
Abstract
Thirty-five percent of patients with Rett syndrome carry nonsense mutations in the MECP2 gene. We have recently shown in transfected HeLa cells that readthrough of nonsense mutations in the MECP2 gene can be achieved by treatment with gentamicin and geneticin. This study was performed to test if readthrough can also be achieved in cells endogenously expressing mutant MeCP2 and to evaluate potentially more effective readthrough compounds. A mouse model was generated carrying the R168X mutation in the MECP2 gene. Transfected HeLa cells expressing mutated MeCP2 fusion proteins and mouse ear fibroblasts isolated from the new mouse model were treated with gentamicin and the novel aminoglycosides NB30, NB54, and NB84. The localization of the readthrough product was tested by immunofluorescence. Readthrough of the R168X mutation in mouse ear fibroblasts using gentamicin was detected but at lower level than in HeLa cells. As expected, the readthrough product, full-length Mecp2 protein, was located in the nucleus. NB54 and NB84 induced readthrough more effectively than gentamicin, while NB30 was less effective. Readthrough of nonsense mutations can be achieved not only in transfected HeLa cells but also in fibroblasts of the newly generated Mecp2 R168X mouse model. NB54 and NB84 were more effective than gentamicin and are therefore promising candidates for readthrough therapy in Rett syndrome patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rett syndrome (RTT) is a severe neurodevelopmental disorder that almost exclusively affects females [1]. After an initial period of normal or near-normal development lasting 8–18 months, loss of hand function and language is noted followed by a long period of stability in the clinical status [2]. RTT is caused by mutations in the X-chromosomal gene MECP2 encoding the methyl-CpG-binding protein 2 (MeCP2) [3]. MeCP2 binds to methylated CpGs in genomic DNA and is therefore part of the epigenetic control of gene expression [4]. Deficient mouse models that completely or partially lack Mecp2 recapture features seen in humans including the delayed onset of symptoms [5–8]. Remarkable results were obtained in a mouse model in which the endogenous Mecp2 gene is silenced by insertion of a lox-Stop cassette and then conditionally activated [9]. The activation of MeCP2 expression leads to improvement of symptoms and has therefore given hope that RTT is a treatable disorder. As 35% of RTT patients carry nonsense mutations [10], a pharmacological induced readthrough of premature stop codons is an attractive treatment approach. In our previous study using transfected HeLa cells, we have demonstrated that the aminoglycosides gentamicin and geneticin suppress RTT-causing nonsense mutations with efficiencies of up to 20% [11]. These findings have since been reproduced by Popescu et al. [12]. They also demonstrated that readthrough is possible in a lymphocyte cell line derived from a Rett syndrome girl. However, readthrough was not achieved when the dosage was reduced to a level safe for human use. Gentamicin and other aminoglycosides when employed as antibiotics in humans have, especially when administered in higher dosages, significant side effects, including nephrotoxicity and ototoxicity, limiting their application for readthrough therapy.
In the present report, we therefore tested three novel aminoglycosides—NB30, NB54, and NB84—that were developed on the paromomycin scaffold by optimization of structure–activity–toxicity relationship. Compared to the parent compound paromomycin and to gentamicin, these compounds display significantly lower toxicity and exhibit higher readthrough activity [13, 14]. We also report on the generation of a knock-in mouse model carrying one of the most frequently occurring nonsense mutations in the Mecp2 gene, R168X, and readthrough studies in ear fibroblasts of this mouse model.
Materials and methods
Gene targeting construct
A targeting vector was designed to generate a knock-in mouse model carrying the most common nonsense mutation associated with Rett syndrome (R168X). The mutation introduces an in-frame UGA stop codon in place of a codon for arginine (AGA). Genomic sequences were derived from a BAC plasmid containing the whole genomic sequence of mouse Mecp2 (BAC-Klon RPCI-23, RZPD). A PCR-amplified 921-bp XmaI genomic fragment (primer 1: MeCP2-LA1-F-(XmaI) 5′-ATACCCGGGTGCCTTGGTTAAAATGGAGG-3′, primer 2: MeCP2-LA1-R-(XmaI) 5′-GTCTCCCGGGTCTTGCGCTTCTTGATG-3′) was subcloned in pGEM-T easy (Promega) and used for site-directed mutagenesis to replace adenine at position 502 within exon 4 by thymine (c.502 A > T, MeCP2-A502Tmut: 5′-AGCCCCTCCAGGTGAGAGCAGAAACCACC-3′). The mutated fragment was fused to a 7.2-kb genomic fragment generated by XmaI-NdeI digestion of the BAC plasmid. This 8.1-kb fragment resulted in the 3′ region of homology. The 5′ region of homology consisted of a PCR amplified 1.1-kb NotI-AgeI Mecp2 fragment (primer 3: MeCP2-KA-F-(NotI) 5′-ATAGCGGCCGCGGGATGAGATTAGCTGCT-3′, primer 4: MeCP2-KA-R-(AgeI) 5′-ATAACCGGTTGGTGTCCAGCCTTTTTGGG-3′) spanning from within intron 2 to intron 3. Surrounded by the homology arms, the targeting vector contains a neomycin-resistance cassette flanked by Flp-recombinase target (FRT) sites for subsequent removal with Flp-recombinase. The vector was linearized for electroporation at the 5′ end using ClaI.
ES cell culture and gene targeting
R1 mouse embryonic stem cells were electroporated with 50 μg of the linearized targeting vector, and colonies were isolated after G418 selection as previously described [15]. Five G418-resistant clones were identified as homologous recombinants by nested PCR amplification of a 1.9-kb genomic fragment. The correct genomic location was verified in the DNA of heat-lysed cells, by using forward primers at positions outside the homology region (primer 5: MeCP-Ki-KV1F 5′-CCAAGACAGGCTTTGACCC-3′, primer 7: MeCP-Ki-KV2F 5′-CCCTAAAGTACATGAAGGAACCC-3′) and reverse primers located in the neo cassette (primer 6: neo2R 5′-GCAATCCATCTTGTTCAATGGC-3′, primer 8: neo3R 5′-CCATCTTGTTCAATGGCCGATC-3′). Both PCR reactions were performed in 25-μl reactions and started with denaturing at 95°C for 30 s. Annealing was at 56°C for 45 s and extension at 72°C for 2 min. For the first PCR step, we used 25 cycles, and 40 cycles were used for the second step. Microinjection of selected ES cells into C57BL/6 blastocysts and embryo transfer to pseudopregnant females was performed by standard procedures [15]. Highly chimeric males were bred to C57BL/6 female, and germ line transmission was confirmed by PCR and sequencing. Heterozygous (Mecp2 +/−) F1 females were crossed with C57BL/6 males to obtain hemizygous (Mecp2 R168X -/Y) male mutants. To remove the neo cassette, heterozygous (Mecp2 +/−) females were mated with FLIP-recombinase-expressing males. The genotype of mutated mice was routinely determined by PCR analysis using primer (primer 9: KI-Geno3F: 5′-GCCTGAAGGTTGGACACGAAAGC-3′) and primer (primer 10: KI-Geno3R: 5′-GGGCTAGACTGAATATCTTTGGTTGGTAC-3′), yielding two fragments. Presence of the wild-type allele was revealed by amplifying a 265-bp fragment. A product of 313-bp was found under mutated conditions. Mice used in the experiments described here were backcrossed through a minimum of 10 generations. Care and handling of the mice were conducted in concordance with Institutional Animal Care and Use Committee-approved protocols.
RNA analysis
Reverse transcriptase PCR (RT-PCR) was used to verify the Mecp2 gene expression in mutant mice. Total RNA was isolated from wild-type and mutant male brain using peqGOLD TriFast™ reagent according to the manufacturer's protocol (peqlab, Erlangen, Germany). After cDNA synthesis with oligo dT primers and AMV-reverse transcriptase (Invitrogen, Darmstadt, Germany) a 1,455-bp MECP2 cDNA fragment was amplified (MeCP2-CDS-F 5′-ATAGAATTCAATGGTAGCTGGGATGTTAGGG-3′ and MeCP2-CDS-R 5′-ATAGGATCCTCAGCTAACTCTCTCGGTCACGG-3′) representing the whole Mecp2 coding region.
Cell culture and fibroblast isolation
HeLa cells and mouse ear fibroblasts were maintained as monolayer cultures growing in Dulbecco's modified Eagle's medium (DMEM/low glucose, Cambrex Bio Science, Verviers, Belgium) supplemented with 10% fetal bovine serum (Biochrom, Berlin, Germany) and 1% l-glutamine (PAA, Pasching, Austria). Cells were incubated at 37°C in an atmosphere of 5% CO2. Primary mouse ear fibroblasts were isolated from both male wild-type and knock-in mice. Using scalpels, pieces of individual ears were cut into small fragments to expose fibroblasts, which were separated on 25-cm2 tissue culture flasks with the aid of a sterile pipette. The fibroblasts were allowed to grow for 8–10 days. HeLa cells and fibroblasts were subcultured using trypsin when they were 85–90% confluent.
Transfection and drug treatment
For the majority of experiments, assaying readthrough HeLa cells were seeded at 3 × 105 cells/well in six-well culture plates and transfected with the appropriate eukaryotic expression vectors as described previously [11] using Effectene reagent (QIAGEN, Hilden, Germany) following manufacturer's protocols. Transfected HeLa cells were cultured in fresh media and media containing 500 μg/ml NB54 or NB84 (provided by Timor Baasov, Haifa, Israel) for 24 h. Mouse ear fibroblasts (1 × 106 cells/10-cm culture plate) were treated with a daily dose of different concentrations (0–800 μg/ml) of gentamicin (SIGMA, Taufkirchen, Germany), NB54, and NB84 for 4 days. During treatment, fibroblasts were refed with growth media containing the desired drug dilution with replacement every 48 h.
Preparation of cell lysates and nuclear proteins
After treatment, cell lysates of transfected HeLa were obtained using RIPA buffer as described previously [11]. For detecting readthrough in drug-treated mouse ear fibroblasts, nuclear proteins were prepared as described previously [16]. Protein concentration was measured using the BC assay (Uptima, Montlucon cedex, France), following manufacturer's protocols.
Western blotting and antibodies
An amount of 30 μg of protein from each sample was assayed via SDS–PAGE followed by western blot analysis. Membranes (Schleicher and Schuell, Dassel, Germany) were blocked with 5% nonfat milk/TBST and probed with a primary antibody overnight (anti-Flag M2 and anti-GAPDH, SIGMA, Taufkirchen, Germany, 1:1000; anti-MeCP2 (D4F3), Cell Signaling Technology, Danvers, MA, USA., 1:1000) followed by secondary antibody incubation for 1 h (horseradish peroxidase-conjugated donkey anti-mouse IgG and goat anti-rabbit IgG, Jackson Immuno-Research Laboratories, West Grove, PA, USA). All antibodies were used at concentrations recommended by the manufacturer. Immunoreactive bands were visualized using enhanced chemiluminescence (Lumi-Light Western Blotting Substrate, Roche Diagnostics, Heidelberg, Germany). The efficiency of the readthrough was estimated by comparing the expression of total MeCP2 to the expression of full-length protein using densitometry (Camera: Fuji LAS-1000plus; Programm: Aida Version 3.10).
Immunofluorescent cell staining
Immunofluorescence experiments of drug-treated transfected HeLa cells were carried out as previously described [11]. For the staining of treated mouse ear fibroblasts, cells were grown on 18-mm cover slips and fixed in 4% paraformaldehyde for 15 min at RT. After washing three times with PBS, the cells were blocked (PBS, 5% normal goat serum, 0.3% Triton X-100) for 1 h at RT. Following an overnight incubation at 4°C with a cocktail of primary antibodies (anti-Tubulin, 1:2000, SIGMA, Taufkirchen, Germany; anti-MeCP2 (D4F3), Cell Signaling Technology, Danvers, MA, USA, 1:200; dilution in 1% BSA/PBS with 0.3% Triton X-100), cells were washed three times with PBS and incubated with the corresponding secondary antibody (goat anti-mouse IgG Cy3, 1:500, Jackson Immuno-Research Laboratories, West Grove, PA, USA; Alexa Fluor 488 goat anti-rabbit IgG, Molecular Probes Inc., Eugene, OR, USA, 1:500; dilution in 1% BSA/PBS with 0.3% Triton X-100) at RT for 1 h. After triple washing with PBS, the prepared cover slips were mounted onto slides with ProLong® Gold antifade reagent with DAPI (Molecular Probes Inc., Eugene, OR, USA). The stained cells were analyzed using fluorescent microscopy (Zeiss, Göttingen, Germany).
Results
NB54 and NB84 show increased readthrough of RTT-causing premature stop mutations
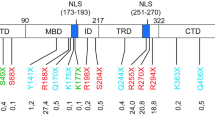
To evaluate the readthrough efficiency of NB30, NB54, and NB84, HeLa cells were transfected with eukaryotic expression vectors carrying N-terminally FLAG-tagged wild-type MECP2 cDNA or mutated MECP2 cDNAs. The mutations introduce an in-frame UGA stop codon in place of a codon for arginine at position 168, 255, 270, and 294 of MeCP2. HeLa cells were grown in the presence of 500 μg/ml gentamicin, NB30, NB54, and NB84 for 24 h. Western blot analysis confirmed the absence of full-length MeCP2 in untreated cells expressing mutated FLAG-MeCP2 isoforms (Fig. 1a, b). After treatment, full-length FLAG-MeCP2 was detected using an anti-FLAG and an anti-MeCP2 C-terminal antibody. The readthrough efficiency of NB30 was significantly lower compared to gentamicin, and this compound was therefore not used for further experiments (Fig. 1a). NB54 and NB84 were found to be more effective than gentamicin. NB54-induced readthrough ranged from 16.8% to 27.6%; NB84 was most effective with readthrough between 24% and 32.1% (Fig. 1b). The highest level of suppression was detected in the R294X mutation, while NB84 and NB54 were less effective in the R168X and R270X mutations. (Fig. 1c).

NB54- and NB84-mediated suppression of premature stop mutations. Western blot analysis of protein samples prepared from aminoglycoside-treated and untreated transiently transfected HeLa cells, using a monoclonal anti-FLAG antibody or a specific antibody detecting the C terminus of MeCP2. Thirty microgram of protein extract was loaded. Arrows denote the full-length FLAG-MeCP2 protein as well as the truncated FLAG-MeCP2 isoforms. As a loading control, GAPDH was detected in all samples. The cells were treated for 24 h with 500 μg/ml of gentamicin (a), NB30 (a), NB54 (a–b), or NB84 (b). The effect of 24-h drug treatment was quantified by densitometric western blot analysis from at least three independent experiments (c)
The subcellular localization of MeCP2 proteins that resulted from readthrough in transfected HeLa cells was investigated by immunofluorescent staining. For this purpose, HeLa cells were transfected with eukaryotic vectors expressing C-terminally FLAG-tagged MeCP2 fusion proteins, whereby only wild-type MeCP2-FLAG and readthrough proteins were detected using a FLAG-specific antibody. In untreated cells, MeCP2-FLAG fusion protein was only detected in the nucleus of the cells transfected with wild-type MeCP2 (Fig. 2a–c), and no signal was detected in cells expressing a mutated MeCP2 isoform (Fig. 2d–f). After treatment with NB54 and NB84, some cells showed again nuclear-localized fusion protein (Fig. 2g–l).

Nuclear localization of readthrough full-length MeCP2-FLAG fusion protein in drug-treated HeLa cells. Immunofluorescence of HeLa cells transfected with MeCP2-WT-FLAG- (a–c) compared to MeCP2-R168X-FLAG-expressing HeLa cells cultured under untreated (d–f) and drug-treated (g–i 500 μg/ml NB54; j–l 500 μg/ml NB84) conditions. Treatment was performed for 24 h. Localization of FLAG fusion proteins (left column) was visualized by using a monoclonal anti-FLAG antibody and immunofluorescence microscopy. Because the fusion protein was C-terminally FLAG-tagged, only full-length MeCP2 proteins were detected. The nuclei were counterstained with DAPI (middle column). Right column MeCP2-WT-FLAG expression as well as treated MeCP2-294-FLAG overlaps with DAPI staining, indicating a nuclear localization. Scale bars are 50 μm
Generation of Mecp2 R168X knock-in mice
To test the efficiency of the novel compounds in a more in vivo-like system, we generated a mouse model for Rett syndrome by introducing the truncating mutation R168X into the murine Mecp2 protein (Mecp2 R168X, Fig. 3a). This engineered truncation retains the methyl-CpG-binding domain but eliminates the C-terminal part of the coding sequence including the transcription repressor domain and nuclear localization signals. The Mecp2 transcript in brain was confirmed by RT-PCR using cDNA prepared from total RNA, and the mutation was detected by sequencing (Fig. 3d). Because it is a C-terminally truncating mouse model, we used an antibody recognizing the amino-terminal half of Mecp2 that should be preserved with the truncation. Western blot analysis showed a 72-kDa full-length protein in male wild-type mice, but failed to detect a wild-type or truncated protein of expected 18-kDa in the mutant males (Fig. 3e). Staining of ear fibroblasts with the same antibody confirmed the absence of Mecp2 protein in mutant cells (Fig. 3f). The phenotype of knock-in mice will be reported elsewhere, and preliminary observations indicate that the mouse model shows characteristics as described in other Mecp2-deficient mice.

Gene targeting and Flp-mediated recombination at the Mecp2 locus. The diagram shows the wild-type Mecp2 genomic locus, the targeting vector, and the targeted locus before and after Flp-mediated recombination. The 5′ and 3′ regions of homology are shown in light gray. Small arrows indicate primers used to confirm gene targeting and Flp-mediated recombination, and to screen the genotype (a). Homologous recombination was verified by nested PCR of genomic DNA of the selected ES cell clones; primers 5/6 and 7/8 were as shown in a. The 1.9-kb band is from the mutated allele. The primer sets were designed not to amplify the wild-type Mecp2 gene. E5, E7, E32, and E38 are names of ES clones, while c and b indicate a plasmid DNA control demonstrating the mutant PCR fragment and blank. Primer set 9 and 10 were used for the genotyping PCR after FLP recombination (c). The 313-bp band represents the mutant allele (Hem hemizygous male, Het heterozygous female) containing the remaining FRT site, while the 265-bp band represents the wild-type allele (WT wild type). To confirm the presence of Mecp2 mRNA in the brain of two wild-type (WT (+/Y)) and two R168X (−/Y) mutant male mice, total RNA was extracted from brain of male wild-type and mutant mice and used for cDNA synthesis and RT-PCR (d). Western blot analysis (e) and immunofluorescent staining (f) demonstrated the absence of wild-type MeCP2 protein in nuclear extracts of hemizygous brains (Sup supernatant, Nuc nuclear extract) and in nuclei of mouse ear fibroblast using an antibody recognizing the N terminus of MeCP2. Note that no additional truncated protein was detected
Readthrough treatment of fibroblasts isolated from the Mecp2 R168X knock-in mice
Isolated mouse ear fibroblasts from male wild-type and Mecp2 R168X knock-in mice were treated for 4 days with different concentrations of gentamicin, NB54, and NB84. Using an anti-MeCP2 antibody detecting a C-terminal epitope of the protein, we found a dose-dependent re-expression of Mecp2 in nuclear extracts (Fig. 4a). Compared to gentamicin (0.6%) and NB54 (2.5%), the highest level of suppression was observed after treatment with NB84 (5%; Fig. 4b). Using NB84, full-length Mecp2 expression was already detected at the lowest dose of 100 μg/ml. Immunofluorescent staining using an antibody directed against the carboxy-terminal epitope of Mecp2 and fluorescent microscopy confirmed the re-expression of endogenous Mecp2 with correct location in the nucleus (Fig. 5).

Drug-mediated re-expression of endogenous MeCP2. Western blot analysis of nuclear extracts prepared from aminoglycoside-treated and untreated mouse ear fibroblasts generated from wild-type and Mecp2 168 male mice: Immunoreactivity was detected by using a monoclonal anti-FLAG antibody and a specific antibody detecting the C terminus of MeCP2. Thirty microgram of protein extract was loaded into each lane. Arrows denote the full-length endogenous MeCP2 protein. The cells were treated for 4 days with 0–800 μg/ml of gentamicin, NB54, or NB84 (a). The effect of drug treatment was quantified by densitometric developed western blot analysis from at least three independent experiments (b)

Nuclear localization of endogenous Mecp2 in drug-treated mouse fibroblasts. Immunofluorescent staining of mouse ear fibroblasts generated from wild-type and Mecp2 168 male mice. Cells were treated 4 days with 800 μg/ml gentamicin (i–l), NB54 (m–p), and NB84 (q–t), respectively. Localization of endogenous MeCP2 protein (first column) was visualized by using a specific MeCP2 antibody detecting the C-terminal part of the protein and immunofluorescent microscopy. The nuclei and the cytoskeleton of the cells were counterstained with DAPI (second column) and tubulin (third column). All three stainings are merged (last column). No MeCP2-specific staining was detected in untreated fibroblasts (e–h). Scale bars are 50 μm
Discussion
Approximately one third of inherited diseases result from premature termination codon mutations. Aminoglycosides have emerged as vanguard pharmacogenetic agents in treating human genetic disorders due to their unique ability to suppress translation termination induced by nonsense mutations. In preclinical and several pilot clinical studies [17–21], this therapeutic approach shows promise in phenotype correction by promoting otherwise defective protein synthesis. However, the most critical factor that largely limits the potential of aminoglycosides for suppression therapy is their high human toxicity [22, 23]. Nevertheless, the clinical drug gentamicin is frequently used for proof-of-concept experiments in various disease models and in clinical trials [18–21], while no systematic study has been performed to tune aminoglycoside structures for better readthrough activity and lower toxicity. Recently, we hypothesized that by separating the structural elements of aminoglycosides that induce readthrough from those that affect toxicity, we might reach potent derivatives with improved readthrough activity and reduced toxicity [13, 24]. By keeping this line of research, we systematically developed the lead compounds NB30, NB54, and NB84 that exhibit significantly reduced toxicity and superior readthrough efficiency than that of gentamicin [13, 24].
Previously, we [11] and others [12] have reported that readthrough of different RTT-causing premature stop codons can be achieved by different aminoglycosides including gentamicin in transient transfection assays. In this study, we provide the first comparative evaluation of novel aminoglycosides NB30, NB54, NB84, and gentamicin in a number of clinically relevant RTT models.
It is noteworthy that the testing of readthrough therapy in fibroblasts from RTT patients was not feasible because MECP2 is an X-linked gene, meaning that due to random X-chromosome inactivation in female patient fibroblast cultures, ∼50% of wild-type MeCP2 expression is found. Reliable detection of low-level readthrough is therefore difficult if not impossible. Since no mouse model suitable for readthrough therapy was available, we generated a knock-in mouse carrying the nonsense mutation that is most frequently found in RTT patients, R168X. The preliminary characterization of the mice showed that the male mice have a phenotype comparable with that reported from other Mecp2-deficient mice including tremor, motor impairments, hypoactivity, anxiety-related behavior, hind limb clasping, and breathing irregularities [25]. In brain tissue of the Mecp2 R168X knock-in mice, we were not able to detect a shortened Mecp2 protein indicating that it is degraded rapidly.
Initially, mouse ear fibroblasts isolated from the new mouse model were treated with gentamicin. Using immunofluorescent and western blot analysis, we were able to detect full-length Mecp2, showing that readthrough of the R168X mutation is possible in fibroblasts. The Mecp2 protein was found to be correctly targeted to the nucleus. However, the efficiency of gentamicin-induced readthrough was low even at concentrations as high as 800 μg/ml, a dose that is above the range that is considered safe for clinical use (Fig. 4) [26].
To search for more efficient drugs, we used our established transient transfection system to test the suppression efficiencies of three novel aminoglycosides that were designed especially for readthrough therapy [13, 14]. NB30, NB54, and NB84 are pseudo-trisaccharide derivatives of the clinical aminoglycoside paromomycin. We found that NB30 induces less readthrough than gentamicin, but NB54 and NB84 were more effective. These results are in agreement with studies using similar assays for other genetic disorders [13, 27]. As in our previous study [11], we found that the efficiencies depend on the stop codon and its surrounding sequence as described in other genetic disorders [11, 28].
Fibroblasts isolated from male Mecp2 R168X knock-in mice were then treated with NB54 and NB84, and the efficiency was compared to that of gentamicin. Both NB54 and NB84 were found to suppress the Mecp2-R168X stop mutation more efficiently than gentamicin. The level of readthrough was dose-dependent and highest with NB84. Thus, NB54 and NB84 are promising compounds for readthrough therapy in RTT and are currently being tested in vivo in male Mecp2 R168X knock-in mice.
References
Rett A (1946) On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr 116:723–726
Hagberg B (1995) Rett syndrome: clinical peculiarities and biological mysteries. Acta Paediatr 84:971–976
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188
D’Esposito M, Quaderi NA, Ciccodicola A, Bruni P, Esposito T, D’Urso M, Brown SD (1996) Isolation, physical mapping, and northern analysis of the X-linked human gene encoding methyl CpG-binding protein, MECP2. Mamm Genome 7:533–535
Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27:322–326
Chen RZ, Akbarian S, Tudor M, Jaenisch R (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27:327–331
Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H (2002) Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35:243–254
Pelka GJ, Watson CM, Radziewic T, Hayward M, Lahooti H, Christodoulou J, Tam PP (2006) Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain 129:887–898
Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science 315:1143–1147
Percy AK, Lane JB, Childers J, Skinner S, Annese F, Barrish J, Caeg E, Glaze DG, MacLeod P (2007) Rett syndrome: North American database. J Child Neurol 22:1338–1341
Brendel C, Klahold E, Gartner J, Huppke P (2009) Suppression of nonsense mutations in Rett syndrome by aminoglycoside antibiotics. Pediatr Res 65:520–523
Popescu AC, Sidorova E, Zhang G and Eubanks JH Aminoglycoside-mediated partial suppression of MECP2 nonsense mutations responsible for Rett syndrome in vitro. Journal of neuroscience research 88: 2316–2324
Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, Chen F, Schacht J, Pilch DS, Ben-Yosef T, Baasov T (2009) Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem 52:2836–2845
Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V and Baasov T Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg Med Chem 18: 3735–3746
Forss-Petter S, Werner H, Berger J, Lassmann H, Molzer B, Schwab MH, Bernheimer H, Zimmermann F, Nave KA (1997) Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J Neurosci Res 50:829–843
Esfandiari F, Green R, Cotterman RF, Pogribny IP, James SJ, Miller JW (2003) Methyl deficiency causes reduction of the methyl-CpG-binding protein, MeCP2, in rat liver. Carcinogenesis 24:1935–1940
Rowe SM, Clancy JP (2009) Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. BioDrugs 23:165–174
Wagner KR, Hamed S, Hadley DW, Gropman AL, Burstein AH, Escolar DM, Hoffman EP, Fischbeck KH (2001) Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol 49:706–711
Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, Lewis S, Shilling CJ, Kota J, Serrano-Munuera C et al (2010) Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol 67:771–780
Clancy JP, Bebok Z, Ruiz F, King C, Jones J, Walker L, Greer H, Hong J, Wing L, Macaluso M et al (2001) Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med 163:1683–1692
Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L et al (2003) Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med 349:1433–1441
Forge A, Schacht J (2000) Aminoglycoside antibiotics. Audiol neuro otol 5:3–22
Mingeot-Leclercq MP, Tulkens PM (1999) Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother 43:1003–1012
Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V, Baasov T (2010) Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg Med Chem 18:3735–3746
Ricceri L, De Filippis B, Laviola G (2008) Mouse models of Rett syndrome: from behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav Pharmacol 19:501–517
Du M, Keeling KM, Fan L, Liu X, Kovacs T, Sorscher E, Bedwell DM (2006) Clinical doses of amikacin provide more effective suppression of the human CFTR-G542X stop mutation than gentamicin in a transgenic CF mouse model. J Mol Med (Berlin, Germany) 84:573–582
Rebibo-Sabbah A, Nudelman I, Ahmed ZM, Baasov T, Ben-Yosef T (2007) In vitro and ex vivo suppression by aminoglycosides of PCDH15 nonsense mutations underlying type 1 Usher syndrome. Hum Genet 122:373–381
Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF (2000) Sequence specificity of aminoglycoside-induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol 48:164–169
Acknowledgments
This study was financially supported by the German Research Foundation (HU 941/2-1) Kennedyallee 40, 53175 Bonn, Germany; the DFG Research Center for Molecular Physiology of the Brain (CMPB/EXC171), Humboldtallee 23, 37073 Göttingen, Germany; the E-rare EuroRett network/BMBF, Hannoversche Strasse 28, 10115 Berlin, Germany; and the US-Israel Binational Science Foundation (grant no. 2006/301; for T.B.). V.B. acknowledged the financial support by the Center of Absorption in Science, the Ministry of Immigration Absorption, and the Ministry of Science and Technology, Israel (Kamea Program).
Conflict of interest
The authors declare no conflict of interests related to this study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s00109-013-1029-x.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Brendel, C., Belakhov, V., Werner, H. et al. Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med 89, 389–398 (2011). https://doi.org/10.1007/s00109-010-0704-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-010-0704-4