
Abstract
Drugs containing carboxylic acid moieties are associated with a range of toxicities, some related directly to their pharmacology and others to the metabolites that they produce on undergoing biotransformations that reduce biological activity and enhance excretion. The reactions involved in metabolism include a wide range of conjugations to the carboxylic acid moiety plus modifications to the spacers used to attach them to the main body of the drug. Here we provide a metabolic perspective on the biotransformations that have been found to occur with drugs that contain a carboxylic acid and discuss the potential of these to cause toxicity. Based on our current understanding of the metabolism of carboxylic acid-containing drugs we then consider approaches that may mitigate toxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carboxylic acids remain important, often essential, functional groups for drugs, conferring both solubility and pharmacological activity to compounds such as the non-steroidal anti-inflammatory drugs (NSAIDs) and many other therapies. Unfortunately, many of these carboxylic-acid containing drugs have also been found to be toxic (usually causing hepatotoxicity), in some cases leading to their withdrawal. In attempting to understand why these drugs cause drug induced liver injury (DILI) attention has often focussed on the carboxylic acid function as being the “common denominator” for toxicity. Indeed, in terms of quantity, both the in vitro and in vivo metabolic fates of carboxylic acid-containing drugs are dominated by a few major biotransformations which involve conjugation reactions to a variety of endogenous metabolites. Two of these types of conjugation, glucuronidation and acyl-CoA conjugation, lead to the formation of reactive metabolites with known ability to form protein adducts. This property has led to attention becoming focussed on these reactive metabolites as being a probable cause of toxicity. However, there are also a whole variety of other metabolic outcomes, including (as a result of CoA conjugation) the formation of amino acid conjugates etc, which can also be major metabolites, depending on structure and species. In addition, a whole host of other, often CoA-related, minor metabolites can be observed. As these minor metabolites are usually detected in the presence of the more abundant potential toxins this has probably led to the fact that little effort has been put into examining their effects, which therefore remain, at best, poorly understood. Metabolism of these acidic drugs is also not limited to direct biotransformations of the carboxylate moiety but is also seen to occur on the side chains often used to anchor it to the rest of the molecule. Thus, as will be discussed, oxidative metabolism of the side chain, used as a spacer between the carboxyl group itself and the main body of the drug, may also provide a site for both metabolism such as chiral inversion in the “profen” NSAIDs (following the formation of acyl-CoA conjugates) and the production of reactive metabolites via oxidative metabolism. Thus, even our current, somewhat limited, knowledge of toxicity vs structure provides suggestions on how the toxicity of carboxylic acid-containing drugs, even if it cannot be completely avoided, could be mitigated by careful drug design. Here we will provide a brief overview and perspective on the metabolism of carboxylic acid containing drugs, and its potential relationship with toxicity, with emphasis on the benzoates, phenylacetic acids, aliphatic and the aryl-2-propionic acids as found in the “profens”.
It has not been possible in this short perspective to go in great detail into the very large, and still evolving, body of knowledge of the involvement of the metabolism of carboxylic acid-containing drugs in toxicity. We have therefore, of necessity, been selective in our use of the literature. However, whilst we do not claim that this article is a substitute for such an in-depth survey it does seem feasible, based on the existing state of our knowledge, to modify some of the metabolic aspects of the toxicity of carboxylic acid-containing drugs by careful drug design.
Glucuronidation and acyl migration as a potential source of toxicity
A very common metabolic fate for acidic drugs (e.g., the NSAIDs), ester prodrugs, or drugs with aliphatic side chains that can be oxidized to acids etc., is ester conjugation to glucuronic acid. This was for a long time viewed as resulting in the detoxication/deactivation of the aglycone and, in addition, by increasing polarity and water solubility glucuronidation acted as an aid to excretion. However, from the late 1970’s this view changed somewhat with the realization that their acyl migration made drug-acyl glucuronides (AGs) reactive metabolites with the potential to react with proteins (e.g., see Faed 1984 [1]). This reactivity has resulted in their “guilt by association” for the liver [2, 3] and gut toxicity [4] often observed with NSAID exposure (giving cause for regulatory concern and guidance e.g., ref. [5]). However, the case for AGs being the toxic agents causing gut toxicity was completely undermined by elegant work in the rat and mouse on NSAIDs such as diclofenac, the AG of which is excreted in significant amounts in bile. This research showed that, if the hydrolysis of the NSAID-acyl glucuronides by bacterial glucuronidase enzymes was inhibited, gut toxicity was eliminated [6, 7]. So, the AG itself was revealed to not the be the cause of gut toxicity but an innocent party that had been falsely accused in a clear case of “guilt by association”. The AG was simply a delivery vehicle for the actual toxin, the parent drug. What is undeniable, however, is that the rate of disappearance in terms of the half-life of the 1-O-β-acyl glucuronide in buffer at pH 7.4 (which is usually dominated by transacylation rather than hydrolysis reactions) can be used as a surrogate for potential toxicity as there does appear to be a clear relationship between the observation of DILI and those acyl-glucuronides with a short half-life (e.g., see ref. [8]). Other studies found that the correlation between the half-life of disappearance of the 1-O-β-AG and toxicity was improved if, instead of using only this value, the “relative half-lives” were obtained by incorporating the data on the production of the other, transacylated, AG-isomers [9]. A major benefit of this, as yet unproven, attribution of toxicity to AGs is that it provided a significant impetus to the development of efficient methods for their synthesis (see ref. [10] and references therein). Another approach that may also be increasingly valuable in the future for drug discovery is the use of computational chemistry to model acyl-glucuronide reactivity (e.g. ref. [10]) without the need for the synthesis of either the compound or its acyl glucuronide. In this way any compounds likely to form AGs that are also predicted to have short half-lives could be avoided altogether.
However, the hypothesis that drug induced liver injury (DILI) is caused by acyl-glucuronides as a result of their reactivity with proteins remains unproven as, almost always, alternative biotransformations producing reactive metabolite occur in parallel, precluding a clear attribution of blame. In addition, some of the alternative biotransformations are not associated directly with the carboxylic acid moiety at all, but result from e.g., CYP450 metabolism elsewhere on the molecule. One example of this additional layer of complexity is provided by an in vitro study in hepatocytes on diclofenac where the results suggested that reducing the covalent binding associated with its acyl-glucuronide actually enhanced toxicity via CYP2C [11]. Whilst P450-related metabolism caused cell death, and the covalent binding to proteins caused by the acyl-glucuronide did not appear to result in acute cytotoxicity, it was suggested that it may “...be toxicologically relevant for the expression of diclofenac hepatitis...” [11].
Some support for the potential of these acyl-glucuronide metabolites to be toxic has been provided by the confirmation of covalent binding to proteins, both in vitro e.g., ref. [11] and in vivo e.g., ref. [12] using radiolabelling studies. In addition, proteomic techniques can be very useful in these studies, particularly where a radiolabel is not available, or for examining ex vivo samples from patients, as it can provide information on both the presence, types and sites of protein modification by acyl-glucuronides [13, 14]. The types of reactions of AGs with proteins are illustrated in Fig. 1 and sites of modification in Fig. 2. From such studies it has become clear that, as might have been anticipated, the structure of the acyl-glucuronide had a significant effect on adduct formation [13]. This was shown e.g., by study on protein adduct formation using the synthetic acyl-glucuronides of ibufenac, R- and S-ibuprofen, a dimethyl analog of ibuprofen (identified as “bibuprofen”), p-bromobenzoic acid and ponalrestat incubated with human serum albumin (HSA) at biologically relevant concentrations [13]. This study revealed that adduct formation via both transacylation and glycosylation occurred but that the balance of these reactions was structure dependent. Thus, particularly in the case of p-bromobenzoic acid acyl-glucuronide, glycosylation was the dominant route rather than transacylation. Not unexpectedly the degree of steric hinderance of the acyl glucuronide had an important effect on transacylation rates and was also correlated with higher HSA adduct formation, thus linking the in vitro kd of compounds in buffer to the amount of protein modification observed. In this study the sites of adduction on HSA were shown to be the lysine residues 137, 195, 199, 436 and 525 (with lysines 137 and 436 showing both glycosylation and transacylation). A further investigation following the in vitro incubation of diclofenac acyl-glucuronide with HSA (at a molar ration of 50:1) also found both glycation and transacylation of lysines 137, 190, 195, 199, 351, 432, 436, 525 and 451 as well as glycation at lysine residue 162 [13] (Fig. 2). When conducted at somewhat lower glucuronide: HSA molar ratios (e.g. 0.1:1) fewer lysine residues were modified (including those at positions 190, 199 and 432).

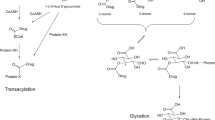
The formation, reactions and putative products of nucleophilic groups on drugs (a) glucuronidation of nucleophilic groups (Nu) in drugs during conjugative metabolism. A production of stable glucuronide glycosidic bonds (e.g., with phenolic groups) (b) glucuronidation to produce acyl-glucuronides which can then react with proteins via transacylation or (c) intramolecular acyl migration and then reaction to form glycated and glycosylated proteins. (Reproduced from ref. [13] with permission)

The reaction sites and types of acyl-glucuronide metabolites with HSA. The sequence map shows the primary amino acid sequence with disulfide bonds shown in yellow. Reaction sites are shown in red for glycosylation, transacylation sites are indicated in blue whilst Lysine 525, which had both glycosylation and transacylation reactivity is green. (Reproduced from ref. [13] with permission)
Such studies highlight the potential reactivity of acyl-glucuronides and pointed to a means of monitoring reactivity in early in vivo studies (both in animals and humans) and subsequently in patients even in the absence of a radiolabelled candidate drug. Indeed, this has been demonstrated in ex vivo samples obtained from diclofenac-treated patients where the analysis of HSA obtained from 6 patients revealed that a total of 7 lysines had been modified (7 N-acylations and 3 glycations) [14]. All 6 patients showed acyl-glucuronide transacylation to lysine 195 with 1 subject having a total of 7 lysine adducts. However, the study also showed that the in vitro studies discussed above were only indicative as no adducts to lysine residues 137 and 351 were found. Perhaps more importantly, despite these circulating adducted proteins in patients none showed evidence of drug-related hypersensitivity. Immune responses, in the form of circulating antibodies were seen for e.g., valproic acid (whose main circulating metabolite is the acyl-glucuronide) in a minority of patients (9/57) where protein adducts were found in the plasma [15]. However, antibody titres were low and the authors suggested that valproic acid adducted-proteins had low immunogenicity [15]. This correlated with the lack of observations of hypersensitivity reported for valproate [15].
However, there are other routes to acyl-glucuronide toxicity that may have little to do with reactivity but are based on transporter inhibition and solubility. For example, in the course of a drug repeat dose toxicology study in rat and dog on the recently withdrawn antidiabetic fasiglifam (structure in Fig. 2) [16]. DILI, exemplified by granulomatous inflammation and the detection of crystals in the liver and bile ducts, was found only in dogs. Investigations by MALDI/MS on sections of the dog liver found both fasiglifam and its AG in the crystals. After 14 days of oral dosing at 200 mg/kg/day to dogs, concentrations of both drug and AG were much higher than their solubility limit of ca. 3.0 mg/ml. This led the authors to suggest that the high concentrations detected “when taken together with lower bile flow rate could cause crystal formation in dog bile” and that its relevance to effects in humans was unclear.
In addition, both fasiglifam and its acyl-glucuronide were also found to inhibit transporters as shown by studies in hepatocytes derived from both rat and dog [16]. Similarly, in vitro and in vivo studies in rat, dog and cynomolgus monkeys and in vitro experiments with human transporters by others also exposed transporter inhibition by fasiglifam and the AG with the latter apparently exceptionally potent against MRP3 [17].
In addition, as can be seen from Fig. 3, based on the circulating metabolites of fasiglifam detected in the rat, a large range of biotransformations were seen [18, 19]. Clearly many of these could, as seen for e.g., metabolite TAK 875-M1, also undergo acyl glucuronidation with similar effects on transporters.
Whilst not investigated as transporter inhibitors, the interactions of both diclofenac-AG (and diclofenac itself) with different human drug transporters have been studied in vitro [20]. Whilst the parent drug was only an OAT substrate its AG was transported by OATs 1-4, two organic anion-transporting polypeptides (OATP1B1and OATP2B1), the breast cancer resistance protein (BCRP) and both MRP2 and MRP3. Given the ability of AGs to adduct proteins it would be interesting to know if the transporter inhibition seen with fasiglifam was reversible, or irreversible due to inactivation of the transporter by covalent binding. It may be significant in this respect that radioactivity has been found to be covalently bound to unidentified membrane proteins from rat hepatocytes incubated with 14C diclofenac although the multiple options for reactive metabolite formation that do not go via acyl glucuronidation clearly make interpretation difficult [21]. It would certainly be of interest to know if transporter inhibition was a general problem for drug-AGs.

The circulating metabolites of fasiglifam (TAK875), shown as semi-localized structures indicating the likely sites of metabolism where these could not be determined with confidence based on MSMS data alone (ref. [18] with permission)
Acyl-CoA conjugate formation as a potential source of toxicity
An alternative form of metabolism that directly involves the carboxylic acid moiety is biotransformation to highly reactive S-acyl-CoA conjugates that can transacylate readily onto other thiol containing biomolecules such as e.g., glutathione (GSH), with the rate depending on structure [22]. These S-acyl-CoA conjugates appear to be much more reactive (40–60 times) than AGs with respect to adduct formation with proteins and as a result these metabolites have been a fertile area of research for those trying to elucidate mechanism(s) of toxicity [23,24,25,26]. However, acyl-CoA-conjugation is a two-step process that first requires adenosine monophosphate (AMP) to be covalently transferred from ATP onto the carboxylic acid moiety of the drug. Only after the biosynthesis of the drug-AMP conjugate is the acyl-CoA thioester formed via the displacement of AMP. This was elegantly shown by Horng and Benet for the drug for mefenamic acid (MFA) who also demonstrated the reactivity of both the acyl-adenylate (MFA-AMP) and the corresponding acyl-CoA (MFA-CoA) conjugates via their ability to transacylate onto GSH, N-acetylcysteine and acyl-CoA [27, 28] (Fig. 4).

The series of biotransformations of mefenamic acid (MFA) leading, via the production of MFA-AMP, to the S-acyl-CoA conjugate (MFA-CoA)
Fortunately, a variety of trapping assays, using soft nucleophile thiols such as cysteine, N-acetyl cysteine, glutathione and homocysteine are available, with a recently published assay using cysteine trapping to detect S-acyl-CoA conjugates [29]. When applied to a number of NSAIDs a correlation between the rate of formation of the cysteine adduct from the conjugates, the drugs’ AUC and DILI was obtained. In addition, in vitro systems that enable determination of the propensity of acyl-CoA drug conjugates to form protein-adducts have been developed based on e.g., liver microsomes supplemented with CoA, NADPH or UDPGA [30]. As well as these microsomal systems the use of cell-based assays such as hepatocytes, HepG2 or HepaRG cells etc., can be employed, e.g. refs. [9, 17, 21, 31]. When the microsomal assay was applied e.g., to the acyl-CoA conjugates of the NSAIDs ibuprofen, ibufenac, fenclozic acid, tienilic acid, suprofen, and zomepirac the CoA conjugates for ibuprofen and ibufenac resulted in covalent binding of 1000 and 8600 pmol drug eq/mg protein, respectively [30]. The same investigation highlighted that, for the NSAIDs suprofen and tienilic acid, the greatest contribution to overall reactivity in the in vitro metabolic protein binding experiments came from oxidative, NADPH dependent, metabolism and not via either AGs or acyl-CoA-conjugates. In a more recent investigation, the DILI caused by MK-8666 (like fasiglifam, an antidiabetic GPR40 agonist), where development was halted because of toxicity, covalent binding studies using liver microsomes and rat and human hepatocytes were used to study reactive metabolite formation [31]. In human hepatocytes the MK-8666-AG was observed, whilst in rat both the acyl-glucuronide and taurine conjugates were detected, (clearly implicating a S-CoA intermediate), along with protein binding (similar for both rat and human hepatocytes). Analysis of protein digests identified both transacylation and glycation adducts (to lysine, serine, and cysteine). When microsomes were provided with either ATP or CoA protein binding increased compared to incubations fortified with UDPGA. In addition, when glutathione was added to the incubations binding due to acyl-CoA thioesters fell by more than 40%.
It is perhaps worth stating the obvious, that acyl-glucuronidation and acyl-CoA conjugation occur in parallel and both result in the formation of reactive metabolites that, to an extent form the same transacylated (but not glycated) protein adducts. However, and equally obvious, acyl-glucuronides are relatively “stable” reactive metabolites that are transported out of cells into the interstitium and systemic circulation post-formation, and are easy to detect, isolate and study. The acyl-CoA-conjugates, on the other hand, are not transported out of cells and are not detected in the circulation. They are relatively unstable and harder to detect, and consequently, must be actively sought. Some indication of the production of acyl-CoA-conjugates can, however, be made by looking for their downstream products such as e.g., the variety of amino acid conjugates that are biosynthesized from them (discussed below). It is also noteworthy that for NSAIDs of the “profen” class of 2-arylpropionic acids, such as e.g., ibuprofen, that are administered as racemic mixtures that there is often a significant amount of chiral inversion from the R- to the S-enantiomer [32,33,34]. So, for example, R-ibuprofen undergoes chiral inversion to produce the pharmacologically active S-ibuprofen which requires the formation of the S-acyl-CoA thioesters [35]. This reaction is essentially unidirectional because the conjugation of CoA to ibuprofen is stereoselective and favors the R-enantiomer [36]. For this class of molecular structure, therefore, chiral inversion may provide a useful indicator of the involvement of CoA-conjugation even when other evidence is not immediately available.
An alternative “signature” of acyl-CoA formation is the production of amino acid conjugates, and the incorporation of acidic xenobiotics into lipids which are considered in more detail below.
Amino acid conjugation
Whilst the reactivity of acyl-CoA conjugates of carboxylic acid-containing drugs and xenobiotics is clearly of major interest it is important to remember that these compounds represent a gateway to a range of further metabolites of several classes (summarized in Fig. 5 below). It seems fair to say that the biological consequences of the production of many of these metabolites have been much less well studied than they perhaps deserve.

Overview of conjugative metabolism of carboxylic acids where acyl-CoA- mediated reactions may also apply to drug metabolism
One of the major downstream biotransformations resulting from the formation of S-acyl-CoA conjugates of carboxylic acids is the biosynthesis of amino acid conjugates (Fig. 6). Indeed, the conversion of benzoic acid to its glycine conjugate hippuric acid represents the first example of human xenobiotic metabolism to be reported [37]. Amino acid conjugation can be a major pathway in some species (e.g., rat, mouse, dog and humans). Such biotransformations have been noted for benzoates, phenylacetic acids, aliphatic and aryl 2-propionic acids. Glycine, taurine and glutamine conjugates are most often encountered [38] but, depending upon the species, conjugation to alanine, arginine, aspartic acid, glutamic acid, histidine, ornithine and serine as well as to peptides such as glycyl-taurine, -glycine and -valine have also been found [39, 40].

Examples of amino acid conjugations of endogenous and drug substrates
Simple benzoates, such as benzoic acid, are converted in mitochondria into the glycine conjugate hippuric acid following formation of the benzoyl-S-CoA thioester and then further mitochondrial metabolism via glycine N-acyltransferase into the conjugate. It has not escaped the notice of those who have studied this reaction that participation of this ligase in both drug and fatty acid metabolism provides an obvious mechanism for drug-acyl-CoA conjugates to interfere with mitochondrial energy production via e.g., β-oxidation as a consequence of CoA sequestration. However, in general, when the formation of amino acid conjugates results in a glycine conjugate it seems to be associated with the detoxication of the acyl-CoA conjugates [40]. The taurine-producing reaction, which is akin to that which produces bile acids such as taurocholic acid, has different substrate specificity to glycine conjugation (also seen for bile acids) and, for example does not seem to target simple benzoates [41]. However, whilst glycine conjugates as such appear largely benign, this area of xenobiotic metabolism seems in general not to have been particularly well explored for other members of the class of carboxylic acid containing drugs [40]. The loose structural similarity to bile acids seems to us at least to suggest the potential for transporter interactions which may contribute to adverse effects on e.g., bile acid homeostasis, and this aspect does not seem to have been afforded any attention. In addition, this type of biotransformation may result in CoA depletion, with consequent effects on mitochondrial energy production etc., if large amounts of drug CoA-conjugates are formed. This is supported by evidence for both e.g., glycine depletion and the saturation of amino acid conjugate production [40]. Depending on the substrate, the CoA conjugate of a drug may also be more stable than the endogenous ones, and when linked to a drug CoA is not available for endogenous processes adding to the problem.
Acyl carnitine conjugation
Acylcarnitine conjugates represent a different type of amino acid conjugate compared to those described above in that the link between carnitine and its acidic substrate is through an ester bond, and not an amide bond.
Carboxylic acid-containing drugs are converted into drug-S-acyl-CoA thioesters on the outer mitochondrial membrane, hijacking the biosynthetic machinery used for fatty acids and can then be converted to carnitine conjugates, to facilitate transport across the inner mitochondrial membrane. For endogenous long chain fatty acids this process is essential for β-oxidation and its potential disruption by drug acylcarnitines has limited but convincing experimental support (e.g. see ref. [42]). Whilst studies have been undertaken to assess metabolism by this type of biotransformation for a limited number of compounds such as e.g., cyclopropanecarboxylic acid [43] and valproic acid [44] etc., the effects of these conjugates on mitochondrial function do not seem to have been subject to serious study. In our opinion, it seems likely that by effectively “hijacking” the CoA-fatty acyl transport system into mitochondria and consuming carnitine it may also deplete the amount of this essential metabolite for other purposes.
Hybrid triglyceride formation
The production of acyl-CoAs from drugs is of concern as they may then mimic the endogenous fatty acid-CoAs. One consequence of this that has been observed is the production of so called “hybrid” mono-, di- and triglycerides [45,46,47,48]. Thus, in a study employing 3T3-L1 adipocytes, the mono- and di-acylglycerol conjugates of the NSAID fenbufen were identified [49]. In the case of chiral profen NSAIDs the formation of these hybrid triglycerides was found to be stereoselective, as might be expected based on the stereoselectivity shown in the biosynthesis of acyl-CoA-profen conjugates [33,34,35,36]. This was shown for R-fenoprofen, which was incorporated into glycerolipids in vivo and in vitro by both hepatocytes and adipocytes but was not the seen for the S-enantiomer [50,51,52]. In addition to fenoprofen this stereospecific incorporation of profens such as e.g., ibuprofen [53] and ketoprofen [54] seems to be selective for the R-form and can lead to the accumulation of these metabolites in tissue. It would also seem that e.g., the R-, but not the S-enantiomer of fenoprofen was responsible for the stereospecific inhibition of triacylglycerol biosynthesis in isolated rat hepatocytes and adipocytes [52].
Another consequence of exposure to drugs such as aspirin (and thus its metabolite salicylic acid), the 2-arylpropionic acids and aliphatic acids such as valproic acid is that mitochondrial β-oxidation can be affected. (e.g., see refs. [55, 56]). As well as hybrid triglyceride formation there have been a few reports of acidic drugs being modified via cholesteryl and bile acid conjugation such as the case of the experimental hypolipidaemic 1-(4-carboxyphenoxy)-10-(4-chlorophenoxy) decane (BRL 24139 or CCD) which formed both a hybrid triglyceride and a novel cholesteryl ester [57]. To our knowledge bile acid conjugation has not yet been reported for drugs, but in the case of an anilino acid metabolite of a pyrethroid insecticide a variety of bile acid conjugates (to e.g., cholic, taurocholic and taurochenodeoxycholic acid etc.,) were formed in significant amounts [58]. Whilst, presumably, if these sterol conjugates were major metabolites of carboxylic acid containing drugs they would have been detected more frequently. However, one can ask the question “even if present as “minor” metabolites, if potent, might they still contribute to the emergence of DILI whilst remaining under the DMPK radar”?
Metabolism centered on the side chain spacer
With drugs based on either ethanoic- or 2-propionic acid side chain-linked carboxylic acids the possibility of biotransformation at the alpha carbon exists and can be important in the metabolism of these the compounds. For ethanoic acids, such as e.g., diclofenac, a reaction first observed in vitro by Grillo et al. results ultimately, via the loss of the linking CH2, in the oxidative decarboxylation of the drug and side chain shortening to a benzoate structure [59]. This reaction was also seen in vivo in the rat [60] and has more recently been observed for the withdrawn NSAIDs lumiracoxib [61] and fenclozic acid [62] and recently, the antidiabetic drug fasiglifam [18].
From the in vitro studies on diclofenac the observed oxidative decarboxylation was found to be catalyzed by a CYP450 3A4-mediated reaction, via an intermediate benzylic carbon-centered free radical [59]. This reaction either resulted in elimination through an O-imine methide (that could be highly reactive and form a GSH conjugate or protein adducts) or alternatively formed a benzyl alcohol. This benzyl alcohol could then be dehydrated to the reactive O-imine methide that could also result in GSH or protein adducts. Irrespective of the mechanism the overall result was side chain shortening and an unexpected route to reactive metabolites. Whilst not apparently a major biotransformation for any of the compounds mentioned above, it clearly has the potential to add to the body burden experienced by preclinical species, human volunteers and patients. As discussed by the authors of the in vitro study on diclofenac this type of biotransformation could easily have “implications with respect to the CYP450-mediated metabolism of structurally related carboxylic acid-containing drugs that may be able to undergo decarboxylation and subsequent elimination- or dehydration-type reactions leading to (for example) imine-, quinone-, or thioquinone-methide chemically reactive, and potentially toxic, intermediates” [59].
We note, however, that oxidative metabolism of this type, resulting in both side chain shortening, and the production of reactive metabolites, has not been reported for the aryl 2-propionic acids. The implications of the small change from an aryl ethanoic to aryl 2-propionic acid seem to us obvious and the routine use of this side chain would also be beneficial in terms of reducing the transacylation rates of any acyl-glucuronides (especially for the S-enantiomer). As an illustration, the effect of methyl substitution on acyl glucuronide t 1/2 on ibufenac, through R- and S-ibuprofen to the dimethyl analog is shown (for both buffer and plasma) in Table 1 [63]. Interestingly, in buffer the main reaction seen at pH 7.4 was transacylation whilst in plasma it was hydrolysis. The clear trend in buffer from ibufenac through R- and S-ibuprofen to “bibuprofen” is toward longer t 1/2 with that of the latter being in excess of 24 h [63].
Strategies for reducing the drug metabolism-related toxicity of carboxylic acid-containing drugs
Whilst little can be done to eliminate toxicity that is specific to the pharmacological properties of drugs it is clearly important to minimize/eliminate any “collateral damage” arising from metabolism wherever possible. As described above, the major, and many of the minor, biotransformations that can occur when organism and cells are exposed to carboxylic acid-containing drugs are by now probably established and their potential consequences highlighted. In addition, understanding of the relationship between the structure of the drug and its metabolism, and the effects on reactivity, have been established, but the underlying structure activity relationships (SARs) remain elusive. For example, both acyl glucuronidation and acyl-CoA conjugation are clearly seen as likely to be the cause of toxicity (prompting the issuing of Regulatory Guidance in the case of the former [5]). However, even where these constitute major pathways for conjugation, toxicity is not guaranteed. For example, the most obvious member of the benzoates, and by far the simplest, is clearly benzoic acid. Whilst benzoic acid is not a drug as such it has been used as topical antiseptic and antifungal, and also as a preservative in food (e.g., sausages) and can be produced from dietary components via microbial and liver co-metabolism before being converted to its glycine conjugate (hippurate). In benzoic acid, we have a compound that forms large amounts of an acyl-CoA-conjugate but which appears to be fairly lacking in toxicity, particularly DILI, even when exposure (from whatever source) is quite high. In the case of the classic “benzoate” drug salicylic acid (dosed as acetylsalicylate) the addition of the phenolic hydroxyl results in quite complex metabolism with both oxidations and conjugation reactions. This “metabolic promiscuity” results in a mixture of metabolites with glucuronidation (acyl and phenolic), glycine conjugation to form salicylhippuric acid, phenolic sulfation and further hydroxylation to gentisic acid (also subject to sulfation) all observed. All of these metabolites are found in variable amounts depending upon a large number of factors such as ethnicity, sex and diet etc [64, 65] Nevertheless, the major product is salicylhippurate accounting for ca. 45+% of the dose, indicating a high degree of acyl-CoA-based metabolism [64]. However, despite large doses, significant acyl-CoA conjugate formation and acyl glucuronidation, there seem to be few suggestions that any of the observed toxicities are due to metabolism leading to covalent binding with e.g., the amount of in vivo covalent binding seen in human plasma after 1.2 g of aspirin/day was less than 100 ng of salicylic acid equivalents/mL [66]. This is not to say that such benzoates are without toxicity as it seems clear e.g., that the accumulation of the benzoyl-CoA conjugates of a variety of non-drug p-substituted benzoates causes reproductive toxicity in the rat [67].
However, whilst the metabolism of benzoates such as salicylic acid might be relatively benign with regard to DILI the same cannot be said of NSAIDs such as ibufenac, fenclozic acid or diclofenac etc. Such phenyl acetic (ethanoic) acids have, for many years, been associated with DILI, leading to both ibufenac and fenclozic acid being withdrawn from clinical use (ibufenac) or late stage clinical development (fenclozic acid). Diclofenac has survived, and remains in widespread clinical use, but like acetaminophen, it is a liver toxin at high dose and remains the subject of much research interest with respect to DILI. Diclofenac represents something of a problem when trying to obtain definitive answers regarding the contribution to covalent binding of the carboxyl group to toxicity as in vivo there is concomitant oxidative, CYP450-related, metabolism which also leads to covalent binding. However, as discussed earlier the detection of acyl-glucuronide adducts to circulating plasma proteins in patients does not seem automatically to result in adverse drug reactions. Fenclozic acid represents another unsolved puzzle, representing an NSAID drug that was not hepatotoxic to any animal species tested but was the cause of DILI in patients [68]. Whilst a problem compound, fenclozic acid is also of interest in terms of the sheer diversity of the metabolism observed in chimeric liver humanized mice (Fig. 6) administered the drug, diversity that was not replicated in conventional mice [62].
From Fig. 7, the expected acyl-glucuronide, glycine and taurine conjugates can be seen, accompanied by glutamine and carnitine conjugates clearly showing the involvement of acyl-CoA-conjugate formation. However, in addition a large number of modifications to the side chain can be seen, both shortening and lengthening, some of which may presumably have required an acyl-CoA intermediate. As well as these highlighted biotransformations there was also evidence of oxidative metabolism leading to GSH conjugation, providing evidence on non-carboxylic acid-dependent reactive metabolism. Based on the differences seen between conventional animals and the liver-humanized mice and the species-specific toxicity of the drug to humans, it is tempting to ascribe the DILI in humans to the differences seen in the metabolic profiles. However, in the absence of the results of further investigations it would be unwise to overinterpret these observations.

The metabolism of fenclozic acid after oral administration to chimeric liver humanized mice. From ref. [62] with permission
Which brings us to the aryl-2-propionic acids exemplified by ibuprofen. The aryl-ethanoic acid ibufenac caused abnormal liver function tests in ca. 20–30% of patients and jaundice in a further 5%, plus high transaminase levels and cases of severe DILI. However, the difference that adding a single methyl group can make is stark [69]. Thus, whilst ibufenac was a potent cause of DILI its aryl-2-propionic acid analog ibuprofen is not and is, in contrast, is an over the counter (OTC) drug in many countries. Where ibuprofen-related toxicity has been observed, it seems in the main to be attributable to pharmacology rather than DILI. Whilst both the acyl-glucuronides and acyl-CoA conjugates of ibuprofen have been shown to be reactive in vitro it is noteworthy that despite the clear occurrence of these “warning” pathways, and a reasonably high therapeutic dose, this does not seem to translate into DILI.
So here, we have a clear “worked example” of a single, simple change to the spacer group attaching the carboxylic acid to the rest of the drug effecting a major change in reducing toxicity but not at the expense of efficacy. It is tempting in this instance to use the term “magic methyl” given the very widespread use of ibuprofen. Had the acyl-CoA binding assay described above been available at the time and ibufenac shown to have covalent binding of 8600 pmol drug eq/mg of protein vs 1000 for ibuprofen respectively it seems reasonable to suggest that the latter would have been taken forward for development.
Presumably, moving to the administration of only the S-enantiomer of ibuprofen would mean that if acyl-CoA conjugation was a problem it would be avoided. There are examples of the marketing of the single enantiomers of two NSAIDs include S-ibuprofen (as dexibuprofen) and S-naproxen. Whilst in the case of dexibuprofen there does not seem to be any evidence of an improved safety profile for S-ibuprofen compared to the racemate positive benefits cannot be excluded for other classes of carboxylic acid containing drugs.
From all of this, admittedly incomplete information, it seems possible to make the following comments that may be relevant to the pragmatic design of carboxylic acid-based drugs:
-
1.
Despite decades of research, we suggest that the evidence that acyl-glucuronides, whilst reactive, are potent toxins by this route remains weak (this does not exclude toxicity by e.g. transporter effects etc).
-
2.
The correlation between the short half-lives of some 1-O-β-AGs and the observation of toxicity does not necessarily reflect causation but shows that the carboxylic acid is a metabolically active group. As such it can also be converted into other, such as e.g., the less easily detected and more reactive S-acyl-CoA thioesters or other reactive metabolites resulting in toxicity.
-
3.
When designing compounds for success, avoid, if at all possible, arylethanoic acids, especially if the drugs are expected to be high dose (greater than 50 mg/day [70, 71]) and instead use a profen-like structure (preferably the S-enantiomer), which will also prevent the formation of reactive metabolites through side chain shortening (by whatever mechanism).
-
4.
If, for e.g., reasons of loss of potency resulting from the insertion of methyl group on the side chain, an arylethanoic-like structure must be used, in vitro assay systems available for studying acyl-CoA conjugate formation may be of value in choosing between candidates for development. Further, proof of the involvement of acyl-CoA conjugates in vivo may then be sought by looking for the formation of glycine and taurine conjugates in species such as, e.g., the mouse.
Conclusions
As pointed out in the introduction to this perspective, we do not claim to give a full account of the vast literature on the metabolism of carboxylic acid-containing drugs and their potential involvement in toxicity. Nevertheless, we hope that this article provides the reader with insight and ideas on approaches to addressing some of the metabolic aspects of carboxylic acid-containing drugs and mitigating drug toxicity risk. Bluntly, based on our current understanding, candidate drugs forming 1-O-β-- acyl-glucuronide with a short half-life in buffer at pH 7.4 (e.g., 1.7 h [8]) as a major metabolite and the detection of acyl-CoA conjugates (or their downstream products) are sufficient warning to trigger further studies. It seems self-evident that, if such a compound has an anticipated high daily dose and long duration of dosing, leading to the potential for a high, and sustained, daily body burden of covalent adducts, then there is already a cause for concern. If further aggravating factors are also present, such as transporter interactions and other biotransformations that result in reactive metabolite formation, problems in clinical use may be expected, and require appropriate risk-benefit assessments to be made before proceeding.
However, we are optimistic, given what we now know, that designing in modest changes in molecular structure may well reduce/prevent some of these problems so that they become manageable risks.
References
Faed EM. Properties of acyl glucuronides: implications for studies of the pharmacokinetics and metabolism of acidic drugs. Drug Metab Rev. 1984;15:1213–49. https://doi.org/10.3109/03602538409033562.
Horng H Spahn-Langguth H Benet LZ. Mechanistic role of acyl glucuronides. In: Kaplowitz N, DeLeve LD, editors. Definition of Drug-Induced Liver Disease. Amsterdam: Elsevier; 2013. pp 35−70. https://doi.org/10.1016/B978-0-12-387817-5.00003-0.
Smith DA, Hammond T, Baillie TA. Safety assessment of acyl glucuronides-a simplified paradigm. Drug Metab Dispos. 2018;46:908–12. https://doi.org/10.1124/dmd.118.080515.
Boelsterli UA, Ramirez-Alcantara V. NSAID acyl glucuronides and enteropathy. Curr Drug Metab. 2011;12:245–52. https://doi.org/10.2174/138920011795101877.
US Department of Health and Human Services FDA, Center for Drug Evaluation and Research. Guidance for industry: safety testing of drug metabolites. Silver Spring, MD: US Department of Health and Human Services FDA, Center for Drug Evaluation and Research; 2008. FDA-2008-D-0065.
LoGuidice A, Wallace BD, Bendel L, Redinbo MR, Boelsterli UA. Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J Pharm Exp Ther. 2012;341:447–54. https://doi.org/10.1124/jpet.111.191122.
Saitta KS, Zhang C, Lee KK, Fujimoto K, Redinbo MR, Boelsterli UA. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: Mode of action and pharmacokinetics. Xenobiotica. 2014;44:28–35. https://doi.org/10.3109/00498254.2013.811314.
Sawamura R, Okudaira N, Watanabe K, Murai T, Kobayashi Y, Tachibana M, Ohnuki T, Masuda K, Honma H, Kurihara A, Okazaki O. Predictability of idiosyncratic drug toxicity risk for carboxylic acid-containing drugs based on the chemical stability of acyl glucuronide. Drug Metab Dispos. 2010;38:1857–64. https://doi.org/10.1124/dmd.110.034173.
Lassila T, Hokkanen J, Aatsinki SM, Mattila S, Turpeinen M, Tolonen A. Toxicity of carboxylic acid-containing drugs: The role of acyl migration and CoA conjugation investigated. Chem Res Toxicol. 2015;28:2292–303. https://doi.org/10.1021/acs.chemrestox.5b00315.
Bradshaw PR, Athersuch TJ, Stachulski AV, Wilson ID. Acyl glucuronide reactivity in perspective. Drug Discov Today. 2020;25:1639–50. https://doi.org/10.1016/j.drudis.2020.07.009.
Kretz-Rommel A, Boelsterli UA. Diclofenac covalent protein binding is dependent on acyl glucuronide formation and is inversely related to P450-mediated acute cell injury in cultured rat hepatocytes. Toxicol Appl Pharm. 1993;120:155–61. https://doi.org/10.1006/taap.1993.1097.
Takakusa H, Masumoto H, Yukinaga H, Makino C, Nakayama S, Okazaki O, Sudo K. Covalent binding and tissue distribution/retention assessment of drugs associated with idiosyncratic drug toxicity. Drug Met Disp. 2008;36:1770–79. https://doi.org/10.1124/dmd.108.021725.
Monrad RN, Errey JC, Barry CS, Iqbal M, Meng X, Iddon L, Perrie JA, Harding JR, Wilson ID, Stachulski AV, Davis BG. Dissecting the reaction of Phase II metabolites of ibuprofen and other NSAIDS with human plasma protein. Chem Sci. 2014;5:3789–94. https://doi.org/10.1039/C4SC01329H.
Hammond G, Meng X, Jenkins RE, Maggs JL, Castelazo AS, Regan SL, Bennett SNL, Earnshaw CJ, Aithal GP, Pande I, Kenna JG, Stachulski AV, Park BK, Williams DP. Mass spectrometric characterization of circulating covalent protein adducts derived from a drug acyl glucuronide metabolite: multiple albumin adductions in diclofenac patients. J Pharm Exp Ther. 2014;350:387–402. https://doi.org/10.1124/jpet.114.215079.
Williams AM, Worrall S, de Jersey J, Dickinson RG. Studies on the reactivity of acyl glucuronides—III: Glucuronide-derived adducts of valproic acid and plasma protein and anti-adduct antibodies in humans. Biochem Pharm. 1992;43:745–55. https://doi.org/10.1016/0006-2952(92)90239-f.
Kogame A, Moriya Y, Mori I, Pan L, Morohashi A, Ebihara T, Fukui H, Tagawa Y, Benet LZ. Characterization of fasiglifam-related liver toxicity in dogs. Drug Metab Dispos. 2019;47:525–34. https://doi.org/10.1124/dmd.118.084889.
Otieno MA, Snoeys J, Lam W, Ghosh A, Player MR, Pocai A, Salter R, Simic D, Skaggs H, Singh B, Lim H-K. Fasiglifam (TAK-875): Mechanistic investigation and retrospective identification of hazards for drug induced liver injury. Toxicological Sci. 2018;163:374–84. https://doi.org/10.1093/toxsci/kfx040.
Molloy BJ, King A, Gethings LA, Plumb RS, Mortishire-Smith RJ. Wilson IDInvestigation of the pharmacokinetics and metabolic fate of Fasiglifam (TAK-875) in male and female rats following oral and intravenous administration. Xenobiotica. 2023;53:93–105. https://doi.org/10.1080/00498254.2023.2179952.
Kogame A, Leeb R, Pan L, Sudoa M, Nonaka M, Moriya Y, Higuchi T, Yoshihiko T. Disposition and metabolism of the G protein-coupled receptor 40 agonist TAK-875 (fasiglifam) in rats, dogs, and humans. Xenobiotica. 2019;49:433–45. https://doi.org/10.1080/00498254.2018.1453100.
Zhang Y, Han Y-H, Putluru SP, Matta MK, Kole P, Mandlekar S, Furlong MT, Liu T, Iyer RA, Marathe P, Yang Z, Lai Y, Rodrigues AD. Diclofenac and its acyl glucuronide: determination of in vivo exposure in human subjects and characterization as human drug transporter substrates in vitro. Drug Metab Dispos. 2016;44:320–8. https://doi.org/10.1124/dmd.115.066944.
Kretz-Rommel A, Boelsterli UA. Selective protein adducts to membrane proteins in cultured rat hepatocytes exposed to diclofenac: radiochemical and immunochemical analysis. Mol Pharm. 1994;45:237–44. PMID: 8114673.
Sidenius U, Skonberg C, Olsen J, Hansen SH. In vitro reactivity of carboxylic acid-CoA thioesters with glutathione. Chem Res Toxicol. 2004;17:75–81. https://doi.org/10.1021/tx034127o.
Li C, Benet LZ, Grillo MP. Enantioselective covalent binding of 2-phenylpropionic acid to protein in vitro in rat hepatocytes. Chem Res Toxicol. 2002;15:1480–87.
Grillo MP, Benet LZ. Studies on the reactivity of clofibryl-S-acyl-CoA thioester with glutathione in vitro. Drug Metab Dispos. 2002;30:55–62. https://doi.org/10.1124/dmd.30.1.55.
Olsen J, Bjørnsdottir I, Tjørnelund J, Hansen SH. Chemical reactivity of the naproxen acyl glucuronide and the naproxen coenzyme A thioester towards bionucleophiles. J Pharm Biomed Anal 2002. 2002;29:7–15. https://doi.org/10.1016/s0731-7085(02)00026-2.
Shore LJ, Fenselau C, King AR, Dickinson RG. Characterization and formation of the glutathione conjugate of clofibric acid. Drug Metab Dispos. 1995;23:119–23. PMID: 7720514.
Horng H, Benet LZ. Characterization of the acyl-adenylate linked metabolite of mefenamic Acid. Chem Res Toxicol. 2013;26:465–76. https://doi.org/10.1021/tx300520j.
Horng H, Benet LZ. The Nonenzymatic Reactivity of the acyl-linked metabolites of mefenamic acid toward amino and thiol functional group bionucleophiles. Drug Metab Dispos. 2013;41:1923–33. https://doi.org/10.1124/dmd.113.053223.
Kakutani N, Kobayashi S, Taniguchi T, Nomura Y. A cysteine trapping assay for risk assessment of reactive acyl CoA metabolites. Xenobiotica. 2022;52:16–25. https://doi.org/10.1080/00498254.2022.2035016.
Darnell M, Breitholtz K, Isin EM, Jurva U, Weidolf L. Significantly different covalent binding of oxidative metabolites, acyl glucuronides, and S-Acyl CoA conjugates formed from xenobiotic carboxylic acids in human liver microsomes. Chem Res Toxicol. 2015;28:886–96. https://doi.org/10.1021/tx500514z.
Cancilla M, Samuel K, Chen Q, Chobanian HR, Thomas A, Tong W, Josien H, Buevich AV, Mitra K, Shang J, Tschirret-Guth R. Bioactivation of GPR40 agonist MK-8666: formation of protein adducts in vitro from reactive acyl glucuronide and acyl CoA thioester. Chem Res Toxicol. 2020;33:191–201. https://doi.org/10.1021/acs.chemrestox.9b00226.
Mills RFN, Adams SS, Cliffe EE, Dickinson W, Nicholson JS. The metabolism of ibuprofen. Xenobiotica. 1973;3:589–98. https://doi.org/10.3109/00498257309151547.
Hutt AJ, Caldwell J. The metabolic chiral inversion of 2-arylpropionic acids. A novel route with pharmacological consequences. J Pharm Pharm. 1983;35:693–704. https://doi.org/10.1111/j.2042-7158.1983.tb02874.x.
Knihinicki RD, Williams KM, Day RO. Chiral inversion of 2-arylpropionic acid non-steroidal anti-inflammatory drugs—1: In vitro studies of ibuprofen and flurbiprofen. Biochemical Pharm. 1989;38:4389–395. https://doi.org/10.1016/0006-2952(89)90647-3.
Grillo MP, Hua F. Enantioselective formation of ibuprofen-S-acyl-glutathione in vitro in incubations of ibuprofen with rat hepatocytes. Chem Res Toxicol. 2008;21:1749–59. https://doi.org/10.1021/tx800098h.
Woodman TJ, Wood PJ, Thompson AS, Hutchings TJ, Steel GR, Jiao P, Threadgill MD, Lloyd MD. Chiral inversion of 2-arylpropionyl-CoA esters by human alpha-methylacyl- CoA racemase 1A (P504S)–a potential mechanism for the anti-cancer effects of ibuprofen. Chem Commun. 2011;47:7332–34. https://doi.org/10.1039/c1cc10763a.
Ure A. On gouty concretions; with a new method of treatment. Pharm J Trans. 1841;24:30–35. PMID: 20895736.
Hutt AJ Caldwell J. Chapter 10, Amino acid conjugation. In: Mulder GJ, editor. Conjugation reactions in drug metabolism. London, UK: Taylor and Francis; 1990. p 34.
Steventon GB Hutt AJ. Chapter 14, The amino acid conjugations. In: Ioannides C, editor. Enzyme systems that metabolize drugs and other xenobiotics. Chichester, UK: John Wiley & Sons Ltd; 2002. pp 501-21.
Knights KM, Sykes MJ, Miners JO. Amino acid conjugation: contribution to the metabolism and toxicity of xenobiotic carboxylic acids. Expert Opin Drug Metab Toxicol. 2007;3:159–68. https://doi.org/10.1517/17425255.3.2.159.
Idle JR, Millburn P, Williams RT. Taurine conjugates as metabolites of arylacetic acids in the ferret. Xenobiotica. 1978;8:253–64. https://doi.org/10.3109/00498257809056147.
Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharm Ther. 1995;67:101–54. PMID: 7494860.
Quistad GB, Staiger LE, Schooley DA. The role of carnitine in the conjugation of acidic xenobiotics. Drug Met Disp. 1986;14:521–25. PMID: 2876856.
Silva MFB, Aires CCP, Luis PBM, Ruiter JPN, IJlst L, Duran M, Wanders RJA, Tavares de Almeida I. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: a review. J Inherit Metab Dis. 2008;31:205–16. PMID: 18392741.
Caldwell J, Marsh MV. Interrelationships between xenobiotic metabolism and lipid biosynthesis. Biochem Pharm. 1983;32:1667–72. https://doi.org/10.1016/0006-2952(83)90107-7.
Caldwell J. Novel xenobiotic-lipid conjugates. Biochem Soc Trans. 1985;13:852–54.
Fears R, Baggaley KH, Alexander R, Morgan B, Hindley RM. The participation of ethyl 4-benzyloxybenzoate (BRL 10894) and other aryl-substituted acids in glycerolipid metabolism. J Lipid Res. 1978;19:3–11. PMID: 621438.
Fears R, Richards DH. Association between lipid lowering activity of aryl-substituted carboxylic acids and formation of substituted glycerolipids in rats. Biochem Soc Trans. 1981;9:572–73.
Dodds PF, Chou SC, Ranasinghe A, Coleman RA. Metabolism of fenbufen by cultured 3T3-L1 adipocytes: synthesis and metabolism of xenobiotic glycerolipids. J Lipid Res 1995;36:2493–503. PMID: 8847476.
Sallustio BC, Meffin PJ, Knights KM. The stereospecific incorporation of fenoprofen into rat hepatocyte and adipocyte triacylglycerols. Biochem Pharm. 1988;37:1919–23. https://doi.org/10.1016/0006-2952(88)90537-0.
Sallustio BC, Meffin PJ, Thompson M. HPLC quantitation of triacylglycerol containing fenoprofen from biological samples. J Chromatogr. 1987;422:33–41. https://doi.org/10.1016/0378-4347(87)80437-1.
Sallustio E, Knights KM, Meffin PJ. The stereospecific inhibition of endogenous triacylglycerol synthesis by fenoprofen in rat isolated adipocytes and hepatocytes. Biochem Pharm. 1990;40:1414–17. https://doi.org/10.1016/0006-2952(90)90412-e.
Williams K, Day R, Knihinicki R, Duffield A. The stereoselective uptake of ibuprofen enantiomers into adipose tissue. Biochem Pharm. 1986;35:3403–5. https://doi.org/10.1016/0006-2952(86)90443-0.
Carabaza A, Suesa N, Tost D, Pascual J, Gomez M, Gutierrez M, Ortega E, Montserrat X, Garcia AM, Mis R, Cabre F, Mauleon D, Carganico G. Stereoselective metabolic pathways of ketoprofen in the rat: incorporation into triacylglycerols and enantiomeric inversion. Chirality 1996;8:163–72. https://doi.org/10.1002/(SICI)1520-636X(1996)8:2.
Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharm Ther. 1995;67:101–54. https://doi.org/10.1016/0163-7258(95)00012-6.
Dodds PF. Xenobiotic lipids: the inclusion of xenobiotic compounds in pathways of lipid biosynthesis. Prog Lipid Res. 1995;34:219–47. https://doi.org/10.1016/0163-7827(95)00007-m.
Fears R, Baggaley KH, Walker P, Hindley RM. Xenobiotic cholesteryl ester formation. Xenobiotica. 1982;12:427–33. https://doi.org/10.3109/00498258209052484.
Quistad G, Staiger L, Schooley D. Xenobiotic conjugation: a novel role for bile acids. Nature. 1982;296:462–64. https://doi.org/10.1038/296462a0.
Grillo MP, Ma J, Teffera Y, Waldon DJ. A novel bioactivation pathway for 2-[2-(2,6-dichlorophenyl)aminophenyl]ethanoic acid (diclofenac) initiated by cytochrome P450-mediated oxidative decarboxylation. Drug Metab Dispos. 2008;36:1740–44. https://doi.org/10.1124/dmd.108.021287.
Sarda S, Page C, Pickup K, Schulz-Utermoehl T, Wilson I. Diclofenac metabolism in the mouse: novel in vivo metabolites identified by high performance liquid chromatography coupled to linear ion trap mass spectrometry. Xenobiotica. 2012;42:179–94. https://doi.org/10.3109/00498254.2011.607865.
Dickie AP, Wilson CE, Schreiter K, Wehr R, Wilson EM, Bial J, Wilson ID, Riley RJ. The pharmacokinetics and metabolism of lumiracoxib in chimeric humanized and murinized FRG mice. Biochem Pharm. 2017;135:139–50. https://doi.org/10.1016/j.bcp.2017.03.015.
Ekdahl A, Weidolf L, Baginski M, Morikawa Y, Thompson RA, Wilson ID. 2018. The metabolic fate of fenclozic acid in chimeric mice with a humanized liver. Arch Toxicol 2018;92:2819–28. https://doi.org/10.1007/s00204-018-2274-0.
Johnson CH, Karlsson E, Sarda S, Iddon L, Iqbal M, Meng X, Harding JR, Stachulski AV, Nicholson JK, Wilson ID, Lindon JC. Integrated HPLC-MS and 1H-NMR spectroscopic studies on acyl migration reaction kinetics of model drug ester glucuronides. Xenobiotica 2009;40:9–23. https://doi.org/10.3109/00498250903348720.
Hutt AJ, Caldwell J, Smith RL. The metabolism of aspirin in man: a population study. Xenobiotica. 1986;16:239–49. https://doi.org/10.3109/00498258609043527.
Navarro SL, Saracino MR, Makar KW, Thomas SS, Li L, Zheng Y, Levy L, Schwarz Y, Bigler J, Potter JD, Lampe JW. Determinants of aspirin metabolism in healthy men and women: effects of dietary inducers of UDP-glucuronosyltransferases. J Nutrigenet Nutrigenomics. 2011;4:110–18. https://doi.org/10.1159/000327782.
Dickinson RG, Baker PV, King AR. Studies on the reactivity of acyl glucuronides -VII. Salicyl acyl glucuronide reactivity in vitro and covalent binding of salicylic acid to plasma protein of humans taking aspirin. Biochem Pharm. 1994;9:469–76. https://doi.org/10.1016/0006-2952(94)90177-5.
Laue H, Badertscher RP, Hostettler L, Weiner-Sekiya Y, Haupt T, Nordone A, Adamson GM, Natsch A. Benzoyl-CoA conjugate accumulation as an initiating event for male reprotoxic effects in the rat? Structure–activity analysis, species specificity, and in vivo relevance. Arch Toxicol. 2020;94:4115–29. https://doi.org/10.1007/s00204-020-02918-9.
Hart FD, Bain LS, Huskisson EC, Littler TR, Taylor RT. Hepatic effects of fenclozic acid. Ann Rheum Dis. 1970;29:684. https://doi.org/10.1136/ard.29.6.684.
Hart FD, Boardman PL. Ibufenac (4-Isobutylphenyl acetic acid). Ann Rheum Dis. 1965;24:61–5. https://doi.org/10.1136/ard.24.1.61.
Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Michael DA. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem Res Toxicol 2011;24:1345–1410.
Kalgutkar AS. Designing around structural alerts in drug discovery. J Med Chem. 2020;63:6276–6302. https://doi.org/10.1021/acs.jmedchem.9b00917.
Author information
Authors and Affiliations
Contributions
Both authors contributed equally to the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weidolf, L., Wilson, I. Minimizing the DILI potential of carboxylic acid-containing drugs: a perspective. Med Chem Res 32, 2034–2047 (2023). https://doi.org/10.1007/s00044-023-03140-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03140-9