
Abstract
The aging process is associated with a remodeling of the immune system involving chronic low-grade inflammation and a gradual decline in the function of the immune system. These processes are also called inflammaging and immunosenescence. The age-related immune remodeling is associated with many clinical changes, e.g., risk for cancers and chronic infections increases, whereas the efficiency of vaccination and immunotherapy declines with aging. On the other hand, there is convincing evidence that chronic inflammatory states promote the premature aging process. The inflammation associated with aging or chronic inflammatory conditions stimulates a counteracting immunosuppression which protects tissues from excessive inflammatory injuries but promotes immunosenescence. Immunosuppression is a driving force in tumors and chronic infections and it also induces the tolerance to vaccination and immunotherapies. Immunosuppressive cells, e.g., myeloid-derived suppressor cells (MDSC), regulatory T cells (Treg), and type M2 macrophages, have a crucial role in tumorigenesis and chronic infections as well as in the tolerance to vaccination and immunotherapies. Interestingly, there is substantial evidence that inflammaging is also associated with an increased immunosuppressive activity, e.g., upregulation of immunosuppressive cells and anti-inflammatory cytokines. Given that both the aging and chronic inflammatory states involve the activation of immunosuppression and immunosenescence, this might explain why aging is a risk factor for tumorigenesis and chronic inflammatory states and conversely, chronic inflammatory insults promote the premature aging process in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
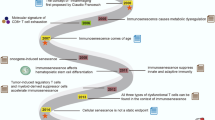
The aging process is associated with a remodeling of the immune system in humans and different animal species [1, 2]. The senescence of both immune and non-immune cells evokes a low-grade pro-inflammatory state, called the senescence-associated secretory phenotype (SASP) [3]. There is substantial evidence that the age-related remodeling of the immune system induces many clinical changes, e.g., the risk for cancers and chronic infections increases, whereas the efficiency of vaccination and immunotherapy decreases with aging (Fig. 1). Moreover, it is known that chronic inflammatory states promote the aging process (see below). The resolution phases of inflammatory insults are associated with counteracting immunosuppressive responses which are intended to protect tissues from excessive inflammatory injuries although immunosuppression also promotes the senescence of immune and non-immune cells [4, 5]. Consequently, this provokes many age-related clinical alterations described below. There is convincing evidence that the activation of immunosuppressive network, e.g., myeloid-derived suppressor cells (MDSC), regulatory T cells (Treg), and type M2 macrophages, has a crucial role in tumorigenesis, chronic infections, and the inefficiencies of vaccination and immunotherapy (see below). It seems that the activation of immunosuppressive network promotes the age-related immunosenescence which enhances the age-related clinical immune changes. Here, it will be examined the role of the age-related remodeling of the immune system in the generation of clinically observed immune alterations with aging. Original and review articles were searched from major databases including PubMed, Scopus, and Google Scholar.

Inflammaging and chronic inflammatory conditions remodel the immune system by inducing a counteracting immunosuppression which promotes immunosenescence. The immunosuppressive network involves MDSCs, Tregs, Bregs, DCregs, NKregs, NKT II cells, and type M2 macrophages. Subsequently, immunosuppression and an inefficient immune system disturb the efficacy of immune responses which increase the risk for cancers and chronic infections as well as decreasing the efficiency of vaccination and immunotherapy. Conversely, the increased immunosuppression associated with aging enhances transplantation tolerance. Abbreviation: Breg, regulatory B cell; DCreg, regulatory dendritic cell; MDSC, myeloid-derived suppressor cell; NKreg, regulatory natural killer cell; NKT II, type II natural killer T cell; Treg, regulatory T cell
Clinical interactions between aging and chronic inflammatory conditions
Aging is a major risk factor for chronic inflammatory diseases
There is extensive evidence that aging is a major risk factor for a wide variety of chronic diseases, especially for chronic inflammatory diseases. Given that the aging process is associated with a low-grade inflammatory state [1, 2], it is not surprising that chronic inflammation is involved in many diseases across the lifespan [6]. For instance, chronic inflammation prevails in cardiovascular diseases [7], chronic kidney diseases [8], and neurodegenerative diseases [9]. Moreover, chronic inflammation has a crucial role in the pathogenesis of many cancers [10], the incidence of which significantly increases with aging [11]. Commonly, multimorbidity and frailty exist in the elderly and mortality increases exponentially in aged-related diseases [12, 13]. Several studies have revealed that inflammatory markers predict frailty status and mortality in older people [14, 15].
Centenarians are living examples of a successful aging process and thus these people have been studied in attempts to understand the mechanisms underpinning healthspan regulation. A survey of several community-based prospective studies indicated that the inflammation score significantly predicted the all-cause mortality, capability, and cognition in individuals of extreme old age [16]. Interestingly, the inflammation index of centenarians’ offspring was lower than that of their age-matched controls. Rubino et al. [17] demonstrated in their study on the offspring of Sicilian centenarians that these individuals were more resistant to the aging-induced immune changes than their counterparts. For instance, the serum level of Tregs and γδ T cells as well as that of senescent T cells were significantly lower in the offspring of centenarians as compared to those values of their non-centenarian counterparts. There are other studies indicating that the offspring of centenarian people have a lower prevalence of several age-related diseases [18]. These results imply that epigenetic factors regulate the healthspan of centenarians and their offspring. The epigenetic clock based on DNA methylation has been used to evaluate the biological aging process [19]. Horwath et al. [20] reported that the DNA methylation level of peripheral blood mononuclear cells (PBMC) from Italian semi-supercentenarians and their offspring revealed a decreased epigenetic age compared to the age-matched controls. Currently, the epigenetic regulation of the healthspan and lifespan needs to be clarified. Interestingly, Jylhävä et al. [21] demonstrated that the mortality-associated methylation sites mapped to genes functionally clustering around the nuclear factor κB (NF-κB) complexes in nonagenarians. NF-κB transcription factor is a major driver of inflammatory responses. There is abundant evidence that NF-κB signaling seems to be the molecular culprit of inflammaging process controlling both healthspan and lifespan [22]. It seems that the age-related inflammatory state predisposes an organism to chronic inflammatory diseases which are also driven by the activation of NF-κB signaling.
Chronic inflammatory disorders promote premature aging in humans
There is convincing evidence that chronic inflammatory insults experienced during childhood or adulthood can enhance the premature aging process in humans. There are examples of both local and systemic inflammatory states which can promote the aging process. For instance, the UV-radiation (UVR)-induced photoaging of the skin is a well-known example of an accelerated aging process driven by repeated inflammatory insults [23, 24]. Recent studies have revealed that UVR and environmental pollution trigger damages in the skin, e.g., oxidative stress and DNA-damage, which evoke a cellular senescent state in skin fibroblasts and keratinocytes [25, 26]. Senescent cells, which possess an SASP phenotype, are secretory cells releasing many inflammatory factors, e.g., cytokines, chemokines, and metalloproteinases. A senescent state is common in aging tissues promoting the aging process and also age-related diseases [27]. Tumorigenesis is another example of an increased chronic inflammatory state which can promote both local and systemic premature aging processes in cancer survivors [28, 29]. The cellular senescence and inflammation occurring in tumor sites enhance the tumorigenesis process. Several epidemiological studies have revealed that either cancer itself or cancer therapies accelerate the biological aging process, e.g., osteoporosis, muscle atrophy, pulmonary fibrosis, cardiotoxicity, and characteristics of frailty [28, 30]. Moreover, cancer survivors suffer from impaired wound healing, increased infections, decreased cognitive status, chronic fatigue, and increased morbidity. These properties indicate that cancers promote premature aging in cancer-survivors.
It is known that chronic infections enhance immunosenescence and are able to promote the aging process in humans (Fig. 1). Cytomegalovirus (CMV) infections increase with aging and in fact, most elderly people are CMV-seropositive. However, CMVs are normally living in a dormant state in the host cells but their reactivation triggers a chronic low-grade inflammation. Several investigators have revealed that CMV infection is able to enhance immunosenescence [31, 32] although this topic is still a matter of some debate. Recently, Hassouneh et al. [33] demonstrated that there are clear differences in the immunosenescent states induced by the aging process or CMV infection, e.g., in alterations of the T cell subsets. Genetic studies have revealed that CMV infection accelerated the epigenetic aging profile of human PBMC cells [34, 35]. Given that CMV infection affects the properties of immune cells and impairs immune responses, it is not surprising that it increases the risk for age-related diseases [31, 36]. The immunosenescence induced by CMV and the ageing process seem to induce a more severe COVID-19 disease in elderly people [37]. There are studies indicating that patients suffering from human immunodeficiency virus (HIV) infection display a premature onset of age-related morbidities, e.g., cardiovascular diseases, osteoporosis, cognitive impairments, and frailty [38, 39]. Gianesin et al. [40] demonstrated that a significant premature aging and immunosenescent phenotype were evident in HIV-infected children. The telomeres of PBMCs were considerably shorter in the HIV-infected than uninfected children. There appeared significant changes in T cell population, e.g., lower thymic output and reduced maturation level of T cell receptors (TCR). It is noteworthy that these alterations were not attributed to antiretroviral therapy, instead these changes indicated the presence of a premature immunosenescent state in the HIV-infected children [40].
There are several other chronic inflammatory states which enhance immunosenescence and promote a premature aging process, e.g., chronic kidney disease (CKD) [8, 41], chronic obstructive pulmonary disease (COPD) [42], and rheumatoid arthritis [43] (Fig. 1). It is known that CKD triggers a persistent uremic inflammation which promotes the appearance of the immunosenescent phenotype involving the loss of thymic function, a decrease in the numbers of naïve B and T cells, the expansion of memory T cell population, and a skewed TCR repertoire [44]. CKD also exposes the individual to the co-morbidity of many age-related diseases. The pathogenesis of CKD involves a typical array of inflammatory changes, such as oxidative stress, mitochondrial dysfunction, and cellular senescence [8]. One specific indicator of CDK is a major decline in the production of α-klotho protein, a well-known anti-aging protein [45]. The reduced secretion of klotho protein from kidney disturbs cellular calcium/phosphate metabolism, thus enhancing co-morbidities, such as atherosclerosis, osteoporosis, and skin atrophy. Klotho protein has many anti-inflammatory properties which confer protection against the immune senescence of human monocytic leukemia cell line-1 (THP-1) [46]. Klotho-deficient mice display many pathological properties which are similar to those appearing during the aging process [47]. Given that the expression of klotho decreases with aging, it seems that the decline in the expression of klotho protein in CKD promotes premature aging processes in the whole body. COPD is also associated with an increase in the hallmarks of aging, e.g., epigenetic changes, telomere shortening, cellular senescence, immunosenescence, and low-grade inflammation [42]. COPD is also accompanied by several co-morbidities. In conclusion, it seems that the persistent inflammatory state, either in the aging process itself or in inflammatory diseases, enhances an accelerated aging process with the appearance of more and more co-morbidities with aging. The mechanisms underlying the inflammation-driven aging process need to be clarified. It is known that chronic inflammation triggers a counteracting activation of the immunosuppressive network which not only induces immunosenescence but also increases cellular senescence in inflamed tissues and in the whole body (see below).
Inflammation stimulates immunosuppression
Acute inflammatory states stimulate compensatory anti-inflammatory responses which counteract excessive inflammatory reactions, promote resolution processes, and thus prevent unneeded damages to the host tissues [48, 49]. In chronic inflammation, immunosuppressive activity has a crucial role in the stabilisation of inflamed microenvironment. Inflammatory mediators, such as colony-stimulating factors (CSF), chemokines (e.g., CCL2), and interferons, stimulate the expansion of the myelopoietic lineage and augment the generation of myeloid cells in the bone marrow (BM). Increased myelopoiesis stimulates the generation of immature myeloid-derived suppressor cells (MDSC), both monocytic M-MDSCs and polymorphonuclear PMN-MDSCs, which are released from the BM and are subsequently recruited into extramedullary immune sites, e.g., spleen and lymph nodes, or directly into inflamed tissues [50, 51]. In the target sites, MDSCs proliferate and enhance their immunosuppressive potentials to suppress pro-inflammatory responses. The immune cells are exceedingly plastid and the phenotype of immune cells can be modulated from a pro-inflammatory to an immunosuppressive state, a process also called polarization [52]. For instance, monocytes and granulocytes can be converted into immunosuppressive M-MDSCs and PMN-MDSCs in an environment where there is persistent inflammation. Moreover, MDSCs can promote the differentiation of resting T cells to immunosuppressive Tregs [53, 54]. Tregs are a heterogeneous group of immunosuppressive T cells, e.g., the thymic Tregs (tTreg) maintain self-tolerance, whereas inducible Tregs (iTreg) prevent excessive inflammation. There are also tissue-resident, specific Treg populations, e.g., in the skin [55] and the adipose tissue [56]. The Forkhead box P3 (FoxP3) protein is the epigenetically-regulated master protein of the Tregs. Tregs are crucial immunosuppressive cells since they suppress the functions of effector T and B cells, inhibit the activity of natural killer (NK) cells, and promote the formation of other immunosuppressive cells, e.g., tolerogenic dendritic cells (DC) and anti-inflammatory M2 macrophages [57,58,59].
The host cells in the inflammatory microenvironment are able to educate immune cells, e.g., to switch pro-inflammatory cells to immunosuppressive phenotypes [60]. For instance, pro-inflammatory macrophages, called M1 macrophages, can be polarized to immunosuppressive M2 phenotypes in chronic inflammation [52]. The tumor microenvironment with persistent inflammation contains immunosuppressive tumor-associated macrophages (TAM) which suppress the antitumor activity of effector immune cells [61]. The immunosuppressive network also includes regulatory subsets of B cells (Breg) [62], DC cells (DCreg) [63], NK cells (NKreg) [64], and type II NKT cells [65] (Fig. 1). These immunosuppressive subsets of each immune cell type are heterogenous with respect to their phenotypes and functions and in addition, there exists a clear tissue-specificity in their properties. These regulatory cells have not been as extensively studied as MDSCs, Tregs, and M2 subsets of macrophages. Recently, I have reviewed the common properties of each member of immunosuppressive network and discussed how their changes are associated with the aging process [59]. Immunosuppressive cells possess a diverse armament of tools with which they not only regulate the immunosuppressive potency of the network but also suppress the functions of effector immune cells, e.g., the properties of T, B, DC, and NK cells. Common immunosuppressive mechanisms include (i) secretion of anti-inflammatory cytokines, such as TGF-β, IL-4, IL-10, IL-1 receptor antagonist (IL-1ra), (ii) release of reactive oxygen and nitrogen species (ROS/RNS), (iii) secretion of immunosuppressive compounds like adenosine and prostaglandin E2 (PGE2), (iv) expression of inhibitory immune checkpoint receptors, e.g., programmed cell death protein-1 (PD-1)/PD-L1 and cytotoxic T-lymphocyte associated protein 4 (CTLA4), and (v) expression of enzymes catabolising amino acids, such as indoleamine 2,3-dioxygenase 1 (IDO1) and arginase 1 (ARG1) [4, 66,67,68]. The remodelling of immune cells toward either an immunosuppressive or an immunosenescent state is underpinned by epigenetic regulation [69, 70].
Immunosuppressive activity induces senescence state in immune and non-immune cells
The purpose of immunosuppression is to prevent excessive inflammatory responses although recent observations have indicated that the immunosuppressive period can be extended to the post-resolution phase since immunosuppressive cells can support the repair processes of host tissues [71, 72]. Indeed, the suppressive armament exploited by immunosuppressive cells (see above) contains many tools, e.g., secretion of IL-10, TGF-β, PGE2, and ROS/RNS as well as the depletion of distinct amino acids, which are not only drivers of immunosenescence but they can also enhance the senescence of non-immune cells [4, 5]. Senescent immune cells display a reduced ability to function as active effector cells which is attributed to the remodeling process in the immune system, especially in persistent inflammatory states [5, 73,74,75,76] (Fig. 1). Interestingly, immunosuppressive cells can induce changes in the phenotypes of immune cells which are very similar to those of immunosenescent cells. For instance, Tregs can induce the senescent phenotype of T cells [76]. Moreover, M-MDSCs and Tregs reduce the efficacy of immunosurveillance and the cytotoxic activities of NK and CD8 T cells [77,78,79]. For instance, an exposure to TGF-β suppressed the cytotoxic activity of human NK and CD8 T cells by inhibiting the expression of natural killer group 2D (NKG2D) receptor [80]. Nagaraj et al. [81] demonstrated that MDSCs were able to nitrate the TCR complex and thus inhibiting the antigen recognition of mouse tolerant CD8 T cells. Furthermore, MDSCs can impair the properties of DCs and B cells [74, 82]. In fact, immunosenescent cells display many of the characteristics encountered in the senescence of non-immune cells, e.g., telomere shortening, DNA damages, augmented expression of SA-β-Gal and cell cycle inhibitors (p16INK4a, p21WAF1, p53) as well as increased oxidative and endoplasmic reticulum stresses [5, 76, 83, 84]. Immunosenescence is not only associated with the aging process but it commonly occurs in chronic inflammatory states, especially those associated with the aging process [85, 86]. The accumulation of immunosenescent cells also occurs in chronic infections and autoimmune diseases, such as rheumatoid arthritis. [87, 88].
As discussed above, inflammatory mediators evoke a counteracting immunosuppression which impairs the function of NK and CD8 T cells. Consequently, the surveillance of senescent cells is inhibited because NK cells and CD8 T cells are the major surveying cells safeguarding tissue homeostasis. This impaired immune surveillance leads to an accumulation of senescent cells in both the immune system and peripheral tissues [89]. It is known that NK cells can recognize senescent fibroblasts and eliminate them through perforin-mediated cytolysis [90]. Ovadya et al. [89] demonstrated that the ablation of the Perforin 1 (Prf1) gene in mice increased the accumulation of senescent cells within tissues, triggered chronic inflammatory state, and finally reduced the survival of the animals. The NKG2D receptors of NK and CD8 T cells recognize the NKG2D ligands, especially the increased levels of MHC class I polypeptide-related sequence A (MICA) and UL16-binding protein 2 (ULBP2) proteins, on the surface of senescent cells and subsequently trigger their clearance [91]. However, inflammaging and chronic inflammatory states can impair the function of the NKG2D/NKG2D ligand axis and thus impair the cytotoxicity of NK and CD8 T cells. For instance, immunosuppressive cells, e.g., MDSCs, Tregs, and M2 macrophages, can inhibit the expression of NKG2D in NK cells through membrane-bound TGF-β1 signaling or the IDO1-mediated kynurenine secretion [92, 93]. Recently, I reviewed the mechanisms which immunosuppressive cells exploit to inhibit the surveillance and clearance of senescent cells [94]. I proposed a feed-forward model in which chronic low-grade inflammation induced a compensatory immunosuppression which subsequently enhanced the accumulation of pro-inflammatory senescent cells. It is well known that senescent cells, both immune and non-immune cells, secrete pro-inflammatory mediators and thus they are able to propagate an inflammatory state both during aging and in chronic inflammatory states [3, 5, 95].
Immunosuppressive activity increases with aging
The aging process is associated with a remodeling of the immune system, both innate and adaptive immunity. It seems that the chronic inflammaging state triggers the remodelling process stimulating a counteracting immunosuppression which leads to immunosenescence (Fig. 1). This pathological highway is well known in many chronic inflammatory states (see below). Currently, although inflammaging and immunosenescence have been widely recognized as the driving forces behind the aging process, these two processes are not able to explain many clinical observations without the inclusion of an immunosuppressive component (see below). There is convincing evidence that inflammaging is associated with increased immunosuppressive activity, as also occurs in other chronic inflammatory conditions. For instance, inflammaging enhanced myelopoiesis in the bone marrow [96], a process which was associated with an age-related increase in the level of immunosuppressive MDSCs in the circulation of mice [97] and humans [98]. Accordingly, the number of Tregs was augmented with aging in human blood [78, 99]. Studies on the bone marrow (BM) and secondary lymphoid organs have revealed that in these tissues the upregulation in the numbers of MDSCs and Tregs with aging was more robust than in the circulation. For instance, Flores et al. [100] demonstrated that the percentage of MDSCs robustly increased with aging in both mouse BM and spleen. There are numerous reports which have revealed that the presence of MDSCs significantly increased with aging in mouse spleen and lymph nodes [97, 101]. Correspondingly, the level of Tregs was also augmented with aging in the spleen and lymph nodes [99, 102]. However, there exist different subsets of Tregs, e.g., thymic (tTreg), peripheral (pTreg), and inducible (iTreg), which have some similar and some distinctive properties [103]. It is known that the levels of natural FoxP3+ Tregs (nTreg), originated either from CD4 T or CD8 T cells, increase with aging, whereas those of iTreg and tTreg decline during the aging process [104, 105]. The decrease in the numbers of tTreg cells was most probably associated with an age-related involution of the thymus. However, Szurek et al. [106] demonstrated that the markers of tTreg and iTreg, i.e., helios and neuropilin-1 (Nrp-1), did not distinguish tTreg cells from iTregs in mice. Interestingly, van der Geest et al. [107] revealed that the proportion of the naïve Tregs declined with aging in human blood, whereas that of the memory Tregs (memTreg) clearly increased. Moreover, the level of memTregs inversely correlated to the human vaccination efficiency [107]. In conclusion, it seems that the remodeling of the immune system involves a significant increase in the accumulation of immunosuppressive MDSCs and Tregs.
There are some technical problems to quantify whether the presence of MDSCs and Tregs increases with aging in non-immune tissues. However, there are observations indicating that the level of MDSCs increased with aging in mouse and human skin [108]. Ruhland et al. [108] reported that the numbers of CD14, CD15, and CD33-positive cells were robustly increased in the skin of elderly people. These proteins are common markers of human MDSCs. They also demonstrated that stromal senescence enhanced the occurrence of both MDSCs and Tregs within mouse skin. The role of Tregs in the aging process is still far from clear since tissues contain several unique populations of tissue-resident Tregs which are crucially involved in the regulation of tissue homeostasis [58, 109]. For instance, the Tregs found in mouse adipose tissue are involved in the regulation of energy metabolism and insulin resistance. The numbers of Treg cells significantly increased with aging in the visceral adipose tissue of lean mice, whereas obesity induced a decline in the numbers of Tregs [110, 111]. However, Bapat et al. [112] demonstrated that the depletion of fat-resident Tregs prevented the development of age-associated insulin resistance (IR). It is known that there exist two types of insulin resistance, i.e., the age-related IR and the obesity-associated IR. Given that obesity increases inflammation, it seems that the obesity-associated IR was induced by the tissue-infiltrated Tregs.
In addition to MDSCs and Tregs, there are clear changes with aging in the phenotypes of other members of the immunosuppressive network [59]. For instance, tissue-resident macrophages displayed an increased polarization toward the anti-inflammatory M2 phenotype not only in BM, spleen, and lymph nodes but also in lungs and skeletal muscles [113, 114]. However, because macrophages are remarkably plastic cells, they can adapt to local disturbances in their microenvironment and thus express the M1 phenotype. Nonetheless, Duong et al. [115] demonstrated that the depletion of macrophages in elderly mice augmented the antitumor activity of T cells and improved the responses of mice to tumor immunotherapy. These observations indicate that macrophages can augment their immunosuppressive activity during the aging process. There is convincing evidence that both MDSCs and Tregs in aged mice/humans are immunosuppressive, i.e., they suppress the antigen-induced proliferation of T cells, increase the production of IL-10, TGF-β, and ROS, inhibit the maturation and function of DCs, decrease the cytotoxic activity of NK and CD8 T cells and increase the susceptibility to infections and cancer formation [78, 97, 101, 102]. It seems that the low-grade inflammation associated with aging is a driving force for immunosuppression and subsequent immunosenescence.
Increased immunosuppression is associated with chronic inflammatory states
Systemic chronic inflammation has a major role in the pathogenesis of many chronic diseases, e.g., cardiovascular diseases (CVD), COPD, CKD, hepatic steatosis, many cancers, autoimmune diseases, and neurodegenerative diseases [6]. There is a multitude of causes for chronic inflammatory conditions; many of these are still unknown such as that of the aging process. Nonetheless, a chronic inflammatory state activates immunosuppressive cells which impair the functional abilities of the immune system, thus promoting immunosenescence (Fig. 1). Studies conducted on cardiovascular diseases, e.g., atherosclerosis, hypertension, acute coronary syndrome, and stroke, have indicated that immunosuppressive cells were involved in the pathology of CVD [116,117,118,119]. For instance, the occurrence of a stroke increased the level of Tregs which attenuated inflammatory responses and enhanced the appearance of post-stroke regeneration [119, 120]. Accordingly, stroke increased the level of circulating M-MDSCs in human stroke patients [121]. It is known that stroke increases the risk for post-stroke infections, probably due to immunosenescence (see below). In COPD patients, the number of circulating MDSCs significantly increased [122], whereas the Th17/Treg balance was upregulated in the acute phase of COPD but significantly decreased when COPD moved to the stable phase [123]. This indicated that there existed an immunosuppressive state in the stable COPD. Chronic inflammation enhanced the progression of non-alcoholic fatty liver disease (NAFLD) which is a world-wide public health problem [124]. Zhou et al. [125] demonstrated that the accumulation of MDSCs into human liver during the progression of NAFLD positively correlated with the clinical parameters of liver disease. There is clear evidence that the balance between Th17 and Treg cells has a significant role in the progression of NAFLD in both mice and humans [126, 127]. An increased level of pro-inflammatory Th17 cells enhanced the progression of NAFLD, whereas Tregs prevented the generation of NAFLD.
Many persistent viral infections, e.g., HIV and herpes simplex virus (HSV), stimulate a chronic low-grade inflammatory state which is associated with increased immunosuppression and immunosenescence [128,129,130]. More severe, pathogen-induced sepsis also triggers both pro-inflammatory and immunosuppressive phases [131, 132]. It seems that MDSCs, especially M-MDSCs, have a key role in the resolution of inflammation in infectious foci induced by bacteria or viruses. In non-resolving infections, MDSCs stimulate the immunosuppressive microenvironment which prevents excessive inflammation [128]. For instance, Sarkar et al. [129] demonstrated that in mouse ocular HSV1 infection, MDSCs suppressed the proliferation and cytokine production of activated CD4 T cells, i.e., MDSCs induced the immunosenescence of CD4 T cells. They also reported that an adoptive transfer of the in vitro-generated MDSCs into infected mice expanded the pool of Tregs and subsequently reduced the severity of the HSV1-induced ocular infection. Currently, there are observations that COVID-19 infection induced a massive expansion of PMN-MDSCs which inhibited the specific T cell responses and increased the fatal outcome in patients infected by COVID-19 [133]. Accordingly, the number of Tregs and the expression level of FoxP3 protein were robustly accentuated in the blood of COVID-19 patients and these changes closely correlated with the severity of the disease [134]. The robust increase in the levels of MDSCs and Tregs in the COVID-19 syndrome indicates that overwhelming inflammation activates immunosuppressive cells which induce immunosenescence and an inefficient immune defence. It seems that the aging-induced immunosuppressive changes leading to immunosenescence in the immune system enhance the severity of the COVID-19 syndrome in older patients [135].
Chronic inflammation is an important player in different phases of tumorigenesis [10]. Inflammation is able to predispose to the development of tumors and subsequently the inflammatory tumor microenvironment (TME) enhances the formation of local immunosuppression [82, 136]. The immunosuppressive state is generated by the presence of both invading and resident immunosuppressive cells, e.g., TAMs, MDSCs, Tregs, and cancer-associated fibroblasts (CAF) [136,137,138]. Immunosuppression induces an immunosenescent state in TME by inhibiting the normal functions of macrophages, T cells, NK cells, and dendritic cells [139]. This local immunosenescence allows tumor cells to evade immune surveillance. The inflammatory state not only triggers the senescence of immune cells but it also elicits cellular senescence in non-immune cells which secrete pro-inflammatory mediators (SASP state), thus aggravating the inflammatory state in TME [140]. This kind of immunosuppression-induced immune senescence poses a challenge for cancer immunotherapies. Given that chronic inflammation, immunosuppression, and immunosenescence are involved in the aging process (see above), it has been claimed that an increased risk for tumors with aging could be attributed to an inflammaging process [141].
Photoaging is another local chronic process in which repeated UV-induced inflammatory insults enhance immunosuppression in the skin and consequently accelerate its aging process [142,143,144]. Skin exposure to UVR not only enhances photoaging but can also make the affected skin susceptible to carcinogenesis [145]. UVR induces both the local and systemic attenuation of the immune system [146, 147]. It has been reported that the induction of Tregs has a key role in the generation of UV-induced immunosuppression [148, 149]. The presence of tissue-resident Tregs is enriched in normal skin involving a heterogeneous population of Treg cells [150]. Treg cells have important functions in the skin, e.g., they augment wound healing, participate in hair follicle regeneration, prevent autoimmunity, and maintain immune tolerance against skin commensal microbes. It seems that there are different mechanisms involved in the activation of Tregs by UVR. For example, Soontrapa et al. [146] demonstrated that the UVB-induced stimulation of Tregs was mediated by PGE2 via signaling through the EP4 receptors in mouse epidermis. Moreover, Navid et al. [151] revealed that UVR stimulated aryl hydrocarbon receptor (AhR) signaling which triggered the Treg-mediated immunosuppression in mouse skin. It is known that UVR promotes the catabolic breakdown of tryptophan to kynurenine and 6-formylindolo [3,2-b]-carbazole (FICZ) which are potent activators of AhR signaling and subsequently they stimulate FoxP3 expression and the activation of Tregs [152, 153]. Several studies have revealed that an increased expression of AhR factor in skin also predisposed to the development of skin cancers [154]. Photoaging is an interesting example of how the immunosuppression induced by repeated inflammatory insults not only accelerates the aging process but is also a risk factor for the development of skin cancers.
Clinical disadvantages of age-related increase of immunosuppression
As stated above, the aging process increases the risk for cancer development and it aggravates infections and several inflammatory diseases. On the other hand, many cancers and chronic inflammatory conditions accelerate the aging process (see above). It seems that inflammation and the subsequent immunosuppression, triggered either by the aging process or tumors, possess similar properties which mutually can promote each other’s pathology. The immunosenescence encountered in both states provides an escape of senescent cells and tumor cells from immune surveillance by NK and CD8 T cells. For instance, Ruhland et al. [108] demonstrated that an experimentally induced senescence of stromal cells in mouse skin increased inflammation and established an immunosuppressive microenvironment which promoted tumorigenesis. The SASP state of senescent cells increased the secretion of IL-6 which triggered the expansion of MDSCs and Tregs in mouse skin. There is convincing evidence that the inflammation-induced recruitment and expansion of MDSCs and Tregs promote the age-related tumor incidence in different tissues [101, 155, 156]. It has been recognized that chronic inflammation and immunosuppression shape the chromatin landscape which might modify the genetic background not only during the aging process but in many other diseases [157, 158]. Interestingly, many investigators have reported that the epigenetic aging profile (see above) predicts the risk of cancer, cardiovascular disease, and mortality in many diseases [159, 160]. Currently, the molecular significance of the aging-associated epigenetic drift in tumorigenesis needs to be clarified.
Aged people are not only susceptible to tumorigenesis but also infections induced by diverse pathogens. There is convincing evidence that the age-related increase in immunosenescence is a major cause of an increased sensitivity to infections in the elderly [135, 161, 162]. An increased susceptibility to infections has also been observed in individuals suffering from many chronic inflammatory diseases and immunocompromised states. These conditions display an enhanced immunosenescence although there are differences in the decline in immune defence. As discussed above, infections are associated with an increased activity of immunosuppressive cells which evoke immunosenescence of effector immune cells (Fig. 1). For instance, the age-related immunosenescence affects infections since it (i) induces defects in the functions of neutrophils, (ii) decreases the responses of T cells, e.g., the affinity of TCR for antigens and the cytotoxicity of CD8 T cells, (iii) diminishes the maturation and antigen presentation by DCs, (iv) disturbs the functions of B cells, (v) down-regulates the activating receptors of NK cells [161, 163,164,165]. The age-related decline in immune host defence increases a person’s susceptibility to infections as well as promoting the persistence of infections. Currently, it seems that cytokines secreted by immunosuppressive cells, e.g., IL-10 and TGF-β, are important epigenetic regulators which suppress the functions of effector immune cells by modifying their chromatin landscape [5, 166, 167].
It is known that vaccination efficiency decreases with aging. This decline has been attributed to the age-related immune deficiency, i.e., immunosenescence [161, 164, 168]. Vaccination efficiency is reduced in cancers and chronic inflammatory states, such as obesity [169], indicating that immunosenescence is involved. In addition, chronic CMV infection reduces the responsiveness to influenza vaccination in aged people [170]. The decline in vaccination efficacy has been observed in different vaccination protocols involving influenza and cancer vaccines. There is convincing evidence that the decrease in vaccination efficiency with aging is caused by an increased level of immunosuppressive cells, e.g., MDSCs and Tregs [169, 171, 172]. Corsini et al. [171] demonstrated that an increased IL-10 level in the elderly was associated with a low antibody response to influenza vaccination. Moreover, a decrease in the level of MDSCs was reported to improve the vaccination efficacy [173]. Currently, there are several strategies aiming to improve vaccination efficiency in elderly people. For instance, attenuating immunosenescence via the inhibition of immunosuppression might be a successful approach.
There are many studies indicating that the efficiency of immunotherapeutic treatments becomes diminished in elderly people [174]. The senescence of the immune system is a major difficulty in the activating immunotherapies, e.g., in the treatments of cancer patients [175, 176]. Given that aging and cancers increase immunosuppression and subsequently promote immunosenescence (Fig. 1), there are different approaches to inhibit the functions of MDSCs and Tregs [177, 178]. It is known that MDSCs mount a major resistance to immune checkpoint inhibitor (ICI) therapies [179]. The clinical efficiency of ICI treatments can be improved by the combination therapies which involve the chemical inhibition of MDSC function. For instance, the combination of gemtuzumab ozogamicin (GO), an immunotoxic conjugate linking CD33 antibody with cytotoxic calicheamicin, has been exploited to target and eliminate MDSCs in human cancer patients [180]. Fultang et al. [180] demonstrated that in the treatment of acute myeloid leukemia, the GO therapy targeted MDSCs and also improved the chimeric antigen receptor T cell (CAR-T) immunotherapy. Moreover, tumor immunosuppression can be attenuated and immunotherapy enhanced by using STAT3 inhibitors [181], all-trans retinoic acid (ATRA) [182], and many phytochemicals [183]. The provision of Treg immunotherapy is a more difficult challenge since Tregs maintain immunological tolerance and immune homeostasis. However, there are some specific surface molecules on Treg cells which can be targeted in cancer immunotherapy [178]. Hurez et al. [184] demonstrated that the depletion of MDSCs improved antitumor immunity in aged mice, whereas a reduction in the numbers of Tregs increased the level of MDSCs in old mice. They also revealed that the exhaustion of MDSCs was an effective treatment in aged but not in young mice bearing B16 tumors. However, combining the depletions of both MDSCs and Tregs improved the therapeutic efficacy in old B16-bearing mice [184]. These few examples clearly indicate that an increased level of immunosuppressive activity is a real problem in exploiting immunotherapy in the elderly.
Increased immunosuppression with aging not only induces immune deficiency but it also disturbs the homeostasis of the host tissues. Immunosuppressive cells possess a molecular armament which inhibits effector immune cells, e.g., anti-inflammatory cytokines, ROS/RNS, and enzymes catabolizing amino acids (see above). These mechanisms also affect the neighboring non-immune cells in host tissues [4]. For instance, TGF-β signaling is able to induce cellular senescence and tissue fibrosis as well as evoking a remodelling of the extracellular matrix (ECM) [185,186,187]. An increase in the level of TGF-β signaling has been associated with many age-related diseases, such as muscle and skin atrophies and cardiovascular diseases. Accordingly, IL-10 signaling can inhibit autophagy [188] which is known to decline with aging. IL-10 and TGF-β cytokines can also modify the chromatin landscape via the STAT3 and SMAD pathways and thus induce epigenetic changes in gene expression profiles with aging. The secretion of ROS/RNS by immunosuppressive cells is an important mechanism to inhibit the activity of effector immune cells and thus generate immunosenescence [189]. Many researchers believe that ROS and oxidative stress play a crucial role in tissue degeneration involved in the aging process. In addition, immunosuppressive cells robustly express ARG1 and IDO1 enzymes which catabolize L-arginine (L-Arg) and L-tryptophan (L-Trp), respectively [67]. Since there is an increase in the activities of ARG1 and IDO1 in inflammatory conditions, this will deplete L-Arg and L-Trp amino acids and thus not only suppress the proliferation of immune cells but also inhibit the protein synthesis in neighboring cells, thus enhancing tissue atrophy [4]. Tissue atrophy is typically encountered in both the aging process and chronic inflammatory diseases.
Clinical benefits of the age-related increase of immunosuppression
Increased immunosuppression and subsequent immunosenescence have not only harmful effects but there are studies indicating that age-related immune deficiency can confer some benefits, e.g., transplantation tolerance is enhanced with aging [190,191,192,193]. It is known that the age of donor and recipient patients determines the outcome of tissue transplantation [194]. There is a common rule indicating that old persons are poor donors but good receivers. However, it is dependent on the tissue being transferred since for instance, kidney transplantation requires a younger donor than that of heart transfer [194]. The immune senescence of the recipient patient improves the acceptance of transplants of different organs [190, 195, 196]. The better tolerance of transplants in older recipients is attributed to the age-related increase in the immunosuppressive activity which inhibits the functions of effector immune cells, such as the surveillance by NK and CD8 T cells (see above). There are many studies indicating that the transplantation tolerance is induced by the expansion of immunosuppressive regulatory myeloid cells, i.e., MDSCs, Tregs, DCregs, and M2 macrophages [192, 193, 197]. In proof of principle, adoptive cell therapies with immunosuppressive cells have significantly improved transplantation tolerance [198].
Immunosenescence is associated with many autoimmune diseases, e.g., rheumatoid arthritis and multiple sclerosis [87, 199]. With respect to the role of aging, there are significant differences between the diverse autoimmune diseases. However, it seems that the incidence of autoimmune diseases increases with aging although the severity of diseases can be reduced [200, 201]. On the other hand, some autoimmune diseases, e.g., rheumatoid arthritis, accelerate the aging process, similarly to the situation in many other chronic inflammatory diseases [43, 200]. There is substantial evidence that MDSCs have a crucial protective role in autoimmune diseases [202,203,204]. Given that MDSCs suppress T cell responses and the activity of NK cells, it is not surprising that MDSCs are able to enhance immune tolerance in autoimmune diseases. However, there are many questions on their specificity in different tissues and diseases, especially related to the function of separate subpopulations of MDSCs [204]. It seems that PMN-MDSCs are more immune suppressive than M-MDSCs, whereas M-MDSCs promote the development of Tregs and inhibit the functions of B cells, e.g., in autoimmune arthritis. Treg cells maintain self-tolerance and thus disturbances in their activity are thought to lead to the development of autoimmune diseases [205]. There are a variety of approaches, either drug-based or cell-based therapies, targeting both MDSCs and Tregs aiming to enhance their immunosuppressive properties in autoimmune diseases [204,205,206]. Currently, adoptive cell therapies exploiting engineering technologies in the production of the antigen-specific Tregs and chimeric antigen receptor (CAR) Tregs have been developed to induce specific immune tolerance in autoimmune diseases [206, 207]. It does seem that the age-related increase in immunosuppressive activity is able to reduce the severity of autoimmune diseases but its level and specificity are insufficient to totally resolve the disease.
Conclusions
Chronic inflammatory states stimulate a counteracting immunosuppression which promotes immunosenescence in an attempt to protect tissues against excessive and detrimental inflammatory processes. There is convincing evidence that the age-related chronic low-grade inflammation also induces immunosuppression which consequently enhances the senescence of the immune system. Interestingly, there are observations that the aging process increases the risk for cancers as well as augmenting the risk for suffering chronic inflammatory diseases and conversely, repeated or persistent inflammatory states accelerate the aging process. This means that inflammatory components are a major driving force in the aging process. On the other hand, an age-related increase in immunosuppressive activity and immunosenescence explain many clinical consequences associated with aging. For instance, the incidence of cancers increases with aging since immunosuppression has a crucial role in tumorigenesis. Moreover, elderly people are susceptible to long-term infections, i.e., increased immunosuppression inhibits the function of effector immune cells and thus the resolution of infections tends to be prolonged in the elderly. It is also known that there are declines in both vaccination efficiency and the efficacy of immunotherapies in aged people. The senescent immune system, associated with either aging, cancers, or chronic inflammatory diseases, is unable to respond effectively to vaccines and antibodies. Currently, there are trials in progress with many drug- or cell-based therapies attempting to reinforce the inefficient immune system not only in cancer and chronic inflammatory states but also in the aging process.
References
Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci 908:244–254. https://doi.org/10.1111/j.1749-6632.2000.tb06651.x
Benayoun BA, Pollina EA, Singh PP, Mahmoudi S, Harel I, Casey KM, Dulken BW, Kundaje A, Brunet A (2019) Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 29:697–709. https://doi.org/10.1101/gr.240093.118
Coppe JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144
Salminen A (2021) Increased immunosuppression impairs tissue homeostasis with aging and age-related diseases. J Mol Med (Berl) 99:1–20. https://doi.org/10.1007/s00109-020-01988-7
Salminen A (2021) Immunosuppressive network promotes immunosenescence associated with aging and chronic inflammatory conditions. J Mol Med (Berl) 99:1553–1569. https://doi.org/10.1007/s00109-021-02123-w
Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, Ferrucci L, Gilroy DW, Fasano A, Miller GW et al (2019) Chronic inflammation in the etiology of disease across the life span. Nat Med 25:1822–1832. https://doi.org/10.1038/s41591-019-0675-0
Wang M, Jiang L, Monticone RE, Lakatta EG (2014) Proinflammation: the key to arterial aging. Trends Endocrinol Metab 25:72–79. https://doi.org/10.1016/j.tem.2013.10.002
Ebert T, Pawelzik SC, Witasp A, Arefin S, Hobson S, Kublickiene K, Shiels PG, Bäck M, Stenvinkel P (2020) Inflammation and premature ageing in chronic kidney disease. Toxins (Basel) 12:227. https://doi.org/10.3390/toxins12040227
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA (2019) Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 15:565–581. https://doi.org/10.1038/s41582-019-0244-7
Greten FR, Grivennikov SI (2019) Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 51:27–41. https://doi.org/10.1016/j.immuni.2019.06.025
Smetana K Jr, Lacina L, Szabo P, Dvorankova B, Broz P, Sedo A (2016) Ageing as an important risk factor for cancer. Anticancer Res 36:5009–5017. https://doi.org/10.21873/anticanres.11069
Marengoni A, Angleman S, Melis R, Mangialasche F, Karp A, Garmen A, Meinow B, Fratiglioni L (2011) Aging with multimorbidity: a systematic review of the literature. Ageing Res Rev 10:430–439. https://doi.org/10.1016/j.arr.2011.03.003
Belikov AV (2019) Age-related diseases as vicious cycles. Ageing Res Rev 49:11–26. https://doi.org/10.1016/j.arr.2018.11.002
De Martinis M, Franceschi C, Monti D, Ginaldi L (2006) Inflammation markers predicting frailty and mortality in the elderly. Exp Mol Pathol 80:219–227. https://doi.org/10.1016/j.yexmp.2005.11.004
Marcos-Perez D, Sanchez-Flores M, Proietti S, Bonassi S, Costa S, Teixeira JP, Fernandez-Tajes J, Pasaro E, Laffon B, Valdiglesias V (2020) Association of inflammatory mediators with frailty status in older adults: results from a systematic review and meta-analysis. Geroscience 42:1451–1473. https://doi.org/10.1007/s11357-020-00247-4
Arai Y, Martin-Ruiz CM, Takayama M, Abe Y, Takebayashi T, Koyasu S, Suematsu M, Hirose N, von Zglinicki T (2015) Inflammation, but not telomere length, predicts successful ageing at extreme old age: A longitudinal study of semi-supercentenarians. EBioMedicine 2:1549–1558. https://doi.org/10.1016/j.ebiom.2015.07.029
Rubino G, Bulati M, Aiello A, Aprile S, Gambino CM, Gervasi F, Caruso C, Accardi G (2019) Sicilian centenarian offspring are more resistant to immune ageing. Aging Clin Exp Res 31:125–133. https://doi.org/10.1007/s40520-018-0936-7
Bucci L, Ostan R, Cevenini E, Pini E, Scurti M, Vitale G, Mari D, Caruso C, Sansoni P, Fanelli F et al (2016) Centenarians’ offspring as a model of healthy aging: a reappraisal of the data on Italian subjects and a comprehensive overview. Aging (Albany NY) 8:510–519. https://doi.org/10.18632/aging.100912
Horvath S, Raj K (2018) DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet 19:371–384. https://doi.org/10.1038/s41576-018-0004-3
Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, Mari D, Arosio B, Monti D, Passarino G et al (2015) Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY) 7:1159–1170. https://doi.org/10.18632/aging.100861
Jylhävä J, Kananen L, Raitanen J, Marttila S, Nevalainen T, Hervonen A, Jylhä M, Hurme M (2016) Methylomic predictors demonstrate the role of NF-κB in old-age mortality and are unrelated to the aging-associated epigenetic drift. Oncotarget 7:19228–19241. https://doi.org/10.18632/oncotarget.8278
Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2008) Activation of innate immunity system during aging: NF-κB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev 7:83–105. https://doi.org/10.1016/j.arr.2007.09.002
Clydesdale GJ, Dandie GW, Muller HK (2001) Ultraviolet light induced injury: immunological and inflammatory effects. Immunol Cell Biol 79:547–568. https://doi.org/10.1046/j.1440-1711.2001.01047.x
Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn-Konijnenberg D, Hömig-Hölzel C et al (2014) Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 507:109–113. https://doi.org/10.1038/nature13111
Fitsiou E, Pulido T, Campisi J, Alimirah F, Demaria M (2021) Cellular senescence and the senescence-associated secretory phenotype as drivers of skin photoaging. J Invest Dermatol 141:1119–1126. https://doi.org/10.1016/j.jid.2020.09.031
Lee YI, Choi S, Roh WS, Lee JH, Kim TG (2021) Cellular senescence and inflammaging in the skin microenvironment. Int J Mol Sci 22:3849. https://doi.org/10.3390/ijms22083849
Campisi J, Andersen JK, Kapahi P, Melov S (2011) Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol 21:354–359. https://doi.org/10.1016/j.semcancer.2011.09.001
Cupit-Link MC, Kirkland JL, Ness KK, Armstrong GT, Tchkonia T, LeBrasseur NK, Armenian SH, Ruddy KJ, Hashmi SK (2017) Biology of premature ageing in survivors of cancer. ESMO Open 2:e000250. https://doi.org/10.1136/esmoopen-2017-000250
Ness KK, Kirkland JL, Gramatges MM, Wang Z, Kundu M, McCastlain K, Li-Harms X, Zhang J, Tchkonia T, Pluijm SMF et al (2018) Premature physiologic aging as a paradigm for understanding increased risk of adverse health across the lifespan of survivors of childhood cancer. J Clin Oncol 36:2206–2215. https://doi.org/10.1200/JCO.2017.76.7467
Armenian SH, Gibson CJ, Rockne RC, Ness KK (2019) Premature aging in young cancer survivors. J Natl Cancer Inst 111:226–232. https://doi.org/10.1093/jnci/djy229
Derhovanessian E, Larbi A, Pawelec G (2009) Biomarkers of human immunosenescence: impact of cytomegalovirus infection. Curr Opin Immunol 21:440–445. https://doi.org/10.1016/j.coi.2009.05.012
Weltevrede M, Eilers R, de Melker HE, van Baarle D (2016) Cytomegalovirus persistence and T-cell immunosenescence in people aged fifty and older: A systematic review. Exp Gerontol 77:87–95. https://doi.org/10.1016/j.exger.2016.02.005
Hassouneh F, Goldeck D, Pera A, van Heemst D, Slagboom PE, Pawelec G, Solana R (2021) Functional changes of T-cell subsets with age and CMV infection. Int J Mol Sci 22:9973. https://doi.org/10.3390/ijms22189973
Kananen L, Nevalainen T, Jylhävä J, Marttila S, Hervonen A, Jylhä M, Hurme M (2015) Cytomegalovirus infection accelerates epigenetic aging. Exp Gerontol 72:227–229. https://doi.org/10.1016/j.exger.2015.10.008
Poloni C, Szyf M, Cheishvili D, Tsoukas CM (2022) Are the healthy vulnerable? Cytomegalovirus seropositivity in healthy adults is associated with accelerated epigenetic age and immune-dysregulation. J Infect Dis 225:443–452. https://doi.org/10.1093/infdis/jiab365
Aiello AE, Chiu YL, Frasca D (2017) How does cytomegalovirus factor into diseases of aging and vaccine responses, and by what mechanisms? Geroscience 39:261–271. https://doi.org/10.1007/s11357-017-9983-9
Kadambari S, Klenerman P, Pollard AJ (2020) Why the elderly appear to be more severely affected by COVID-19: The potential role of immunosenescence and CMV. Rev Med Virol 30:e2144. https://doi.org/10.1002/rmv.2144
Hearps AC, Martin GE, Rajasuriar R, Crowe SM (2014) Inflammatory co-morbidities in HIV+ individuals: learning lessons from healthy ageing. Curr HIV/AIDS Rep 11:20–34. https://doi.org/10.1007/s11904-013-0190-8
Nasi M, De Biasi S, Gibellini L, Bianchini E, Pecorini S, Bacca V, Guaraldi G, Mussini C, Pinti M, Cossarizza A (2017) Ageing and inflammation in patients with HIV infection. Clin Exp Immunol 187:44–52. https://doi.org/10.1111/cei.12814
Gianesin K, Noguera-Julian A, Zanchetta M, Del Bianco P, Petrara MR, Freguja R, Rampon O, Fortuny C, Camos M, Mozzo E et al (2016) Premature aging and immune senescence in HIV-infected children. AIDS 30:1363–1373. https://doi.org/10.1097/QAD.0000000000001093
Kooman JP, Dekker MJ, Usvyat LA, Kotanko P, van der Sande FM, Schalkwijk CG, Shiels PG, Stenvinkel P (2017) Inflammation and premature aging in advanced chronic kidney disease. Am J Physiol Renal Physiol 313:F938–F950. https://doi.org/10.1152/ajprenal.00256.2017
Barnes PJ (2017) Senescence in COPD and its comorbidities. Annu Rev Physiol 79:517–539. https://doi.org/10.1146/annurev-physiol-022516-034314
Chalan P, van den Berg A, Kroesen BJ, Brouwer L, Boots A (2015) Rheumatoid arthritis, immunosenescence and the hallmarks of aging. Curr Aging Sci 8:131–146. https://doi.org/10.2174/1874609808666150727110744
Betjes MG (2020) Uremia-associated ageing of the thymus and adaptive immune responses. Toxins (Basel) 12:224. https://doi.org/10.3390/toxins12040224
Zou D, Wu W, He Y, Ma S, Gao J (2018) The role of klotho in chronic kidney disease. BMC Nephrol 19:285. https://doi.org/10.1186/s12882-018-1094-z
Mytych J, Romerowicz-Misielak M, Koziorowski M (2018) Klotho protects human monocytes from LPS-induced immune impairment associated with immunosenescent-like phenotype. Mol Cell Endocrinol 470:1–13. https://doi.org/10.1016/j.mce.2017.05.003
Kuro-o M (2009) Klotho and aging. Biochim Biophys Acta 1790:1049–1058. https://doi.org/10.1016/j.bbagen.2009.02.005
Kanterman J, Sade-Feldman M, Baniyash M (2012) New insights into chronic inflammation-induced immunosuppression. Semin Cancer Biol 22:307–318. https://doi.org/10.1016/j.semcancer.2012.02.008
Amodio G, Cichy J, Conde P, Matteoli G, Moreau A, Ochando J, Oral BH, Pekarova M, Ryan EJ, Roth J et al (2019) Role of myeloid regulatory cells (MRCs) in maintaining tissue homeostasis and promoting tolerance in autoimmunity, inflammatory disease and transplantation. Cancer Immunol Immunother 68:661–672. https://doi.org/10.1007/s00262-018-2264-3
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174. https://doi.org/10.1038/nri2506
Millrud CR, Bergenfelz C, Leandersson K (2017) On the origin of myeloid-derived suppressor cells. Oncotarget 8:3649–3665. https://doi.org/10.18632/oncotarget.12278
Murray PJ (2017) Macrophage polarization. Annu Rev Physiol 79:541–566. https://doi.org/10.1146/annurev-physiol-022516-034339
Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH (2006) Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 66:1123–1131. https://doi.org/10.1158/0008-5472.CAN-05-1299
Wang L, Zhao J, Ren JP, Wu XY, Morrison ZD, Elgazzar MA, Ning SB, Moorman JP, Yao ZQ (2016) Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory T cells in HIV-1+ individuals. AIDS 30:1521–1531. https://doi.org/10.1097/QAD.0000000000001083
Clark RA (2010) Skin-resident T cells: the ups and downs of on site immunity. J Invest Dermatol 130:362–370. https://doi.org/10.1038/jid.2009.247
Zeng Q, Sun X, Xiao L, Xie Z, Bettini M, Deng T (2018) A unique population: Adipose-resident regulatory T cells. Front Immunol 9:2075. https://doi.org/10.3389/fimmu.2018.02075
Vignali DA, Collison LW, Workman CJ (2008) How regulatory T cells work. Nat Rev Immunol 8:523–532. https://doi.org/10.1038/nri2343
Sharma A, Rudra D (2018) Emerging functions of regulatory T cells in tissue homeostasis. Front Immunol 9:883. https://doi.org/10.3389/fimmu.2018.00883
Salminen A (2020) Activation of immunosuppressive network in the aging process. Ageing Res Rev 57:100998. https://doi.org/10.1016/j.arr.2019.100998
Kim R, Emi M, Tanabe K, Arihiro K (2006) Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res 66:5527–5536. https://doi.org/10.1158/0008-5472.CAN-05-4128
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555. https://doi.org/10.1016/s1471-4906(02)02302-5
Mauri C, Menon M (2015) The expanding family of regulatory B cells. Int Immunol 27:479–486. https://doi.org/10.1093/intimm/dxv038
Schmidt SV, Nino-Castro AC, Schultze JL (2012) Regulatory dendritic cells: there is more than just immune activation. Front Immunol 3:274. https://doi.org/10.3389/fimmu.2012.00274
Lünemann A, Lünemann JD, Münz C (2009) Regulatory NK-cell functions in inflammation and autoimmunity. Mol Med 15:352–358. https://doi.org/10.2119/molmed.2009.00035
Singh AK, Tripathi P, Cardell SL (2018) Type II NKT cells: An elusive population with immunoregulatory properties. Front Immunol 9:1969. https://doi.org/10.3389/fimmu.2018.01969
Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA (2006) Transforming growth factor-β regulation of immune responses. Annu Rev Immunol 24:99–146. https://doi.org/10.1146/annurev.immunol.24.021605.090737
Murray PJ (2016) Amino acid auxotrophy as a system of immunological control nodes. Nat Immunol 17:132–139. https://doi.org/10.1038/ni.3323
Curdy N, Lanvin O, Laurent C, Fournie JJ, Franchini DM (2019) Regulatory mechanisms of inhibitory immune checkpoint receptors expression. Trends Cell Biol 29:777–790. https://doi.org/10.1016/j.tcb.2019.07.002
Sidler C, Woycicki R, Ilnytskyy Y, Metz G, Kovalchuk I, Kovalchuk O (2013) Immunosenescence is associated with altered gene expression and epigenetic regulation in primary and secondary immune organs. Front Genet 4:211. https://doi.org/10.3389/fgene.2013.00211
Gimenez JLG, Carbonell NE, Mateo CR, Lopez EG, Palacios L, Chova LP, Berenguer E, Garzo CG, Pallardo FV, Blanquer J (2016) Epigenetics as the driving force in long-term immunosuppression. J Clin Epigenet 2:2. https://doi.org/10.21767/2472-1158.100017
Motwani MP, Newson J, Kwong S, Richard-Loendt A, Colas R, Dalli J, Gilroy DW (2017) Prolonged immune alteration following resolution of acute inflammation in humans. PLoS ONE 12:e0186964. https://doi.org/10.1371/journal.pone.0186964
Newson J, Motwani MP, Kendall AC, Nicolaou A, Muccioli GG, Alhouayek M, Bennett M, Van De Merwe R, James S, De Maeyer RPH et al (2017) Inflammatory resolution triggers a prolonged phase of immune suppression through COX-1/mPGES-1-derived prostaglandin E2. Cell Rep 20:3162–3175. https://doi.org/10.1016/j.celrep.2017.08.098
Solana R, Pawelec G (1998) Molecular and cellular basis of immunosenescence. Mech Ageing Dev 102:115–129. https://doi.org/10.1016/s0047-6374(98)00029-3
Salminen A, Kaarniranta K, Kauppinen A (2019) Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci 76:1901–1918. https://doi.org/10.1007/s00018-019-03048-x
Fulop T, Larbi A, Hirokawa K, Cohen AA, Witkowski JM (2020) Immunosenescence is both functional/adaptive and dysfunctional/maladaptive. Semin Immunopathol 42:521–536. https://doi.org/10.1007/s00281-020-00818-9
Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y, Varvares MA, Hoft DF, Peng G (2012) Human regulatory T cells induce T-lymphocyte senescence. Blood 120:2021–2031. https://doi.org/10.1182/blood-2012-03-416040
Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, Puig PE, Novault S, Escudier B, Vivier E et al (2005) CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-β-dependent manner. J Exp Med 202:1075–1085. https://doi.org/10.1084/jem.20051511
Trzonkowski P, Szmit E, Mysliwska J, Mysliwski A (2006) CD4+CD25+ T regulatory cells inhibit cytotoxic activity of CTL and NK cells in humans - impact of immunosenescence. Clin Immunol 119:307–316. https://doi.org/10.1016/j.clim.2006.02.002
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, Lehner F, Manns MP, Greten TF, Korangy F (2009) Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 50:799–807. https://doi.org/10.1002/hep.23054
Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT (2010) TGF-β downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol 12:7–13. https://doi.org/10.1093/neuonc/nop009
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI (2007) Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13:828–835. https://doi.org/10.1038/nm1609
Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK (2012) Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 22:275–281. https://doi.org/10.1016/j.semcancer.2012.01.011
Chou JP, Ramirez CM, Ryba DM, Koduri MP, Effros RB (2014) Prostaglandin E2 promotes features of replicative senescence in chronically activated human CD8+ T cells. PLoS ONE 9:e99432. https://doi.org/10.1371/journal.pone.0099432
Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I et al (2016) Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 8:1294–1315. https://doi.org/10.18632/aging.100991
Fulop T, Dupuis G, Witkowski JM, Larbi A (2016) The role of immunosenescence in the development of age-related diseases. Rev Invest Clin 68:84–91
Barbe-Tuana F, Funchal G, Schmitz CRR, Maurmann RM, Bauer ME (2020) The interplay between immunosenescence and age-related diseases. Semin Immunopathol 42:545–557. https://doi.org/10.1007/s00281-020-00806-z
Bauer ME (2020) Accelerated immunosenescence in rheumatoid arthritis: impact on clinical progression. Immun Ageing 17:6. https://doi.org/10.1186/s12979-020-00178-w
Monneret G, Gossez M, Venet F (2021) Sepsis and immunosenescence: closely associated in a vicious circle. Aging Clin Exp Res 33:729–732. https://doi.org/10.1007/s40520-019-01350-z
Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, Yosef R, Sagiv A, Agrawal A, Shapira A et al (2018) Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 9:5435. https://doi.org/10.1038/s41467-018-07825-3
Sagiv A, Biran A, Yon M, Simon J, Lowe SW, Krizhanovsky V (2013) Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 32:1971–1977. https://doi.org/10.1038/onc.2012.206
Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, Golani O, Polic B, Krizhanovsky V (2016) NKG2D ligands mediate immunosurveillance of senescent cells. Aging (Albany NY) 8:328–344. https://doi.org/10.18632/aging.100897
Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, Moretta L, Moretta A, Vitale M (2006) The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 108:4118–4125. https://doi.org/10.1182/blood-2006-03-006700
Lazarova M, Steinle A (2019) Impairment of NKG2D-mediated tumor immunity by TGF-β. Front Immunol 10:2689. https://doi.org/10.3389/fimmu.2019.02689
Salminen A (2021) Feed-forward regulation between cellular senescence and immunosuppression promotes the aging process and age-related diseases. Ageing Res Rev 67:101280. https://doi.org/10.1016/j.arr.2021.101280
Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN, Henson SM (2018) Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 17:e12675. https://doi.org/10.1111/acel.12675
Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL (2011) Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A 108:20012–20017. https://doi.org/10.1073/pnas.1116110108
Enioutina EY, Bareyan D, Daynes RA (2011) A role for immature myeloid cells in immune senescence. J Immunol 186:697–707. https://doi.org/10.4049/jimmunol.1002987
Verschoor CP, Johnstone J, Millar J, Dorrington MG, Habibagahi M, Lelic A, Loeb M, Bramson JL, Bowdish DM (2013) Blood CD33+HLA-DR- myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol 93:633–637. https://doi.org/10.1189/jlb.0912461
Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, Belkaid Y, Chougnet C (2008) Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J Immunol 181:1835–1848. https://doi.org/10.4049/jimmunol.181.3.1835
Flores RR, Clauson CL, Cho J, Lee BC, McGowan SJ, Baker DJ, Niedernhofer LJ, Robbins PD (2017) Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-κB-dependent mechanism. Aging Cell 16:480–487. https://doi.org/10.1111/acel.12571
Grizzle WE, Xu X, Zhang S, Stockard CR, Liu C, Yu S, Wang J, Mountz JD, Zhang HG (2007) Age-related increase of tumor susceptibility is associated with myeloid-derived suppressor cell mediated suppression of T cell cytotoxicity in recombinant inbred BXD12 mice. Mech Ageing Dev 128:672–680. https://doi.org/10.1016/j.mad.2007.10.003
Garg SK, Delaney C, Toubai T, Ghosh A, Reddy P, Banerjee R, Yung R (2014) Aging is associated with increased regulatory T-cell function. Aging Cell 13:441–448. https://doi.org/10.1111/acel.12191
Shevach EM, Thornton AM (2014) tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev 259:88–102. https://doi.org/10.1111/imr.12160
Jagger A, Shimojima Y, Goronzy JJ, Weyand CM (2014) Regulatory T cells and the immune aging process: a mini-review. Gerontology 60:130–137. https://doi.org/10.1159/000355303
Darrigues J, van Meerwijk JPM, Romagnoli P (2018) Age-dependent changes in regulatory T lymphocyte development and function: A mini-review. Gerontology 64:28–35. https://doi.org/10.1159/000478044
Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, Ignatowicz L (2015) Differences in expression level of Helios and Neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ regulatory T cells. PLoS ONE 10:e0141161. https://doi.org/10.1371/journal.pone.0141161
van der Geest KS, Abdulahad WH, Tete SM, Lorencetti PG, Horst G, Bos NA, Kroesen BJ, Brouwer E, Boots AM (2014) Aging disturbs the balance between effector and regulatory CD4+ T cells. Exp Gerontol 60:190–196. https://doi.org/10.1016/j.exger.2014.11.005
Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, Belt BA, Alspach E, Leahy K, Luo J et al (2016) Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 7:11762. https://doi.org/10.1038/ncomms11762
Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S et al (2009) Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15:930–939. https://doi.org/10.1038/nm.2002
Cipolletta D, Cohen P, Spiegelman BM, Benoist C, Mathis D (2015) Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPARγ effects. Proc Natl Acad Sci U S A 112:482–487. https://doi.org/10.1073/pnas.1423486112
Kalathookunnel Antony A, Lian Z, Wu H (2018) T cells in adipose tissue in aging. Front Immunol 9:2945. https://doi.org/10.3389/fimmu.2018.02945
Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, Zhou C, Liang Y, LeBlanc M, Liddle C et al (2015) Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528:137–141. https://doi.org/10.1038/nature16151
Jackaman C, Radley-Crabb HG, Soffe Z, Shavlakadze T, Grounds MD, Nelson DJ (2013) Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell 12:345–357. https://doi.org/10.1111/acel.12062
Cui CY, Driscoll RK, Piao Y, Chia CW, Gorospe M, Ferrucci L (2019) Skewed macrophage polarization in aging skeletal muscle. Aging Cell 18:e13032. https://doi.org/10.1111/acel.13032
Duong L, Radley-Crabb HG, Gardner JK, Tomay F, Dye DE, Grounds MD, Pixley FJ, Nelson DJ, Jackaman C (2018) Macrophage depletion in elderly mice improves response to tumor immunotherapy, increases anti-tumor T cell activity and reduces treatment-induced cachexia. Front Genet 9:526. https://doi.org/10.3389/fgene.2018.00526
Shah KH, Shi P, Giani JF, Janjulia T, Bernstein EA, Li Y, Zhao T, Harrison DG, Bernstein KE, Shen XZ (2015) Myeloid suppressor cells accumulate and regulate blood pressure in hypertension. Circ Res 117:858–869. https://doi.org/10.1161/CIRCRESAHA.115.306539
Wang YG, Xiong X, Chen ZY, Liu KL, Yang JH, Wen Q, Wu FQ, Hu XF, Peng YD, Wu JJ et al (2015) Expansion of myeloid-derived suppressor cells in patients with acute coronary syndrome. Cell Physiol Biochem 35:292–304. https://doi.org/10.1159/000369696
Kuan R, Agrawal DK, Thankam FG (2021) Treg cells in atherosclerosis. Mol Biol Rep 48:4897–4910. https://doi.org/10.1007/s11033-021-06483-x
Wang H, Wang Z, Wu Q, Yuan Y, Cao W, Zhang X (2021) Regulatory T cells in ischemic stroke. CNS Neurosci Ther 27:643–651. https://doi.org/10.1111/cns.13611
Santamaria-Cadavid M, Rodriguez-Castro E, Rodriguez-Yanez M, Arias-Rivas S, Lopez-Dequidt I, Perez-Mato M, Rodriguez-Perez M, Lopez-Loureiro I, Hervella P, Campos F et al (2020) Regulatory T cells participate in the recovery of ischemic stroke patients. BMC Neurol 20:68. https://doi.org/10.1186/s12883-020-01648-w
Achmus L, Ruhnau J, Grothe S, von Sarnowski B, Bröker BM, Dressel A, Schulze J, Vogelgesang A (2020) Stroke-induced modulation of myeloid-derived suppressor cells (MDSCs) and IL-10-producing regulatory monocytes. Front Neurol 11:577971. https://doi.org/10.3389/fneur.2020.577971
Scrimini S, Pons J, Agusti A, Clemente A, Sallan MC, Bauca JM, Soriano JB, Cosio BG, Lopez M, Crespi C et al (2015) Expansion of myeloid-derived suppressor cells in chronic obstructive pulmonary disease and lung cancer: potential link between inflammation and cancer. Cancer Immunol Immunother 64:1261–1270. https://doi.org/10.1007/s00262-015-1737-x
Li XN, Pan X, Qiu D (2014) Imbalances of Th17 and Treg cells and their respective cytokines in COPD patients by disease stage. Int J Clin Exp Med 7:5324–5329
Hijona E, Hijona L, Arenas JI, Bujanda L (2010) Inflammatory mediators of hepatic steatosis. Mediators Inflamm 2010:837419. https://doi.org/10.1155/2010/837419
Zhou Z, Lai P, Zhang S, Wang Y, Qu N, Lu D, Gao L, Xu L, Yang Y, Zhang T et al (2021) The relationship between hepatic myeloid-derived suppressor cells and clinicopathological parameters in patients with chronic liver disease. Biomed Res Int 2021:6612477. https://doi.org/10.1155/2021/6612477
He B, Wu L, Xie W, Shao Y, Jiang J, Zhao Z, Yan M, Chen Z, Cui D (2017) The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol 18:33. https://doi.org/10.1186/s12865-017-0215-y
Cairoli V, De Matteo E, Rios D, Lezama C, Galoppo M, Casciato P, Mullen E, Giadans C, Bertot G, Preciado MV et al (2021) Hepatic lymphocytes involved in the pathogenesis of pediatric and adult non-alcoholic fatty liver disease. Sci Rep 11:5129. https://doi.org/10.1038/s41598-021-84674-z
Dorhoi A, Du Plessis N (2018) Monocytic myeloid-derived suppressor cells in chronic infections. Front Immunol 8:1895. https://doi.org/10.3389/fimmu.2017.01895
Sarkar R, Mathew A, Sehrawat S (2019) Myeloid-derived suppressor cells confer infectious tolerance to dampen virus-induced tissue immunoinflammation. J Immunol 203:1325–1337. https://doi.org/10.4049/jimmunol.1900142
Rosado-Sanchez I, De Pablo-Bernal R, Rull A, Gonzalez J, Moreno S, Vinuesa D, Estrada V, Munoz-Fernandez MA, Vidal F, Leal M et al (2020) Increased frequencies of myeloid-derived suppressor cells precede immunodiscordance in HIV-infected subjects. Front Immunol 11:581307. https://doi.org/10.3389/fimmu.2020.581307
Hotchkiss RS, Monneret G, Payen D (2013) Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13:862–874. https://doi.org/10.1038/nri3552
Schrijver IT, Theroude C, Roger T (2019) Myeloid-derived suppressor cells in sepsis. Front Immunol 10:327. https://doi.org/10.3389/fimmu.2019.00327
Sacchi A, Grassi G, Bordoni V, Lorenzini P, Cimini E, Casetti R, Tartaglia E, Marchioni L, Petrosillo N, Palmieri F et al (2020) Early expansion of myeloid-derived suppressor cells inhibits SARS-CoV-2 specific T-cell response and may predict fatal COVID-19 outcome. Cell Death Dis 11:921. https://doi.org/10.1038/s41419-020-03125-1
Galvan-Pena S, Leon J, Chowdhary K, Michelson DA, Vijaykumar B, Yang L, Magnuson AM, Chen F, Manickas-Hill Z, Piechocka-Trocha A et al (2021) Profound Treg perturbations correlate with COVID-19 severity. Proc Natl Acad Sci USA 118:e2111315118. https://doi.org/10.1073/pnas.2111315118
Cunha LL, Perazzio SF, Azzi J, Cravedi P, Riella LV (2020) Remodeling of the immune response with aging: Immunosenescence and its potential impact on COVID-19 immune response. Front Immunol 11:1748. https://doi.org/10.3389/fimmu.2020.01748
Wang D, DuBois RN (2015) Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 36:1085–1093. https://doi.org/10.1093/carcin/bgv123
Elliott LA, Doherty GA, Sheahan K, Ryan EJ (2017) Human tumor-infiltrating myeloid cells: Phenotypic and functional diversity. Front Immunol 8:86. https://doi.org/10.3389/fimmu.2017.00086
Monteran L, Erez N (2019) The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol 10:1835. https://doi.org/10.3389/fimmu.2019.01835
Lian J, Yue Y, Yu W, Zhang Y (2020) Immunosenescence: a key player in cancer development. J Hematol Oncol 13:151. https://doi.org/10.1186/s13045-020-00986-z
Yang J, Liu M, Hong D, Zeng M, Zhang X (2021) The paradoxical role of cellular senescence in cancer. Front Cell Dev Biol 9:722205. https://doi.org/10.3389/fcell.2021.722205
Derhovanessian E, Solana R, Larbi A, Pawelec G (2008) Immunity, ageing and cancer. Immun Ageing 5:11. https://doi.org/10.1186/1742-4933-5-11
Fisher GJ, Kang S, Varani J, Bata-Csorgo Z, Wan Y, Datta S, Voorhees JJ (2002) Mechanisms of photoaging and chronological skin aging. Arch Dermatol 138:1462–1470. https://doi.org/10.1001/archderm.138.11.1462
Halliday GM, Damian DL, Rana S, Byrne SN (2012) The suppressive effects of ultraviolet radiation on immunity in the skin and internal organs: implications for autoimmunity. J Dermatol Sci 66:176–182. https://doi.org/10.1016/j.jdermsci.2011.12.009
Prasad R, Katiyar SK (2017) Crosstalk among UV-induced inflammatory mediators, DNA damage and epigenetic regulators facilitates suppression of the immune system. Photochem Photobiol 93:930–936. https://doi.org/10.1111/php.12687
Hart PH, Norval M (2018) Ultraviolet radiation-induced immunosuppression and its relevance for skin carcinogenesis. Photochem Photobiol Sci 17:1872–1884. https://doi.org/10.1039/c7pp00312a
Soontrapa K, Honda T, Sakata D, Yao C, Hirata T, Hori S, Matsuoka T, Kita Y, Shimizu T, Kabashima K et al (2011) Prostaglandin E2-prostaglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proc Natl Acad Sci U S A 108:6668–6673. https://doi.org/10.1073/pnas.1018625108
Skobowiat C, Postlethwaite AE, Slominski AT (2017) Skin exposure to ultraviolet B rapidly activates systemic neuroendocrine and immunosuppressive responses. Photochem Photobiol 93:1008–1015. https://doi.org/10.1111/php.12642
Schwarz T (2005) Regulatory T cells induced by ultraviolet radiation. Int Arch Allergy Immunol 137:187–193. https://doi.org/10.1159/000086330
Maeda A, Beissert S, Schwarz T, Schwarz A (2008) Phenotypic and functional characterization of ultraviolet radiation-induced regulatory T cells. J Immunol 180:3065–3071. https://doi.org/10.4049/jimmunol.180.5.3065
Ali N, Rosenblum MD (2017) Regulatory T cells in skin Immunology 152:372–381. https://doi.org/10.1111/imm.12791
Navid F, Bruhs A, Schuller W, Fritsche E, Krutmann J, Schwarz T, Schwarz A (2013) The aryl hydrocarbon receptor is involved in UVR-induced immunosuppression. J Invest Dermatol 133:2763–2770. https://doi.org/10.1038/jid.2013.221
Fritsche E, Schäfer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, Hübenthal U, Cline JE, Hajimiragha H, Schroeder P et al (2007) Lightening up the UV response by identification of the aryl hydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci U S A 104:8851–8856. https://doi.org/10.1073/pnas.0701764104
Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA (2010) An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185:3190–3198. https://doi.org/10.4049/jimmunol.0903670
Hidaka T, Fujimura T, Aiba S (2019) Aryl hydrocarbon receptor modulates carcinogenesis and maintenance of skin cancers. Front Med (Lausanne) 6:194. https://doi.org/10.3389/fmed.2019.00194
Okuma A, Hanyu A, Watanabe S, Hara E (2017) p16Ink4a and p21Cip1/Waf1 promote tumour growth by enhancing myeloid-derived suppressor cells chemotaxis. Nat Commun 8:2050. https://doi.org/10.1038/s41467-017-02281-x
Ladomersky E, Scholtens DM, Kocherginsky M, Hibler EA, Bartom ET, Otto-Meyer S, Zhai L, Lauing KL, Choi J, Sosman JA et al (2019) The coincidence between increasing age, immunosuppression, and the incidence of patients with glioblastoma. Front Pharmacol 10:200. https://doi.org/10.3389/fphar.2019.00200
Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A (2014) Histone demethylase Jumonji D3 (JMJD3/KDM6B) at the nexus of epigenetic regulation of inflammation and the aging process. J Mol Med (Berl) 92:1035–1043. https://doi.org/10.1007/s00109-014-1182-x
Zhao Y, Forst CV, Sayegh CE, Wang IM, Yang X, Zhang B (2016) Molecular and genetic inflammation networks in major human diseases. Mol Biosyst 12:2318–2341. https://doi.org/10.1039/c6mb00240d
Perna L, Zhang Y, Mons U, Holleczek B, Saum KU, Brenner H (2016) Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin Epigenetics 8:64. https://doi.org/10.1186/s13148-016-0228-z
Zheng C, Berger NA, Li L, Xu R (2020) Epigenetic age acceleration and clinical outcomes in gliomas. PLoS ONE 15:e0236045. https://doi.org/10.1371/journal.pone.0236045
Pera A, Campos C, Lopez N, Hassouneh F, Alonso C, Tarazona R, Solana R (2015) Immunosenescence: Implications for response to infection and vaccination in older people. Maturitas 82:50–55. https://doi.org/10.1016/j.maturitas.2015.05.004
Channappanavar R, Perlman S (2020) Age-related susceptibility to coronavirus infections: role of impaired and dysregulated host immunity. J Clin Invest 130:6204–6213. https://doi.org/10.1172/JCI144115
Uyemura K, Castle SC, Makinodan T (2002) The frail elderly: role of dendritic cells in the susceptibility of infection. Mech Ageing Dev 123:955–962. https://doi.org/10.1016/s0047-6374(02)00033-7
Frasca D, Blomberg BB (2020) Aging induces B cell defects and decreased antibody responses to influenza infection and vaccination. Immun Ageing 17:37. https://doi.org/10.1186/s12979-020-00210-z
Simmons SR, Bhalla M, Herring SE, Tchalla EYI, Bou Ghanem EN (2021) Older but not wiser: the age-driven changes in neutrophil responses during pulmonary infections. Infect Immun 89:e00653-e720. https://doi.org/10.1128/IAI.00653-20
Goronzy JJ, Hu B, Kim C, Jadhav RR, Weyand CM (2018) Epigenetics of T cell aging. J Leukoc Biol 104:691–699. https://doi.org/10.1002/JLB.1RI0418-160R
Rodriguez RM, Saiz ML, Suarez-Alvarez B, Lopez-Larrea C (2022) Epigenetic networks driving T cell identity and plasticity during immunosenescence. Trends Genet 38:120–123. https://doi.org/10.1016/j.tig.2021.08.014
Aspinall R, Del Giudice G, Effros RB, Grubeck-Loebenstein B, Sambhara S (2007) Challenges for vaccination in the elderly. Immun Ageing 4:9. https://doi.org/10.1186/1742-4933-4-9
Chen S, Akbar SM, Miyake T, Abe M, Al-Mahtab M, Furukawa S, Bunzo M, Hiasa Y, Onji M (2015) Diminished immune response to vaccinations in obesity: role of myeloid-derived suppressor and other myeloid cells. Obes Res Clin Pract 9:35–44. https://doi.org/10.1016/j.orcp.2013.12.006
Merani S, Pawelec G, Kuchel GA, McElhaney JE (2017) Impact of aging and cytomegalovirus on immunological response to influenza vaccination and infection. Front Immunol 8:784. https://doi.org/10.3389/fimmu.2017.00784
Corsini E, Vismara L, Lucchi L, Viviani B, Govoni S, Galli CL, Marinovich M, Racchi M (2006) High interleukin-10 production is associated with low antibody response to influenza vaccination in the elderly. J Leukoc Biol 80:376–382. https://doi.org/10.1189/jlb.0306190
Batista-Duharte A, Pera A, Alino SF, Solana R (2021) Regulatory T cells and vaccine effectiveness in older adults. Challenges and prospects Int Immunopharmacol 96:107761. https://doi.org/10.1016/j.intimp.2021.107761
Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D (2013) Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother 62:909–918. https://doi.org/10.1007/s00262-013-1396-8
Hamilton JAG, Henry CJ (2020) Aging and immunotherapies: New horizons for the golden ages. Cancer Aging 1:30–44. https://doi.org/10.1002/aac2.12014
Elias R, Karantanos T, Sira E, Hartshorn KL (2017) Immunotherapy comes of age: Immune aging & checkpoint inhibitors. J Geriatr Oncol 8:229–235. https://doi.org/10.1016/j.jgo.2017.02.001
Hegde PS, Chen DS (2020) Top 10 challenges in cancer immunotherapy. Immunity 52:17–35. https://doi.org/10.1016/j.immuni.2019.12.011
Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, Utikal J, Umansky V (2018) Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol 9:398. https://doi.org/10.3389/fimmu.2018.00398
Kim JH, Kim BS, Lee SK (2020) Regulatory T cells in tumor microenvironment and approach for anticancer immunotherapy. Immune Netw 20:e4. https://doi.org/10.4110/in.2020.20.e4
Hou A, Hou K, Huang Q, Lei Y, Chen W (2020) Targeting myeloid-derived suppressor cell, a promising strategy to overcome resistance to immune checkpoint inhibitors. Front Immunol 11:783. https://doi.org/10.3389/fimmu.2020.00783
Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, Booth S, Lenton R, Noyvert B, Shannon-Lowe C et al (2019) MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine 47:235–246. https://doi.org/10.1016/j.ebiom.2019.08.025
Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X (2020) Targeting STAT3 in cancer immunotherapy. Mol Cancer 19:145. https://doi.org/10.1186/s12943-020-01258-7
Bauer R, Udonta F, Wroblewski M, Ben-Batalla I, Santos IM, Taverna F, Kuhlencord M, Gensch V, Päsler S, Vinckier S et al (2018) Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res 78:3220–3232. https://doi.org/10.1158/0008-5472.CAN-17-3415
Salminen A, Kaarniranta K, Kauppinen A (2018) Phytochemicals inhibit the immunosuppressive functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and age-related chronic inflammatory disorders. Int Immunopharmacol 61:231–240. https://doi.org/10.1016/j.intimp.2018.06.005
Hurez V, Daniel BJ, Sun L, Liu AJ, Ludwig SM, Kious MJ, Thibodeaux SR, Pandeswara S, Murthy K, Livi CB et al (2012) Mitigating age-related immune dysfunction heightens the efficacy of tumor immunotherapy in aged mice. Cancer Res 72:2089–2099. https://doi.org/10.1158/0008-5472.CAN-11-3019
Krstic J, Santibanez JF (2014) Transforming growth factor-β and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. ScientificWorldJournal 2014:521754. https://doi.org/10.1155/2014/521754
Meng XM, Nikolic-Paterson DJ, Lan HY (2016) TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 12:325–338. https://doi.org/10.1038/nrneph.2016.48
Tominaga K, Suzuki HI (2019) TGF-β signaling in cellular senescence and aging-related pathology. Int J Mol Sci 20:5002. https://doi.org/10.3390/ijms20205002
Wu TT, Li WM, Yao YM (2016) Interactions between autophagy and inhibitory cytokines. Int J Biol Sci 12:884–897. https://doi.org/10.7150/ijbs.15194
Ohl K, Tenbrock K (2018) Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol 9:2499. https://doi.org/10.3389/fimmu.2018.02499
Heinbokel T, Elkhal A, Liu G, Edtinger K, Tullius SG (2013) Immunosenescence and organ transplantation Transplant Rev (Orlando) 27:65–75. https://doi.org/10.1016/j.trre.2013.03.001
Krenzien F, ElKhal A, Quante M, Rodriguez Cetina Biefer H, Hirofumi U, Gabardi S, Tullius SG (2015) A rationale for age-adapted immunosuppression in organ transplantation. Transplantation 99:2258–2268. https://doi.org/10.1097/TP.0000000000000842
Cao P, Sun Z, Feng C, Zhang J, Zhang F, Wang W, Zhao Y (2020) Myeloid-derived suppressor cells in transplantation tolerance induction. Int Immunopharmacol 83:106421. https://doi.org/10.1016/j.intimp.2020.106421
Lee WC, Wang YC, Hsu HY, Hsu PY, Cheng CH, Lee CF, Wu TJ, Chan KM (2021) Immunological discrepancy in aged mice facilitates skin allograft survival. Aging (Albany NY) 13:16219–16228. https://doi.org/10.18632/aging.203152
Dayoub JC, Cortese F, Anzic A, Grum T, de Magalhaes JP (2018) The effects of donor age on organ transplants: A review and implications for aging research. Exp Gerontol 110:230–240. https://doi.org/10.1016/j.exger.2018.06.019
Trzonkowski P, Debska-Slizien A, Jankowska M, Wardowska A, Carvalho-Gaspar M, Hak L, Moszkowska G, Bzoma B, Mills N, Wood KJ et al (2010) Immunosenescence increases the rate of acceptance of kidney allotransplants in elderly recipients through exhaustion of CD4+ T-cells. Mech Ageing Dev 131:96–104. https://doi.org/10.1016/j.mad.2009.12.006
Martins PN, Tullius SG, Markmann JF (2014) Immunosenescence and immune response in organ transplantation. Int Rev Immunol 33:162–173. https://doi.org/10.3109/08830185.2013.829469
Ferrer IR, Hester J, Bushell A, Wood KJ (2014) Induction of transplantation tolerance through regulatory cells: from mice to men. Immunol Rev 258:102–116. https://doi.org/10.1111/imr.12158
Rickert CG, Markmann JF (2019) Current state of organ transplant tolerance. Curr Opin Organ Transplant 24:441–450. https://doi.org/10.1097/MOT.0000000000000670
Thewissen M, Linsen L, Somers V, Geusens P, Raus J, Stinissen P (2005) Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann NY Acad Sci 1051:255–262. https://doi.org/10.1196/annals.1361.066
Goronzy JJ, Li G, Yang Z, Weyand CM (2013) The janus head of T cell aging - autoimmunity and immunodeficiency. Front Immunol 4:131. https://doi.org/10.3389/fimmu.2013.00131
Watad A, Bragazzi NL, Adawi M, Amital H, Toubi E, Porat BS, Shoenfeld Y (2017) Autoimmunity in the elderly: Insights from basic science and clinics - A mini-review. Gerontology 63:515–523. https://doi.org/10.1159/000478012
Barnie PA, Zhang P, Lv H, Wang D, Su X, Su Z, Xu H (2017) Myeloid-derived suppressor cells and myeloid regulatory cells in cancer and autoimmune disorders. Exp Ther Med 13:378–388. https://doi.org/10.3892/etm.2016.4018
Wegner A, Verhagen J, Wraith DC (2017) Myeloid-derived suppressor cells mediate tolerance induction in autoimmune disease. Immunology 151:26–42. https://doi.org/10.1111/imm.12718
Li M, Zhu D, Wang T, Xia X, Tian J, Wang S (2018) Roles of myeloid-derived suppressor cell subpopulations in autoimmune arthritis. Front Immunol 9:2849. https://doi.org/10.3389/fimmu.2018.02849
Eggenhuizen PJ, Ng BH, Ooi JD (2020) Treg enhancing therapies to treat autoimmune diseases. Int J Mol Sci 21:7015. https://doi.org/10.3390/ijms21197015
Selck C, Dominguez-Villar M (2021) Antigen-specific regulatory T cell therapy in autoimmune diseases and transplantation. Front Immunol 12:661875. https://doi.org/10.3389/fimmu.2021.661875
Zhang Q, Lu W, Liang CL, Chen Y, Liu H, Qiu F, Dai Z (2018) Chimeric antigen receptor (CAR) Treg: A promising approach to inducing immunological tolerance. Front Immunol 9:2359. https://doi.org/10.3389/fimmu.2018.02359
Acknowledgements
The author thanks Dr. Ewen MacDonald for checking the language of the manuscript.
Funding
Open access funding provided by University of Eastern Finland (UEF) including Kuopio University Hospital. The author declares that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
This article is the sole work of the author.
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salminen, A. Clinical perspectives on the age-related increase of immunosuppressive activity. J Mol Med 100, 697–712 (2022). https://doi.org/10.1007/s00109-022-02193-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-022-02193-4