
Abstract
Purpose
Recent research identified histone H3 K27M mutations to be associated with a dismal prognosis in pediatric diffuse midline glioma (pDMG); however, data on detailed MRI characteristics with respect to H3 K27 mutation status and molecular subgroups (H3.1 and H3.3 K27M mutations) are limited.
Methods
Standardized magnetic resonance imaging (MRI) parameters and epidemiologic data of 68 pDMG patients (age <18 years) were retrospectively reviewed and compared in a) H3 K27M mutant versus H3 K27 wildtype (WT) tumors and b) H3.1 versus H3.3 K27M mutant tumors.
Results
Intracranial gliomas (n = 58) showed heterogeneous phenotypes with isointense to hyperintense signal in T2-weighted images and frequent contrast enhancement. Hemorrhage and necrosis may be present. Comparing H3 K27M mutant to WT tumors, there were significant differences in the following parameters: i) tumor localization (p = 0.001), ii) T2 signal intensity (p = 0.021), and iii) T1 signal homogeneity (p = 0.02). No significant imaging differences were found in any parameter between H3.1 and H3.3 K27M mutant tumors; however, H3.1 mutant tumors occurred at a younger age (p = 0.004). Considering spinal gliomas (n = 10) there were no significant imaging differences between the analyzed molecular groups.
Conclusion
With this study, we are the first to provide detailed MR imaging data on H3 K27M mutant pDMG with respect to molecular subgroup status in a large patient cohort. Our findings may support diagnosis and future targeted therapeutic trials of pDMG within the framework of the radiogenomics concept.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Molecular characteristics of pediatric brain tumors have become an integral aspect of risk-adapted treatment strategies [1, 2]. Based on the revised classification of tumors of the central nervous system (CNS) by the World Health Organization (WHO) in 2016, the novel entity diffuse midline glioma, H3 K27M-mutant has been defined [3,4,5]. It includes the majority of diffuse high-grade gliomas (HGG) that emerge in midline structures of the CNS and typically affect children and young adults [5].
The leading molecular characteristic is a mutation of genes encoding histone H3 with replacement of a key lysine residue at position 27 by a methionine residue (K27M); this occurs in the HIST1H3B/C and H3F3A genes, among others, which encode for the variants H3.1 and H3.3, respectively [6,7,8,9]. The H3 K27M mutation is suggested to drive gliomagenesis, inhibit physiological cell differentiation [10,11,12,13,14], and maintain tumor growth [15]. The variants H3.1 and H3.3 K27M mutant tumors define distinct subgroups with differing prognosis and potentially phenotypes [16].
Although H3 K27M mutant diffuse midline gliomas (DMG) also occur in adults, differences in tumor biology of pediatric and adult tumors have been suggested [17,18,19,20]. In children particularly, H3 K27M mutations have been associated with infiltrative growth and a dismal prognosis, corresponding to WHO grade IV, regardless of tumor histology [11, 21, 22]; however, tumor biopsy is often challenging due to the location in midline CNS structures (mainly pons, thalamus, and spinal cord) [7, 13, 23]. To develop new therapeutic interventions that target the described histone mutations or underlying epigenetic pathways, a clinical/imaging surrogate marker of molecular subgroup status in DMG is desirable.
Only very limited data have been published using magnetic resonance imaging (MRI) to characterize H3 K27M mutant pediatric DMG (pDMG). Aboian et al. first described MRI features with variable contrast enhancement, edema, and necrosis but without significant differences from H3 K27 wildtype (WT) pDMG [24]. Another study by Rodriguez Gutierrez et al. in non-brainstem pediatric HGG further suggested a higher proportion of strongly enhancing H3 K27M mutant compared to WT tumors in the HERBY cohort [25]. In spinal pDMG, hemorrhage may be the only significantly discriminating feature between H3 K27M mutants and their WT counterparts in children and adults [26].
Some authors described MRI characteristics in H3 K27M subgroups (including H3.1 and H3.3 mutation) in diffuse intrinsic pontine glioma (DIPG) and found differences in the apparent diffusion coefficient (ADC) distribution and contrast enhancement patterns between subgroups [16, 27]; however, detailed data on MRI phenotypes of pDMG considering H3 K27 mutation status including molecular subgroups are still lacking.
In this study we describe and compare MRI of pDMG with known H3 K27 mutation status including H3.1 and H3.3 K27M subgroups in a large patient cohort. Radiological, pathological, and clinical data were retrieved from the HIT-HGG database of the Society of Pediatric Oncology and Hematology (Gesellschaft für Pädiatrische Onkologie und Hämatologie, GPOH). Our results may support diagnosis and future therapeutic trials of pDMG.
Methods
Patient Characteristics
A total of 85 pediatric patients with DMG were retrieved from the HIT-HGG registry (C.M.K., M.H., M.K.) of the GPOH. Patients were diagnosed between 1999 and 2016. Corresponding histopathological and molecular genetic data including H3 K27 mutation status were obtained from the German Neuropathological Reference Center for Pediatric Brain Tumor (HIT) Studies (T.P., G.H.G.). The MRI studies at diagnosis were obtained from the imaging database of the German Neuroradiological Reference Center for Pediatric Brain Tumor (HIT) Studies (M.W.-M., J.N., A.S.). Clinical and epidemiological data of the patient cohort have already been published [22].
Inclusion criteria of the presented study were patient age < 18 years at diagnosis, known molecular genetic H3 K27 mutation status (for HIST1H3B/C and H3F3A, resulting in H3.1 K27M or H3.3 K27M mutants, respectively, or H3 K27 WT), and availability of MRI scans at diagnosis with at least T1-weighted and T2-weighted images as well as T1-weighted images after gadolinium injection. Finally, MRI scans of 68 patients were analyzed.
The analysis of patient and MRI data was approved by the local and central ethics committees of the HIT studies and performed in accordance with the Declaration of Helsinki. Informed consent for data storage and statistical analyses was given by all patients and/or their parents at the time of registration and treatment within the HIT-HGG studies.
Image Analysis
The MRI scans were acquired with MR scanners of different manufacturers operating at field strengths between 1.0 and 3.0 T. Retrospective imaging review was performed separately by two experienced radiologists (M.W‑M. and J.N.) together with a medical student (A.H.). In cases of initial differences between both readers, cases were further discussed in a consensus meeting.
For intracranial tumors the following MRI characteristics were analyzed: tumor localization (thalamus or basal ganglia, midbrain/tectum, pons, spinal cord, others), tumor volume (length in coronal, sagittal, and axial planes; approximated by a * b * c * 0.5 cm3), and characteristics of tumor margins (well-defined, i.e. more than 90% of the tumor margin/moderately well-defined, more than 50% and up to 90% of the tumor margin/ill-defined, 50% or less of the tumor margin). The T1 and T2 tumor signals were characterized by intensity (hyperintense/isointense/hypointense) in relation to normal cortex and by homogeneity (homogeneous/predominantly homogeneous/predominantly inhomogeneous/inhomogeneous). Further criteria included contrast enhancement with respect to intensity (none/mild/intermediate/strong), enhancement pattern (homogeneous/predominantly homogeneous/predominantly inhomogeneous/inhomogeneous), volume of the enhancing tumor part in relation to the whole tumor mass (< 25%, 25–50%, 50–75%, 75–100%), and the presence of ring enhancement. Furthermore, the presence of necrosis, peritumoral edema (including edema width in cm), and cysts (including cyst signal intensity compared to cerebrospinal fluid, CSF) were evaluated. Intratumoral necrosis was defined as intratumoral fluid accumulation with peripheral (ring) enhancement. In addition, multifocality of the tumor, CSF dissemination (M0 or M1, M2, according to Chang et al. [28]), intracranial metastases at diagnosis, and extent of hydrocephalus (none/low grade/intermediate/high grade, defined by HIT criteria) were assessed.
Additional criteria (when available within the MRI dataset) included cellularity within the tumor mass suggested by diffusion-weighted imaging (DWI; hyperintense/isointense/hypointense, compared to normal tissue) and ADC (diffusion restriction/no diffusion restriction). Presence of tumor hemorrhage was evaluated in all MRI and, if available, computed tomography (CT) sequences, including susceptibility weighted imaging (SWI) or T2*-weighted imaging (signal loss/no signal loss in SWI/T2*). Calcifications were assessed in CT scans (none/fine/gross), when available.
The MRI morphology of spinal tumors was characterized separately by assessing tumor expansion in relation to anatomic localization (cervical/cervicothoracic/thoracic/thoracolumbar), tumor length (in the sagittal plane in cm and in vertebral segment count, following recommendations of a previous study [26]), swelling of the cord (yes/no), gadolinium enhancement (yes/no/unclear), and presence of necrosis or hemorrhage.
Statistical Analysis
We compared epidemiological and imaging data of a) H3.1 versus H3.3 K27M mutant tumors versus WT tumors and b) H3.1 versus H3.3 mutant tumors. Imaging features of spinal tumors were studied separately as tumor imaging characteristics are different from intracranial tumors. Statistical two-group comparisons were conducted using Fisher’s exact test of independence for parameters with two categories. For parameters with three or more categories, the two-sided χ2-test or Fisher-Freeman-Halton test was applied. Continuous parameters were analyzed using Mann-Whitney U tests. Statistical three-group comparisons were conducted in order to test for possible discrimination of all three genotypes (H3.3, H3.1, WT) by MRI, using the two-sided χ2-test or Fisher-Freeman-Halton test for categorial and Kruskal-Wallis test for continuous variables. P-values < 0.05 were considered statistically significant. For subsequent intergroup comparisons of H3.1 and H3.3 mutant pDMG, the significance level was adjusted to p < 0.017 after applying Bonferroni correction. Analysis was performed using IBM SPSS Statistics software, RRID:SCR_019096, versions 24–26 (IBM, Armonk, NY, USA).
Results
Patient Cohort and Molecular Status
The MRI studies of 68 pediatric patients with DMG were analyzed for this study (n = 58 intracranial, n = 10 spinal). A total of 17/85 patients were excluded due to missing or incomplete MRI data at diagnosis (n = 16) and age ≥ 18 years at diagnosis (n = 1). Molecular analysis revealed H3.1 K27M mutant (n = 6, 8.8%), H3.3 K27M mutant (n = 46, 67.6%), and WT tumors (n = 16, 23.5%). There was a slight difference in median manifestation age between H3 K27M mutant (10.2 years, range 1.25–17.75 years) and WT (9.6 years, range 0.33–16.17 years) tumors (U = 367.00, z = −0.71, p = 0.48); however, the median age at diagnosis differed significantly between H3.1 (3.9 years, range 1.25–13.25 years) and H3.3 (10.4 years, range 4.00–17.75 years) mutant tumors (U = 38.00, z = −2.87, p = 0.004), an effect also seen when comparing age between H3.1 K27M vs. H3.3 K27M vs. H3 K27 WT (H(2) = 6.84; p = 0.03). Sex was equally distributed with 51.5% males and 48.5% females across our patient cohort without any significant differences comparing H3 K27 mutation status (p = 0.78, Fisher’s exact test).
Tumor Localization
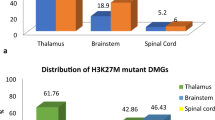
The vast majority of H3 K27M mutants were found in the pons (51.9%) and thalamus/basal ganglia (36.5%) and further arose from spinal (9.6%) and other localizations (1.9%, including 1 tumor originating from the medulla oblongata). In contrast, H3 K27 WT tumors were more widely distributed (31.3% thalamus/basal ganglia, 31.3% spinal cord, 12.5% pons, 12.5% midbrain/tectum, 12.5% other intracranial localizations, including 1 tumor originating from the medulla oblongata and 1 tumor with relation to the lateral ventricle), thereby significantly differing from mutants (p = 0.001, Fisher-Freeman-Halton test). Tumor localization also differed significantly between H3.1 K27M vs. H3.3 K27M vs. H3 K27 WT (p = 0.01, Fisher-Freeman-Halton test) whereas molecular subgroups H3.1 and H3.3 did not differ (p = 1.00, Fisher-Freeman-Halton test). A detailed overview on tumor localization is given in Supplementary Table S1 (Online Resource 1). Tumors in typical localizations are exemplarily shown in Fig. 1. The variety of tumor distribution in pDMG is illustrated in Fig. 2.

Typical localizations of pDMG in the pons (a, b) and thalamus (c, d) in axial MR images. Different H3 K27 genetic subgroups may show very similar MRI phenotypes with hyperintense, heterogeneous T2 signal (a and b, c and d). However, T2 signal may also appear very different even within the same genetic group (e, f)

Other localizations of H3 K27M mutant and WT tumors in the midbrain/tectum (a, sagittal images) and medulla oblongata (b, axial images). Note that bithalamic gliomas (c, axial images) typically harbor epidermal growth factor receptor (EGFR) mutation rather than histone H3 mutation [29]. Rare MRI phenotypes with diffuse infiltrative growth (arrowheads in d, axial images) and with leptomeningeal spread at diagnosis (arrowheads in e, axial images)
MR Imaging
H3 K27M versus H3 K27 WT
The majority of intracranial H3 K27M mutant tumors showed hyperintense T2 signal (93.6%), compared to only 63.6% in WT tumors (p = 0.02, Fisher’s exact test; remaining tumors: isointense). In T2-weighted images, H3 K27M mutant tumors appeared rather inhomogeneous (48.9% predominantly inhomogeneous, 8.5% inhomogeneous), whereas WT tumors displayed rather homogeneous T2 signal (63.6% predominantly homogeneous, 9.1% homogeneous) but not reaching significance (p = 0.06, Fisher-Freeman-Halton test). Fig. 1 shows selected T2-weighted MRI phenotypes. There was a statistically significant difference in T1 signal homogeneity, with H3 K27M mutants showing more inhomogeneous tumors (39.1% predominantly inhomogeneous, 2.2% inhomogeneous), compared to WT tumors (9.1% inhomogeneous, 0.0% predominantly inhomogeneous; p = 0.02, Fisher-Freeman-Halton test). Intratumoral hemosiderin deposits were more frequent in H3 K27M mutants (39.1%) than in WT tumors (9.1%, p = 0.08, Fisher-Freeman-Halton test). We did not see significant differences in any other parameter. Fig. 3 illustrates the spectrum of contrast enhancement in pDMG. Supplementary Table S2 (Online Resource 2) gives a detailed overview on all assessed imaging parameters.

H3 K27M mutant tumors may show solid, enhancing, and necrotic portions (a, sagittal image)*, patchy enhancement (b, axial image), solid enhancing and non-enhancing areas (c, axial image), or necrosis only (d, axial image). Note that ring enhancement usually demarcates necrotic tumor portions (e, f; axial images). *Same patient as in Fig. 2a. CE+ contrast enhanced
H3.1 K27M versus H3.3 K27M
We did not find any MR imaging characteristic that differed significantly between H3.1 and H3.3 K27M mutant tumors. All assessed imaging parameters are displayed in Supplementary Table S2 (Online Resource 2). The MRI phenotypes of molecular subgroups are shown in Figs. 1 and 2.
H3.1 K27M versus H3.3 K27M versus H3 K27 WT
Testing for possible differentiation of all three genotypes (H3.1 K27M, H3.3 K27M, H3 K27 WT), we found significant differences regarding T2 signal intensity (p < 0.05, Fisher-Freeman-Halton test): H3.1 and H3.3 mutant tumors showed higher signal intensity (hyperintense: 100.0% and 92.7%, respectively) compared to WT tumors (hyperintense: 63.6%), hence complementing our findings with respect to H3 K27 mutation status (H3 K27M vs. H3 K27 WT). The MRI phenotypes are shown in Figs. 1 and 2, and additional imaging parameters and post hoc test information are listed in Supplementary Table S2 (Online Resource 2).
Spinal Tumors
Molecular analysis of spinal pDMG revealed H3.3 K27M mutant (n = 5) and WT tumors (n = 5). There were no spinal H3.1 K27M mutant tumors in our cohort. The majority of H3.3 mutant tumors originated in cervical (40.0%) and cervicothoracic segments (40.0%), thereby differing from WT cases which were located in lower spinal segments (60.0% thoracolumbar, 40.0% thoracic) but not reaching statistical significance (p = 0.06, Fisher-Freeman-Halton test). There were no imaging characteristics that significantly differentiated the two molecular groups. Imaging parameters of spinal pDMG are displayed in Supplementary Table S3 (Online Resource 3) and characteristic MR phenotypes are shown in Fig. 4.

Spinal H3.3 K27M mutant tumor in the cervicothoracic region with cystoid necrosis and contrast enhancement (a, b; sagittal images). Spinal H3 K27 WT tumor with marked swelling of the conus medullaris and strong peripheral contrast enhancement (c, d; sagittal images). CE+ contrast enhanced
Discussion
With the genesis of the novel entity diffuse midline glioma, H3 K27M mutant in 2016 [3,4,5], various studies have been conducted to characterize and compare H3 K27M mutant tumors to their WT counterparts. In addition, biological and neuroanatomical differences between HGG of children and adults have been suggested [6, 7, 9, 15, 17, 18, 30, 31]. In our study, we, for the first time, systematically analyzed MR imaging features of 68 pDMG patients with known H3 K27 mutation status including H3.1 and H3.3 K27M molecular subgroups. We found that H3 K27M mutant and H3 K27 WT tumors may be differentiated by tumor localization, T2 signal intensity, and T1 signal homogeneity. The MRI phenotypes between H3.1 and H3.3 K27M subgroup tumors did not differ significantly, although differences in age of onset were identified. The clinical data of our pediatric cohort, including H3.1 and H3.3 subgroup-specific survival data, have already been published [22] and are now complemented by detailed MR imaging characteristics. The authors found that H3 K27M mutation was associated with shorter overall survival compared to H3 K27 WT and that H3 K27 mutation status was the only independent parameter to predict overall survival.
Regarding MR imaging in the pediatric population, few studies have aimed to characterize pDMG, DIPG, and non-brainstem HGG with respect to H3 K27 mutation status [16, 24, 25]. In a first study, Aboian et al. did not find any significant imaging differences between H3 K27M mutant pDMG and their WT counterparts [24]; however, data were based on a relatively small cohort, and there was no differentiation of mutant tumors into H3.1 and H3.3 mutant subgroups. Regarding H3.1 and H3.3 K27M subgroups in DIPG, Castel et al. suggested that H3.1 subgroup tumors show distinct contrast enhancement patterns (more frequent tumors with large necrotic areas and ring or nodular enhancement) and present lower ADC values [16]. H3.1 mutant patients presented at a younger age, had better clinical treatment response and fewer metastatic recurrences compared to patients with H3.3 K27M subgroup DIPG. In contrast, in a recent study H3.1 K27M subgroup DIPG presented higher ADC values [27]. A radiological evaluation of pediatric non-brainstem HGG within the HERBY trial revealed increased contrast enhancement in H3 K27M mutant tumors, not differentiating between H3.1 and H3.3 subgroups [25]; however, a study on pDMG with and without H3 K27M mutations did not find differences regarding the presence of contrast enhancement and necrotic areas with respect to mutation status [32]. Furthermore, a recent study performed by Thust et al. in a relatively small cohort of H3 K27M mutant pediatric and young adult DMG (n = 15) did not find characteristic MR morphologic parameters including diffusivity for this tumor entity; however, a control group of WT tumors was absent [33].
Using quantitative imaging techniques, no differences were found in ADC histogram analysis comparing H3 K27M mutant or WT pDMG but H3.1 and H3.3 K27M subgroups were not addressed [34]. A study applying advanced MR imaging revealed H3 K27M mutant tumors to exhibit higher relative arterial spin labeling-derived maximum cerebral blood flows, lower DWI-derived relative minimum ADC values and higher choline to N‑acetyl aspartate peak area ratios in 1H‑MR spectroscopy. In positron emission tomography (PET) analysis, higher 18F‑dihydroxyphenylalanine (DOPA) uptake was found in H3 K27M mutants compared to WT tumors [32].
In our study, we confirmed the typical localization of H3 K27M mutant pDMG, as previously reported [5, 23]: 88.5% of H3 K27M mutant tumors arose in a pontine or thalamic and 9.6% in a spinal localization, whereas H3 K27 WT tumors were more widely distributed (see Supplementary Table S1, Online Resource 1, and also detailed clinical data of our cohort as published by Karremann et al. [22]). We first reported significant differences between H3 K27M mutant tumors and their WT counterparts in terms of T2 signal intensity and T1 signal homogeneity. From a radiological perspective, higher T2 signals may point to lower cellularity (in H3 K27M mutant tumors) and in MR imaging of brain tumors there is a trend to inhomogeneous signal (more frequently seen in H3 K27M mutant pDMG) being linked with more aggressive growth; however, these findings are also subject to interobserver bias and as we did not find correlating differences in other standard MR sequences (such as DWI and contrast-enhanced T1), our findings alone may not differentiate H3 K27M mutant from WT tumors. Interestingly, Jansen et al. linked ring enhancement in DIPG (not addressing mutation status at that time) in MRI with a shorter overall survival [35]. For pDMG, a worse clinical course and shorter survival was hence linked to an H3 K27M mutant status [22]. In our imaging cohort, ring enhancement was exhibited more frequently by H3 K27M mutant (53.2%) compared to H3 K27 WT (36.4%) tumors but this finding did not reach statistical significance.
We are the first to describe that MRI phenotypes of H3.1 and H3.3 K27M subgroups of pDMG may not differ but we do see that H3.1 subgroup pDMG occurred at a significantly younger age. This finding is in line with previously described findings in DIPG and pDMG [16, 22], pointing to differences in tumor biology between the subgroups. Interestingly, a single case of our cohort presenting with metastases at diagnosis carried an H3.3 K27M mutation, potentially supporting previous observations of a higher tendency towards metastatic relapse in H3.3 K27M mutant DIPG [16].
Few studies show single MRI cases of spinal H3 K27M mutant DMG in different age groups, not differentiating between H3.1 and H3.3 subgroups [24, 31, 36,37,38,39]. With our study, we, for the first time, systematically analyzed MRI parameters of spinal DMG in the pediatric population with respect to H3 K27 mutation status but there was no H3.1 mutant spinal tumor in our cohort. In a study by Jung et al. in pediatric and adult spinal DMG, hemorrhage was the only imaging parameter that differed significantly (more frequently in H3 K27M mutants compared to no hemorrhage in WT tumors) [26]. In our study, H3.3 K27M and WT pDMG did not differ significantly regarding the presence of hemorrhage, with a single WT case showing clear signs of hemorrhage; however, the localization differed slightly, with H3.3 K27M pDMG arising in higher spinal segments than WT tumors but not reaching significance (p = 0.06). In summary, a differentiation of molecular status in spinal pDMG by means of MRI may not be feasible at present.
This study is limited by its retrospective design and heterogeneous MRI data provided by the referring centers within the HIT trials. Therefore, quantitative analysis of MR parameters (such as diffusivity) or analysis of advanced imaging techniques (such as MR perfusion or MR spectroscopy) and machine learning-based approaches could not be investigated comparatively. Only a few studies applying machine learning-based models to predict H3 K27M mutation in spinal glioma, brainstem glioma, or midline glioma based on conventional MRI parameters have been published [26, 40,41,42]. Furthermore, reliability of data evaluation based on consensus reading of two experienced experts was not statistically validated and patients of our cohort were not evenly distributed along the assessed molecular groups, impeding statistical analysis.
Nevertheless, the multicenter design (referring to a network of reference centers for oncology, neuropathology, and neuroradiology) provides a comparatively large cohort of pDMG cases with known H3 K27 mutation status, including a WT control group.
Conclusion
A differentiation of molecular status in diffuse midline glioma, H3 K27M mutant using conventional MRI seems challenging at present. We are the first to report that this may also apply for H3.1 and H3.3 K27M subgroups in children, as well as for their discrimination from WT tumors. Further investigations applying machine learning-based approaches (implementing imaging, clinical, and molecular data) are needed to design a non-invasive prediction model for H3 K27 mutation status of these rare tumors with a dismal prognosis.
References
Jones C, Karajannis MA, Jones DTW, Kieran MW, Monje M, Baker SJ, Becher OJ, Cho YJ, Gupta N, Hawkins C, Hargrave D, Haas-Kogan DA, Jabado N, Li XN, Mueller S, Nicolaides T, Packer RJ, Persson AI, Phillips JJ, Simonds EF, Stafford JM, Tang Y, Pfister SM, Weiss WA. Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro Oncol. 2017;19:153–61.
Northcott PA, Pfister SM, Jones DT. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol. 2015;16:e293–302.
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–20.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO classification of tumours of the central nervous system. 4th ed. Vol. 1. Lyon: International Agency for Research on Cancer; 2016. p. 408.
Hawkins C, Ellison D, Sturm D. Diffuse midline glioma, H3 K27M-mutant. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification of tumours of the central nervous system. Lyon: International Agency for Research on Cancer; 2016. pp. 57–9.
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jäger N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Frühwald MC, Roggendorf W, Kramm C, Dürken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31.
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu XY, Fontebasso AM, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann MU, van Meter T, Frühwald MC, Hauch H, Pekrun A, Radlwimmer B, Niehues T, von Komorowski G, Dürken M, Kulozik AE, Madden J, Donson A, Foreman NK, Drissi R, Fouladi M, Scheurlen W, von Deimling A, Monoranu C, Roggendorf W, Herold-Mende C, Unterberg A, Kramm CM, Felsberg J, Hartmann C, Wiestler B, Wick W, Milde T, Witt O, Lindroth AM, Schwartzentruber J, Faury D, Fleming A, Zakrzewska M, Liberski PP, Zakrzewski K, Hauser P, Garami M, Klekner A, Bognar L, Morrissy S, Cavalli F, Taylor MD, van Sluis P, Koster J, Versteeg R, Volckmann R, Mikkelsen T, Aldape K, Reifenberger G, Collins VP, Majewski J, Korshunov A, Lichter P, Plass C, Jabado N, Pfister SM. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37.
Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–61.
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Zhang J, Baker SJ; St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–3.
Kasper LH, Baker SJ. Invited Review: Emerging functions of histone H3 mutations in paediatric diffuse high-grade gliomas. Neuropathol Appl Neurobiol. 2020;46:73–85.
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–47.
Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, Radlwimmer B, Højfeldt JW, Truffaux N, Castel D, Schubert S, Ryzhova M, Seker-Cin H, Gronych J, Johann PD, Stark S, Meyer J, Milde T, Schuhmann M, Ebinger M, Monoranu CM, Ponnuswami A, Chen S, Jones C, Witt O, Collins VP, von Deimling A, Jabado N, Puget S, Grill J, Helin K, Korshunov A, Lichter P, Monje M, Plass C, Cho YJ, Pfister SM. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24:660–72.
Fontebasso AM, Liu XY, Sturm D, Jabado N. Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain Pathol. 2013;23:210–6.
Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, Sarkaria J, Zhang Z. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27:985–90.
Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, Deshmukh S, Chen CCL, Belle J, Mikael LG, Marchione DM, Li R, Nikbakht H, Hu B, Cagnone G, Cheung WA, Mohammadnia A, Bechet D, Faury D, McConechy MK, Pathania M, Jain SU, Ellezam B, Weil AG, Montpetit A, Salomoni P, Pastinen T, Lu C, Lewis PW, Garcia BA, Kleinman CL, Jabado N, Majewski J. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun. 2019;10:1262.
Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, Pagès M, Taylor KR, Saulnier P, Lacroix L, Mackay A, Jones C, Sainte-Rose C, Blauwblomme T, Andreiuolo F, Puget S, Grill J, Varlet P, Debily MA. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015;130:815–27.
Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer. 2014;14:10.1038/nrc3811.
Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, Temelso S, Popov S, Molinari V, Raman P, Waanders AJ, Han HJ, Gupta S, Marshall L, Zacharoulis S, Vaidya S, Mandeville HC, Bridges LR, Martin AJ, Al-Sarraj S, Chandler C, Ng HK, Li X, Mu K, Trabelsi S, Brahim DH, Kisljakov AN, Konovalov DM, Moore AS, Carcaboso AM, Sunol M, de Torres C, Cruz O, Mora J, Shats LI, Stavale JN, Bidinotto LT, Reis RM, Entz-Werle N, Farrell M, Cryan J, Crimmins D, Caird J, Pears J, Monje M, Debily MA, Castel D, Grill J, Hawkins C, Nikbakht H, Jabado N, Baker SJ, Pfister SM, Jones DTW, Fouladi M, von Bueren AO, Baudis M, Resnick A, Jones C. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell. 2017;32:520–7.e5.
Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, Broniscer A, Ellison DW, Grundy RG, Baker SJ. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28:3061–8.
Rood BR, MacDonald TJ. Pediatric high-grade glioma: molecular genetic clues for innovative therapeutic approaches. J Neurooncol. 2005;75:267–72.
Ryall S, Krishnatry R, Arnoldo A, Buczkowicz P, Mistry M, Siddaway R, Ling C, Pajovic S, Yu M, Rubin JB, Hukin J, Steinbok P, Bartels U, Bouffet E, Tabori U, Hawkins C. Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol Commun. 2016;4:93.
Karremann M, Gielen GH, Hoffmann M, Wiese M, Colditz N, Warmuth-Metz M, Bison B, Claviez A, van Vuurden DG, von Bueren AO, Gessi M, Kühnle I, Hans VH, Benesch M, Sturm D, Kortmann RD, Waha A, Pietsch T, Kramm CM. Diffuse high-grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro Oncol. 2018;20:123–31.
Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJ, Perry A. Diffuse Midline Gliomas with Histone H3-K27M Mutation: A Series of 47 Cases Assessing the Spectrum of Morphologic Variation and Associated Genetic Alterations. Brain Pathol. 2016;26:569–80.
Aboian MS, Solomon DA, Felton E, Mabray MC, Villanueva-Meyer JE, Mueller S, Cha S. Imaging Characteristics of Pediatric Diffuse Midline Gliomas with Histone H3 K27M Mutation. AJNR Am J Neuroradiol. 2017;38:795–800.
Rodriguez Gutierrez D, Jones C, Varlet P, Mackay A, Warren D, Warmuth-Metz M, Aliaga ES, Calmon R, Hargrave DR, Cañete A, Massimino M, Azizi AA, Le Deley MC, Saran F, Rousseau RF, Zahlmann G, Garcia J, Vassal G, Grill J, Morgan PS, Jaspan T. Radiological Evaluation of Newly Diagnosed Non-Brainstem Pediatric High-Grade Glioma in the HERBY Phase II Trial. Clin Cancer Res. 2020;26:1856–65.
Jung JS, Choi YS, Ahn SS, Yi S, Kim SH, Lee SK. Differentiation between spinal cord diffuse midline glioma with histone H3 K27M mutation and wild type: comparative magnetic resonance imaging. Neuroradiology. 2019;61:313–22.
Calmon R, Dangouloff-Ros V, Varlet P, Deroulers C, Philippe C, Debily MA, Castel D, Beccaria K, Blauwblomme T, Grevent D, Levy R, Roux CJ, Purcell Y, Saitovitch A, Zilbovicius M, Dufour C, Puget S, Grill J, Boddaert N. Radiogenomics of diffuse intrinsic pontine gliomas (DIPGs): correlation of histological and biological characteristics with multimodal MRI features. Eur Radiol. 2021;31:8913–24.
Chang CH, Housepian EM, Herbert C Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology. 1969;93:1351–9.
Mondal G, Lee JC, Ravindranathan A, Villanueva-Meyer JE, Tran QT, Allen SJ, Barreto J, Gupta R, Doo P, Van Ziffle J, Onodera C, Devine P, Grenert JP, Samuel D, Li R, Metrock LK, Jin LW, Antony R, Alashari M, Cheshier S, Whipple NS, Bruggers C, Raffel C, Gupta N, Kline CN, Reddy A, Banerjee A, Hall MD, Mehta MP, Khatib Z, Maher OM, Brathwaite C, Pekmezci M, Phillips JJ, Bollen AW, Tihan T, Lucas JT Jr, Broniscer A, Berger MS, Perry A, Orr BA, Solomon DA. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020;139:1071–88.
Jones C, Perryman L, Hargrave D. Paediatric and adult malignant glioma: close relatives or distant cousins? Nat Rev Clin Oncol. 2012;9:400–13.
Schulte JD, Buerki RA, Lapointe S, Molinaro AM, Zhang Y, Villanueva-Meyer JE, Perry A, Phillips JJ, Tihan T, Bollen AW, Pekmezci M, Butowski N, Oberheim Bush NA, Taylor JW, Chang SM, Theodosopoulos P, Aghi MK, Hervey-Jumper SL, Berger MS, Solomon DA, Clarke JL. Clinical, radiologic, and genetic characteristics of histone H3 K27M-mutant diffuse midline gliomas in adults. Neurooncol Adv. 2020;2:vdaa142.
Piccardo A, Tortora D, Mascelli S, Severino M, Piatelli G, Consales A, Pescetto M, Biassoni V, Schiavello E, Massollo M, Verrico A, Milanaccio C, Garrè ML, Rossi A, Morana G. Advanced MR imaging and 18F-DOPA PET characteristics of H3K27M-mutant and wild-type pediatric diffuse midline gliomas. Eur J Nucl Med Mol Imaging. 2019;46:1685–94.
Thust S, Micallef C, Okuchi S, Brandner S, Kumar A, Mankad K, Wastling S, Mancini L, Jäger HR, Shankar A. Imaging characteristics of H3 K27M histone-mutant diffuse midline glioma in teenagers and adults. Quant Imaging Med Surg. 2021;11:43–56.
Aboian MS, Tong E, Solomon DA, Kline C, Gautam A, Vardapetyan A, Tamrazi B, Li Y, Jordan CD, Felton E, Weinberg B, Braunstein S, Mueller S, Cha S. Diffusion Characteristics of Pediatric Diffuse Midline Gliomas with Histone H3-K27M Mutation Using Apparent Diffusion Coefficient Histogram Analysis. AJNR Am J Neuroradiol. 2019;40:1804–10.
Jansen MH, Veldhuijzen van Zanten SE, Sanchez Aliaga E, Heymans MW, Warmuth-Metz M, Hargrave D, van der Hoeven EJ, Gidding CE, de Bont ES, Eshghi OS, Reddingius R, Peeters CM, Schouten-van Meeteren AY, Gooskens RH, Granzen B, Paardekooper GM, Janssens GO, Noske DP, Barkhof F, Kramm CM, Vandertop WP, Kaspers GJ, van Vuurden DG. Survival prediction model of children with diffuse intrinsic pontine glioma based on clinical and radiological criteria. Neuro Oncol. 2015;17:160–6.
Kumar A, Rashid S, Singh S, Li R, Dure LS. Spinal Cord Diffuse Midline Glioma in a 4-Year-Old Boy. Child Neurol Open. 2019;6:2329048X19842451.
Song D, Xu D, Gao Q, Hu P, Guo F. Intracranial Metastases Originating From Pediatric Primary Spinal Cord Glioblastoma Multiforme: A Case Report and Literature Review. Front Oncol. 2020;10:99.
Meyronet D, Esteban-Mader M, Bonnet C, Joly MO, Uro-Coste E, Amiel-Benouaich A, Forest F, Rousselot-Denis C, Burel-Vandenbos F, Bourg V, Guyotat J, Fenouil T, Jouvet A, Honnorat J, Ducray F. Characteristics of H3 K27M-mutant gliomas in adults. Neuro Oncol. 2017;19:1127–34.
Gao Y, Feng YY, Yu JH, Li QC, Qiu XS, Wang EH. Diffuse midline gliomas with histone H3-K27M mutation: A rare case with PNET-like appearance and neuropil-like islands. Neuropathology. 2018;38:165–70.
Pan CC, Liu J, Tang J, Chen X, Chen F, Wu YL, Geng YB, Xu C, Zhang X, Wu Z, Gao PY, Zhang JT, Yan H, Liao H, Zhang LW. A machine learning-based prediction model of H3K27M mutations in brainstem gliomas using conventional MRI and clinical features. Radiother Oncol. 2019;130:172–9.
Su X, Chen N, Sun H, Liu Y, Yang X, Wang W, Zhang S, Tan Q, Su J, Gong Q, Yue Q. Automated machine learning based on radiomics features predicts H3 K27M mutation in midline gliomas of the brain. Neuro Oncol. 2020;22:393–401.
Kandemirli SG, Kocak B, Naganawa S, Ozturk K, Yip SSF, Chopra S, Rivetti L, Aldine AS, Jones K, Cayci Z, Moritani T, Sato TS. Machine Learning-Based Multiparametric Magnetic Resonance Imaging Radiomics for Prediction of H3K27M Mutation in Midline Gliomas. World Neurosurg. 2021;151:e78–85.
Acknowledgements
We thank Dr. Carolin Malsch for her help regarding statistical analyses. Further, we want to thank all colleagues, patients, and their parents who contributed their data to the HIT-HGG registry.
Funding
This work was supported by the German Childhood Cancer Foundation. Annika Hohm was supported by the German Academic Foundation Cusanuswerk and the Max Weber Programme of the State of Bavaria (student scholarship).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation and data collection were performed by Christof M. Kramm, Marion Hoffmann, Gerrit H. Gielen, Torsten Pietsch, Michael Karremann, Monika Warmuth-Metz, Annika Stock, Johannes Nowak, and Annika Hohm. Image analysis was performed by Monika Warmuth-Metz, Johannes Nowak, and Annika Hohm. Statistical analysis was conducted by Annika Hohm, data interpretation was performed by Johannes Nowak, Annika Hohm, Monika Warmuth-Metz, Annika Stock, Brigitte Bison, Mirko Pham, and Michael Karremann. The first draft of the manuscript was written by Annika Hohm and Johannes Nowak and proofread by Lindsey A. Vandergrift. All authors critically revised and commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
A. Hohm, M. Karremann, G.H. Gielen, T. Pietsch, M. Warmuth-Metz, L.A. Vandergrift, B. Bison, A. Stock, M. Hoffmann, M. Pham, C.M. Kramm and J. Nowak declare that they have no competing interests.
Ethical standards
The analysis of patient and MRI data was approved by the local and central ethics committees of the HIT studies and performed in accordance with the Declaration of Helsinki. Consent to participate: informed consent for data storage and statistical analyses was given by all patients and/or their parents at the time of registration and treatment within the HIT-HGG studies. Consent for publication: not applicable.
Additional information
The authors C.M. Kramm and J. Nowak contributed equally to this work.
Trial Registration Number and Date of Registration
Pediatric Brain Tumor (HIT-HGG) Studies of the Society of Pediatric Oncology and Hematology:
HIT-HGG-2013: NCT03243461; Aug 9, 2017
HIT-HGG-2007: ISRCTN19852453; May 25, 2009
HIT-GBM-D: NCT00278278; Jan 18, 2006
Availability of Data and Material
All authors declare that all data and material as well as software applications support their published claims and comply with field standards.
Code Availability
Not applicable.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hohm, A., Karremann, M., Gielen, G.H. et al. Magnetic Resonance Imaging Characteristics of Molecular Subgroups in Pediatric H3 K27M Mutant Diffuse Midline Glioma. Clin Neuroradiol 32, 249–258 (2022). https://doi.org/10.1007/s00062-021-01120-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00062-021-01120-3