. Amplification products were separated on 2–4% agarose gel. Molecular markers were detected by size polymorphism. Otherwise, after purification, PCR products (QiAGEN, QIAquick PCR purification Kit) were sent for sequencing to Genopôle Languedoc-Rousillon or to Genome Express (http://www.genome-express.com). Sequence polymorphism (restriction site, InDel) between the two parents was then identified by alignment with Lasergene software (DNASTAR) or with BLAST (http://www.infobiogen.fr) and used to design CAPS markers and PCR-specific primers for Azucena or IR64.
Analysis of RILs by ecotilling
The Tilling experiment was performed according to the procedure described by Nieto et al. (2007). A pair of primers, ADH3F2 (5′-GCATTTACTGGGAGTTATG-3′) and ADH4R (5′-CCACCAAGTTCCAGTGAAAC-3′) were designed on the Nipponbare sequence to amplify a 235 bp fragment, including the mutation observed by Bradbury et al. (2005a) in the badh2 aroma allele sequence of Basmati. PCR amplifications were performed on genomic DNA of the nine independent RILs and of the two parents, Azucena and IR64, as well as on Nipponbare genomic DNA as control. Amplified genomic DNA of each RIL was mixed with IR64 or Azucena genomic DNA and then heated and renatured. Heteroduplexes were cleaved at the mismatched site using CelI enzyme (kindly provided by Abdelhafid Bendahmane, URGV Evry, France). After digestion, the samples were separated on 4% agarose gel.
Azucena BAC library synthesis and screening of BAC clone 05K17
A small Azucena BAC library of 6,000 clones was synthesised with nuclei extracted from 2-week-old Azucena leaves according to a previously described protocol (Noir et al. 2004). Plants were grown in soil in darkness at 28°C and 70% humidity. The Azucena BAC library was screened for the aroma region by PCR amplification with the molecular markers used for fine mapping and specific primers for the BADH2 gene. The insert size of the BAC clones was analysed by pulse-field gel electrophoresis after digestion with NotI. DNA of the positive BAC clone (05K17) was isolated with a large-construct kit (QIAGEN) and 5 μg were mechanically sheared (HydroShear, Genemachines). A sub-cloning library was constructed using the Zero Blunt TOPO PCR cloning kit (Invitrogen). Plasmids were extracted and sequenced with the ABI 3100 automated Capillary DNA Sequencer (Applied Biosystems, Courtaboeuf, France). Gaps between contigs were filled by PCR amplifications and direct sequencing of PCR products. PCR-primers were designed with Primer 3 software.
Sequence analysis and comparison
After shotgun sequencing, the sequences were assembled using PHRED/PHRAP software (Ewing et al. 1998). A total of 560 sequences were processed, giving an average of 4.12 time coverage. BAC sequence analysis was performed using BLAST algorithms, the EMBOSS package (Rice et al. 2000) and by dot plot (DOTTER) (Sonnhammer and Durbin 1995). Putative genes were identified using a combination of prediction software available through the RiceGAAS website (http://ricegaas.dna.affrc.go.jp). Transposable elements (TEs) were identified using RepeatMasker (http://www.repeatmasker.org) with the TIGR Repeat database and the RetrOryza database (Chaparro et al. 2007) (http://www.retroryza.org). Final annotation was performed using the Artemis annotation tool (Rutherford et al. 2000). Comparative sequence analyses were performed by dot plot using pseudomolecules (V.05) of O. sativa ssp. japonica Cv. Nipponbare available at TIGR (http://www.tigr.org) and O. sativa ssp. indica Cv. 93-11 available at BGI (http://rice.genomics.org.cn). Sequence alignments and IndDels and SNP detections were carried out using stretcher and diffseq programs, respectively.
Estimation of haplotype combinations in aromatic and non-aromatic rice varieties
The presence or absence of the mutation in the BADH 2 gene as well as MITE insertion were tested by PCR experiments on a collection of traditional rice varieties. This collection contains aromatic and non-aromatic rice varieties that are representative of Asian rice diversity (Tables 5, 6). The same collection was previously used for isozyme classification of rice varieties and represents the two major groups, namely indica and japonica varieties corresponding to isozyme groups I and VI, respectively. Additional groups V (including Basmati varieties) and II (Aus varieties) were also proportionally represented. The two major aroma genetic resources of economic importance were represented by Khao Dawk Mali (indica variety representative of Thai rice varieties) and Basmati 370 as the reference variety of the aromatic Basmati rice family.
PCR experiments were performed on 25 ng of genomic DNA in 15 μl containing 1 unit of Taq DNA polymerase, 1X reaction buffer, 0.66 mm dNTPs and 0.66 μm of primers with the following primers to determine on one hand aromatic and non-aromatic varieties on (AR_5 5′-TTGTTTGGAGCTTGCTGATG-3′ and AR_3 5′-ACCAGAGCAGCTGAAATAT-3′; AR_5 and NAR_3 5′-GGAGCAGCTGAAGCCATAATC-3′), and on the other hand the presence of the MITE (MITE_5 5′-GCAGACAAACTTAAAAACCGACT-3′ and MITE_3 5′ -TCAGAAATTTTATCATTTTTGTTACG-3′).
GenBank sequence submission
The sequence of the Azucena BAC clone 05K17 was registered under GenBank accession number EU155083. Sequences around the 12 bp sequence variation observed in Azucena were submitted for the following varieties: Arias, Khao- Kag_Xang, JC120 under the GenBank accession numbers: EU357907, EU357913 and EU357910, respectively; as well as for JC157, KDM105, Basmati370: EU357911, EU357912, and EU357908, respectively.
References
Ahn SN, Bollisch CN, Tanksley SD (1992) RFLP tagging of a gene for aroma in rice. Theor Appl Genet 84:825–828
Albar L, Bangratz-Reyser M, Hebrard E, Ndjiondjop MN, Jones M, Ghesquière A (2006) Mutations in the eIF(iso) 4G translation initiation factor confer high resistance of rice to Rice yellow mottle virus. Plant J 47:417–426
Amarawathi Y, Singh R, Sing AK, Singh VP, Mohapatra T, Sharma TR, Singh NK (2007) Mapping ofd quantitative trait loci for basmati quality traits in rice (Oryza sativa L.). Mol Breed. doi:10.1007/s11032-007-9108-8
Bautista NS, Solis R, Kamijima O, Ishii T (2001) RAPD, RFLP and SSLP analyses of phylogenetic relationships between cultivated and wild species of rice. Genes Genet Syst 76:71–79
Bradbury LM, Fitgerald TL, Henry RJ, Jin Q, Waters DLE (2005a) The gene for fragrance in rice. Plant Biotech J 3:363–370
Bradbury LMT, Henry RJ, Jin Q, Reinke RF, Waters DLE (2005b) A perfect marker for fragrance genotyping in rice. Mol Breed 16:279–283
Buttery RG, Ling LC, Juliano BO, Turnbaugh JG (1983) Cooked rice aroma and 2-acetyl–1-pyroline in rice. J Agric Food Chem 31:823–826
Chaparro C, Guyot R, Zuccolo A, Piegu B, Panaud O (2007) RetrOryza: a database of the rice LTR-retrotransposons. Nucleic Acids Res 35:D66–D70
Cordeiro GM, Christopher MJ, Henry RJ, Reineke RF (2002) Identification of microsatellite markers for fragrance in rice by analysis of the rice genome sequence. Mol Breed 9:245–250
Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA minipreparation: version II. Plant Mol Biol Rep 1:19–21
Edwards K, Johnstone C, Thompson C (1991) A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res 9:1349
Ewing B, Hillier L, Wendl MC, Greene P (1998) Base-calling of automated sequencer traces using phred I: accuracy assessment. Genome Res 8:175–185
Garland S, Lewin L, Blakeney A, Reineke R, Henry R (2000) PCR-based molecular markers for the fragrance gene in rice (Oryza sativa L.). Theor Appl Genet 101:364–371
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch SR (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169:1631–1638
Glaszmann JC (1987) Isozymes and classification of Asian rice varieties. Theor Appl Genet 74:21–30
Guiderdoni E, Galinato E, Luistro J, Vergara G (1992) Anther culture of tropical japonica x indica hybrids of rice (Oryza sativa L.). Euphytica 62:219S–224S
Jin Q, Waters D, Cordeiro GM, Henry RJ, Reineke RF (2003) A single nucleotide polymorphism (SNP) marker linked to the fragrance gene in rice (Oryza sativa L.). Plant Sci 165:359–364
Khush GS (1997) Origin, dispersal, cultivation and variation of rice. Plant Mol Biol 35:25–34
Konishi S, Izawa T, Lin SY, Ebana K, Fukuta Y, Sasaki T, Yano M (2006) An SNP caused loss of seed shattering during rice domestication. Science 312:1392–1396
Kovach MJ, Sweeney MT, McCouch SR (2007) New insights into the history of rice domestication. Trends Genet 11:578–587
Li C, Zhou A, Sang T (2006a) Rice domestication by reducing shattering. Science 311:1936–1939
Li C, Zhou A, Sang T (2006b) Genetic analysis of rice domestication syndrome with the wild annual species, Oryza nivara. New Phytol 170:185–194
Livingstone JR, Maruo T, Yoshida I, Tarui Y, Hirooka, Yamamoto Y, Tstui N, Hirasawa E (2003) Purification and properties of betaine aldehyde dehydrogenase from Avena sativa. J Plant Res 116:133–140
Londo JP, Chiang YC, Hung KH, Chiang TY, Schaal BA (2006) Phylogeography of Asian wild rice, Oryza rufipogon reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc Natl Acad Sci 103:9578–9583
Lorieux M, Petrov M, Huang N, Guiderdoni E, Ghesquière A (1996) Aroma in rice: genetic analysis of a quantitative trait. Theor App Genet 93:1145–1151
Ma J, Bennetzen JL (2004) Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci 101:12404–12410
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Scheider D, Carthinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
McCouch SR, Sweeney M, Li J, Jiang H, Thomson M, Septiningsih E, Edwards J, Moncada P, Xiao J, Garris A, Tai T, Martinez C, Thome J, Sugiono M, McClung A, Yuan LP, Ahn SN (2007) Through the genetic bottleneck: O rufipogon as a source of trait-enhancing alleles for O. sativa. Euphytica 154:317–339
Nieto C, Piron F, Dalmais M, Marco CF, Moriones E, Gomez-Guillamon ML, Truniger V, Gomez P, Garcias-Mas J, Aranda MA, Bendahmane A (2007) EcoTILLING for the identification of allelic variants of melon eIF4E, a factor that controls virus susceptibility. BMC Plant Biol 7:34
Noir S, Patheyron S, Combes MC, Lashermes P, Chalhoub B (2004) Construction and characterisation of a BAC library for genome analysis of the allotetraploid coffee species (Coffea arabica L.). Theor Appl Genet 109:225–230
Oka HI (1988) Weedy forms of rice. In: Origin of cultivated rice. Elsevier, Amsterdam, pp 107–114
Paule CM, Powers JJ (1989) Sensory and chemical examination of aromatic and non aromatic rices. J Food Sci 54:343–346
Petrov M, Danzart M, Giampaoli P, Faure J, Richard H (1996) Rice aroma analysis Discrimination between a scented and a non scented rice. Sci Aliments 16:347–360
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barell B (2000) Artemis: sequence visualization and annotation. Bioinformatics 16:944–945
Sakamoto, Murata (2002) The role of glycine betaine in the protection of plants from stress: clues from transgenic plants. Plant Cell Environ 25:163–171
Salse J, Bolot S, Throude M, Jouffe V, Piegu B, Masood U, Calcagno T, Cooke R, Delseny M, Feuillet C (2008) Identification and characterization of conserved duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell (in press)
Sang T, Ge S (2007a) Genetics and phylogenetics of rice domestication. Curr Opin Genet Dev 17:1–6
Sang T, Ge S (2007b) The puzzle of rice domestication. J Integr Plant Biol 49:760–768
Second G (1982) Origin of the genic diversity of cultivated rice (Oryza ssp.) study of the polymorphism scored at 40 isozymes loci. Jpn J Genet 57:25–57
Shen Y, Jiang H, Jin JP, Zhang ZB, Xi B, He YY, Wang C, Qian L, Li X, Yu QB, Niu HJ, Chen DH, Gao JH, Huang H, Shi TL, Yang ZN (2004) Development of genome-wide DNA polymorphism database for map-based cloning of rice genes. Plant Physiol 135:1198–1205
Shi W, Yang Y, Chen S. Xu M (2008) Discovery of a new frangance allele and the development of functional marker for the breeding of fragrant rice varieties. Mol Breed. doi:10.1007/s11032-008-9165-7 (in press)
Sonnhammer EL, Durbin R (1995) A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:1–2
Sood BC, Siddiq EA (1978) A rapid technique for scent determination in rice. Indian J Genet Plant Breed 38:268–271
Sweeney MT, McCouch S (2007) The complex history of the domestication of rice. Ann Bot 100:951–957
Sweeney MT, Thomson MJ, Pfeil BE, McCouch S (2006) Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 18:283–294
Sweeney MT, Thomson MJ, Cho YG, Park YJ, Williamson SH, Bustamante CD, McCouch SR (2007) Global dissemination of a single mutation conferring white pericarp in rice. PLoS Genet 8:e133
Tang T, Lu J, Huang J, He J, McCouch SR, Shen Y, Kai Z, Purrugganan MD, Shi S, Wu C (2007) Genomic variation in rice: genesis of highly polymorphic linkage blocks during domestication. PLoS Genet 2:e199
Temnykh S, Park WD, Ayres N, Cartinhour S, Hauck N, Lipovich L, Cho YG, Isshii T, McCouch SR (2000) Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor Appl Genet 100:697–712
Tranchant C, Ahmadi N, Courtois B, Lorieux M, McCouch SR, Glaszman JC, Ghesquière A (2005) New resources and integrated map for IR64 × Azucena, a reference population in rice. Poster presented to generation challenge programme 2005 annual research meeting, Sept 29th-Oct 1st, 2005, Roma, Italy
Trossat C, Rathinasabapathi B, Hansdon AD (1997) Transgenically expressed betaine aldehyde dehydrogenase efficiently catalyzes oxidation of dimethylsulfonipropionaldehyde and omega-aminoaldehydes. Plant Physiol 113:1457–1461
Tuteja (2007) Mechanisms of high salinity tolerance in plants. Methods Enzymol 428:419–438
Vanavichit A, Yoshihashi T, Wanchana S, Areekit S, Saengsraku D, Kamolsukyunyong W, Lanceras J, Toojinda T, Tragoonrung S (2004) Positional cloning of Os2AP, the aromatic gene controlling the biosynthetic switch of 2-acetyl-1-pyrroline and gamma aminobutyric acid (GABA) in rice. In: Proceedings of the 1st international conference on rice for the future, Aug 31–Sep 3, 2004, Bangkok
Verbruggen N, Hua XJ, May M, Van Montagu M (1996) Environmental and developmental signals modulate proline homeostasis: evidence fort a negative transcriptional regulator. Proc Natl Acad Sci USA 93:8787–8791
Vitte C, Ishii T, Lamy F, Brar D, Panaud O (2004) Genomic paleontology provides evidence for two distinct origins of Asian rice (Oryza sativa L.). Mol Genet Genomics 272:504–511
Wanchana S, Kamolsukyunyong W, Ruengphayak S, Toojinda T, Tragoonrung S, Vanavichit A (2005) A rapid construction of a physical contig across a 4.5 cM region for rice grain aroma facilitates marker enrichment for positional cloning. Sci Asia 31:299–306
Weretilnyk E, Hanson AD (1990) Molecular cloning of a plant betaine-aldehyde dehydrogenase, an enzyme implicated in adaptation to salinity and drought. Proc Natl Acad Sci USA 87:2745–2749
Yoshihashi T, Huong NTT, Inatomi H (2002) Precursors of 2-acetyl-1-pyrroline, a potent flavour compound of an aromatic rice variety. J Agric Food Chem 50:2001–2004
Acknowledgments
This work was partly supported by the French Agency for Innovation (ANVAR). FB was financed by a postdoctoral fellowship from the French Research Ministry, and ET by ANVAR. We thank C. Mestres and his group (CIRAD-AMIS, Montpellier) for gas chromatography aroma evaluation, T. Matthieu for his technical help in the greenhouse and M. Rondeau. We wish to acknowledge R. Cooke, M. Laudié and C. Berger for the sequencing of the Azucena BAC clone as well as the Génopôle Languedoc-Roussillon.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Lizhong Xiong.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bourgis, F., Guyot, R., Gherbi, H. et al. Characterization of the major fragance gene from an aromatic japonica rice and analysis of its diversity in Asian cultivated rice. Theor Appl Genet 117, 353–368 (2008). https://doi.org/10.1007/s00122-008-0780-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-008-0780-9