
Abstract
The accumulation of senescent cells within tissues is a hallmark of the aging process. Senescent cells are also commonly present in many age-related diseases and in the cancer microenvironment. The escape of abnormal cells from immune surveillance indicates that there is some defect in the function of cytotoxic immune cells, e.g., CD8+ T cells and natural killer (NK) cells. Recent studies have revealed that the expression of programmed death-ligand 1 (PD-L1) protein is abundantly increased in senescent cells. An increase in the amount of PD-L1 protein protects senescent cells from clearance by the PD-1 checkpoint receptor in cytotoxic immune cells. In fact, the activation of the PD-1 receptor suppresses the cytotoxic properties of CD8+ T and NK cells, promoting a state of immunosenescence. The inhibitory PD-1/PD-L1 checkpoint pathway acts in cooperation with immunosuppressive cells; for example, activation of PD-1 receptor can enhance the differentiation of regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), and M2 macrophages, whereas the cytokines secreted by immunosuppressive cells stimulate the expression of the immunosuppressive PD-L1 protein. Interestingly, many signaling pathways known to promote cellular senescence and the aging process are crucial stimulators of the expression of PD-L1 protein, e.g., epigenetic regulation, inflammatory mediators, mTOR-related signaling, cGAS-STING pathway, and AhR signaling. It seems that the inhibitory PD-1/PD-L1 immune checkpoint axis has a crucial role in the accumulation of senescent cells and thus it promotes the aging process in tissues. Thus, the blockade of the PD-1/PD-L1 checkpoint signaling might be a potential anti-aging senolytic therapy.
Key messages
-
Senescent cells accumulate within tissues during aging and age-related diseases.
-
Senescent cells are able to escape immune surveillance by cytotoxic immune cells.
-
Expression of programmed death-ligand 1 (PD-L1) markedly increases in senescent cells.
-
Age-related signaling stimulates the expression of PD-L1 protein in senescent cells.
-
Inhibitory PD-1/PD-L1 checkpoint pathway suppresses clearance of senescent cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There are certain traits typically encountered in the aging process which are associated with functional disturbances of the immune system. For instance, a chronic low-grade inflammation, commonly called inflammaging, is a challenge for the maintenance of homeostasis in aged tissues [1]. The low-grade inflammatory state with aging has been linked with an accumulation of pro-inflammatory senescent cells into tissues [2]. The senescence-associated secretory phenotype (SASP) is not only associated with aging but also cells of the tissues with several aged-related diseases display the specific markers of the SASP state [3]. An acute or chronic inflammatory process without a counteracting anti-inflammatory response leads to destructive effects in affected tissues. Thus, there exist diverse anti-inflammatory/immunosuppressive mechanisms which are not only able to suppress inflammatory responses but can also enhance the resolution of the inflammatory state and trigger tissue repair processes. The NF-κB signaling pathway is a major inducer of pro-inflammatory responses [4] and its inhibition, e.g., via endotoxin tolerance [5] or an increased level of the heat-shock proteins HSP70 and HSP90 [6, 7], can suppress inflammatory responses and accordingly enhance immunosuppression. On the other hand, the immune system possesses an armoury of immunosuppressive cells, e.g., myeloid-derived suppressor cells (MDSC), regulatory T cells (Treg), and M2 macrophages, which secrete many immunosuppressive cytokines, such as IL-6, IL-10, IFN-γ, and TGF-β [8,9,10]. These immunosuppressive mediators act to impair the functions of many immune effector cells and thus promote immunosuppression which subsequently induces the immunosenescence state, i.e., a decline in the activity of the immune system is evident not only in cancer and chronic inflammatory diseases but also in the aging process [11,12,13].
Moreover, there are many inhibitory immune checkpoint proteins which provide a crucial mechanism for the maintenance of self-tolerance and the regulation of immune responses, e.g., in chronic inflammatory conditions. For example, the programmed cell death protein-1 (PD-1) and programmed death-ligand 1 (PD-L1) establish an important inhibitory checkpoint axis which has a crucial role in the suppression of T cell activation and thus it can alleviate many immune responses [14] (Fig. 1). In fact, the PD-1/PD-L1 pathway represents a co-inhibitory mechanism which inhibits the activation of the T cell receptor (TCR), thus suppressing antigen presentation via the major histocompatibility complex class II (MHC-II) proteins. Mizuno et al. [15] demonstrated that the binding of the PD-L1 protein to the PD-1 receptor on T cells primarily suppressed the TCR signaling pathway and thus it inhibited cytokine production by mouse CD4+ and CD8+ T cells. Currently, the inhibitory mechanisms still need to be clarified. Interestingly, the presence of immunosenescence with aging and during age-related diseases is associated with a significant decline in the numbers and functions of CD4+ and CD8+ T cells as well as in the activity of B, NK, and dendritic cells and tissue macrophages [13, 16, 17]. These age-related changes indicate that the function of the PD-1/PD-L1 signaling pathway might be enhanced during aging and in age-related diseases. In fact, it is known that the expression of the PD-L1 protein is robustly increased in aged tissues and senescent cells [18,19,20]. This may represent an important mechanism of underpinning both immunosenescence and many age-related immune deficiencies. Here, I will briefly elucidate the properties of the PD-1/PD-L1 checkpoint pathway and then examine in detail the age-related mechanisms promoting the activation of the PD-L1 checkpoint and its role in the aging process and age-related diseases.

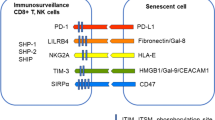
A schematic illustration on the function of PD-1/PD-L1 signaling. The membrane-bound PD-L1 protein activates the PD-1 receptor in a context-dependent manner. The activation of the PD-1 receptor in T, B, or NK cells induces functional disturbances, e.g., a decrease in cytotoxic activity, cellular exhaustion, and apoptosis. PD-1 signaling can also induce the differentiation of immune cells into immunosuppressive Tregs, MDSCs, and M2 macrophages. Moreover, immunosuppressive cells secrete many cytokines which further decrease the activity of the effector immune cells. Abbreviations: MDSC, myeloid-derived suppressor cell; NK, natural killer, PD-1, programmed cell death protein-1; PD-L1, programmed death-ligand 1
The immunosuppressive PD-1/PD-L1 checkpoint pathway
The inhibitory PD-1/PD-L1 checkpoint pathway confers a significant immunosuppressive protection in autoimmune diseases and many chronic inflammatory states although in cancers and fibrotic diseases it is associated with major detrimental effects [14, 21, 22]. The PD-1 receptor (CD279) is expressed on the surface of the activated T and B cells as well as on macrophages, monocytes, and innate lymphoid cells (ILC) including natural killer (NK) cells [14, 23,24,25] (Fig. 1). The PD-1 protein is a membrane-bound glycoprotein containing an extracellular IgV domain, a transmembrane sequence, and a cytoplasmic tail which contains a tyrosine-based inhibitory motif (ITIM) and a tyrosine-based switch motif (ITSM). The binding of the PD-L1 protein to the PD-1 receptor induces the phosphorylation of these two inhibitory motifs providing docking sites for the Src-homology-2 (SH2) protein phosphatases. Subsequently, the SHP-1 and SHP-2 phosphatases dephosphorylate the signaling components of the TCR and the costimulatory CD28 pathway, thus suppressing the activation, proliferation, and cytokine production of T cells [23, 26,27,28]. Stanford et al. [26] have reviewed the function of diverse protein tyrosine phosphatases (PTP) which are involved in the activation and inhibition of the TCR pathway thus regulating the immune homeastasis of T cells. The cytosolic SHP-1, SHP-2, and SHIP1 are major protein phosphatases suppressing TCR signaling associated with the inhibitory function of the PD-1/PD-L1 axis. It is known that PD-1/SHP-1 signaling was able to attenuate the cytotoxic activity of mouse CD8+ T cells [29]. Given that NK cells also display PD-1 signaling [25], it seems plausible that cancer cells with their extensive expression of PD-L1 proteins can exploit the PD-1/PD-L1 pathway to suppress the anticancer armoury of the CD4+ and CD8+ T cells and NK cells, thus escaping the surveillance by the immune system. For instance, Niu et al. [30] demonstrated that the expression of PD-1 protein was significantly increased in the NK cells of human lung cancer. These NK cells exhibited a decline in antitumor activity as compared to the PD-1-negative NK cells. Moreover, it is known that the PD-1 receptor robustly inhibited the activation of human B cells [31]. In 2001, Nishimura et al. [32] demonstrated that the PD-1 receptor-deficient mice developed dilated cardiomyopathy, an autoimmune disorder. Subsequent studies have revealed that many human immunodeficiency diseases are associated with single nucleotide polymorphism (SNP) on human PD-1 gene [33, 34] although the role of the CD-1 receptor in autoimmune diseases still needs to be clarified.
The PD-L1 (CD274 or B7-H1) protein is a type 1 integral membrane glycoprotein and it is expressed in several immune cells, especially in antigen presenting and immunosuppressive cells, as well as in many stromal cells, such as fibroblasts, epithelial and endothelial cells, pericytes, and smooth muscle cells [14, Human Protein Atlas]. There is also an extensive expression of PD-L1 protein in diverse cancer cells and in cancer-associated fibroblasts (CAF), i.e., the PD-1/PD-L1 axis is a major player in the maintenance of the immunosuppressive microenvironment in tumors [35, 36]. Dong et al. [37] cloned the human B7-H1/CD274 gene (PD-L1) from the human placenta cDNA library. They revealed that the B7-H1/PD-L1 protein did not bind to the CD28 protein, a crucial co-stimulator of T cells, but IL-2 interaction was required to induce T cell proliferation and secretion of immunosuppressive IL-10 cytokine. There also exists the PD-L2 protein (CD273 or B7-DC), mostly expressed in dendritic cells, but the regulation of the PD-1/PD-L2 axis has been less extensively studied than that of the PD-1/PD-L1 counterpart [38]. As discussed above, the PD-L1 protein acts as a ligand which activates PD-1 signaling in T cells and consequently leads to their exhaustion and even apoptosis (Fig. 1). In human NK cells, there are also several other checkpoint proteins, e.g., the HLA-E:CD94-NKG2A complex, which inhibit the immune surveillance by NK cells [39]. It is known that many inflammatory mediators, such as IFNγ, IL-6, IL-10, and TGF-β, are potent inducers of the expression of the PD-L1 protein, e.g., via epigenetic regulation [40, 41]. In inflammatory conditions, the PD-L1-positive cells can activate PD-1 signaling not only through the contact-dependent manner but also by secreting PD-L1-containing extracellular vesicles (EV) which subsequently activate PD-1 signaling in T cells [42]. This EV-mediated signaling is an especially important inducer of immunosuppression in tumors and chronic inflammatory diseases. The regulation of PD-L1 expression will be reviewed more thoroughly in the following sections with respect to cellular senescence and the aging process.
Interestingly, emerging studies have revealed that the PD-L1 protein can also trigger intrinsic effects not only in cancer cells but also in other PD-L1-containing cells, such as in fibroblasts [43,44,45]. The cell-intrinsic (either cytoplasmic or nuclear) signaling of the PD-L1 protein can regulate many cellular activities, e.g., cellular differentiation, autophagic flux, energy metabolism, and inflammatory responses [45, 46]. For instance, Guo et al. [44] demonstrated that the PD-L1 protein interacted with the Smad3 transcription factor and via β-catenin signaling induced the differentiation of human pulmonary fibroblasts into fibrogenic myofibroblasts. These investigators also reported that the expression of PD-L1 was clearly increased in the lungs of patients with idiopathic pulmonary fibrosis (IPF). It is known that the PD-1/PD-L1 signaling pathway has a crucial profibrotic role in the development of IPF disease [22, 47], although it is not known whether it is caused by intrinsic or extrinsic regulation. Interestingly, Gao et al. [48] demonstrated that the acetylation of the PD-L1 protein at Lys263 within its cytoplasmic tail regulated the translocation of the PD-L1 protein from the cell-surface into the nucleus. The acetylation of the PD-L1 protein by the p300 acetyltransferase blocked its translocation into the nucleus, whereas the deacetylation by histone deacetylase 2 (HDAC2) enhanced its nuclear translocation. The relocation of the PD-L1 protein into the nucleus was driven by clathrin-dependent endocytosis. Gao et al. [48] also revealed results indicating that the nuclear PD-L1 might trigger the expression of those genes which are involved in the immune-response pathways and thus could facilitate the immune evasion of tumor cells. They also reported that the inhibition of the PD-L1 nuclear translocation enhanced the efficacy of the PD-1 blockade therapy.
The PD-1/PD-L1 signaling pathway is also able to enhance the differentiation of immune cells towards the immunosuppressive state (Fig. 1). For instance, there is robust evidence that an increase in PD-1 signaling can promote the differentiation of CD4+ T cells into immunosuppressive Treg cells in diverse experimental models [49, 50]. It seems that cancer chemotherapy with the anti-PD-L1 antibody can alleviate both the inhibition of T cells and suppress the differentiation of Tregs. Interestingly, the expression level of PD-1 receptor controls the polarization of macrophages, i.e., a deficiency of PD-1 expression promoted the development of the pro-inflammatory M1 phenotype, whereas its overexpression enhanced the appearance of the immunosuppressive M2 phenotype, especially in tumor-associated macrophages (TAM) [51, 52]. Wei et al. [52] reported that the M2 polarization was induced via the activation of the ERK/AKT/mTOR signaling pathway in human macrophages. TAMs have an important role in the maintenance of an immunosuppressive state in the tumor microenvironment. In addition, it is known that PD-1 signaling enhanced the proliferation and immunosuppressive activity of MDSC, especially in the microenvironment of tumors [53] and sepsis [54]. It seems that the PD-L1-induced activation of PD-1 enhances immunosuppression either by suppressing effector immune cells, such as T, B, and NK cells, or by stimulating the differentiation of immunosuppressive Tregs, MDSCs, and M2 macrophages (Fig. 1). On the other hand, immunosuppressive cells secrete many cytokines, such as IL-6, IL-10 and TGF-β [55, 56], which are potent inducers of the expression of PD-L1, i.e., the PD-1/PD-L1 checkpoint axis acts in cooperation with immunosuppressive cells (Fig. 2).

The role of PD-1/PD-L1 signaling in the accumulation of senescent cells with aging. Several aging-associated signaling pathways can stimulate the expression of PD-L1 protein which via PD-1/PD-L1 signaling suppresses immune surveillance of senescent cells and thus promotes their accumulation within aging tissues. The collaboration with immunosuppressive cells also enhances immunosenescence in aged tissues and thus helps senescent cells to evade immune surveillance and subsequent elimination. Abbreviations: AhR, aryl hydrocarbon receptor; cGAS-STING, cyclic GMP-AMP synthase stimulator of interferon response cGAMP interactor; mTOR, mechanistic target of rapamycin
The aging process upregulates the expression of the PD-L1 protein
Aging tissues
There is abundant evidence that the aging process is associated with the activation of an immunosuppressive network [10, 57]. The numbers of MDSCs, Tregs, and M2 macrophages increase not only in the secondary lymphoid organs but also in many peripheral tissues [58,59,60,61,62]. However, less is known about the expression of PD-1/PD-L1 components in aging tissues although it represents a potent immunosuppressive mechanism in many pathological conditions. Several investigators have reported that the expression levels of PD-1 and PD-L1 proteins were robustly increased in many tissues with aging, e.g., bone marrow, heart, kidney, liver, lung, cerebellum, and spleen [18,19,20, 63, 64]. Currently, the cellular source of the age-related increase in the expression of PD-L1 protein needs to be clarified in different tissues although Yousefzadeh et al. [2] reported that the expression levels of the p16 and p21, markers of cellular senescence, were significantly increased in the brain, heart, kidney, liver, lung, and spleen in aged mice.
Cellular senescence
There are several hallmarks of cellular senescence, e.g., irreversible cell-cycle arrest, impaired proteostasis and energy metabolism, and the SASP state with the secretion of many inflammatory mediators, such as cytokines and chemokines [65]. Interestingly, senescent cells also secrete exosomal vesicles which have immunosuppressive properties, e.g., carrying the membrane-bound PD-L1 proteins [66]. In their seminal study, Onorati et al. [18] demonstrated that the expression of PD-L1 protein was robustly upregulated in human IMR90 and BJ fibroblasts after the cells were exposed to different inducers of cellular senescence. They also reported that the induction of PD-L1 expression was mediated by secreted inflammatory factors via the activation of the JAK/STAT3 signaling pathway. Accordingly, Wang et al. [20] demonstrated that PD-L1-positive senescent cells accumulated into mouse lung, liver, and kidney with aging. Single-cell transcriptome analysis revealed that the expression of PD-L1 correlated with an increased level of inflammatory factors in senescent cells.
Given that the accumulation of senescent cells promotes the aging process, it is clearly important to clarify whether treatment with an anti-PD-L1 antibody would be able to enhance the elimination of senescent cells in aging tissues. Wang et al. [20] demonstrated in vitro that those mouse pulmonary fibroblats which expressed a high level of PD-L1 protein were more resistant to the surveillance of cytotoxic CD8+ T cells than their PD-L1-negative counterparts. This confirms that the PD-L1-positive senescent fibroblasts were able to suppress the activity of cytotoxic PD-1-positive T cells and thus evaded immune clearance. Wang et al. [20] also revealed that the treatment of aging mice with an anti-PD-1 antibody reduced the numbers of both senescent cells and PD-L1-positive cells in mouse liver indicating that senescent cells were not able to evade immune elimination after immunotherapy.
On the other hand, it is known that the expression of PD-1 protein increases in senescent human T cells and thus it exposes them to the exhaustion/apoptosis by the PD-L1-positive stromal/immune cells [67, 68]. For instance, Kasamatsu et al. [68] demonstrated that sublethal doses of chemotherapeutic agents induced a senescent phenotype in human T cells and significantly increased the expression of PD-1 protein in these cells. Interestingly, it seems that with aging or in age-related diseases, senescent stromal cells possess a dual defence against immune elimination, i.e., an increased expression of PD-L1 protein improves their immunodefence against cytotoxic immune cells and moreover, the senescence/exhaustion of T cells with aging increase their expression of PD-1 protein which exposes them to the elimination by PD-L1-positive senescent stromal cells. Currently, it seems that in cancer therapies there are no significant differences between young adults and elderly people in the efficacy of the blocking treatments utilizing PD-L1 and PD-1 antibodies [69].
PD-1/PD-L1 signaling is augmented in many age-related diseases
The age-related diseases are progressive disorders which commonly involve both inflammatory changes and fibrotic degeneration. Typically, these diseases also are associated with the accumulation of senescent cells with inflammatory and degenerative changes, such as in atherosclerosis, chronic obstructive lung disease (COPD), coronary artery disease (CAD), and Alzheimer’s disease (AD) [70,71,72,73] as well as in fibrotic diseases, e.g., in idiopathic pulmonary disease (IPF), cardiac fibrosis, and systemic sclerosis [70, 74, 75]. There exists a significant crosstalk between senescent cells and inflammatory cells in the pathogenesis of many age-related diseases, e.g., via the control of the expression of inhibitory checkpoint proteins. For instance, the PD-1/PD-L1 axis has an important role in the pathogenesis of human AD and COPD [76,77,78]. Kummer et al. [79] demonstrated that the expression of PD-L1 was robustly increased in the brain of transgenic AD mice with clear positive staining in the astrocytes surrounding amyloid-β plaques. Moreover, a deficiency of microglial PD-1 protein increased the inflammatory response and the deposition of amyloid-β plaques within the brain. Given that inflammatory mediators are potent inducers of the expression of PD-L1 protein, it seems that in many age-related diseases, the activation of PD-1/PD-L1 signaling is related to defence against chronic inflammation. The pro-inflammatory SASP state of senescent stromal cells maintains the inflammatory microenvironment and aggravates the pathology of age-related diseases. It seems plausible that the induction of PD-L1 expression in senescent cells has a crucial role in the pathogenesis of chronic age-related diseases since it promotes immunosenescence and prevents the clearance of senescent cells.
Fibrosis in cardiac muscle, kidney, liver, and lung as well as in cases of systemic sclerosis are common age-related fibrotic diseases. The differentiation of tissue fibroblasts into fibrogenic myofibroblasts and an increase in the numbers of senescent fibroblasts are the major hallmarks of fibrotic lesions [75, 80]. Emerging studies have revealed that the function of the PD-1/PD-L1 axis has a crucial role in the accumulation of fibrotic lesions [22, 47]. There are a number of investigations indicating that in mouse and human fibrotic tissues, there exists a robust increase in the expression of PD-L1 protein not only in fibroblasts but also in endothelial and epithelial cells [18, 22, 44, 47, 81]. Onorati et al. [18] utilized a single-cell transcriptome technique to determine in human interstitial lung fibrosis that the high expression of PD-L1 protein was localized into senescent cells which also displayed a proinflammatory phenotype. There are studies indicating that the PD-L1 protein might promote fibrosis by enhancing the differentiation of tissue fibroblasts into fibrogenic myofibroblasts. For example, Guo et al. [44] demonstrated that the PD-L1 protein was required for the TGF-β-induced differentiation of myofibroblasts both from human primary lung fibroblasts or fibroblasts isolated from the IPF patients. They revealed that TGF-β exposure induced the interaction between the PD-L1 protein and the SMAD3 transcription factor to induce the transcription of α-smooth muscle actin (α-SMA). It seems that the PD-L1 protein can enhance tissue fibrosis via some form of intrinsic regulation. There are several reports that immunosuppressive CAFs express a high level of PD-L1 protein [82, 83]. For instance, Li et al. [82] demonstrated that CAFs were able to promote the expression of PD-L1 protein in mouse cancer cells by secreting the CXCL5 chemokine. Given that the inhibitors of PD-1/PD-L1 signaling are effective in many cancers, the checkpoint inhibitors may also represent promising therapeutic choices for treating fibrotic diseases [84]. Currently, there are some animal studies in experimental disease models which have revealed that the PD-1/PD-L1 inhibitors are able to attenuate the severity of the fibrosis [85, 86].
The aging-associated signaling stimulates the expression of the PD-L1 protein
Given that the PD-1/PD-L1 pathway has an important role in the generation of an immunosuppressive microenvironment in tumors, there has been a significant interest in examining the regulation mechanisms underpinning the expression of the PD-L1 protein [43, 87, 88]. It seems that there exists a complex network of regulatory processes from epigenetic to post-transcriptional mechanisms which not only control the expression level of PD-L1 but also the resistance to the PD-1/PD-L1 blocking therapies. Currently, the signaling mechanisms which regulate the age-related increase in the expression of PD-L1 still need to be clarified although many of them are known to be associated with the regulation of the aging process. Next, I will briefly examine some of those signaling pathways and emphasize that the upregulation of the PD-1/PD-L1 pathway seems to be involved in the regulation of the aging process (Fig. 2).
Epigenetic regulation
There is robust evidence from cancer studies that different forms of epigenetic regulation, such as DNA methylation and histone modifications, control the chromatin landscape around the PD-L1 gene and affect its transcription [89, 90]. For instance, the PD-L1 gene contains many CpG methylation sites on its 5’-untranslated region as well as on exon 1. The hypomethylation of the CpG clusters on the promoter regions of the PD-L1 gene is a major hallmark encountered in different tumors, e.g., in melanoma and colorectal cancer [91, 92]. It is generally recognized that a high methylation level of the PD-L1 promoter is associated with a low expression of PD-L1. For example, exposure to 5-azacytidine, an inhibitor of DNA methyltransferase, increased the degree of hypomethylation and elevated the expression of the PD-L1 gene [93]. However, there are reports indicating that certain CpG loci in the PD-L1 promoter were actually hypermethylated in some cancers, e.g., in advanced gastric cancer [94], thus it seems that not all methylation sites are involved in the regulation of PD-L1. It is not only in cancers where it is believed that DNA methylation controls the expression of the PD-L1 gene. For instance, Chang et al. [95] demonstrated that the demethylation of the PD-L1 promoter enhanced the expression of PD-L1 and consequently inhibited the experimental autoimmune encephalomyelitis in mice.
The regulation of histone acetylation and deacetylation is another epigenetic mechanism which regulates the transcription of the PD-L1 gene and thus it is an important therapeutic target [96]. The acetylation of histones facilitates the opening of chromatin and consequently enhances the transcription of genes. Histone acetyltransferases (HAT) increases the acetylation of histones, whereas HDACs remove acetyl groups from histones, thus decreasing transcription. Wang et al. [97] demonstrated that the activation of JNK/c-Jun signaling stimulated the expression of PD-L1 in diverse drug-resistant cancer cell lines. Subsequently, they revealed that the accumulation of c-Jun protein inhibited the expression of HDAC3 and thereby increased the acetylation of histone H3 on the PD-L1 promoter in cancer cells. Accordingly, the overexpression of HDAC3 reduced H3 acetylation and inhibited the expression of PD-L1. There are many studies indicating that the inhibition of HDACs increased the expression of PD-L1 protein in human and murine cancer cells and consequently augmented the efficacy of anti-PD-L1 therapy, e.g., in B-cell lymphomas [98] and mouse experimental tumor models [99]. Currently, certain HDAC inhibitors are clinically used as anticancer agents. Moreover, microRNAs (miR) are important players which can control the expression level of PD-L1 [100] and subsequently inhibit tissue immunosuppression. Wang et al. [100] categorised a number of miRs which can directly or indirectly inhibit the expression of PD-L1.
The remodelling of the chromatin landscape is a hallmark of both cellular senescence and the aging process [101,102,103]. Currently, there is some debate whether this represents a cause or consequence of the aging process. Shortly, the following changes have been observed, (i) an increase in global DNA hypomethylation although certain CpG islands may be hypermethylated, (ii) a reduction in the level of heterochromatin and nucleosomes, and (iii) clear changes in histone structure and epigenetic marks. Benayoun et al. [64] demonstrated that the age-related increase in an inflammatory phenotype was associated with both epigenetic remodelling and changes in tissue transcriptome profiles. For instance, many genes of the immune pathways displayed an enrichment of the activating epigenetic marks, such as greater intensities of the H3K4me3 and H3K27ac, and accordingly an increase in their mRNA expression. Interestingly, the IFNγ response, a major inducer of PD-L1 transcription [40], displayed an age-related increase in activating epigenetic marks around the gene as well as an increased transcription in mouse heart, liver, and cerebellum. Erbe et al. [104] surveyed the multi-omics databases focusing on the age-related transcription of immune checkpoint genes, also that of the PD-L1 gene. They reported that an increased transcription of the PD-L1 gene was associated with a clear decrease in the degree of promoter methylation. Currently, the aging-associated changes in different CpG loci in the promoter of the PD-L1 gene need to be clarified. However, it seems that an age-related increase in the expression of PD-L1 and accordingly the enhanced tissue immunosuppression can be partly attributed to the global DNA hypomethylation that takes place with aging.
Inflammatory mediators
The aging process is associated with an accumulation of senescent cells as well as an increase in pro-inflammatory changes, not only in the organs of immune system but also in peripheral tissues. As described above, Benayoun et al. [64] demonstrated that the age-related inflammatory signature in many tissues was associated with epigenetically driven alterations in the transcription of inflammatory genes involving cytokines, chemokines, complements, and the response to interferons. Moreover, the age-related changes were very similar in the tissues of mice, rats, and humans. Interestingly, recent single-cell transcriptome experiments on aged tissues have revealed that the inflammatory response is induced not only by infiltrating immune cells but that there is also a robust increase in the markers of the inflammatory phenotype in the stromal cells of the tissues, such as fibroblasts, endothelial, and epithelial cells [105,106,107,108]. Given that most of the inflammatory markers are probably derived from senescent cells, many single-cell transcriptome screening trials have examined the coordination of different type of senescent genes (SnG), e.g., cell-cycle inhibitors and the SASP genes, in aging tissues of human and mice [109, 110]. For instance, Xu et al. [110] reported that many SnGs were enriched in distinct clusters in several human tissues. Fibroblasts were the most prominent senescent cell type across the tissues although several other stromal cells displayed many properties of senescence. In particular, diverse cytokine regulators, e.g., the interferon regulator STAT3 and the cytokine regulator RelA from the NF-κB pathway, were enriched with common cell cycle inhibitors in the examined aging tissues. Currently, the high-throughput screening and multiomic studies have tended to focus on assessing the common age-related alterations rather than examining more specific immune pathways.
Currently, it is known that a diverse set of inflammatory mediators, associated both with acute and chronic inflammatory conditions, are able to stimulate the expression of PD-L1. However, it seems that the NF-κB and JAK/STAT pathways are the major inflammation-related inducers of transcription of the PD-L1 gene [21, 40, 111,112,113]. For instance, many studies have revealed that IFN-γ is a major inducer of PD-L1 and PD-L2 expression via the JAK/STAT pathways in several cancer models [40, 111]. Garcia-Diaz et al. [111] demonstrated that the STAT1/3 signaling targeted the interferon regulatory factor 1 (IRF1) which subsequently transactivated the promoter of the PD-L1 gene in human melanoma cells. Not only cytokines, such as IL-1β, IL-17, TNF-α, and GM-CSF, but also reactive oxygen species (ROS), UV irradiation, and lipopolysaccharide triggered the expression of PD-L1 via NF-κB signaling under many experimental conditions [114,115,116,117,118,119]. Recently, Lu et al. [120] revealed that the NLRP3 inflammasome, which is primed by NF-κB signaling, upregulated the expression of PD-L1 in mouse lymphoma. They also reported that a blockade of NLRP3 signaling (i) inhibited the expression of PD-L1, (ii) reduced the level of immunosuppressive cells, and (iii) subsequently blocked the growth of mouse lymphoma. Moreover, it is known that the non-canonical RelB pathway of NF-κB signaling increased the expression of PD-L1 protein and allowed mouse prostate cancer cells to evade surveillance by the immune system [121]. The complement system might also be involved in the regulation of PD-L1 expression. For example, An et al. [122] demonstrated that treatment of human monocytes with the complement factor C5a induced the expression of PD-L1 protein which was mediated via the activation of the complement receptor C5aR1. Accordingly, Zha et al. [123] reported that the inhibition of C5aR signaling promoted the efficiency of PD-1/PD-L1 inhibitor therapies in mouse tumors. These studies indicate that diverse pro-inflammatory treatments are able to stimulate the expression of PD-L1 and thus evoke tissue immunosuppression through the PD-1/PD-L1 pathway.
It is known that TGF-β and prostaglandin E2 (PGE2) are important immunosuppressive factors and enhancers of immune suppressor cells, such as MDSCs and M2 macrophages. Interestingly, there are studies indicating that PGE2 and TGF-β are also potent inducers of PD-L1 expression [124, 125]. For example, Kang et al. [125] demonstrated that TGF-β exposure not only stimulated the expression of PD-L1 in human fibroblasts but it also triggered an exosomal secretion of PD-L1 proteins. They also reported that TGF-β treatment induced the transactivation of the PD-L1 gene either via the SMAD2/3 pathway or through YAP/TAZ signaling. There are clinical observations indicating that the simultaneous inhibition of TGF-β and PD-1/PD-L1 signaling conferred synergistic benefits in cancer therapy [126]. Prima et al. [124] revealed that PGE exposure increased the expression level of PD-L1 in mouse macrophages and accordingly enhanced their immunosuppressive activity in tumor microenvironment. Interestingly, it is known that the synthesis of PGE2 was significantly upregulated in the macrophages isolated from aged mice, a phenotype which was attributable to an increase in the expression of cyclo-oxygenase 2 (COX-2) [127]. Moreover, Li et al. [128] demonstrated that the expression levels of COX-2 and PGE synthase 1 (PTGES1) were robustly augmented in the skin fibroblasts of aged humans, concurrently with an increased concentration of PGE2. These observations might be connected to the accumulation of senescent cells and the immunosenescence found in the aged skin. In summary, there is abundant evidence that many pro-inflammatory factors stimulate the expression of PD-L1 protein and that this property can even be promoted by immunosuppressive mediators, such as PGE2 and TGF-β.
mTOR-related signaling
The mechanistic target of rapamycin (mTOR) is a key protein kinase which regulates a number of vital cellular functions involving, e.g., cell growth and survival, protein synthesis, protein degradation via autophagy, metabolism, and many immune responses [129, 130]. Thus, it is generally believed that mTOR signaling is a major regulator of the aging process and many age-related diseases [131]. It is known that (i) mTOR signaling inhibits autophagy and (ii) autophagic activity declines with aging, i.e., cleansing of cellular components is reduced with aging which disturbs the maintenance of homeostasis [132, 133]. However, currently this paradigm contains many open questions since both mTOR signaling and autophagy are heavily regulated upstream and furthermore they are driving many diverse functions which seem to be cell-type specific. With respect to the upstream regulation, many growth factors, such as insulin and IGF-1, and many cytokines are connected to the PI-3 K/AKT/mTOR signaling pathway which can be inhibited by the PTEN protein phosphatase. This pathway is a key regulator of mTOR-driven immune responses, such as the activation and differentiation of T and B cells as well as the antigen presenting cells [130] and moreover, mTOR signaling has a crucial role in the development of immune evasion in the tumor microenvironment [134]. In addition, there exist two kinds of autophagy, i.e., a nonselective bulk macroautophagy and a chaperone-assisted selective autophagy for specific targets ranging from distinct molecules to whole organelles [135]. For instance, it is known that selective autophagy controls many innate immune responses [136].
There is evidence emerging from different experimental models that the inhibition of autophagy by mTOR signaling can stimulate the expression level of PD-L1 protein [137,138,139]. The early observations indicated that the activation of the PI-3 K/AKT/mTOR signaling pathway induced the expression of PD-L1. For instance, Parsa et al. [140] demonstrated that the activation of the PI-3 K/AKT/mTOR pathway by inhibiting the PTEN phosphatase stimulated the expression of PD-L1 protein in glioma cells. They also revealed that the level of PD-L1 protein increased through translational regulation and consequently enhanced the extent of immunoresistance in an experimental mouse glioma. Subsequently, Wang et al. [137] made a seminal observation indicating that the inhibition of autophagy, either with pharmacological inhibitors or after knockout of autophagic genes, robustly increased the expression of PD-L1 in mouse and human gastric cancer cells. They also reported that the induction of autophagy by rapamycin, an inhibitor of mTOR, reduced the expression levels of PD-L1 in cancer cells. This indicates that mTOR signaling inhibits autophagy and consequently augments the expression of PD-L1 protein and conversely the activation of autophagy decreases the level of PD-L1 protein. Wang et al. [137] also demonstrated that the inhibition of autophagy induced the accumulation of p62/SQSTM1 protein, a common receptor of selective autophagy [141], indicating that the PD-L1 protein is degraded by a process of selective autophagy. This is an interesting observation since a decline in the autophagic flux with aging might explain why the aging process is associated with an increase in the expression of PD-L1.
For many years, it has been known that the PD-L1 protein also possesses many cell-intrinsic, nonimmune functions [43, 142]. For example, Gao et al. [143] demonstrated that the PD-L1 protein activated the AKT/mTOR signaling pathway and increased the proliferation of human ovarian cancer cells. Accordingly, Clark et al. [144] reported that the PD-L1 protein mediated a suppression of autophagy via a cell-intrinsic, immune-independent pathway in murine ovarian cancer and melanoma cells. They revealed that the PD-L1 of tumor cells promoted mTOR signaling which inhibited autophagic flux and thus maintained the expression of PD-L1 protein upregulated in tumor cells. However, it seems that there are some cell-type dependent regulatory differences in this bidirectional interplay between the PD-L1 protein and the phenomenon of autophagy [43, 142].
cGAS-STING signaling
The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) is a cytoplasmic sensor for double-stranded DNA breaks which can originate from either nuclear, mitochondrial, or microbial DNA [145, 146]. The primary cause of aging is still unknown although many investigators have suggested that a loss of DNA integrity, e.g., genome instability, telomere attrition, and dysfunctions in mitochondrial DNA (mtDNA), is a hallmark of the aging process [147]. There is robust evidence that the activation of cGAS-STING signaling is associated with cellular senescence and inflammatory states driven by DNA damage [148,149,150]. Recently, Gulen et al. [151] demonstrated that cGAS-STING signaling promoted both age-related inflammation and neurodegeneration in mice. They also reported that the inhibition of STING activity blocked the inflammatory SASP response in cultured senescent human fibroblasts. Next, they revealed that if old mice were administered an inhibitor of STING activity, this significantly attenuated the inflammatory signature in their brain, kidney, and liver. Gulen et al. [151] demonstrated that the levels of the phosphorylated STING, a marker of its activation, were enriched in the hippocampus of aged mice, especially in microglia. They also reported that mtDNA triggered the activation of cGAS-STING signaling in aged microglia and thus induced an inflammatory state in the aged mouse brain.
It is known that STING signaling stimulates the activation of IRF3 and NF-κB signaling pathways as well as promoting autophagic degradation [145, 146]. There is clear evidence from many clinical experiments that the activation of STING (i) induced the expression of PD-L1 protein, (ii) inhibited T cell function, and (iii) promoted immune evasion in many clinical experiments [152, 153]. For instance, Cheng et al. [152] demonstrated that oxidative stress in cancer cells activated the mitochondrial Lon protease, subsequently promoting the release of mtDNA into the cytoplasm where it activated cGAS-STING signaling and stimulated the expression of PD-L1 protein. Interestingly, they revealed that an increased Lon activity in M2 macrophages promoted the secretion of EVs which carried a cargo of mtDNA and the PD-L1 protein capable of inhibiting the function of T cells. Moreover, it is known that ionizing radiation therapy triggered the activation of cGAS-STING signaling and induced the upregulation of PD-L1 expression in the cell-surface of human and mouse liver cancer cells and subsequently it enhanced immune evasion in mouse tumor model [153]. On the other hand, Lee et al. [154] demonstrated that via its intrinsic regulation, the nuclear PD-L1 protein inhibited the transcription of STING protein by binding to the promoter region of the STING gene in cancer cells. Accordingly, silencing of PD-L1 expression induced a robust upregulation of STING expression and subsequently inducing cellular senescence of mouse cancer cells. It seems that there is a negative feedback regulation between the cGAS-STING and PD-L1 signaling pathways and an increase in PD-L1 expression with aging might inhibit autophagy and aggravate the maintenance of genome integrity.
AhR signaling
The aryl hydrocarbon receptor (AhR) is an evolutionarily conserved, ligand-regulated transcription factor which is not only a major sensor of environmental toxins but it is also activated by a number of metabolites ranging from heme, arachidonic acid, and kynurenine pathways as well as indoles from microbiota [155, 156]. AhR signaling cooperates closely with indoleamine 2,3-dioxygenase 1 (IDO1), a common immune checkpoint, in the regulation of the aging process [157, 158]. For instance, AhR signaling can remodel the immune system by stimulating immunosuppressive changes, e.g., through the induction of FoxP3 and the differentiation of Tregs [157, 159]. Interestingly, the promoter of the PD-L1 gene contains several response elements capable of binding the AhR factor (AhRE) [160]. Kenison et al. [160] demonstrated that the activation of AhR signaling by TCDD exposure transactivated the PD-L1 gene in cancer cells, whereas the AhR knockout robustly reduced the induction of the PD-L1 gene. It has been demonstrated in many experimental models that AhR signaling can stimulate the expression of PD-L1 protein [161,162,163]. For example, Wang et al. [161] revealed that cigarette smoke, a potent activator of AhR signaling, induced in vivo the expression of PD-L1 protein in the epithelial cells of mouse lungs. They also verified that the induction of PD-L1 was caused by AhR signaling. Moreover, Wang et al. [161] reported that the inhibition of AhR signaling potentiated the therapeutic response induced with anti-PD-L1 antibody treatment in mouse lung cancer model. Tryptophan metabolites, e.g., compounds produced via the kynurenine, indole, serotoinin, or tryptamine pathways, are activators of AhR signaling as well as immune modulators [156, 164]. Currently, there is clear evidence that the gut microbiota can modulate the efficacy of anti-PD-1/PD-L1 therapy both in humans and mice [165]. The gut microbiota generates several indole derivatives which are potent agonists of the AhR protein in many tissues via the circulation [156]. For example, oxidative stress triggered the formation of 2-oxindole which stimulated the expression of PD-L1 in human keratinocytes [162]. It does seem that the robust expression of the AhR protein in the barrier tissues, such as vascular tissues, has an important role in the regulation of the aging-associated immunosuppression and immunosenescence [157].
The anti-aging signaling suppresses the expression of the PD-L1 protein
Currently, there is a debate if it will be possible to develop anti-aging therapies which might increase health span and even extend the lifespan of elderly humans. Recently, Guarente et al. [166] summarized the present clinical knowledge and ongoing trials on the eight most promising drug candidates for anti-aging medicines. Two of these drugs, i.e., metformin and rapamycin, are clinically utilized drugs which have been able to extend the lifespan of rodents [167, 168]. Metformin, a drug used in the clinical treatments of type 2 diabetes, has multiple targets in the body, e.g., it activates the AMP-activated kinase (AMPK) which controls many aging-associated signaling pathways [169]. Interestingly, metformin has displayed diverse therapeutic properties in many immune-mediated diseases [170] and also alleviated many of the hallmarks of cancers [171]. For instance, metformin exposure inhibited the function of immunosuppressive MDSCs in tumor-bearing mice [172]. This is probably induced by the activation of AMPK signaling which is known to inhibit the functions of MDSCs [173]. There is abundant evidence that metformin can reduce the expression of PD-L1 protein through diverse mechanisms, although mainly via AMPK signaling [174,175,176]. Cha et al. [174] demonstrated that metformin via AMPK activation phosphorylated the PD-L1 protein at the Ser195 site in cancer cells. The phosphorylation of the PD-L1 protein induced its glycosylation and consequently proteasomal degradation via the endoplasmic-reticulum-associated degradation (ERAD) pathway. Cha et al. [174] also reported that the metformin-induced downregulation of the amount of PD-L1 protein present in cancer cells enhanced their surveillance by cytotoxic T lymphocytes. They also verified that metformin exposure reduced the level of PD-L1 in human tumors. It seems that there are other mechanisms which metformin can exploit to inhibit the expression of PD-L1 protein. Lu et al. [175] revealed that metformin treatment inhibited the expression of PD-L1 by suppressing the IL-6/JAK2/STAT3 pathway in human esophageal squamous carcinoma cells. This inhibition was an AMPK-independent process. However, it seems likely that many metformin-induced responses are mediated by AMPK-induced autophagy, either by inhibiting mTOR or activating ULK1 signaling [177].
Rapamycin, an inhibitor of mTOR signaling, is a potent enhancer of autophagy. As discussed above in the section ‘mTOR-related signaling’, the signaling pathways via the activation of mTOR stimulate the expression of PD-L1 protein in many experimental conditions. Given that rapamycin has been able to extend the lifespan of rodents [167], the inhibitors of mTOR protein, such as rapalogs, have been claimed to be promising candidates for anti-aging therapeutics [178]. Khedri et al. [179] demonstrated that exposure of mice to rapamycin significantly reduced the expression of PD-L1 in the brain but not in the spleen. They proposed that rapamycin might have beneficial effects on CNS autoimmune diseases, such as those encountered in multiple sclerosis [180]. Onorati et al. [18] reported that rapamycin exposure was able to downregulate the expression of PD-L1 protein, especially on the cell surface of senescent human fibroblasts. Rapamycin treatment also reduced the expression of IL-6 in senescent cells, an effect associated with a decline in the SASP state. This is an interesting observation since a decrease in the expression of PD-L1 in senescent cells might enhance their immune surveillance and thus rapamycin could possess senolytic properties.
Traditional medicine and phytochemicals have long been exploited in health care for diverse diseases and even as anti-aging remedies. Many clinically used drugs, such as aspirin and metformin, have their roots in herbal medicine. Phytochemicals, mostly polyphenols, are multitarget molecules, like metformin, and they affect many age-related signaling pathways. In fact, it is known that several phytochemicals possess immunomodulatory properties [181]. For instance, there is clear evidence that phytochemicals can improve the efficacy of many cancer therapies based on PD-1/PD-L1 blockade [182, 183]. Currently, the molecular mechanisms need to be clarified before phytochemicals can be administered as checkpoint inhibitor therapies. Guo et al. [184] revealed in in silico experiments that several polyphenols, such as curcumin and resveratrol, were able directly to inhibit the interaction between the PD-1 and PD-L1 proteins. Accordingly, Jing et al. [185] reported that quercetin also inhibited the interaction between the PD-1/PD-L1 proteins and thus promoted the ability of T cells to destroy human cancer cells. Phytochemicals can also enhance the efficacy of immunotherapy with PD-1/PD-L1 inhibitors by downregulating the expression of PD-L1 in cancer cells. For instance, Liu et al. [186] demonstrated that curcumin exposure suppressed the expression of PD-L1 and PD-L2 proteins in human head and neck squamous carcinoma cells (HNSCC). They also reported that curcumin potentiated the cytotoxic activity of CD8+ T cells when administered as a combination treatment with PD-L1 antibody. Recently, Liu et al. [187] reviewed the molecular mechanisms describing how different phytochemicals alter the function of the PD-1/PD-L1 pathway in tumor immunotherapy.
The PD-1/PD-L1 checkpoint signaling promotes aging-related immunosuppression and immunosenescence
There is convincing evidence that the expression of PD-L1 protein is significantly increased in many tissues with aging. This age-related response seems to be attributable to an accumulation of senescent cells which are known to exhibit a robust expression of the PD-L1 protein. This increased expression level of the PD-L1 protein indicates that senescent cells can evade immune clearance by inhibiting the activity of cytotoxic CD8+ T and NK cells. This might explain why senescent cells accumulate with aging and disturb the maintenance of tissue homeostasis. Senescent cells not only express the PD-L1 protein but they can also secrete EVs coated with PD-L1 proteins. It is known that the shedding of EVs is robustly increased from the senescent cells of diverse cell types, e.g., fibroblasts and endothelial cells [188, 189]. The secretion of EVs enhances the paracrine spreading of senescence into neighboring cells within tissues [190, 191]. Borghesan et al. [190] reported that the IFN-γ inducible transmembrane 3 protein (IFITM3) was able to increase the secretion of EVs and thus promote the paracrine senescence in aged tissues. Interestingly, Benayoun et al. [64] demonstrated that in mice, the gene transcription of the IFN-γ-driven genes was strongly enriched in different aging tissues. Moreover, Kang et al. [125] demonstrated that TGF-β exposure of human and mouse fibroblasts also induced the secretion of the PD-L1-coating EVs which inhibited the function of T cells. There is mounting evidence that the PD-L1-coated EVs, secreted either from cancer cells or CAFs, have a key role in the maintenance of immunosuppressive state in the microenvironment surrounding tumors [192, 193]. It is known that chronic inflammatory conditions also increase the secretion of immunosuppressive EVs [66]. It seems that an excessive expression of PD-L1 protein and their packaging into EVs in senescent cells not only induces an accumulation of senescent cells into aged tissues but also promotes the generation of immunosenescence with aging by inhibiting the functions of T and NK cells (Fig. 2).
The immunosenescence associated with aging involves a substantial decline in the functional properties of the immune system [13, 16, 194]. Furthermore, the immunosenescence present in tumors displays more or less similar alterations in immune cells as those encountered in the aging process [11]. Currently, it is known that the PD-1/PD-L1 checkpoint axis impairs the functions of T and B cells as well as NK cells and macrophages, inducing relatively similar changes as those observed in immunosenescence, i.e., an activation of the PD-1/PD-L1 pathway with aging might promote the aging of the immune system. However, it needs to be clarified whether other inhibitory immune checkpoints, such as CTLA-4, LAG-3, TIM-3, or VISTA, affect the age-related immune deficiency. In addition to the contact-mediated inhibition, immunosuppressive cells, such as MDSCs, Tregs, and M2 macrophages, secrete cytokines which evoke a decline in the functions of both adaptive and innate immunity [9, 13, 55, 58]. Interestingly, immunosuppressive cells are able to induce alterations in immune cells which are very reminiscent to those observed in the immunosenescent phenotype [13, 195]. In fact, it is known that the contact-dependent PD-1/PD-L1 checkpoints and immunosuppressive cells cooperate in pathological states since as described above, the activation of the PD-1 protein can enhance the differentiation of immunosuppressive cells, and vice versa, the cytokines secreted by immunosuppressive cells can stimulate the expression of the PD-1/PD-L1 proteins (Figs. 1 and 2).
Intriguingly, many crucial enhancers of the aging process are inducers of PD-L1 expression. Cellular senescence might represent the final outcome since many SASP components are important enhancers of PD-L1 expression, whereas an increased expression of PD-L1 inhibits the immune surveillance of senescent cells. Moreover, many inflammatory SASP factors, such as chemokines and complements, promote the recruitment of immunosuppressive cells into aged tissues, thus enhancing the development of an immunosuppressive microenvironment as well as a more systemic immunosenescence. It is likely that many inflammatory mediators strive to stimulate the PD/PD-L1 pathway in order to prevent excessive inflammatory injuries in tissues. Another interesting observation indicates that a decline in the autophagy stimulates the accumulation of PD-L1 proteins because the turnover of PD-L1 proteins is regulated by selective autophagy. In this situation, the signaling pathways activating mTOR signaling have a crucial role since they can also stimulate the expression of the immunosuppressive PD-L1 protein in non-senescent cells. Many investigators have claimed that mTOR signaling has a key role in the aging process. For example, activation of the insulin/IGF-1 pathway stimulates mTOR signaling and consequently can activate STAT3 signaling which promotes the expression of PD-L1 and enhances immunosuppression [196,197,198]. There is clear evidence that insulin/IGF-1 signaling can promote the aging process although it has critical functions in protein synthesis and energy metabolism [199]. Interestingly, current anti-aging therapies, i.e., metformin and rapamycin, are inhibitors of mTOR signaling and accordingly potent stimulators of autophagy. It seems that the decline in autophagy not only disturbs the cellular proteostasis and tissue homeostasis but via the activation of PD-L1 signaling, it can enhance the accumulation of senescent cells and thus promote the aging process.
Conclusions
The presence of a chronic low-grade inflammation with aging remodels the properties of the immune system, inducing a permanent decline in its functional activities, a state called immunosenescence. It is not only during the aging process where one encounters immunosenescence, it also exists in many chronic age-related inflammatory diseases and cancer microenvironment. It is known that a defect in immune surveillance has a key role in allowing tumor cells to evade immune clearance. In tumors, the activation of immunosuppressive cells and many inhibitory immune checkpoints, such as the PD-1/PD-L1 pathway, disturb the function of immune cells, especially that of the cytotoxic CD8+ T and NK cells. Interestingly, it was recently demonstrated that the expression of the immunosuppressive PD-L1 protein was robustly upregulated in senescent cells and that this phenomenon suppressed the cytotoxicity of CD8+ T cells [20]. This is an important observation since it indicates that senescent cells can be protected against immune surveillance and subsequent clearance, i.e., leading to their accumulation within aged tissues. The PD-1/PD-L1 axis has a crucial role in tumor growth but it seems that it also enhances the aging process. Intriguingly, the signaling pathways associated with the promotion of the aging process are crucial inducers of the expression of the immunosuppressive PD-L1 protein in senescent cells. This seems paradoxical but currently, the exact role of senescent cells is unknown although most likely they are enhancers of the aging process. Currently, there is intense research to develop senolytic drugs aiming to destroy senescent cells, so-called senolytic therapy [200]. The PD-1/PD-L1 checkpoint inhibitor therapy might represent a promising approach, given that it is being clinically exploited in cancer immunotherapy.
References
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A (2018) Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14:576–590. https://doi.org/10.1038/s41574-018-0059-4
Yousefzadeh MJ, Zhao J, Bukata C, Wade EA, McGowan SJ, Angelini LA, Bank MP, Gurkar AU, McGuckian CA, Calubag MF, Kato JI, Burd CE, Robbins PD, Niedernhofer LJ (2020) Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 19:e13094. https://doi.org/10.1111/acel.13094
Campisi J, Robert L (2014) Cell senescence: role in aging and age-related diseases. Interdiscip Top Gerontol 39:45–61. https://doi.org/10.1159/000358899
Liu T, Zhang L, Joo D, Sun SC (2017) NF-κB signaling in inflammation. Signal Transduct Target Ther 2:17023. https://doi.org/10.1038/sigtrans.2017.23
Chan C, Li L, McCall CE, Yoza BK (2005) Endotoxin tolerance disrupts chromatin remodeling and NF-κB transactivation at the IL-1β promoter. J Immunol 175:461–468. https://doi.org/10.4049/jimmunol.175.1.461
Salminen A, Paimela T, Suuronen T, Kaarniranta K (2008) Innate immunity meets with cellular stress at the IKK complex: regulation of the IKK complex by HSP70 and HSP90. Immunol Lett 117:9–15. https://doi.org/10.1016/j.imlet.2007.12.017
van Eden W, Jansen MAA, Ludwig I, van Kooten P, van der Zee R, Broere F (2017) The enigma of heat shock proteins in immune tolerance. Front Immunol 8:1599. https://doi.org/10.3389/fimmu.2017.01599
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ (2013) The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 138:105–115. https://doi.org/10.1111/imm.12036
Amodio G, Cichy J, Conde P, Matteoli G, Moreau A, Ochando J, Oral BH, Pekarova M, Ryan EJ, Roth J et al (2019) Role of myeloid regulatory cells (MRCs) in maintaining tissue homeostasis and promoting tolerance in autoimmunity, inflammatory disease and transplantation. Cancer Immunol Immunother 68:661–672. https://doi.org/10.1007/s00262-018-2264-3
Salminen A (2020) Activation of immunosuppressive network in the aging process. Ageing Res Rev 57:100998. https://doi.org/10.1016/j.arr.2019.100998
Lian J, Yue Y, Yu W, Zhang Y (2020) Immunosenescence: a key player in cancer development. J Hematol Oncol 13:151. https://doi.org/10.1186/s13045-020-00986-z
Monneret G, Gossez M, Venet F (2021) Sepsis and immunosenescence: closely associated in a vicious circle. Aging Clin Exp Res 33:729–732. https://doi.org/10.1007/s40520-019-01350-z
Salminen A (2021) Immunosuppressive network promotes immunosenescence associated with aging and chronic inflammatory conditions. J Mol Med (Berl) 99:1553–1569. https://doi.org/10.1007/s00109-021-02123-w
Ghosh C, Luong G, Sun Y (2021) A snapshot of the PD-1/PD-L1 pathway. J Cancer 12:2735–2746. https://doi.org/10.7150/jca.57334
Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki IM, Okazaki T (2019) PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front Immunol 10:630. https://doi.org/10.3389/fimmu.2019.00630
Pawelec G, Remarque E, Barnett Y, Solana R (1998) T cells and aging. Front Biosci 3:d59–d99. https://doi.org/10.2741/a266
Rodriguez IJ, Lalinde Ruiz N, Llano Leon M, Martinez Enriquez L, Montilla Velasquez MDP, Ortiz Aguirre JP, Rodriguez Bohorquez OM, Velandia Vargas EA, Hernandez ED, Parra Lopez CA (2021) Immunosenescence study of T cells: a systematic review. Front Immunol 11:604591. https://doi.org/10.3389/fimmu.2020.604591
Onorati A, Havas AP, Lin B, Rajagopal J, Sen P, Adams PD, Dou Z (2022) Upregulation of PD-L1 in senescence and aging. Mol Cell Biol 42:e0017122. https://doi.org/10.1128/mcb.00171-22
Pippin JW, Kaverina N, Wang Y, Eng DG, Zeng Y, Tran U, Loretz CJ, Chang A, Akilesh S, Poudel C et al (2022) Upregulated PD-1 signaling antagonizes glomerular health in aged kidneys and disease. J Clin Invest 132:e156250. https://doi.org/10.1172/JCI156250
Wang TW, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K, Hatakeyama S, Yamazaki S, Shimizu E, Imoto S et al (2022) Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 611:358–364. https://doi.org/10.1038/s41586-022-05388-4
Beenen AC, Sauerer T, Schaft N, Dörrie J (2022) Beyond cancer: regulation and function of PD-L1 in health and immune-related diseases. Int J Mol Sci 23:8599. https://doi.org/10.3390/ijms23158599
Zhao Y, Qu Y, Hao C, Yao W (2023) PD-1/PD-L1 axis in organ fibrosis. Front Immunol 14:1145682. https://doi.org/10.3389/fimmu.2023.1145682
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC et al (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192:1027–1034. https://doi.org/10.1084/jem.192.7.1027
Mariotti FR, Quatrini L, Munari E, Vacca P, Moretta L (2019) Innate lymphoid cells: expression of PD-1 and other checkpoints in normal and pathological conditions. Front Immunol 10:910. https://doi.org/10.3389/fimmu.2019.00910
Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L (2020) The immune checkpoint PD-1 in natural killer cells: expression, function and targeting in tumour immunotherapy. Cancers (Basel) 12:3285. https://doi.org/10.3390/cancers12113285
Stanford SM, Rapini N, Bottini N (2012) Regulation of TCR signalling by tyrosine phosphatases: from immune homeostasis to autoimmunity. Immunology 137:1–19. https://doi.org/10.1111/j.1365-2567.2012.03591.x
Baldanzi G (2022) Immune checkpoint receptors signaling in T cells. Int J Mol Sci 23:3529. https://doi.org/10.3390/ijms23073529
Wang Q, Bardhan K, Boussiotis VA, Patsoukis N (2021) The PD-1 interactome. Adv Biol (Weinh) 5:e2100758. https://doi.org/10.1002/adbi.202100758
Hou B, Hu Y, Zhu Y, Wang X, Li W, Tang J, Jia X, Wang J, Cong Y, Quan M et al (2024) SHP-1 regulates CD8+ T cell effector function but plays a subtle role with SHP-2 in T cell exhaustion due to a stage-specific nonredundant functional relay. J Immunol 212:397–409. https://doi.org/10.4049/jimmunol.2300462
Niu C, Li M, Zhu S, Chen Y, Zhou L, Xu D, Xu J, Li Z, Li W, Cui J (2020) PD-1-positive natural killer cells have a weaker antitumor function than that of PD-1-negative natural killer cells in lung cancer. Int J Med Sci 17:1964–1973. https://doi.org/10.7150/ijms.47701
Thibult ML, Mamessier E, Gertner-Dardenne J, Pastor S, Just-Landi S, Xerri L, Chetaille B, Olive D (2013) PD-1 is a novel regulator of human B-cell activation. Int Immunol 25:129–137. https://doi.org/10.1093/intimm/dxs098
Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N et al (2001) Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291:319–322. https://doi.org/10.1126/science.291.5502.319
Okazaki T, Wang J (2005) PD-1/PD-L pathway and autoimmunity. Autoimmunity 38:353–357. https://doi.org/10.1080/08916930500124072
Hassani N, Salmaninejad A, Aslani S, Kamali-Sarvestani E, Vessal M (2023) The association between PD-1 gene polymorphisms and susceptibility to multiple sclerosis. Immunol Med 46:69–76. https://doi.org/10.1080/25785826.2022.2137967
Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, Wu X, Ma J, Zhou M, Li X et al (2019) Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer 18:10. https://doi.org/10.1186/s12943-018-0928-4
Pei L, Liu Y, Liu L, Gao S, Gao X, Feng Y, Sun Z, Zhang Y, Wang C (2023) Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer 22:29. https://doi.org/10.1186/s12943-023-01731-z
Dong H, Zhu G, Tamada K, Chen L (1999) B7–H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 5:1365–1369. https://doi.org/10.1038/70932
Solinas C, Aiello M, Rozali E, Lambertini M, Willard-Gallo K, Migliori E (2020) Programmed cell death-ligand 2: a neglected but important target in the immune response to cancer? Transl Oncol 13:100811. https://doi.org/10.1016/j.tranon.2020.100811
Liu X, Song J, Zhang H, Liu X, Zuo F, Zhao Y, Zhao Y, Yin X, Guo X, Wu X et al (2023) Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell 41:272–287.e9. https://doi.org/10.1016/j.ccell.2023.01.001
Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, Smoot DT, Ashktorab H, Oike T, Suzuki Y et al (2018) PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci 109:43–53. https://doi.org/10.1111/cas.13424
Shen X, Zhang L, Li J, Li Y, Wang Y, Xu ZX (2019) Recent findings in the regulation of programmed death ligand 1 expression. Front Immunol 10:1337. https://doi.org/10.3389/fimmu.2019.01337
Zhou K, Guo S, Li F, Sun Q, Liang G (2020) Exosomal PD-L1: new insights into tumor immune escape mechanisms and therapeutic strategies. Front Cell Dev Biol 8:569219. https://doi.org/10.3389/fcell.2020.569219
Hudson K, Cross N, Jordan-Mahy N, Leyland R (2020) The extrinsic and intrinsic roles of PD-L1 and its receptor PD-1: implications for immunotherapy treatment. Front Immunol 11:568931. https://doi.org/10.3389/fimmu.2020.568931
Guo X, Sunil C, Adeyanju O, Parker A, Huang S, Ikebe M, Tucker TA, Idell S, Qian G (2022) PD-L1 mediates lung fibroblast to myofibroblast transition through Smad3 and β-catenin signaling pathways. Sci Rep 12:3053. https://doi.org/10.1038/s41598-022-07044-3
Kornepati AVR, Vadlamudi RK, Curiel TJ (2022) Programmed death ligand 1 signals in cancer cells. Nat Rev Cancer 22:174–189. https://doi.org/10.1038/s41568-021-00431-4
Perrichet A, Ghiringhelli F, Rebe C (2020) Understanding inflammasomes and PD-1/PD-L1 crosstalk to improve cancer treatment efficiency. Cancers (Basel) 12:3550. https://doi.org/10.3390/cancers12123550
Jiang A, Liu N, Wang J, Zheng X, Ren M, Zhang W, Yao Y (2022) The role of PD-1/PD-L1 axis in idiopathic pulmonary fibrosis: friend or foe? Front Immunol 13:1022228. https://doi.org/10.3389/fimmu.2022.1022228
Gao Y, Nihira NT, Bu X, Chu C, Zhang J, Kolodziejczyk A, Fan Y, Chan NT, Ma L, Liu J et al (2020) Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol 22:1064–1075. https://doi.org/10.1038/s41556-020-0562-4
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206:3015–3029. https://doi.org/10.1084/jem.20090847
DiDomenico J, Lamano JB, Oyon D, Li Y, Veliceasa D, Kaur G, Ampie L, Choy W, Lamano JB, Bloch O (2018) The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 7:e1448329. https://doi.org/10.1080/2162402X.2018.1448329
Zhu Z, Zhang H, Chen B, Liu X, Zhang S, Zong Z, Gao M (2020) PD-L1-mediated immunosuppression in glioblastoma is associated with the infiltration and M2-polarization of tumor-associated macrophages. Front Immunol 11:588552. https://doi.org/10.3389/fimmu.2020.588552
Wei Y, Liang M, Xiong L, Su N, Gao X, Jiang Z (2021) PD-L1 induces macrophage polarization toward the M2 phenotype via Erk/Akt/mTOR. Exp Cell Res 402:112575. https://doi.org/10.1016/j.yexcr.2021.112575
Nam S, Lee A, Lim J, Lim JS (2019) Analysis of the expression and regulation of PD-1 protein on the surface of myeloid-derived suppressor cells (MDSCs). Biomol Ther (Seoul) 27:63–70. https://doi.org/10.4062/biomolther.2018.201
Ruan WS, Feng MX, Xu J, Xu YG, Song CY, Lin LY, Li L, Lu YQ (2020) Early activation of myeloid-derived suppressor cells participate in sepsis-induced immune suppression via PD-L1/PD-1 axis. Front Immunol 11:1299. https://doi.org/10.3389/fimmu.2020.01299
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P (2010) The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol 10:554–567. https://doi.org/10.1038/nri2808
Haist M, Stege H, Grabbe S, Bros M (2021) The functional crosstalk between myeloid-derived suppressor cells and regulatory T cells within the immunosuppressive tumor microenvironment. Cancers (Basel) 13:210. https://doi.org/10.3390/cancers13020210
Salminen A (2022) Clinical perspectives on the age-related increase of immunosuppressive activity. J Mol Med (Berl) 100:697–712. https://doi.org/10.1007/s00109-022-02193-4
Enioutina EY, Bareyan D, Daynes RA (2011) A role for immature myeloid cells in immune senescence. J Immunol 186:697–707. https://doi.org/10.4049/jimmunol.1002987
Garg SK, Delaney C, Toubai T, Ghosh A, Reddy P, Banerjee R, Yung R (2014) Aging is associated with increased regulatory T-cell function. Aging Cell 13:441–448. https://doi.org/10.1111/acel.12191
Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, Belt BA, Alspach E, Leahy K, Luo J et al (2016) Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 7:11762. https://doi.org/10.1038/ncomms11762
Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P, Nelson DJ (2017) Aging and cancer: the role of macrophages and neutrophils. Ageing Res Rev 36:105–116. https://doi.org/10.1016/j.arr.2017.03.008
Salminen A, Kaarniranta K, Kauppinen A (2018) The role of myeloid-derived suppressor cells (MDSC) in the inflammaging process. Ageing Res Rev 48:1–10. https://doi.org/10.1016/j.arr.2018.09.001
Lages CS, Lewkowich I, Sproles A, Wills-Karp M, Chougnet C (2010) Partial restoration of T-cell function in aged mice by in vitro blockade of the PD-1/ PD-L1 pathway. Aging Cell 9:785–798. https://doi.org/10.1111/j.1474-9726.2010.00611.x
Benayoun BA, Pollina EA, Singh PP, Mahmoudi S, Harel I, Casey KM, Dulken BW, Kundaje A, Brunet A (2019) Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 29:697–709. https://doi.org/10.1101/gr.240093.118
Hernandez-Segura A, Nehme J, Demaria M (2018) Hallmarks of cellular senescence. Trends Cell Biol 28:436–453. https://doi.org/10.1016/j.tcb.2018.02.001
Salminen A, Kaarniranta K, Kauppinen A (2020) Exosomal vesicles enhance immunosuppression in chronic inflammation: impact in cellular senescence and the aging process. Cell Signal 75:109771. https://doi.org/10.1016/j.cellsig.2020.109771
Janelle V, Neault M, Lebel ME, De Sousa DM, Boulet S, Durrieu L, Carli C, Muzac C, Lemieux S, Labrecque N et al (2021) p16INK4a regulates cellular senescence in PD-1-expressing human T cells. Front Immunol 12:698565. https://doi.org/10.3389/fimmu.2021.698565
Kasamatsu T, Awata-Shiraiwa M, Ishihara R, Murakami Y, Masuda Y, Gotoh N, Oda T, Yokohama A, Matsumura I, Handa H et al (2023) Sub-lethal doses of chemotherapeutic agents induce senescence in T cells and upregulation of PD-1 expression. Clin Exp Med 23:2695–2703. https://doi.org/10.1007/s10238-023-01034-z
Elias R, Giobbie-Hurder A, McCleary NJ, Ott P, Hodi FS, Rahma O (2018) Efficacy of PD-1 & PD-L1 inhibitors in older adults: a meta-analysis. J Immunother Cancer 6:26. https://doi.org/10.1186/s40425-018-0336-8
Kuwano K, Araya J, Hara H, Minagawa S, Takasaka N, Ito S, Kobayashi K, Nakayama K (2016) Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir Investig 54:397–406. https://doi.org/10.1016/j.resinv.2016.03.010
Guerrero A, De Strooper B, Arancibia-Carcamo IL (2021) Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci 44:714–727. https://doi.org/10.1016/j.tins.2021.06.007
Hu C, Zhang X, Teng T, Ma ZG, Tang QZ (2022) Cellular senescence in cardiovascular diseases: a systematic review. Aging Dis 13:103–128. https://doi.org/10.14336/AD.2021.0927
Molnar AA, Pasztor DT, Tarcza Z, Merkely B (2023) Cells in atherosclerosis: focus on cellular senescence from basic science to clinical practice. Int J Mol Sci 24:17129. https://doi.org/10.3390/ijms242417129
Tsou PS, Shi B, Varga J (2022) Role of cellular senescence in the pathogenesis of systemic sclerosis. Curr Opin Rheumatol 34:343–350. https://doi.org/10.1097/BOR.0000000000000898
Osorio JM, Espinoza-Perez C, Rimassa-Tare C, Machuca V, Bustos JO, Vallejos M, Vargas H, Diaz-Araya G (2023) Senescent cardiac fibroblasts: a key role in cardiac fibrosis. Biochim Biophys Acta Mol Basis Dis 1869:166642. https://doi.org/10.1016/j.bbadis.2023.166642
Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Farina E, Alberoni M, Clerici M (2012) A potential role for the PD1/PD-L1 pathway in the neuroinflammation of Alzheimer’s disease. Neurobiol Aging 33:624.e11–22. https://doi.org/10.1016/j.neurobiolaging.2011.03.004
Weyand CM, Berry GJ, Goronzy JJ (2018) The immunoinhibitory PD-1/PD-L1 pathway in inflammatory blood vessel disease. J Leukoc Biol 103:565–575. https://doi.org/10.1189/jlb.3MA0717-283
Polverino F, Mirra D, Yang CX, Esposito R, Spaziano G, Rojas-Quintero J, Sgambato M, Piegari E, Cozzolino A, Cione E et al (2022) Similar programmed death ligand 1 (PD-L1) expression profile in patients with mild COPD and lung cancer. Sci Rep 12:22402. https://doi.org/10.1038/s41598-022-26650-9
Kummer MP, Ising C, Kummer C, Sarlus H, Griep A, Vieira-Saecker A, Schwartz S, Halle A, Brückner M, Händler K et al (2021) Microglial PD-1 stimulation by astrocytic PD-L1 suppresses neuroinflammation and Alzheimer’s disease pathology. EMBO J 40:e108662. https://doi.org/10.15252/embj.2021108662
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G (2007) The myofibroblast: one function, multiple origins. Am J Pathol 170:1807–1816. https://doi.org/10.2353/ajpath.2007.070112
Ahmadvand N, Carraro G, Jones MR, Shalashova I, Noori A, Wilhelm J, Baal N, Khosravi F, Chen C, Zhang JS et al (2022) Cell-surface programmed death ligand-1 expression identifies a sub-population of distal epithelial cells enriched in idiopathic pulmonary fibrosis. Cells 11:1593. https://doi.org/10.3390/cells11101593
Li Z, Zhou J, Zhang J, Li S, Wang H, Du J (2019) Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int J Cancer 145:1946–1957. https://doi.org/10.1002/ijc.32278
Teramoto K, Igarashi T, Kataoka Y, Ishida M, Hanaoka J, Sumimoto H, Daigo Y (2019) Clinical significance of PD-L1-positive cancer-associated fibroblasts in pN0M0 non-small cell lung cancer. Lung Cancer 137:56–63. https://doi.org/10.1016/j.lungcan.2019.09.013
Duitman J, van den Ende T, Spek CA (2019) Immune checkpoints as promising targets for the treatment of idiopathic pulmonary fibrosis? J Clin Med 8:1547. https://doi.org/10.3390/jcm8101547
Lu Y, Zhong W, Liu Y, Chen W, Zhang J, Zeng Z, Huang H, Qiao Y, Wan X, Meng X et al (2022) Anti-PD-L1 antibody alleviates pulmonary fibrosis by inducing autophagy via inhibition of the PI3K/Akt/mTOR pathway. Int Immunopharmacol 104:108504. https://doi.org/10.1016/j.intimp.2021.108504
Zhao Y, Hao C, Li M, Qu Y, Guo Y, Deng X, Si H, Yao W (2022) PD-1/PD-L1 inhibitor ameliorates silica-induced pulmonary fibrosis by maintaining systemic immune homeostasis. Biomed Pharmacother 148:112768. https://doi.org/10.1016/j.biopha.2022.112768
Liu K, Sun Q, Liu Q, Li H, Zhang W, Sun C (2022) Focus on immune checkpoint PD-1/PD-L1 pathway: new advances of polyphenol phytochemicals in tumor immunotherapy. Biomed Pharmacother 154:113618. https://doi.org/10.1016/j.biopha.2022.113618
Zhou YJ, Li G, Wang J, Liu M, Wang Z, Song Y, Zhang X, Wang X (2023) PD-L1: expression regulation. Blood Sci 5:77–91. https://doi.org/10.1097/BS9.0000000000000149
Fabrizio FP, Trombetta D, Rossi A, Sparaneo A, Castellana S, Muscarella LA (2018) Gene code CD274/PD-L1: from molecular basis toward cancer immunotherapy. Ther Adv Med Oncol 10:1758835918815598. https://doi.org/10.1177/1758835918815598
Kumar S, Sharawat SK (2018) Epigenetic regulators of programmed death-ligand 1 expression in human cancers. Transl Res 202:129–145. https://doi.org/10.1016/j.trsl.2018.05.011
Chatterjee A, Rodger EJ, Ahn A, Stockwell PA, Parry M, Motwani J, Gallagher SJ, Shklovskaya E, Tiffen J, Eccles MR et al (2018) Marked global DNA hypomethylation is associated with constitutive PD-L1 expression in melanoma. iScience 4:312–325. https://doi.org/10.1016/j.isci.2018.05.021
Elashi AA, Sasidharan Nair V, Taha RZ, Shaath H, Elkord E (2018) DNA methylation of immune checkpoints in the peripheral blood of breast and colorectal cancer patients. Oncoimmunology 8:e1542918. https://doi.org/10.1080/2162402X.2018.1542918
Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, Fang Z, Nguyen M, Pierce S, Wei Y et al (2014) Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 28:1280–1288. https://doi.org/10.1038/leu.2013.355
Lv D, Xing C, Cao L, Zhuo Y, Wu T, Gao N (2020) PD-L1 gene promoter methylation represents a potential diagnostic marker in advanced gastric cancer. Oncol Lett 19:1223–1234. https://doi.org/10.3892/ol.2019.11221
Chang CB, Lee SP, Chen WM, Wang CM, Song YC, Chan MW, Wu SF (2020) Dendritic cell upregulation of programmed death ligand-1 via DNA demethylation inhibits experimental autoimmune encephalomyelitis. J Autoimmun 107:102362. https://doi.org/10.1016/j.jaut.2019.102362
Ma X, Wu J, Wang B, Liu C, Liu L, Sun C (2022) Epigenetic modifications: critical participants of the PD-L1 regulatory mechanism in solid tumors (Review). Int J Oncol 61:134. https://doi.org/10.3892/ijo.2022.5424
Wang H, Fu C, Du J, Wang H, He R, Yin X, Li H, Li X, Wang H, Li K et al (2020) Enhanced histone H3 acetylation of the PD-L1 promoter via the COP1/c-Jun/HDAC3 axis is required for PD-L1 expression in drug-resistant cancer cells. J Exp Clin Cancer Res 39:29. https://doi.org/10.1186/s13046-020-1536-x
Deng S, Hu Q, Zhang H, Yang F, Peng C, Huang C (2019) HDAC3 inhibition upregulates PD-L1 expression in B-cell lymphomas and augments the efficacy of anti-PD-L1 therapy. Mol Cancer Ther 18:900–908. https://doi.org/10.1158/1535-7163.MCT-18-1068
Zhang P, Du Y, Bai H, Wang Z, Duan J, Wang X, Zhong J, Wan R, Xu J, He X et al (2022) Optimized dose selective HDAC inhibitor tucidinostat overcomes anti-PD-L1 antibody resistance in experimental solid tumors. BMC Med 20:435. https://doi.org/10.1186/s12916-022-02598-5
Wang Q, Lin W, Tang X, Li S, Guo L, Lin Y, Kwok HF (2017) The roles of microRNAs in regulating the expression of PD-1/PD-L1 immune checkpoint. Int J Mol Sci 18:2540. https://doi.org/10.3390/ijms18122540
Kane AE, Sinclair DA (2019) Epigenetic changes during aging and their reprogramming potential. Crit Rev Biochem Mol Biol 54:61–83. https://doi.org/10.1080/10409238.2019.1570075
Unnikrishnan A, Freeman WM, Jackson J, Wren JD, Porter H, Richardson A (2019) The role of DNA methylation in epigenetics of aging. Pharmacol Ther 195:172–185. https://doi.org/10.1016/j.pharmthera.2018.11.001
Crouch J, Shvedova M, Thanapaul RJRS, Botchkarev V, Roh D (2022) Epigenetic regulation of cellular senescence Cells 11:672. https://doi.org/10.3390/cells11040672
Erbe R, Wang Z, Wu S, Xiu J, Zaidi N, La J, Tuck D, Fillmore N, Giraldo NA, Topper M et al (2021) Evaluating the impact of age on immune checkpoint therapy biomarkers. Cell Rep 36:109599. https://doi.org/10.1016/j.celrep.2021.109599
Salzer MC, Lafzi A, Berenguer-Llergo A, Youssif C, Castellanos A, Solanas G, Peixoto FO, Stephan-Otto Attolini C, Prats N, Aguilera M et al (2018) Identity noise and adipogenic traits characterize dermal fibroblast aging. Cell 175:1575–1590.e22. https://doi.org/10.1016/j.cell.2018.10.012
Vidal R, Wagner JUG, Braeuning C, Fischer C, Patrick R, Tombor L, Muhly-Reinholz M, John D, Kliem M, Conrad T et al (2019) Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight 4:e131092. https://doi.org/10.1172/jci.insight.131092
Tabula Muris Consortium (2020) A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583:590–595. https://doi.org/10.1038/s41586-020-2496-1
Zou Z, Long X, Zhao Q, Zheng Y, Song M, Ma S, Jing Y, Wang S, He Y, Esteban CR et al (2021) A single-cell transcriptomic atlas of human skin aging. Dev Cell 56:383–397.e8. https://doi.org/10.1016/j.devcel.2020.11.002
Uyar B, Palmer D, Kowald A, Murua Escobar H, Barrantes I, Möller S, Akalin A, Fuellen G (2020) Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev 64:101156. https://doi.org/10.1016/j.arr.2020.101156
Xu P, Wang M, Song WM, Wang Q, Yuan GC, Sudmant PH, Zare H, Tu Z, Orr ME, Zhang B (2022) The landscape of human tissue and cell type specific expression and co-regulation of senescence genes. Mol Neurodegener 17:5. https://doi.org/10.1186/s13024-021-00507-7
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X et al (2017) Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 19:1189–1201. https://doi.org/10.1016/j.celrep.2017.04.031
Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F (2020) Regulation of PD-L1 expression by NF-κB in cancer. Front Immunol 11:584626. https://doi.org/10.3389/fimmu.2020.584626
Betzler AC, Theodoraki MN, Schuler PJ, Döscher J, Laban S, Hoffmann TK, Brunner C (2020) NF-κB and its role in checkpoint control. Int J Mol Sci 21:3949. https://doi.org/10.3390/ijms21113949
Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, You Z (2017) Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett 184:7–14. https://doi.org/10.1016/j.imlet.2017.02.006
Li H, Xia JQ, Zhu FS, Xi ZH, Pan CY, Gu LM, Tian YZ (2018) LPS promotes the expression of PD-L1 in gastric cancer cells through NF-κB activation. J Cell Biochem 119:9997–10004. https://doi.org/10.1002/jcb.27329
Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, Chu MF, Hodgson K, Berger T, Wakeham A et al (2019) Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci U S A 116:4326–4335. https://doi.org/10.1073/pnas.1819473116
Dickinson SE, Khawam M, Kirschnerova V, Vaishampayan P, Centuori SM, Saboda K, Calvert VS, Petricoin EF 3rd, Curiel-Lewandrowski C (2021) Increased PD-L1 expression in human skin acutely and chronically exposed to UV irradiation. Photochem Photobiol 97:778–784. https://doi.org/10.1111/php.13406
Rong QX, Wang F, Guo ZX, Hu Y, An SN, Luo M, Zhang H, Wu SC, Huang HQ, Fu LW (2021) GM-CSF mediates immune evasion via upregulation of PD-L1 expression in extranodal natural killer/T cell lymphoma. Mol Cancer 20:80. https://doi.org/10.1186/s12943-021-01374-y
Nawas AF, Solmonson A, Gao B, DeBerardinis RJ, Minna JD, Conacci-Sorrell M, Mendelson CR (2023) IL-1β mediates the induction of immune checkpoint regulators IDO1 and PD-L1 in lung adenocarcinoma cells. Cell Commun Signal 21:331. https://doi.org/10.1186/s12964-023-01348-1
Lu F, Zhao Y, Pang Y, Ji M, Sun Y, Wang H, Zou J, Wang Y, Li G, Sun T et al (2021) NLRP3 inflammasome upregulates PD-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett 497:178–189. https://doi.org/10.1016/j.canlet.2020.10.024
Zhang Y, Zhu S, Du Y, Xu F, Sun W, Xu Z, Wang X, Qian P, Zhang Q, Feng J et al (2022) RelB upregulates PD-L1 and exacerbates prostate cancer immune evasion. J Exp Clin Cancer Res 41:66. https://doi.org/10.1186/s13046-022-02243-2
An LL, Gorman JV, Stephens G, Swerdlow B, Warrener P, Bonnell J, Mustelin T, Fung M, Kolbeck R (2016) Complement C5a induces PD-L1 expression and acts in synergy with LPS through Erk1/2 and JNK signaling pathways. Sci Rep 6:33346. https://doi.org/10.1038/srep33346
Zha H, Han X, Zhu Y, Yang F, Li Y, Li Q, Guo B, Zhu B (2017) Blocking C5aR signaling promotes the anti-tumor efficacy of PD-1/PD-L1 blockade. Oncoimmunology 6:e1349587. https://doi.org/10.1080/2162402X.2017.1349587
Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S (2017) COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U S A 114:1117–1122. https://doi.org/10.1073/pnas.1612920114
Kang JH, Jung MY, Choudhury M, Leof EB (2020) Transforming growth factor β induces fibroblasts to express and release the immunomodulatory protein PD-L1 into extracellular vesicles. FASEB J 34:2213–2226. https://doi.org/10.1096/fj.201902354R
Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, Lan Y, Dussault I, Moustakas A (2022) Dual inhibition of TGF-β and PD-L1: a novel approach to cancer treatment. Mol Oncol 16:2117–2134. https://doi.org/10.1002/1878-0261.13146
Wu D, Meydani SN (2004) Mechanism of age-associated up-regulation in macrophage PGE2 synthesis. Brain Behav Immun 18:487–494. https://doi.org/10.1016/j.bbi.2004.05.003
Li Y, Lei D, Swindell WR, Xia W, Weng S, Fu J, Worthen CA, Okubo T, Johnston A, Gudjonsson JE et al (2015) Age-associated increase in skin fibroblast-derived prostaglandin E2 contributes to reduced collagen levels in elderly human skin. J Invest Dermatol 135:2181–2188. https://doi.org/10.1038/jid.2015.157
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12:21–35. https://doi.org/10.1038/nrm3025
Powell JD, Pollizzi KN, Heikamp EB, Horton MR (2012) Regulation of immune responses by mTOR. Annu Rev Immunol 30:39–68. https://doi.org/10.1146/annurev-immunol-020711-075024
Johnson SC, Rabinovitch PS, Kaeberlein M (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493:338–345. https://doi.org/10.1038/nature11861
Salminen A, Kaarniranta K (2009) Regulation of the aging process by autophagy. Trends Mol Med 15:217–224. https://doi.org/10.1016/j.molmed.2009.03.004
Carosi JM, Fourrier C, Bensalem J, Sargeant TJ (2022) The mTOR-lysosome axis at the centre of ageing. FEBS Open Bio 12:739–757. https://doi.org/10.1002/2211-5463.13347
Mafi S, Mansoori B, Taeb S, Sadeghi H, Abbasi R, Cho WC, Rostamzadeh D (2022) mTOR-mediated regulation of immune responses in cancer and tumor microenvironment. Front Immunol 12:774103. https://doi.org/10.3389/fimmu.2021.774103
Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T (2023) The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol 24:167–185. https://doi.org/10.1038/s41580-022-00542-2
Tsapras P, Petridi S, Chan S, Geborys M, Jacomin AC, Sagona AP, Meier P, Nezis IP (2022) Selective autophagy controls innate immune response through a TAK1/TAB2/SH3PX1 axis. Cell Rep 38:110286. https://doi.org/10.1016/j.celrep.2021.110286
Wang X, Wu WKK, Gao J, Li Z, Dong B, Lin X, Li Y, Li Y, Gong J, Qi C et al (2019) Autophagy inhibition enhances PD-L1 expression in gastric cancer. J Exp Clin Cancer Res 38:140. https://doi.org/10.1186/s13046-019-1148-5
Duan Y, Tian X, Liu Q, Jin J, Shi J, Hou Y (2021) Role of autophagy on cancer immune escape. Cell Commun Signal 19:91. https://doi.org/10.1186/s12964-021-00769-0
Gao L, Chen Y (2021) Autophagy controls programmed death-ligand 1 expression on cancer cells. Biomed Rep 15:84. https://doi.org/10.3892/br.2021.1460
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC et al (2007) Loss of tumor suppressor PTEN function increases B7–H1 expression and immunoresistance in glioma. Nat Med 13:84–88. https://doi.org/10.1038/nm1517
Kumar AV, Mills J, Lapierre LR (2022) Selective autophagy receptor p62/SQSTM1, a pivotal player in stress and aging. Front Cell Dev Biol 10:793328. https://doi.org/10.3389/fcell.2022.793328
Garcia-Perez BE, Perez-Torres C, Baltierra-Uribe SL, Castillo-Cruz J, Castrejon-Jimenez NS (2023) Autophagy as a target for non-immune intrinsic functions of programmed cell death-ligand 1 in cancer. Int J Mol Sci 24:15016. https://doi.org/10.3390/ijms241915016
Gao H, Zhang J, Ren X (2019) PD-L1 regulates tumorigenesis and autophagy of ovarian cancer by activating mTORC signaling. Biosci Rep 39:BSR20191041. https://doi.org/10.1042/BSR20191041
Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, Drerup JM, Padron A, Conejo-Garcia J, Murthy K et al (2016) Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res 76:6964–6974. https://doi.org/10.1158/0008-5472.CAN-16-0258
Hopfner KP, Hornung V (2020) Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 21:501–521. https://doi.org/10.1038/s41580-020-0244-x
Chen C, Xu P (2023) Cellular functions of cGAS-STING signaling. Trends Cell Biol 33:630–648. https://doi.org/10.1016/j.tcb.2022.11.001
Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ (2021) The central role of DNA damage in the ageing process. Nature 592:695–703. https://doi.org/10.1038/s41586-021-03307-7
Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19:1061–1070. https://doi.org/10.1038/ncb3586
Yang H, Wang H, Ren J, Chen Q, Chen ZJ (2017) cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 114:E4612–E4620. https://doi.org/10.1073/pnas.1705499114
Li T, Chen ZJ (2018) The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215:1287–1299. https://doi.org/10.1084/jem.20180139
Gulen MF, Samson N, Keller A, Schwabenland M, Liu C, Glück S, Thacker VV, Favre L, Mangeat B, Kroese LJ et al (2023) cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 620:374–380. https://doi.org/10.1038/s41586-023-06373-1
Cheng AN, Cheng LC, Kuo CL, Lo YK, Chou HY, Chen CH, Wang YH, Chuang TH, Cheng SJ, Lee AY (2020) Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1-mediated immunoescape via STING-IFN signaling and extracellular vesicles. J Immunother Cancer 8:e001372. https://doi.org/10.1136/jitc-2020-001372
Du SS, Chen GW, Yang P, Chen YX, Hu Y, Zhao QQ, Zhang Y, Liu R, Zheng DX, Zhou J et al (2022) Radiation therapy promotes hepatocellular carcinoma immune cloaking via PD-L1 upregulation induced by cGAS-STING activation. Int J Radiat Oncol Biol Phys 112:1243–1255. https://doi.org/10.1016/j.ijrobp.2021.12.162
Lee JJ, Kim SY, Kim SH, Choi S, Lee B, Shin JS (2022) STING mediates nuclear PD-L1 targeting-induced senescence in cancer cells. Cell Death Dis 13:791. https://doi.org/10.1038/s41419-022-05217-6
Nebert DW (2017) Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog Lipid Res 67:38–57. https://doi.org/10.1016/j.plipres.2017.06.001
Salminen A (2023) Activation of aryl hydrocarbon receptor (AhR) in Alzheimer’s disease: role of tryptophan metabolites generated by gut host-microbiota. J Mol Med (Berl) 101:201–222. https://doi.org/10.1007/s00109-023-02289-5
Salminen A (2022) Aryl hydrocarbon receptor (AhR) reveals evidence of antagonistic pleiotropy in the regulation of the aging process. Cell Mol Life Sci 79:489. https://doi.org/10.1007/s00018-022-04520-x
Salminen A (2022) Role of indoleamine 2,3-dioxygenase 1 (IDO1) and kynurenine pathway in the regulation of the aging process. Ageing Res Rev 75:101573. https://doi.org/10.1016/j.arr.2022.101573
Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA (2010) An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185:3190–3198. https://doi.org/10.4049/jimmunol.0903670
Kenison JE, Wang Z, Yang K, Snyder M, Quintana FJ, Sherr DH (2021) The aryl hydrocarbon receptor suppresses immunity to oral squamous cell carcinoma through immune checkpoint regulation. Proc Natl Acad Sci U S A 118:e2012692118. https://doi.org/10.1073/pnas.2012692118
Wang GZ, Zhang L, Zhao XC, Gao SH, Qu LW, Yu H, Fang WF, Zhou YC, Liang F, Zhang C et al (2019) The aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat Commun 10:1125. https://doi.org/10.1038/s41467-019-08887-7
Kou Z, Yang R, Lee E, Cuddapah S, Choi BH, Dai W (2022) Oxidative stress modulates expression of immune checkpoint genes via activation of AhR signaling. Toxicol Appl Pharmacol 457:116314. https://doi.org/10.1016/j.taap.2022.116314
Han SC, Wang GZ, Yang YN, Fang WF, Sun BB, Zhang JD, Zhou HQ, Zhang L, Wang Y, Zhou GB (2023) Nuclear AhR and membranous PD-L1 in predicting response of non-small cell lung cancer to PD-1 blockade. Signal Transduct Target Ther 8:191. https://doi.org/10.1038/s41392-023-01416-5
Gargaro M, Manni G, Scalisi G, Puccetti P, Fallarino F (2021) Tryptophan metabolites at the crossroad of immune-cell interaction via the aryl hydrocarbon receptor: implications for tumor immunotherapy. Int J Mol Sci 22:4644. https://doi.org/10.3390/ijms22094644
Zhou Y, Liu Z, Chen T (2022) Gut microbiota: a promising milestone in enhancing the efficacy of PD1/PD-L1 blockade therapy. Front Oncol 12:847350. https://doi.org/10.3389/fonc.2022.847350
Guarente L, Sinclair DA, Kroemer G (2024) Human trials exploring anti-aging medicines. Cell Metab. https://doi.org/10.1016/j.cmet.2023.12.007
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS et al (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460(7253):392–395. https://doi.org/10.1038/nature08221
Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ et al (2013) Metformin improves healthspan and lifespan in mice. Nat Commun 4:2192. https://doi.org/10.1038/ncomms3192
Salminen A, Kaarniranta K (2012) AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 11:230–241. https://doi.org/10.1016/j.arr.2011.12.005
Nojima I, Wada J (2023) Metformin and its immune-mediated effects in various diseases. Int J Mol Sci 24:755. https://doi.org/10.3390/ijms24010755
Hua Y, Zheng Y, Yao Y, Jia R, Ge S, Zhuang A (2023) Metformin and cancer hallmarks: shedding new lights on therapeutic repurposing. J Transl Med 21:403. https://doi.org/10.1186/s12967-023-04263-8
Xu P, Yin K, Tang X, Tian J, Zhang Y, Ma J, Xu H, Xu Q, Wang S (2019) Metformin inhibits the function of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Biomed Pharmacother 120:109458. https://doi.org/10.1016/j.biopha.2019.109458
Salminen A, Kauppinen A, Kaarniranta K (2019) AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): impact on cancer and aging. J Mol Med (Berl) 97:1049–1064. https://doi.org/10.1007/s00109-019-01795-9
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, Li CW, Kim T, Chang SS, Lee HH et al (2018) Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell 71:606–620.e7. https://doi.org/10.1016/j.molcel.2018.07.030
Lu Y, Xin D, Guan L, Xu M, Yang Y, Chen Y, Yang Y, Wang-Gillam A, Wang L, Zong S, Wang F (2021) Metformin downregulates PD-L1 expression in esophageal squamous cell carcinoma by inhibiting IL-6 signaling pathway. Front Oncol 11:762523. https://doi.org/10.3389/fonc.2021.762523
Munoz LE, Huang L, Bommireddy R, Sharma R, Monterroza L, Guin RN, Samaranayake SG, Pack CD, Ramachandiran S, Reddy SJC et al (2021) Metformin reduces PD-L1 on tumor cells and enhances the anti-tumor immune response generated by vaccine immunotherapy. J Immunother Cancer 9:e002614. https://doi.org/10.1136/jitc-2021-002614
Alers S, Löffler AS, Wesselborg S, Stork B (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 32:2–11. https://doi.org/10.1128/MCB.06159-11
Lamming DW, Ye L, Sabatini DM, Baur JA (2013) Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest 123:980–989. https://doi.org/10.1172/JCI64099
Khedri M, Kooshki H, Taheri RA (2021) Rapamycin attenuates gene expression of programmed cell death protein-ligand 1 and Foxp3 in the brain; a novel mechanism proposed for immunotherapy in the brain. Res Pharm Sci 16:165–172. https://doi.org/10.4103/1735-5362.310523
Bagherpour B, Salehi M, Jafari R, Bagheri A, Kiani-Esfahani A, Edalati M, Kardi MT, Shaygannejad V (2018) Promising effect of rapamycin on multiple sclerosis. Mult Scler Relat Disord 26:40–45. https://doi.org/10.1016/j.msard.2018.08.009
Paudel S, Mishra N, Agarwal R (2023) Phytochemicals as immunomodulatory molecules in cancer therapeutics. Pharmaceuticals (Basel) 16:1652. https://doi.org/10.3390/ph16121652
Zhang Q, Yang C, Gao X, Dong J, Zhong C (2024) Phytochemicals in regulating PD-1/PD-L1 and immune checkpoint blockade therapy. Phytother Res 38:776–796. https://doi.org/10.1002/ptr.8082
Huang Y, Yang Z, Zhang L (2024) Polyphenol supplementation enhances the efficacy of PD-1/PD-L1 inhibitors against cancer: a meta-analysis of animal studies. Nutr Cancer 76:17–30. https://doi.org/10.1080/01635581.2023.2277477
Guo Y, Liang J, Liu B, Jin Y (2021) Molecular mechanism of food-derived polyphenols on PD-L1 dimerization: a molecular dynamics simulation study. Int J Mol Sci 22:10924. https://doi.org/10.3390/ijms222010924
Jing L, Lin J, Yang Y, Tao L, Li Y, Liu Z, Zhao Q, Diao A (2021) Quercetin inhibiting the PD-1/PD-L1 interaction for immune-enhancing cancer chemopreventive agent. Phytother Res 35:6441–6451. https://doi.org/10.1002/ptr.7297
Liu L, Lim MA, Jung SN, Oh C, Won HR, Jin YL, Piao Y, Kim HJ, Chang JW, Koo BS (2021) The effect of curcumin on multi-level immune checkpoint blockade and T cell dysfunction in head and neck cancer. Phytomedicine 92:153758. https://doi.org/10.1016/j.phymed.2021.153758
Liu Z, Yu X, Xu L, Li Y, Zeng C (2022) Current insight into the regulation of PD-L1 in cancer. Exp Hematol Oncol 11:44. https://doi.org/10.1186/s40164-022-00297-8
Terlecki-Zaniewicz L, Lämmermann I, Latreille J, Bobbili MR, Pils V, Schosserer M, Weinmüllner R, Dellago H, Skalicky S, Pum D et al (2018) Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging (Albany NY) 10:1103–1132. https://doi.org/10.18632/aging.101452
Riquelme JA, Takov K, Santiago-Fernandez C, Rossello X, Lavandero S, Yellon DM, Davidson SM (2020) Increased production of functional small extracellular vesicles in senescent endothelial cells. J Cell Mol Med 24:4871–4876. https://doi.org/10.1111/jcmm.15047
Borghesan M, Fafian-Labora J, Eleftheriadou O, Carpintero-Fernandez P, Paez-Ribes M, Vizcay-Barrena G, Swisa A, Kolodkin-Gal D, Ximenez-Embun P, Lowe R, Martin-Martin B et al (2019) Small extracellular vesicles are key regulators of non-cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Rep 27:3956–3971.e6. https://doi.org/10.1016/j.celrep.2019.05.095
da Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, Ishaq A, Saretzki G, Nagaraja-Grellscheid S, Nelson G et al (2019) The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 18:e12848. https://doi.org/10.1111/acel.12848
Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, Montabana E, Lang UE, Fu Q, Fong L et al (2019) Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 177:414–427.e13. https://doi.org/10.1016/j.cell.2019.02.016
Dou D, Ren X, Han M, Xu X, Ge X, Gu Y, Wang X (2020) Cancer-associated fibroblasts-derived exosomes suppress immune cell function in breast cancer via the miR-92/PD-L1 pathway. Front Immunol 11:2026. https://doi.org/10.3389/fimmu.2020.02026
Ventura MT, Casciaro M, Gangemi S, Buquicchio R (2017) Immunosenescence in aging: between immune cells depletion and cytokines up-regulation. Clin Mol Allergy 15:21. https://doi.org/10.1186/s12948-017-0077-0
Salminen A, Kaarniranta K, Kauppinen A (2019) Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci 76:1901–1918. https://doi.org/10.1007/s00018-019-03048-x
Zerdes I, Wallerius M, Sifakis EG, Wallmann T, Betts S, Bartish M, Tsesmetzis N, Tobin NP, Coucoravas C, Bergh J et al (2019) STAT3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers (Basel) 11:1479. https://doi.org/10.3390/cancers11101479
Heckl SM, Mau F, Senftleben A, Daunke T, Beckinger S, Abdullazade S, Schreiber S, Röcken C, Sebens S, Schäfer H (2021) Programmed death-ligand 1 (PD-L1) expression is induced by insulin in pancreatic ductal adenocarcinoma cells pointing to its role in immune checkpoint control. Med Sci (Basel) 9:48. https://doi.org/10.3390/medsci9030048
Salminen A, Kaarniranta K, Kauppinen A (2021) Insulin/IGF-1 signaling promotes immunosuppression via the STAT3 pathway: impact on the aging process and age-related diseases. Inflamm Res 70:1043–1061. https://doi.org/10.1007/s00011-021-01498-3
Rincon M, Rudin E, Barzilai N (2005) The insulin/IGF-1 signaling in mammals and its relevance to human longevity. Exp Gerontol 40:873–877. https://doi.org/10.1016/j.exger.2005.06.014
Kirkland JL, Tchkonia T (2020) Senolytic drugs: from discovery to translation. J Intern Med 288:518–536. https://doi.org/10.1111/joim.13141
Acknowledgements
The author thanks Dr. Ewen MacDonald for checking the language of the manuscript.
Funding
Open access funding provided by University of Eastern Finland (including Kuopio University Hospital).
Author information
Authors and Affiliations
Contributions
This article is the sole work of the author.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salminen, A. The role of the immunosuppressive PD-1/PD-L1 checkpoint pathway in the aging process and age-related diseases. J Mol Med 102, 733–750 (2024). https://doi.org/10.1007/s00109-024-02444-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-024-02444-6