
Abstract
Thrombin is the protease involved in blood coagulation. Its deregulation can lead to hemostatic abnormalities, which range from subtle subclinical to serious life-threatening coagulopathies, i.e., during septicemia. Additionally, thrombin plays important roles in many (patho)physiological conditions that reach far beyond its well-established role in stemming blood loss and thrombosis, including embryonic development and angiogenesis but also extending to inflammatory processes, complement activation, and even tumor biology. In this review, we will address thrombin’s broad roles in diverse (patho)physiological processes in an integrative way. We will also discuss thrombin as an emerging major target for novel therapies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The serine protease thrombin: from blood coagulation to far beyond
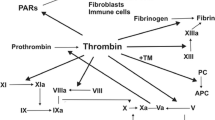
Thrombin is the key effector protease of the blood coagulation system. Although it is best known for this role, it directly contributes to other processes including embryonic development, angiogenesis, organ regeneration [1, 2], innate immunity, acute and chronic inflammatory processes [3], atherosclerosis [4], neuropathology [5], and tumor biology [6, 7] (synopsis, Fig. 1).

Thrombin and thrombin functions in development, physiology, and pathophysiology
The wide spectrum of thrombin functions and its role in physiology and pathophysiology are generally explained by its activity as a serine protease (acting both on soluble and membrane-bound substrates, Table 1).
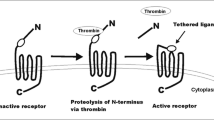
To promote blood coagulation, thrombin converts circulating fibrinogen into fibrin, but it can also serve as a signaling molecule to cells through protease-activated receptors (PARs, Fig. 2) [11]. PARs are G protein-coupled receptors that carry their own ligands, which remain cryptic until unmasked by receptor cleavage. Upon binding of thrombin, an extracellular proteolytic cleavage event is converted into a transmembrane signal, a principle that can account for the vast majority of known thrombin functions on cells.

Mechanisms of thrombin action. Thrombin is a multifunctional serine protease involved in blood coagulation, complement activation, and numerous cellular functions mediated via G protein-coupled prote'ase-activated receptor (PAR) signaling pathways (for further details, see [11]). Thrombin is antagonized by binding to thrombomodulin, a multi-domain proteoglycan found primarily on endothelial cells (see natural inhibitors of thrombin, Table 1). However, thrombin bound to thrombomodulin augments the ability to activate protein C, a natural anticoagulant, which in a negative feedback loop represses the generation of thrombin (protein C itself also has antiapoptotic and anti-inflammatory activity and increases activation of thrombin-activatable fibrinolysis inhibitor (TAFI), an enzyme which blocks the activation of plasminogen and inactivates vasoactive peptides like complement C5a, not shown)
When acting on cells, thrombin triggers a wide spectrum of responses such as cell proliferation, cell division, and changes of the cell morphology and motility. It induces downstream signal transduction cascades thereby affecting electrophysiology, metabolic processes, and global gene expression. In addition, thrombin has a crucial function during acute and chronic inflammatory processes, e.g., by activating the complement cascade, or as a mitogen for immune effector cells [3, 11] (further detailed below). Thrombin also plays an important role for the initiation, formation, and propagation of atherogenesis and thereby collectively illustrates the whole plethora and wide (patho)physiological relevance in many systems including hemostasis, inflammation, proliferation, and vasomotor regulation. Thrombin, however, also induces tumor growth, metastasis, and angiogenesis and might serve to preserve dormant tumor cells in individuals, preventing host eradication [6]. Thus, thrombin plays an important role in an unforeseen dimension of various (patho)physiological processes with significant incidence, prevalence, and mortality—apart from the well-established role in stemming blood loss and its perturbances.
More general information concerning thrombin and its role in blood coagulation is provided in earlier reviews [12–15]. Here, we will focus on some of the most important (patho)physiological processes mediated by thrombin and illustrate how recently uncovered regulatory mechanisms governing thrombin gene expression might explain previously enigmatic links between blood coagulation and cancer.
Thrombin in adaptive and innate immunity
Thrombin is perhaps the most effective agonist for platelet activation; upon binding to PARs, it triggers a shape change of platelets and the release of the platelet activators ADP, serotonin, thromboxane, as well as a variety of chemokines and growth factors. Furthermore, it liberates the major fibrinogen receptor GPIIb-IIIa integrin complex and P-selectin, and mobilizes the CD40 ligand to the platelet surface. While the first two enhance platelet aggregation [16], CD40L induces endothelial cells to secrete chemokines and to express adhesion molecules, thereby generating signals for the recruitment and extravasation of leukocytes [17]. Thrombin also elicits responses in the vascular endothelium including shape and permeability changes, mobilization of adhesive molecules such as vWF and P-selectin, and the production of various cytokines.
Crucially, PAR expression is found on many immune cells, including macrophages, monocytes, dendritic cells, lymphocytes, and mast cells. Thus, is it not surprising that thrombin influences many cellular functions with important roles in immunity; thrombin is chemotactic for monocytes, regulates cytokine production in fibroblasts, and is mitogenic for lymphocytes and mesenchymal cells [11]. However, it also regulates a plethora of further responses in immune cells via activation of PAR signaling [18, 19]. In the complement system, activated thrombin can directly generate C5a and thereby bypass the classical, the alternative, and the lectin pathway to trigger complement activation [20]. Yet, this is not limited to thrombin—almost all serine proteases in the coagulation system signal into the complement system, and vice versa, as descendants of a common ancestral pathway, proteolytic components from the complement system feed into the blood coagulation system [3] (Fig. 3). Thus, thrombin belongs to a complex network in which mutual connections between these two pathways dictate the activity of the entire “coagulo-complementome”. Finally, thrombin promotes the activation of various pro-inflammatory pathways including the production of pro-inflammatory cytokines (such as TNF, IL-1β, and IL-6)—and cytokines, in turn, can stimulate coagulation [3, 20–22].
In summary, apart from generating fibrin to promote hemostasis, thrombin has a host of direct actions on different cell types including platelets and endothelial cells and also various effector cells of the immune system. Moreover, thrombin belongs to the “plasma serine protease system” in which the coagulation and complement systems are tightly connected through multiple direct interactions of serine proteases. In the setting of sepsis, for instance, the extensive cross talk between the coagulation pathways and the complement system is of particular importance, as their uncontrolled activation essentially contributes to and further perpetuates the detrimental pathogenesis of the disease (see for further details [3]).
Thrombin in acute and chronic inflammatory processes
As thrombin acts both on cellular and soluble effectors of the immune system, it has pivotal roles in acute and chronic inflammatory processes [23, 24, 166], many of which are regulated by activation of PARs on respective effector cells [18, 19]. Thus, apart from physiological wound healing where thrombin helps attracting effector cells to organize and repair damaged tissues [25], overwhelming inflammatory responses triggered by thrombin can also cause detrimental responses involved in the pathophysiology of rheumatoid arthritis [26]. Accordingly, thrombin inhibition down-modulates synovial inflammation and has been shown to ameliorate even established arthritis [27]. Interestingly, in the pathogenesis of arthritis, thrombin activation (induced by collagen) exerts a dual function: it leads to an increased expression of PAR-1 in the inflamed joint and it serves, at the same time, as “ligand” for PAR-1-mediated activation of synovial hyperproliferation and an inflammatory destruction [28, 29]. Furthermore, thrombin is implicated in the pathogenesis of inflammatory brain diseases such as multiple sclerosis [30] and possibly Alzheimer’s disease [31–33]. In both cases, uncontrolled inflammatory processes that are triggered by thrombin (predominantly via activation of PARs) are suspected to contribute to the progression of inflammatory brain diseases and neuronal tissue damage by NMDA receptor response potentiation, apoptosis, and inappropriate glial proliferation. Aberrant induction of thrombin can also compromise neuronal function by disturbing the electrophysiology resulting in conduction blocks or seizures [30, 34]. Despite its detrimental role also in edema formation (as a result of thrombin-mediated permeability changes of endothelia) following intracerebral hemorrhage [35] or in vascular dementia [36] and memory impairment [37], thrombin has a neuroprotective function, especially at lower concentrations [38, 39]. Although cell death and a protective function of thrombin share initial signaling components, differences in the amplitude as well as the duration of the signal may result in different final pathways thus explaining the functional dichotomy of thrombin at different concentrations. Finally, rather reflecting its direct function in blood coagulation, the generation of thrombin resulting in local thrombosis and/or fibrin deposition limits the survival and dissemination of some microbial pathogens (by generating a “mechanical” barrier) and might thereby affect host susceptibility to a variety of infectious diseases [40, 41] (for review, [167]). However, although thrombin is also known to be upregulated in various chronic disease entities, the underlying mechanism and exact pathogenetic relevance, whether or not thrombin functions as a driver or passenger in these disease processes, is still poorly understood [31, 32, 42]. Yet, it needs to be noted that (selective) PAR-1 expression and activation, i.e., on dendritic cells, play a critical role in both chronic and acute lethal inflammatory processes [43, 44], putting blood coagulation (and thrombin specifically) also on center stage for an active immune modulatory cellular function.
Thrombin and atherosclerosis
Thrombin is generated at the site of vascular injury and has been proposed to play a crucial role in the pathogenesis of atherosclerosis by activating platelets and promoting a pro-inflammatory response [4, 25, 45]. This is characterized by an increased production of diverse chemokines and cytokines, cell adhesion molecules, enhanced vascular permeability, migration and proliferation of vascular smooth muscle cells, wall thickening, and vasoconstriction. Thus, thrombin is considered to contribute to both the initiation and also the propagation of atherosclerotic lesions. This eventually results in a vicious circle, where progressing endothelial injuries cause further thrombin conversion with detrimental self-sustaining qualities. These findings are further corroborated by mouse models, where the deletion of the natural thrombin inhibitor (heparin cofactor II, see Table 1) promotes an accelerated atherogenic state. In contrast, reduction of thrombin activity attenuates plaque progression and promotes stability in advanced atherosclerotic lesions [46]. Thus, with the advent of novel selective anticoagulants such as direct thrombin inhibitors [47] or PAR inhibition [48], great hope accompanies basic research to find potentially new therapeutic strategies to interfere with thrombin’s role in atherosclerosis. In preclinical models, selective PAR-1 blockade resulted in potent inhibition of thrombin-induced platelet aggregation but appeared to preserve primary hemostatic function [49]. These findings clearly put selective PAR-1 inhibition on center stage as a promising target to interfere with atherosclerosis. Interestingly, while PAR-1 inhibition reduces the risk of cardiovascular death or ischemic events with stable atherosclerosis [50], such effects have not been witnessed so far for acute coronary syndromes [51]. Both studies revealed that PAR-1 inhibition leads to an increased risk of moderate and severe bleeding, including intracranial hemorrhage, thus highlighting the need to optimize the therapeutic regimen to specifically interfere with thrombin’s contribution to the initiation, formation, progression, and destabilization of atherosclerotic plaques. Possibly, key to that might be that thrombin itself has earlier been observed to be regulated in response to (chronic) inflammatory events [42] (further detailed below).
Thrombin in embryonic development and angiogenesis
One of the most striking observations regarding “non-classical” thrombin functions has been made in knockout animals: Predictably, a functional null allele results in severe coagulation abnormalities leading to embryonic and neonatal lethality [52, 53]. However, thrombin is also implicated in maintaining vascular integrity during development as well as postnatal life [41, 52, 53], which is mainly driven by the activation of PARs [11]. Interestingly, thrombin expression at levels of as little as 5–10 % of the norm is still compatible with normal embryonic development, but the resulting animals are hemophilic without showing spontaneous bleeding [54]. This indicates that low levels of thrombin expression are indispensible for normal embryonic development and higher levels are required to control bleeding. Ultimately, thrombin is highly expressed and extensively regulated in muscles during (neonatal) synapse remodeling [55], after muscle denervation, and during brain development [56, 57], suggesting a role of thrombin in neuronal plasticity. This is highlighted by findings of thrombin being produced in the brain either immediately after cerebral hemorrhage or after breakdown of the blood–brain barrier, which occurs in response to many kinds of brain injury [58]. Furthermore, transient global ischemia up-modulates thrombin gene expression in the brain [59].
Thrombin in tissue and organ regeneration and differentiation
The importance of thrombin in tissue regeneration is highlighted by studies of the vertebrate lens [1, 2]. Here, selective thrombin activation has been discovered to control the cell cycle reentry at the site of tissue injury and thereby initiates the process of vertebrate lens regeneration. Thrombin also counteracts the postmitotic arrest in newt myotubes and thereby plays an important role in plasticity and reprogramming of differentiated cells in amphibian regeneration [60]. Yet, also in humans, it regulates (hematopoietic) stem and progenitor cell functions [61], it stimulates various differentiation processes [62, 63], and it has been reported to be up-modulated after spinal cord injuries [64] and other neurotraumas [56]. On the other hand, elevated thrombin production is associated with aging [65] and has been reported to contribute to the development of age-related (neuronal) deficits [37] or an increased propensity for developing blood clots at old age [65].
Thrombin in cancer and tumor biology
Blood coagulation factors in general and thrombin in particular have recently been found to play an important role in cancer biology [6, 7, 66–70]: In tumor patients, increased pro-coagulatory activities are almost inevitably seen at some point during tumor progression, where tumor procoagulants are released into the blood stream and thus give rise to the development of thrombosis with serious—often life-threatening—consequences. However, thrombosis can also represent a forewarning of an as yet undiagnosed “occult” malignancy (so called Trousseau’s syndrome) [71]. Thus, hypercoagulabilities have not only serious therapeutic but also important diagnostic implications. Although considered to be the consequence of an underlying tumor disease for almost 150 years, recent evidence suggests that this syndrome is not a mere paraneoplastic effect, but the result of mechanisms that provide a selective advantage to cancer cells [72–77]—with a striking impact on tumor initiation, tumor progression, and patient prognosis [72, 74, 75, 77–79]. In line with these findings, hyperactivation of blood coagulation is associated with more rapid tumor progression [72–74]. Conversely, impaired blood coagulation reduces the incidence of cancer [75] and inhibits the invasive growth of tumor cells and metastasis in patients treated with anticoagulants [77, 78] or in mice defective for coagulation factors such as fibrinogen [80] or thrombin [81].
How does thrombin influence tumor biology?
In 1986, Harold Dvorak described parallels between wound healing and tumor disease, where hemostasis (local fibrin deposition) is an inherent part of physiological regeneration processes, which are also engaged during tumorigenesis [82]. This includes almost all factors of primary and secondary hemostasis. It involves the direct activation of thrombin and fibrin synthesis by production of pro-coagulatory substances by tumor cells and/or indirectly via the activation of endothelial cells, thrombocytes, and leukocytes by production of cytokines, proteases, glycoproteins, and tissue factor-loaded microparticles [83]. Eventually, this creates a protumorigenic micromilieu, which drives cellular programs promoting cell growth, motility, angiogenesis, and invasiveness [7].
Tissue factor is one of the most important tumor-associated determinants for tumor progression and metastasis (i.e., by the induction of tissue factor signaling promoting tumor growth and angiogenesis; see [84, 85] for review). In addition, tissue factor mediates (local) thrombin generation (Fig. 3), which is crucial for various protumorigenic processes [6, 70, 86, 87]. The critical role of thrombin in augmenting protumorigenic cellular programs reflects the whole plethora of thrombin functions—including its dual role in fibrin formation and platelet activation, the activation of PAR signaling, the proteolytic breakdown of extracellular matrix, and/or direct oncogenic mechanisms (i.e., via induction of c-myc or co-activation of the hepatocyte growth factor) [6, 69, 84, 88–90]. Specifically, thrombin generation is crucial for metastasis not only through fibrin and platelet deposition but also via thrombin-dependent PAR-1 signaling [86, 91–95]. Thrombin stimulates tumor adhesion [91, 92, 94, 96, 97], growth [98], DNA synthesis, and cellular proliferation either directly or in synergy with other mitogens [86, 99]. Thrombin is an effective activator of angiogenesis by clotting-dependent mechanisms involving platelet activation and fibrin deposition. However, thrombin also induces tumor angiogenesis via clotting-independent mechanisms mediated by PAR activation, which leads to an upregulation of various growth factors, including VEGF [100], angiopoetin-1 [101] and angiopoietin-2 [102], the major VEGF receptor KDR, as well as MMP1 and MMP2 in endothelial cells [103, 104]. Furthermore, activated platelets augment the pro-angiogenic process by releasing VEGF and platelet-derived growth factors [100, 105]. Finally, thrombin-dependent fibrin formation and platelet activation create a niche protecting the emerging tumor against natural killer cell attacks [106–108].

Cross talk between the coagulation and complement system. The coagulation cascade, the complement system, and fibrinolysis (simplified) communicate through many direct and bidirectional interactions (indicated). Activated clotting Factor XII can activate the classical complement pathway through cleavage of the complement component C1. Similarly, thrombin, kallikrein, and plasmin directly cleave complement component C3, as well as its activation fragments (not shown). Moreover, thrombin can cleave C5 into C5a, which occurs independently of C3 and therefore represents a bypass of the three traditional complement activation pathways (the classical, the lectin, and alternative pathways) [3]. Thrombin-activatable fibrinolysis inhibitor (TAFI) inactivates C3a and C5a in a negative feedback loop. The complement system also amplifies coagulation through the C5a-mediated induction of expression of tissue factor (TF) and plasminogen activator inhibitor 1 by leukocytes (not shown), the latter of which inhibits fibrinolysis. In addition, mannan-binding lectin serine protease 2 (MASP2) of the lectin complement activation pathway triggers coagulation by converting prothrombin to thrombin. MAC, membrane attack complex (C5b–C9); see also [3, 161]
In patients, the appropriate control of thrombin expression and activation therefore not only determines the delicate balance of pro- and anticoagulatory activities [109–112] but also effects tumor dissemination and metastasis [81, 113]. Accordingly, the prevalence of mutations increasing thrombin expression (such as F2 20210 G>A) are higher in some cancer patients compared to controls [76], and tumor cells treated with thrombin display increased metastatic potential [81, 91]. These observations are highlighted by our own data showing that thrombin gene expression is specifically upregulated in metastatic prostate and colon cancer (Fig. 4; [113]), which ultimately leads to PAR activation and induction of genes involved in thrombin-mediated invasion and angiogenesis.

Stage-dependent induction of ectopic (i.e., extrahepatic) thrombin (F2) gene expression in metastatic prostate cancer. Normalized mRNA expression of the thrombin (F2) gene, of the F2 Receptor (F2R/PAR1), of V-SRC and ARHGEF2 (surrogate for activated F2R signaling), and of cathepsin D and angiopoetin 2 (for invasion and angiogenesis [6, 137]), obtained from gene expression profiling after extraction, normalization, and reassembly of 171 human samples (see [162] GEO GDS2545 record) including metastatic prostate cancer tissues (n = 24; GSE6605), nonmetastatic primary prostate tumors (n = 60; GSE6606), prostate tissues adjacent to the tumor (n = 63; GSE6608), and normal donor prostate tissues (n = 18, GSE6604) [163, 164] (median, horizontal line; 25th through 75th percentile, box; range, standard error of the mean (SEM); *p < 0.05; **p < 0.01; ***p < 0.001)
Inversely, low-level thrombin expression [81] or the specific inhibition of thrombin by sulfohirudin or thrombostatin reduces tumor growth and metastasis in vivo [114, 115]. This prometastatic function of thrombin is further corroborated by findings demonstrating that the endogenous generation and/or activity of thrombin—by altering the thrombomodulin system—plays a crucial role for spontaneous metastasis in vivo [116]. Finally, the expression of thrombin-activated receptors (PAR-1) is frequently up-modulated in highly metastatic tumors [88], which correlates with negative prognosis [117]. Interestingly, numerous reports document beneficial effects of pharmaceutical thrombin inhibition for cancer patient survival (for reviews, see [6, 70, 118] and special issue, Journal of Clinical Oncology [77, 83]).
In summary, thrombin contributes to various hallmark processes directly associated with tumor dissemination and progression including (1) cellular proliferation and tumor growth, (2) tumor adhesion to subendothelial matrix, (3) tissue invasion and extravasation, (4) tumor-associated angiogenesis, (5) tumor-associated pro-inflammatory processes, and (6) the colonization of a metastatic niche (fibrin net encapsulation and platelet activation; Table 2).
Whether thrombin also plays a role for tumor initiation and early events in tumorigenesis is yet to be determined. Nevertheless, it is noteworthy that the prevalence of prothrombotic mutations increasing thrombin gene expression is higher in some cohorts of cancer patients compared to controls [76]. This might either reflect a nonfunctional association or display a potential selective disadvantage and predisposition for carriers of F2 20210 G>A for developing specific cancer entities—although this association clearly does not apply to all tumor types [74, 79].
The functional role of thrombin for tumor initiation might therefore differ with regard to specific tumor entities. Therapeutic approaches targeting thrombin may thus help to interfere with its protumorigenic properties during tumor progression, but possibly also in tumor initiation. Although several hallmark studies document the detrimental effect of disordered hemostasis for cancer onset, these observations have so far eluded mechanistic explanation [72–76, 80, 140]. A possible key to that might be that thrombin is upregulated during inflammatory events [20, 32, 42, 113, 141]. Understanding potentially underlying mechanisms could therefore help to disentangle the enigmatic relationship between blood coagulation and cancer biology, and potentially contribute to the development of novel therapeutic strategies.
When and where is thrombin generated? A p38 MAPK dependent switch controls F2 expression
In order to become biologically active, prothrombin is cleaved into thrombin. This proteolytic activation step is catalyzed by activated Factor X (Fig. 3) and controlled by various negative feedback mechanisms to prevent overwhelming pro-coagulatory activities or other uncontrolled thrombin-dependent activities (see natural thrombin inhibitors, Table 1). Nevertheless, mutations that merely increase thrombin expression (such as F2 20210 G>A) already shift the well-balanced equilibrium of pro- and anticoagulatory activities [109, 142, 143]. This indicates that the activation step of prothrombin cleavage becomes secondary. As a consequence, prothrombin gene expression needs to be tightly controlled: even subtle changes (1.5- to 1.7-fold) of its gene expression [143, 144] can result in a clinically relevant thrombophilia [13, 145].
Although primarily synthesized in hepatocytes (in adults), thrombin is also expressed in the brain [57] and is induced in neurons after cerebral ischemia [59], during embryonic development [52, 53, 55, 57], and in various acute and chronic inflammatory processes [20, 32, 42, 56, 64, 141, 146]. Hence, cells must have evolved mechanism(s) that fine-tune thrombin expression and thereby ensure proper execution of thrombin-mediated cellular programs. Yet—as highlighted above—the underlying molecular principles governing thrombin expression regulation have remained enigmatic until recently.
Inflammatory processes represent possible triggers to induce thrombin gene expression [20, 32, 42, 113, 141, 147, 148]. In addition, the crucial role of thrombin for angiogenesis [11] suggests that a regulatory mechanism controlling thrombin expression might have evolved a sensor for low oxygen pressure. This could explain why thrombin is hyperexpressed in response to ischemic events [59] or in the tumor micromilieu of growing tumors [113], which notoriously suffer from low oxygen pressure.
We recently discovered a gene regulatory mechanism affecting RNA 3′ end processing that operates in response to and integrates environmental stimuli to fine-tune thrombin mRNA expression via p38 MAPK activation [113] (Fig. 5). In turn, p38 MAPK up-modulates the RNA 3′ end processing machinery and directly induces the phosphorylation of inhibitory proteins, which bind to a highly conserved sequence motif in the F2 3′UTR. Upon phosphorylation, these inhibitory proteins fail to bind the prothrombin mRNA, making it accessible to proteins that stimulate 3′ end processing (many of which play important roles in cancer [149]). This eventually results in a higher (pro)thrombin expression under inflammatory conditions such as septicemia. Interestingly, p38 MAPK activation also directs thrombin overexpression in the tumor microenvironment of metastasized colon carcinoma. This, in turn, activates PARs, which induces the expression of genes with crucial roles in neoangiogenesis and tumor dissemination [113] (Figs. 4 and 5), highlighting the physiological importance of novel layers of gene expression control [150–153].

Extracellular stimuli induce thrombin gene expression by p38 MAPK activation. Extracellular stimuli activate p38 MAPK and thereby phosphorylate regulatory proteins (red), which “catalyzes” the remodeling of a stimulatory ribonucleoprotein (RNP) complexes (green) to up-modulate the efficiency of thrombin mRNA 3′ end processing. This mechanism allows modulating cellular functions, such as blood coagulation by controlling the amount of thrombin protein produced. Yet activation of this mechanism also appears to play an important role in other pathophysiological processes (such as tumorigenesis) and drives cellular programs involved in tumor invasion and neoangiogenesis by the activation of thrombin receptor signaling (F2R, PAR-1) and degradation of extracellular matrix (figure adopted from Cell Press [113])
The identification of p38 MAPK controlling thrombin represents a hypothetically interesting “building block” for a model, whereby (tumor-associated) hypoxia and/or ischemia might trigger thrombin expression: hypoxia represents a potent inducer of p38 MAPK [154], and its ablation results in defects of placental angiogenesis [155]. Partly, this phenocopies the lethal vessel-malformation phenotype of mice lacking thrombin [53]. Interestingly, the functional importance of p38 MAPK for the regulation of thrombin gene expression is further corroborated in prostate cancer patients, where a stage-specific induction of p38 MAPK activity correlates with an up-modulation of thrombin gene expression in metastatic prostate cancer (Fig. 6).

Induction of thrombin (F2) gene expression correlates with a stage-dependent activation of p38 MAPK signaling in metastatic prostate cancer. Normalized mRNA expression of p38 MAPK (upper diagram) and of MYO1B (lower left diagram), which reflects activation of p38 MAPK signaling [165], obtained from gene expression profiling of 171 human samples [162] (median, horizontal line; 25th through 75th percentile, box; range, standard error of the mean (SEM); *p < 0.05; **p < 0.01; ***p < 0.001). Correlation of p38 MAPK signaling activation (MYO1B gene expression) with F2 gene expression is shown in the lower right diagram (Spearman’s rank correlation)
Thrombin in blood coagulation, inflammation, cancer and beyond: back to Virchow?
Importantly, an estimated 40–50 % of all human cancers are linked to chronic inflammation [156], which can induce p38 MAPK. But also other environmentally triggered programs such as the DNA damage or oxidative stress response (with important roles during tumorigenesis) can directly activate p38 MAPK [154], and deregulated p38 MAPK signaling itself is associated with cancers in humans and mice [155, 157]. Therefore, protumorigenic signals such as inflammation, DNA damage, or reactive oxygen species might hypothetically represent the long-sought-after common risk factor between deregulated blood coagulation and the increased incidence of cancer [73] with detrimental bidirectional self-sustaining qualities [7, 82] (Fig. 7).

Model for inflammation as a unifying trigger predisposing to deregulated blood coagulation (thrombin gene expression) and tumor formation. Inflammatory stimuli can induce both tumor formation (simplified) and thrombin (F2) gene expression. This in turn leads to a disequilibrium of pro- and anticoagulatory activities (and thereby promotes tumor-associated thrombus formation) and drives protumorigenic cellular programs (in an autocrine and/or paracrine manner; SD unpublished). Tumor formation will thus be supported by the tumor-promoting properties of thrombin; vice versa, tumor formation elicits detrimental inflammatory responses [159], which in turn further promote tumorigenesis and p38 MAPK (p38)-mediated induction of thrombin gene expression. (Extracellular matrix (ECM), Reactive oxygen species (ROS), epithelial–mesenchymal transformation, mesenchymal–epithelial transformation (EMT/MET))
Remarkably, such an association is also corroborated by earlier studies, which demonstrated an inflammatory tumor microenvironment to be associated with the induction of thrombin expression [113]. This regulatory mechanism would provide a molecular basis by which protumorigenic environmental stimuli such as inflammation [158–160] can be directly linked to the hyperexpression of thrombin (and its detrimental consequences on tumor biology). This mechanism might also help to explain thrombin’s role at the center of numerous inflammatory and noninflammatory (patho)physiological processes.
Future directions
Although we unquestionably have broadened our understanding of thrombin and its cellular functions within the past 20 years, we are just beginning to understand that thrombin plays a previously underappreciated role in (patho)physiology—ranging from autoimmunity, blood coagulation, cancer to wound healing, but possibly also regenerative medicine.
What remains puzzling is the contribution of thrombin to many of these processes on a systems’ level in living organisms. This is mainly due to the fact that the complete lack of thrombin is lethal. In fact, most of the aforementioned roles of thrombin’s contribution in diverse (patho)physiologies have been explored in cell culture studies and/or been deduced from animal studies where thrombin activity was reduced by pharmaceutical inhibitors. What continues to be challenging is the multifunctionality of thrombin: it can act systemically but also locally in different, though (patho)physiologically meaningful “sub-compartments.” Thus, systemic determinations of thrombin activity do not necessarily reflect its local activity. In addition, there are numerous tissues in which thrombin is dynamically (re)expressed and modulated in various (patho)physiological conditions (see above). We currently do not understand to what extent (de)regulated thrombin expression and its activation represent cause or consequence of (patho)physiological processes. Therefore, studying the role of thrombin with spatial and temporal resolution could pave the way to much better dissect the roles of this multifunctional serine protease in various tissues and disease entities. This would also help elucidating the potential therapeutic dimension of strategies targeting thrombin gene expression and/or activation.
References
Imokawa Y, Brockes JP (2003) Selective activation of thrombin is a critical determinant for vertebrate lens regeneration. Curr Biol 13:877–881
Maden M (2003) Regeneration: every clot has a thrombin lining. Curr Biol 13:R517–R518
Rittirsch D, Flierl MA, Ward PA (2008) Harmful molecular mechanisms in sepsis. Nat Rev Immunol 8:776–787
Borissoff JI, Spronk HMH, Heeneman S, ten Cate H (2009) Is thrombin a key player in the ‘coagulation-atherogenesis’ maze? Cardiovasc Res 82:392–403
Turgeon VL, Houenou LJ (1997) The role of thrombin-like (serine) proteases in the development, plasticity and pathology of the nervous system. Brain Res Rev 25:85–95
Nierodzik ML, Karpatkin S (2006) Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 10:355–362
Joyce JA, Pollard JW (2009) Microenvironmental regulation of metastasis. Nat Rev Cancer 9:239–252
Rau JC, Beaulieu LM, Huntington JA, Church FC (2007) Serpins in thrombosis, hemostasis and fibrinolysis. J Thromb Haemost 5:102–115
Esmon CT (1989) The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem 264:4743–4746
Andrew M, Vegh P, Johnston M, Bowker J, Ofosu F, Mitchell L (1992) Maturation of the hemostatic system during childhood. Blood 80:1998–2005
Coughlin SR (2000) Thrombin signalling and protease-activated receptors. Nature 407:258–264
Schenone M, Furie BC, Furie B (2004) The blood coagulation cascade. Curr Opin Hematol 11:272–277
Danckwardt S, Hartmann K, Gehring NH, Hentze MW, Kulozik AE (2006) 3′ end processing of the prothrombin mRNA in thrombophilia. Acta Haematol 115:192–197
Tanaka KA, Key NS, Levy JH (2009) Blood coagulation: hemostasis and thrombin regulation. Anesth Analg 108:1433–1446
Becker RC (2005) Understanding the dynamics of thrombin in cardiovascular disease: pathobiology and biochemistry for the clinician. Am Heart J 149:S2–S8
Subramaniam M, Frenette P, Saffaripour S, Johnson R, Hynes R, Wagner D (1996) Defects in hemostasis in P-selectin-deficient mice. Blood 87:1238–1242
Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA (1998) CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 391:591–594
Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD (2005) Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocrine Rev 26:1–43
Shpacovitch V, Feld M, Hollenberg MD, Luger TA, Steinhoff M (2008) Role of protease-activated receptors in inflammatory responses, innate and adaptive immunity. J Leukoc Biol 83:1309–1322
Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM et al (2006) Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 12:682–687
Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA (1986) Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterization and comparison with the actions of interleukin 1. Proc Natl Acad Sci 83:4533–4537
Stouthard JM, Levi M, Hack CE, Veenhof CH, Romijn HA, Sauerwein HP, van der Poll T (1996) Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb Haemost 76:738–742
Chen D, Dorling A (2009) Critical roles for thrombin in acute and chronic inflammation. J Thromb Haemost 7:122–126
Chan C-P, Chang M-C, Wang Y-J, Chen L-I, Tsai Y-L, Lee J-J, Jia H-W, Jeng J-H (2008) Thrombin activates Ras-CREB/ATF-1 signaling and stimulates c-fos, c-jun, and c-myc expression in human gingival fibroblasts. J Periodont 79:1248–1254
Popović M, Smiljanić K, Dobutović B, Syrovets T, Simmet T, Isenović E (2012) Thrombin and vascular inflammation. Mol Cell Biochem 359:301–313
Morris R, Winyard PG, Blake DR, Morris CJ (1994) Thrombin in inflammation and healing: relevance to rheumatoid arthritis. Ann Rheum Dis 53:72–79
Marty I, Péclat V, Kirdaite G, Salvi R, So A, Busso N (2001) Amelioration of collagen-induced arthritis by thrombin inhibition. J Clin Invest 107:631–640
Shin H, Nakajima T, Kitajima I, Shigeta K, Abeyama K, Imamura T, Okano T, Kawahara K, Nakamura T, Maruyama I (1995) Thrombin receptor-mediated synovial proliferation in patients with rheumatoid arthritis. Clin Immunol Immunopathol 76:225–233
Ellis CA, Malik AB, Gilchrist A, Hamm H, Sandoval R, Voyno-Yasenetskaya T, Tiruppathi C (1999) Thrombin induces proteinase-activated receptor-1 gene expression in endothelial cells via activation of Gi-linked Ras/mitogen-activated protein kinase pathway. J Biol Chem 274:13718–13727
Chapman J (2006) Thrombin in inflammatory brain diseases. Autoimmun Rev 5:528–531
Grammas P, Samany PG, Thirumangalakudi L (2006) Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alzheimers Dis 9:51–58
Yin X, Wright J, Wall T, Grammas P (2010) Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol 176:1600–1606
Arai T, Miklossy J, Klegeris A, Guo J-P, McGeer PL (2006) Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J Neuropathol Exp Neurol 65:19–25
Lee KR, Drury I, Vitarbo E, Hoff JT (1997) Seizures induced by intracerebral injection of thrombin: a model of intracerebral hemorrhage. J Neurosurg 87:73–78
Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT (1996) Edema from intracerebral hemorrhage: the role of thrombin. J Neurosurg 84:91–96
Kario K, Matsuo T, Hoshide S, Umeda Y, Shimada K (1999) Effect of thrombin inhibition in vascular dementia and silent cerebrovascular disease. Stroke 30:1033–1037
Mhatre M, Nguyen A, Kashani S, Pham T, Adesina A, Grammas P (2004) Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol Aging 25:783–793
Donovan FM, Cunningham DD (1998) Signaling pathways involved in thrombin-induced cell protection. J Biol Chem 273:12746–12752
Vaughan P, Pike C, Cotman C, Cunningham D (1995) Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J of Neurosc 15:5389–5401
Sun H, Wang X, Degen JL, Ginsburg D (2009) Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood 113:1358–1364
Mullins ES, Kombrinck KW, Talmage KE, Shaw MA, Witte DP, Ullman JM, Degen SJ, Sun W, Flick MJ, Degen JL (2009) Genetic elimination of prothrombin in adult mice is not compatible with survival and results in spontaneous hemorrhagic events in both heart and brain. Blood 113:696–704
Murakami H, Okazaki M, Amagasa H, Oguchi K (2003) Increase in hepatic mRNA expression of coagulant factors in type 2 diabetic model mice. Thromb Res 111:81–87
Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, Derian CK, Andrade-Gordon P, Rosen H, Ruf W (2008) Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 452:654–658
Li X, Syrovets T, Paskas S, Laumonnier Y, Simmet T (2008) Mature dendritic cells express functional thrombin receptors triggering chemotaxis and CCL18/pulmonary and activation-regulated chemokine induction. J Immunol 181:1215–1223
Tracy R (2003) Thrombin, inflammation, and cardiovascular disease: an epidemiologic perspective. Chest 124:49–57
Borissoff JI, Spronk HMH, ten Cate H (2011) The hemostatic system as a modulator of atherosclerosis. N Engl J Med 364:1746–1760
Di Nisio M, Middeldorp S, Büller HR (2005) Direct thrombin inhibitors. N Engl J Med 353:1028–1040
Ramachandran R, Noorbakhsh F, DeFea K, Hollenberg MD (2012) Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov 11:69–86
Members WG, Wright RS, Anderson JL, Adams CD, Bridges CR, Casey DE, Ettinger SM, Fesmire FM, Ganiats TG, Jneid H et al (2011) 2011 ACCF/AHA focused update of the guidelines for the management of patients with unstable angina/ non–ST-elevation myocardial infarction (updating the 2007 guideline). Circulation 123:2022–2060
Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KAA, Lipka LJ, Liu X, Nicolau JC et al (2012) Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med 366:1404–1413
Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E et al (2012) Thrombin-receptor antagonist vorapaxar in acute coronary Syndromes. N Engl J Med 366:20–33
Sun WY, Witte DP, Degen JL, Colbert MC, Burkart MC, Holmback K, Xiao Q, Bugge TH, Degen SJ (1998) Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci 95:7597–7602
Xue J, Wu Q, Westfield LA, Tuley EA, Lu D, Zhang Q, Shim K, Zheng X, Sadler JE (1998) Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc Natl Acad Sci 95:7603–7607
Sun WY, Coleman MJ, Witte DP, Degen SJF (2002) Rescue of prothrombin-deficiency by transgene expression in mice. Thromb Haemost 88:984–991
Zoubine MN, Ma JY, Smirnova IV, Citron BA, Festoff BW (1996) A molecular mechanism for synapse elimination: novel inhibition of locally generated thrombin delays synapse loss in neonatal mouse muscle. Dev Biol 179:447–457
Kim S, Buonanno A, Nelson PG (1998) Regulation of prothrombin, thrombin receptor, and protease nexin-1 expression during development and after denervation in muscle. J Neurosci Res 53:304–311
Dihanich M, Kaser M, Reinhard E, Cunningham D, Monard D (1991) Prothrombin mRNA is expressed by cells of the nervous system. Neuron 6:575–581
Xi G, Reiser G, Keep RF (2003) The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem 84:3–9
Riek-Burchardt M, Striggow F, Henrich-Noack P, Reiser G, Reymann KG (2002) Increase of prothrombin-mRNA after global cerebral ischemia in rats, with constant expression of protease nexin-1 and protease-activated receptors. Neurosci Lett 329:181–184
Brockes JP, Kumar A (2002) Plasticity and reprogramming of differentiated cells in amphibian regeneration. Nature Rev 3:566–574
Grassinger J, Haylock DN, Storan MJ, Haines GO, Williams B, Whitty GA, Vinson AR, Be CL, Li S, Sørensen ES et al (2009) Thrombin-cleaved osteopontin regulates hemopoietic stem and progenitor cell functions through interactions with α9β1 and α4β1 integrins. Blood 114:49–59
Martin K, Weiss S, Metharom P, Schmeckpeper J, Hynes B, O’Sullivan J, Caplice N (2009) Thrombin stimulates smooth muscle cell differentiation from peripheral blood mononuclear cells via protease-activated receptor-1, RhoA, and myocardin. Circulation Res 105:214–218
Tarzami ST, Wang G, Li W, Green L, Singh JP (2006) Thrombin and PAR-1 stimulate differentiation of bone marrow-derived endothelial progenitor cells. J Thromb Haemost 4:656–663
Citron BA, Smirnova IV, Arnold PM, Festoff BW (2000) Upregulation of neurotoxic serine proteases, prothrombin, and protease-activated receptor 1 early after spinal cord injury. J Neurotrauma 17:1191–1203
Bauer KA, Weiss LM, Sparrow D, Vokonas PS, Rosenberg RD (1987) Aging-associated changes in indices of thrombin generation and protein C activation in humans. Normative Aging Study. J Clin Invest 80:1527–1534
Degen JL, Palumbo JS (2012) Hemostatic factors, innate immunity and malignancy. 129. Thromb Res 1:S1–S5, 129 Supplement
Green D, Karpatkin S (2010) Role of thrombin as a tumor growth factor. Cell cycle 9:656–661
Boccaccio C, Comoglio PM (2005) A functional role for hemostasis in early cancer development. Cancer Res 65:8579–8582
Boccaccio C, Comoglio PM (2006) Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer 6:637–645
Franchini M, Mannucci PM (2012) Thrombin and cancer: from molecular basis to therapeutic implications. Semin Thromb Hemost 38:95–101
Varki A (2007) Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 110:1723–1729
Prandoni P, Lensing AW, Buller HR, Cogo A, Prins MH, Cattelan AM, Cuppini S, Noventa F, ten Cate JW (1992) Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 327:1128–1133
Baron JA, Gridley G, Weiderpass E, Nyren O, Linet M (1998) Venous thromboembolism and cancer. Lancet 351:1077–1080
Miller GJ, Bauer KA, Howarth DJ, Cooper JA, Humphries SE, Rosenberg RD (2004) Increased incidence of neoplasia of the digestive tract in men with persistent activation of the coagulant pathway. J Thromb Haemost 2:2107–2114
Schulman S, Lindmarker P (2000) Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. N Engl J Med 342:1953–1958
Blom JW, Doggen CJ, Osanto S, Rosendaal FR (2005) Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 293:715–722
Kuderer NM, Ortel TL, Francis CW (2009) Impact of venous thromboembolism and anticoagulation on cancer and cancer survival. J Clin Oncol 27:4902–4911
Petralia GA, Lemoine NR, Kakkar AK (2005) Mechanisms of disease: the impact of antithrombotic therapy in cancer patients. Nat Clin Pract Oncol 2:356–363
Vossen CY, Hoffmeister M, Chang-Claude JC, Rosendaal FR, Brenner H (2011) Clotting factor gene polymorphisms and colorectal cancer risk. J Clin Oncol 29:1722–1727
Palumbo JS, Potter JM, Kaplan LS, Talmage K, Jackson DG, Degen JL (2002) Spontaneous hematogenous and lymphatic metastasis, but not primary tumor growth or angiogenesis, is diminished in fibrinogen-deficient mice. Cancer Res 62:6966–6972
Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Hu Z, Barney KA, Degen JL (2007) Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and -independent mechanisms. Blood 110:133–141
Dvorak HF (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315:1650–1659
Lyman GH, Khorana AA (2009) Cancer, clots and consensus: new understanding of an old problem. J Clin Oncol 27:4821–4826
Rickles FR, Patierno S, Fernandez PM (2003) Tissue factor, thrombin, and cancer. Chest 124:58S–68S
Ruf W, Disse J, Carneiro-Lobo TC, Yokota N, Schaffner F (2011) Tissue factor and cell signalling in cancer progression and thrombosis. J Thromb Haemost 9:306–315
Ruf W, Mueller BM (2006) Thrombin generation and the pathogenesis of cancer. Semin Thromb Hemost 32:061–068
Palumbo JS (2008) Mechanisms linking tumor cell-associated procoagulant function to tumor dissemination. Semin Thromb Hemost 34:154–160
Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y, Reich R, Vlodavsky I, Bar-Shavit R (1998) Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med 4:909–914
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120:303–313
Kataoka H, Hamasuna R, Itoh H, Kitamura N, Koono M (2000) Activation of hepatocyte growth factor/scatter factor in colorectal carcinoma. Cancer Res 60:6148–6159
Nierodzik ML, Chen K, Takeshita K, Li JJ, Huang YQ, Feng XS, D’Andrea MR, Andrade-Gordon P, Karpatkin S (1998) Protease-activated receptor 1 (PAR-1) is required and rate-limiting for thrombin-enhanced experimental pulmonary metastasis. Blood 92:3694–3700
Nierodzik MLR, Kajumo F, Karpatkin S (1992) Effect of thrombin treatment of tumor cells on adhesion of tumor cells to platelets in vitro and tumor metastasis in vivo. Cancer Res 52:3267–3272
Hu L, Lee M, Campbell W, Perez-Soler R, Karpatkin S (2004) Role of endogenous thrombin in tumor implantation, seeding, and spontaneous metastasis. Blood 104:2746–2751
Wojtukiewicz MZ, Tang DG, Nelson KK, Walz DA, Diglio CA, Honn KV (1992) Thrombin enhances tumor cell adhesive and metastatic properties via increased alpha IIb beta 3 expression on the cell surface. Thromb Res 68:233–245
Wojtukiewicz MZ, Tang DG, Ciarelli JJ, Nelson KK, Walz DA, Diglio CA, Mammen EF, Honn KV (1993) Thrombin increases the metastatic potential of tumor cells. Int J Cancer 54:793–806
Nierodzik MLR, Bain RM, Liu L-X, Shivj i M, Takesh ita K, Karpatki n S (1996) Presence of the seven transmembrane thrombin receptor on human tumour cells: effect of activation on tumour adhesion to platelets and tumour tyrosine phosphorylation. B J Haematol 92:452–457
Klepfish A, Greco MA, Karpatkin S (1993) Thrombin stimulates melanoma tumor-cell binding to endothelial cells and subendothelial matrix. Int J Cancer 53:978–982
Zain J, Huang YQ, Feng X, Nierodzik ML, Li JJ, Karpatkin S (2000) Concentration-dependent dual effect of thrombin on impaired growth/apoptosis or mitogenesis in tumor cells. Blood 95:3133–3138
Franchini M, Montagnana M, Targher G, Manzato F, Lippi G (2007) Pathogenesis, clinical and laboratory aspects of thrombosis in cancer. J Thromb Thrombol 24:29–38
Möhle R, Green D, Moore MAS, Nachman RL, Rafii S (1997) Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci 94:663–668
Li J-J, Huang Y-C, Basch R, Karpatkin S (2001) Thrombin induces the release of angiopoietin-1 from platelets. Thromb Haemost 85:204–206
Huang Y-Q, Li J-J, Hu L, Lee M, Karpatkin S (2002) Thrombin induces increased expression and secretion of angiopoietin-2 from human umbilical vein endothelial cells. Blood 99:1646–1650
Tsopanoglou NE, Maragoudakis ME (1999) On the mechanism of thrombin-induced angiogenesis: potentiation of vascular endothelial growth factor activity on endothelial cells by up-regulation of its receptors. J Biol Chem 274:23969–23976
Duhamel-Clérin E, Orvain C, Lanza F, Cazenave J-P, Klein-Soyer C (1997) Thrombin receptor-mediated increase of two matrix metalloproteinases, MMP-1 and MMP-3, in human endothelial cells. Arterioscler Thromb Vasc Biol 17:1931–1938
Pinedo HM, Verheul HMW, D’Amato RJ, Folkman J (1998) Involvement of platelets in tumour angiogenesis? Lancet 352:1775–1777
Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Jirousková M, Degen JL (2005) Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 105:178–185
Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR (2004) Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 104:397–401
Nieswandt B, Hafner M, Echtenacher B, Männel DN (1999) Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 59:1295–1300
Poort SR, Rosendaal FR, Reitsma PH, Bertina RM (1996) A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 88:3698–3703
Danckwardt S, Hartmann K, Katz B, Hentze M, Levy Y, Eichele R, Deutsch V, Kulozik A, Ben-Tal O (2006) The prothrombin 20209 C>T mutation in Jewish-Moroccan Caucasians: molecular analysis of gain-of-function of 3′ end processing. J Thromb Haemost 4:1078–1085
Danckwardt S, Kaufmann I, Gentzel M, Foerstner KU, Gantzert AS, Gehring NH, Neu-Yilik G, Bork P, Keller W, Wilm M et al (2007) Splicing factors stimulate polyadenylation via USEs at non-canonical 3′ end formation signals. EMBO J 26:2658–2669
Miyawaki Y, Suzuki A, Fujita J, Maki A, Okuyama E, Murata M, Takagi A, Murate T, Kunishima S, Sakai M et al (2012) Thrombosis from a prothrombin mutation conveying antithrombin resistance. N Engl J Med 366:2390–2396
Danckwardt S, Gantzert AS, Macher-Goeppinger S, Probst HC, Gentzel M, Wilm M, Grone HJ, Schirmacher P, Hentze MW, Kulozik AE (2011) p38 MAPK controls prothrombin expression by regulated RNA 3′ end processing. Mol Cell 41:298–310
Esumi N, Fan D, Fidler IJ (1991) Inhibition of murine melanoma experimental metastasis by recombinant desulfatohirudin, a highly specific thrombin inhibitor. Cancer Res 51:4549–4556
Nieman MT, LaRusch G, Fang C, Zhou Y, Schmaier AH (2010) Oral thrombostatin FM19 inhibits prostate cancer. Thromb Haemost 104:1044–1048
Horowitz NA, Blevins EA, Miller WM, Perry AR, Talmage KE, Mullins ES, Flick MJ, Queiroz KCS, Shi K, Spek CA et al (2011) Thrombomodulin is a determinant of metastasis through a mechanism linked to the thrombin binding domain but not the lectin-like domain. Blood 118:2889–2895
Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A et al (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14:518–527
Snyder KM, Kessler CM (2008) The pivotal role of thrombin in cancer biology and tumorigenesis. Sem Thromb Hemost 34:734–741
Dardik R, Savion N, Kaufmann Y, Varon D (1998) Thrombin promotes platelet-mediated melanoma cell adhesion to endothelial cells under flow conditions: role of platelet glycoproteins P-selectin and GPIIb-IIIA. Br J Cancer 77:2069–2075
Karpatkin S (2004) Does hypercoagulability awaken dormant tumor cells in the host? J Thromb Haemost 12:2103–2106
Chen LB, Buchanan JM (1975) Mitogenic activity of blood components. I Thrombin and prothrombin PNAS 72:131–135
Gospodarowicz D, Brown KD, Birdwell CR, Zetter BR (1978) Control of proliferation of human vascular endothelial cells. Characterization of the response of human umbilical vein endothelial cells to fibroblast growth factor, epidermal growth factor, and thrombin. J Cell Biol 77:774–788
Carney DH, Stiernberg J, Fenton JW (1984) Initiation of proliferative events by human alpha-thrombin requires both receptor binding and enzymic activity. J Cell Biochem 26:181–195
Hu L, Ibrahim S, Liu C, Skaar J, Pagano M, Karpatkin S (2009) Thrombin induces tumor cell cycle activation and spontaneous growth by down-regulation of p27Kip1, in association with the up-regulation of Skp2 and MiR-222. Cancer Res 69:3374–3381
Chang M-C, Jeng J-H, Lin C-P, Lan W-H, Tsai W, Hsieh C-C (1999) Thrombin activates the growth, cell-cycle kinetics, and clustering of human dental pulp cells. J Endodont 25:118–122
Lockard MM, Witkowski S, Jenkins NT, Spangenburg EE, Obisesan TO (1985) Hagberg JM (2010) Thrombin and exercise similarly influence expression of cell cycle genes in cultured putative endothelial progenitor cells. J Appl Physiol 108:1682–1690
Xue YH, Zhang XF, Dong QZ, Sun J, Dai C, Zhou HJ, Ren N, Jia HL, Ye QH, Qin LX (2010) Thrombin is a therapeutic target for metastatic osteopontin-positive hepatocellular carcinoma. Hepatol 52:2012–2022
Yin Y-J, Salah Z, Grisaru-Granovsky S, Cohen I, Even-Ram SC, Maoz M, Uziely B, Peretz T, Bar-Shavit R (2003) Human protease-activated receptor 1 expression in malignant epithelia. Arterioscl Thromb Vasc Biol 23:940–944
Konstantoulaki M, Kouklis P, Malik AB (2003) Protein kinase C modifications of VE-cadherin, p120, and beta-catenin contribute to endothelial barrier dysregulation induced by thrombin. Am J Physiol Lung Cell Mol Physiol 285:L434–L442
Radjabi AR, Sawada K, Jagadeeswaran S, Eichbichler A, Kenny HA, Montag A, Bruno K, Lengyel E (2008) Thrombin induces tumor invasion through the induction and association of matrix metalloproteinase-9 and1-integrin on the cell surface. J Biol Chem 283:2822–2834
Zhang T, Ma Z, Wang R, Wang Y, Wang S, Cheng Z, Xu H, Jin X, Li W, Wang X (2010) Thrombin facilitates invasion of ovarian cancer along peritoneum by inducing monocyte differentiation toward tumor-associated macrophage-like cells. Cancer Immunol Immunother 59:1097–1108
Wysoczynski M, Liu R, Kucia M, Drukala J, Ratajczak MZ (2010) Thrombin regulates the metastatic potential of human rhabdomyosarcoma cells: distinct role of PAR1 and PAR3 signaling. Mol Cancer Res 8:677–690
Caunt M, Hu L, Tang T, Brooks PC, Ibrahim S, Karpatkin S (2006) Growth-regulated oncogene is pivotal in thrombin-induced angiogenesis. Cancer Res 66:4125–4132
Maloney JP, Silliman CC, Ambruso DR, Wang J, Tuder RM, Voelkel NF (1998) In vitro release of vascular endothelial growth factor during platelet aggregation. Am J Physiol 275:H1054–H1061
Haralabopoulos GC, Grant DS, Kleinman HK, Maragoudakis ME (1997) Thrombin promotes endothelial cell alignment in Matrigel in vitro and angiogenesis in vivo. Am J Physiol 273:239–245
Herbert JM, Dupuy E, Laplace MC, Zini JM, Bar Shavit R, Tobelem G (1994) Thrombin induces endothelial cell growth via both a proteolytic and a non-proteolytic pathway. Biochem J 303:227–231
Hu L, Roth JM, Brooks P, Luty J, Karpatkin S (2008) Thrombin up-regulates cathepsin D which enhances angiogenesis, growth, and metastasis. Cancer Res 68:4666–4673
Hu L, Roth JM, Brooks P, Ibrahim S, Karpatkin S (2008) Twist is required for thrombin-induced tumor angiogenesis and growth. Cancer Res 68:4296–4302
Ollivier V, Chabbat J, Herbert JM, Hakim J, de Prost D (2000) Vascular endothelial growth factor production by fibroblasts in response to factor VIIa binding to tissue factor involves thrombin and factor Xa. Arterioscler Thromb Vasc Biol 20:1374–1381
Rak J, Yu JL, Luyendyk J, Mackman N (2006) Oncogenes, Trousseau syndrome, and cancer-related changes in the coagulome of mice and humans. Cancer Res 66:10643–10646
Boven LA, Vergnolle N, Henry SD, Silva C, Imai Y, Holden J, Warren K, Hollenberg MD, Power C (2003) Up-regulation of proteinase-activated receptor 1 expression in astrocytes during HIV encephalitis. J Immunol 170:2638–2646
Ceelie H, Bertina RM, van Hylckama Vlieg A, Rosendaal FR, Vos HL (2001) Polymorphisms in the prothrombin gene and their association with plasma prothrombin levels. Thromb Haemost 85:1066–1070
Gehring NH, Frede U, Neu-Yilik G, Hundsdoerfer P, Vetter B, Hentze MW, Kulozik AE (2001) Increased efficiency of mRNA 3′ end formation: a new genetic mechanism contributing to hereditary thrombophilia. Nat Genet 28:389–392
Danckwardt S, Gehring NH, Neu-Yilik G, Hundsdoerfer P, Pforsich M, Frede U, Hentze MW, Kulozik AE (2004) The prothrombin 3′ end formation signal reveals a unique architecture that is sensitive to thrombophilic gain-of-function mutations. Blood 104:428–435
Soria JM, Almasy L, Souto JC, Tirado I, Borell M, Mateo J, Slifer S, Stone W, Blangero J, Fontcuberta J (2000) Linkage analysis demonstrates that the prothrombin G20210A mutation jointly influences plasma prothrombin levels and risk of thrombosis. Blood 95:2780–2785
VanLandingham JW, Cekic M, Cutler SM, Hoffman SW, Washington ER, Johnson SJ, Miller D, Stein DG (2008) Progesterone and its metabolite allopregnanolone differentially regulate hemostatic proteins after traumatic brain injury. J Cereb Blood Flow Metab 28:1786–1794
Levi M, van der Poll T, Büller HR (2004) Bidirectional relation between inflammation and coagulation. Circulation 109:2698–2704
Esmon CT (2005) The interactions between inflammation and coagulation. Br J Haematol 131:417–430
Grosso AR, Martins S, Carmo-Fonseca M (2008) The emerging role of splicing factors in cancer. EMBO rep 9:1087–1093
Danckwardt S, Hentze MW, Kulozik AE (2008) 3′ end mRNA processing: molecular mechanisms and implications for health and disease. EMBO J 27:482–498
David CJ, Manley JL (2010) Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24:2343
Proudfoot NJ (2011) Ending the message: poly(A) signals then and now. Genes Dev 25:1770–1782
Di Giammartino DC, Nishida K, Manley JL (2011) Mechanisms and consequences of alternative polyadenylation. Mol Cell 43:653–866
Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ (1995) Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem 270:7420–7426
Wagner EF, Nebreda AR (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9:537–549
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899
Gilbert LA, Hemann MT (2010) DNA damage-mediated induction of a chemoresistant niche. Cell 143:355–366
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–545
Lin WW, Karin M (2007) A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 117:1175–1183
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454:436–444
Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Brückner UB, Nilsson B, Gebhard F, Lambris JD et al (2010) Molecular intercommunication between the complement and coagulation systems. J Immunol 185:5628–5636
Danckwardt S, Kulozik A (2011) A p38 MAP kinase controlled novel gene regulatory switch links blood coagulation with tumor progression and outcome. Hamostaseologie 1:A75
Chandran UR, Dhir R, Ma C, Michalopoulos G, Becich M, Gilbertson J (2005) Differences in gene expression in prostate cancer, normal appearing prostate tissue adjacent to cancer and prostate tissue from cancer free organ donors. BMC Cancer 5:45
Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon FA (2007) Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 7:64
Briata P, Forcales SV, Ponassi M, Corte G, Chen CY, Karin M, Puri PL, Gherzi R (2005) p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol Cell 20:891–903
Hoppe B, Dorner T (2012) Coagulation and the fibrin network in rheumatic disease: a role beyond haemostasis. Nat Rev Rheumatol 8:738–746
Engelmann B, Massberg S (2013) Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 13:34–45
Acknowledgments
Work in the laboratory of SD is supported by the DFG (DA 1189/2-1), the GRK 1591, by the Federal Ministry of Education and Research (BMBF 01EO1003), by the Hella Bühler Prize for Cancer Research, and the Institute of Clinical Chemistry, University Medical Center Mainz. The work in the labs of AEK and MWH is supported by the DFG SFB 1036 “Cellular Surveillance and Damage Response.” We thank Vanessa Rau, Yulia Kargapolova, and Nikolaos Pazaitis for helpful input and critical comments on the manuscript, and Nikolaos Pazaitis for preparation of Fig. 1. We apologize to all colleagues whose work could not be discussed or cited here because of space constraints.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Danckwardt, S., Hentze, M.W. & Kulozik, A.E. Pathologies at the nexus of blood coagulation and inflammation: thrombin in hemostasis, cancer, and beyond. J Mol Med 91, 1257–1271 (2013). https://doi.org/10.1007/s00109-013-1074-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-013-1074-5