
Abstract
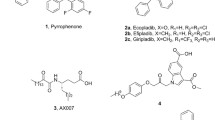
The N,N-disubstituted 4-sulfamoylbenzoic acid derivative 3, which was found to be an inhibitor of cytosolic phospholipase A2α (cPLA2α) with micromolar activity in a ligand-based virtual screening approach, was structurally modified to increase its enzyme inhibitory potency. Replacing the substituents on the sulfonamide nitrogen with other residues such as naphthyl, naphthylmethyl, indolylalkyl and differently substituted phenyl moieties did not lead to a significant increase in activity. Only strong structural convergence to the potent known benzhydrylindole-substituted benzoic acid derivatives that had served as templates in the virtual screening resulted in compounds with considerable potency. Thus, the sulfamoyl benzoic acid derivatives 85 and 88 showed submicromolar IC50 values against cPLA2α.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The arachidonic acid cascade is a biochemical pathway that leads to the formation of oxidized arachidonic acid products such as prostaglandins and leukotrienes, which exert a variety of physiological effects [1]. The excessive stimulation of this metabolic route by inflammatory cytokines, however, results in high concentrations of these compounds, known as eicosanoids, being involved in the pathogenesis of inflammatory diseases. The prostaglandin E2 (PGE2) formed via the cyclooxygenase (COX) pathway and the leukotrienes produced via the 5-lipoxygenase (5-LOX) pathway are particularly responsible for this. Thus, PGE2 mediates the cardinal symptoms of acute inflammation: rubor (redness), calor (heat), tumor (swelling) and dolor (pain) [2]. Certain leukotrienes are potent chemotractants leading to leukocyte recruitment and activation enhancing the inflammatory process [3]. The therapeutically used COX inhibitors like diclofenac and ibuprofen can amorilate the effect of PGE2, but they do not have an impact on leukotriene formation. The inflammatory mediators produced via both pathways could be inhibited simultaneously by blocking the enzyme responsible for releasing the common precursor arachidonic acid from phospholipids, namely cytosolic phospholipase A2α (cPLA2α). Thus, inhibitors of this enzyme are expected to provide new treatment options for inflammatory conditions [4, 5].
Although several highly active inhibitors of cPLA2α have been developed, no such compound is available for clinical use today. Among the most effective of the published substances are thiazolidinediones from Shionogi [6], phenylpropanoic acid derivatives from Asahi Kasei Pharma [7, 8], benzhydrylindoles from Wyeth (now Pfizer) [9,10,11] and propan-2-ones from AstraZeneca [12]. We have synthesized a series of heteroaryl propan-2-ones structurally related to the latter substance class, which also possess a high enzyme inhibitory potency [13, 14]. Unfortunately, these compounds are not suitable for peroral in vivo application, especially due to their rapid metabolic inactivation and their property of being substrates of gastrointestinal efflux transporters. In search of new molecular entities for cPLA2α inhibitors, we recently have performed a molecular-modeling approach [15] inspired by the works of Gianella-Borradori et al. and Naylor et al. [16, 17]. From a series of six structurally related cPLA2α inhibitors of Wyeth with high potency [10, 11], such as WAY-196025 (1) (Fig. 1), energetically favorable conformers were calculated and used for a ligand-based virtual screening method including both three-dimensional shape and electrostatic comparison applying the tools OMEGA (conformer generation), ROCS (screening), EON (ranking) and VIDA (visualization) from OpenEye Scientific (Santa Fee, New Mexico, USA). As the database source, the Screening Compound Database (SCD) from MolPort (Riga, Latvia) with around 6 million compounds in 2017 was chosen. Of the compounds with the closest match in terms of shape and electrostatic similarity expressed in terms of the electrostatic shape-Tanimoto (ET) combo score [18], 47 were purchased from MolPort and tested for inhibition of isolated porcine cPLA2α. Eight of these substances showed an IC50 value between 10 µM and 33 µM, among them the 4-sulfamoylbenzoic acid derivative 3 [19] (IC50: 19 µM) and the indole-substituted 4-(methylenehydrazineyl)benzoic acid 4 (IC50: 22 µM) (Fig. 1). For the commercially available benzhydrylindole-substituted cPLA2α inhibitor 2 from Wyeth [9], which is marketed under the name Axon-1609, an IC50 of 0.21 µM was determined with the same assay. Thus, the hits found with the molecular modeling were about 100-fold less active than 2. Taken into account that the IC50 values of the six cPLA2α inhibitors used as queries in our ligand-based virtual screening had been reported to be even lower than that of 2 [9,10,11], we had to assume that the found hits were substantially less effective than these substances. Nevertheless, we started some structural variations of 3 in order to increase its potency.

Structures of cPLA2α inhibitors of Wyeth (1 and 2) and cPLA2α inhibitor hits (3 and 4) found in a ligand-based virtual screening approach
Results and discussion
Chemistry
For the synthesis of the 4-sulfamoylbenzoic acid derivative 6, bearing a phenyl and a benzyl substituent at the sulfonamide nitrogen, methyl 4-(chlorosulfonyl)benzoate was reacted with N-benzylaniline and the methyl ester group of obtained compound 5 was saponified with aqueous KOH in methanol (Scheme 1). The target compound 11 with a naphthalen-2-yl group instead of the phenyl residue was synthesized analogously using the secondary amine N-benzylnaphthalen-2-amine in the first step. All other N-aryl-N-arylmethyl substituted 4-sulfamoylbenzoic acids were prepared in a similar way as outlined for the synthesis of compound 14 in Scheme 2. First, methyl 4-(chlorosulfonyl)benzoate was reacted with aniline or a substituted aniline derivate in dichloromethane and/or THF in the presence of pyridine or triethylamine. Then the benzyl or naphthylmethyl substituent was introduced at the nitrogen by treatment with benzyl chloride or the appropriate (chloromethyl)naphthalene in DMF using K2CO3 as base. Finally, the methylester group was hydrolyzed with aqueous KOH in methanol. Compound 53, substituted at the sulfonamide nitrogen with an indol-3-ylethyl and a naphthalen-2-yl-methyl group, was similarly synthesized using tryptamine instead of an aniline derivative in the first step (Scheme 3). Substitution of the indole nitrogen of 53 with 2,4-dichlorobenzyl or benzyhydryl residues was carried out by reacting the ester intermediate 52 with 2,4-dichlorobenzyl bromide or benzyhydryl bromide in DMF with sodium hydride as base. Under the selected conditions, saponification of the methyl ester groups occurred simultaneously with the formation of the target compounds 54 and 55, respectively (Scheme 3).

Reagents and conditions: a Pyridine, dichloromethane, THF, room temperature, overnight; b aqueous KOH (10%), methanol, room temperature, overnight

Reagents and conditions: a Pyridine, dichloromethane, THF, room temperature, overnight; b 1-(chloromethyl)naphthalene, K2CO3, KI, DMF, 85 °C, 6 h; c aqueous KOH (5 M), methanol, 60 °C, 4 h

Reagents and conditions: a Triethylamine, dichloromethane, room temperature, 16 h; b 2-(chloromethyl)naphthalene, K2CO3, DMF, 80 °C, 6 h; c aqueous KOH (20 %), methanol, 70 °C, 4 h; d 1. 2,4-dichlorobenzyl bromide (in case of 54) or benzhydryl bromide (in case of 55), NaH, DMF, room temperature, 10 min; 2. water, room temperature, 15 min
The derivatives of 54, in which the N-naphthalen-2-yl-methyl group was replaced by (phenylsulfonylamino)- or (benzylsulfonylamino)alkyl residues were synthesized as shown for compound 63 in Scheme 4. Boc-protected tryptamine was substituted at the indole nitrogen with a 2,4-dichlorobenzyl residue by treatment with the corresponding benzyl bromide in DMF using sodium hydride to deprotonate the indole nitrogen. Deprotection of the alkylamine group of the obtained intermediate 56 with trifluoroacetic acid in dichloromethane followed by its reaction with methyl 4-(chlorosulfonyl)benzoate led to the intermediate 58, which was alkylated at the sulfonamide nitrogen with 2-(Boc-amino)ethyl bromide to yield 60. After cleavage of the Boc-protecting group of the alkyl chain amine, a sulfonamide functionality was formed by reaction with benzenesulfonyl chloride in dichloromethane in the presence of triethylamine. Finally, saponification with aqueous KOH in methanol yielded the target compound 63. The corresponding derivative without sulfonylaminoalkyl residue at the sulfamoyl group of the benzoic acid (59) was obtained by alkaline hydrolysis of the intermediate 58.

Reagents and conditions: a 1. NaH, DMF, room temperature, 30 min; 2. 2,4-dichlorobenzyl bromide, room temperature, 10 min; b trifluoroacetic acid, dichloromethane, room temperature, 3 h; c methyl 4-(chlorosulfonyl)benzoate, pyridine, dichloromethane, room temperature, 16 h; d aqueous KOH (20%), methanol, 70 °C, 5 h; e 2-(Boc-amino)ethyl bromide, Cs2CO3, dimethylacetamide, 60 °C, 16 h; f trifluoroacetic acid, dichloromethane, room temperature, 4 h; g benzenesulfonyl chloride, triethylamine, dichloromethane, room temperature, 16 h
The derivative of Axon-1609 (2) (Fig. 1), in which the ether oxygen at the benzoic acid residue is replaced by a sulfamoyl group (79), and the corresponding compound, which is additionally methylated at the sulfonamide group (81), were synthesized from the appropriately substituted tryptamine derivative as shown in Scheme 5.

Reagents and conditions: a Di-tert-butyl dicarbonate, triethylamine, dioxane, room temperature, 4 h; b 1. NaH, DMF, room temperature, 30 min; 2. benzhydryl bromide, room temperature, 10 min; c 1. HCl in dioxane, room temperature, 4 h; 2. methyl 4-(chlorosulfonyl)benzoate, triethylamine, dichloromethane, THF, room temperature, 16 h; d aqueous KOH (20%), methanol, 70 °C, 5 h; e 1. LiAlH4, THF, reflux, 2 h; 2. methyl 4-(chlorosulfonyl)benzoate, triethylamine, dichloromethane, THF, room temperature, 16 h; f 1. benzhydryl bromide, NaH, DMF, room temperature, 10 min; 2. water, room temperature, 15 min
For the synthesis of the two derivatives with 2-hydroxyethyl- and 5-hydroxypentyl-side chain, respectively, at the sulfonamide nitrogen (83 and 85), the N-monosubstituted sulfonamide 78 was reacted with the corresponding ω-bromoalkyl acetate in the presence of K2CO3 (Scheme 6). The ester groups of the intermediates 82 and 84 obtained were saponified with aqueous KOH in methanol to yield the desired target compounds. The preparation of 4-sulfamoyl benzoic acid 88 bearing a more polar hydroxyethylethoxy substituent at the sulfonamide nitrogen is also outlined in Scheme 6. In this case, the sulfonamide 78 was treated with 2-(2-chlororethoxy)ethanol, with the hydroxy group protected as a silyl ether. The alcohol functionality of the substitution product 86 was deprotected with tetrabutyammonium fluoride in THF, and the ester moiety was again cleaved under alkaline conditions to give the test compound 88.

Reagents and conditions: a 2-Bromoethyl acetate or 5-bromopentyl acetate, K2CO3, DMF, room temperature, 4 h or 16 h; b aqueous KOH (10%), methanol, THF, room temperature, overnight; c tert-butyl[2-(2-chloroethoxy)ethoxy]diphenylsilane, K2CO3, DMF, 100 oC, 2 h and room temperature 6 h; d tetrabutylammonium fluoride, THF, room temperature, overnight; e aqueous KOH (10%), methanol, THF, room temperature, overnight
Biochemical activity
The cPLA2α used in the biological assay was isolated from porcine platelets. The cPLA2α inhibitory activity of the target compounds was evaluated by determining the arachidonic acid release by the enzyme from the substrate 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine in the presence of the vesicle forming lipid 1,2-dioleoyl-sn-glycerol by HPLC and MS detection [20]. Deuterated arachidonic acid was used as an internal standard.
The structure of the hit 3 (Fig. 1) was first simplified by replacing the benzothiophene residue by a phenyl ring and omitting the two substituents of the benzyl residue. In contrast to 3, for which an IC50 of 19 µM was measured, the derivative thus obtained (6) showed no cPLA2α inhibition at the highest test concentration of 33 µM (Table 1). Fusing a benzene ring to the phenyl or benzyl moiety led to the compounds 9, 11, 14 and 16, respectively, which exhibited some activity against the enzyme at 33 µM (35% – 48% inhibition).
Next, in the two derivatives with naphthalen-1-ylmethyl and naphthalen-2-ylmethyl moieties (14 and 16), various substituents such as chlorine, methyl, methoxy and phenyl were introduced into the phenyl group at position 4 or the phenyl group was replaced by a benzyl residue. For the compound with the naphthalen-1-ylmethyl substituent (14), these structural changes did not significantly improve the potency (Table 2). In contrast, more pronounced effects occurred in case of the analogous naphthalen-2-ylmethyl derivative 16. Thus, the cPLA2α inhibitory potency increased by a factor of 3 when a chlorine atom was introduced in position 4 of the phenyl residue (IC50 of 33: 12 µM). A phenyl and a benzyl radical at this position had an analogous effect, as shown by the IC50 values of the compounds 42 and 45. Since Wyeth’s potent cPLA2α inhibitors, which were the starting point for our ligand-based virtual screening for new cPLA2α inhibitors, contain a 3-alkyl-substituted indole ring, a derivative of 16 was prepared that beared a 2-(indol-2-yl)ethyl residue in place of the phenyl ring. However, with an IC50 value of 27 µM this compound (53) was not significantly more potent than 16. The introduction of a 2,4-dichlorophenyl moiety, which was present in one of our other screening hits (4) (Fig. 1), at the indole-1 position of 53 resulted in a negligible increase in potency by a factor of about 1.5 (IC50 of 54: 18 µM). In contrast, compound 55, which like the Wyeth compounds carries a benzhydryl residue at position 1 of the indole, showed a more pronounced inhibitory effect. With an IC50 value of 5.8 µM, it was the most active inhibitor in this series.
In Wyeth’s benzhydrylindoles, a significant increase in cPLA2α inhibition was achieved by introducing benzylsulfonylaminoethyl residues in position 2 of the indole [9,10,11]. Therefore, we next attempted to address the binding site of this residue in the enzyme also with our compounds. We began these studies not with the benzyhydryl-substituted derivative 55, but with the slightly less active derivative 54, since we did not want to get too close structurally to the Wyeth compounds. Moreover, we wanted to introduce this residue not at the indole 2-position but at the sulfonamide nitrogen instead of the naphthalen-2-ylmethyl unit (Table 3). This latter residue apparently does not have a positive effect on the cPLA2α inhibition of compound 54, since its replacement by hydrogen did not significantly change the activity as shown by the inhibition data of 59. Unfortunately, this structural variation also did not lead to more effective compounds despite the variation in the length of the alkyl spacer between the two sulfonylamino groups and the replacement of the terminal benzyl by a phenyl residue, respectively (Table 3). Thus, the IC50s of the investigated compounds 69, 71 and 75 as well as those of the corresponding phenylsulfonylaminoalkyl derivatives 63 and 67 were between 14 µM and 18 µM.
Therefore, we finally decided to align the structure of our 4-sulfamoylbenzoic acid derivatives with that of the Wyeth substances more closely. For this purpose, the benzhydryl-substituted 5-chloro-2-methylindole derivative 79 and its N-methylated analogue 81 were synthesized. With an IC50 of 1.8 µM, compound 79 was significantly more active than the 4-sulfamoylbenzoic acid derivatives synthesized so far (Table 4). The corresponding compound 81 with additional methyl group on the sulfonamide nitrogen had approximately the same activity (IC50: 1.4 µM).
For the 4-sulfamoylbenzoic acid derivatives with N-(dichlorobenzyl)indolylethyl residues, it was shown that the introduction of a second large substituent on the sulfonamide nitrogen, such as naphthylmethyl or benzylsulfonylaminoalkyl, did not significantly affect the cPLA2α inhibitory activity of the molecules (Table 3). Therefore, it could be assued that in this area of the molecules, without loss of activity functional groups could be attached with which the physicochemical or pharmacokinetic properties of the inhibitors can be controlled. A disadvantage of the investigated compounds is their extremely high lipophilicity. For example, compounds 79 and 81 have log P values of 5.0 and 5.8, and that of the reference substance Axon-1609 is even 6.2 (Table 4). Based on these observations, the polarity of the compounds should be increased by adding a polar residue to the sulfonamide nitrogen. By introducing a hydroxyethyl moiety to the sulfonamide nitrogen, the log P decreased to 4.6, while the enzyme-inhibitory effect of the obtained compound 83 was maintained (IC50: 1.5 µM). With a slightly more polar hydroxyethylethoxy radical (88), the inhibitory effect could even be improved by a factor of 2 (IC50: 0.66 µM). The replacement of the ether oxygen atom of this compound by a carbon led, as expected, to a more lipophilic substance (log P of 85: 5.0). At the same time, however, surprisingly, the inhibitory potency of this compound against cPLA2α increased markedly. With an IC50 of 0.25 µM, compound 85 was about as effective as the reference substance Axon-1609. Accordingly, in the case of the N-indolylethyl-substituted 4-sulfamoylbenzoic acid derivatives, it is nevertheless possible to increase the inhibitory effect on cPLA2α by adding a second substituent at the sulfonamide nitrogen.
Conclusion
In conclusion, in the case of the hit 3 (Fig. 1), which was discovered by ligand-based virtual screening, both replacement of the benzothiophene residue with substituted phenyl moieties and substitution of the benzyl residue by naphthylmethyl groups did not significantly improve the compound’s cPLA2α inhibitory activity. Only a clear structural approximation to the benzhydrylindole-substituted benzoic acids such as WAY-196025 [9, 10], on which the virtual screening was based, brought about some increase in activity. The most potent derivatives were approximately as active as the reference inhibitor Axon-1609 (2). In this context, however, it should be noted that this compound is reported to be significantly less active than its highly potent derivatives such as WAY-196025 [8, 10]. Future studies will attempt to further improve the potency and especially the physicochemical properties of the 4-sulfamoylbenzoic acid derivatives investigated, e.g., by introducing acidic or basic residues on the sulfonamide nitrogen.
Experimental
Chemistry
General
Column chromatography was performed on silica gel 60, particle size 0.040–0.063 mm, from Macherey & Nagel (Düren, Germany). Melting points were determined on a Büchi B-540 apparatus (Essen, Germany) and are uncorrected. 1H NMR spectra were recorded on a DD2 spectrometer (400 MHz) or a DD2 spectrometer (600 MHz) from Agilent (Santa Clara, USA). Electron ionization (EI) mass spectra were obtained on a Finnigan (Bremen, Germany) GCQ apparatus. The high resolution mass spectra (HR-MS) were recorded on a Bruker (Bremen, Germany) micrOTOF-Q II spectrometer applying atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI). Purity of the target compounds was determined by reversed phase HPLC with UV-detection. The samples were prepared by mixing 20 μL of a 5 mM solution of the respective compound in DMSO with 180 μL of acetonitrile. 5 μL of the solutions was injected into the HPLC-system. Separation was performed using a Nucleosil 100 RP18 3 µm column (3 mm (I.D.) x 125 mm) at a flow rate of 0.4 mL/min with a gradient consisting of acetonitrile/water/trifluoroacetic acid (18:82:0.1 to 86:14:0.1, v/v/v). UV-absorbance was measured at 254 nm. Under these conditions, most compounds had a purity of 95 % or more, with the exception of compounds 86, 88 and 91, whose purity levels were between 90% and 94%.
For the syntheses of the target compounds not described below: see Supplementary Material.
Methyl 4-(N-benzyl-N-phenylsulfamoyl)benzoate (5)
A solution of benzylaniline (187 mg, 1.02 mmol) was treated with a mixture of dry dichloromethane/dry THF (4:1, 5 mL) followed by pyridine (1 mL). Then methyl 4-(chlorosulfonyl)benzoate (200 mg, 0.85 mmol) was added portionwise and the obtained mixture was stirred overnight at room temperature. After dilution with aqueous HCl (1 M), the organic phase was separated. The aqueous phase was extracted exhaustively with ethyl acetate. The combined organic phases were washed with water and brine, dried over anhydrous Na2SO4 and concentrated in vacuo to yield 5 as a solid (180 mg, 55%). C21H19NO4S (381.4); mp 143 °C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 8.15 (d, J = 8.7 Hz, 2H), 7.79 (d, J = 8.7 Hz, 2H), 7.31–7.18 (m, 8H), 7.06–7.02 (m, 2H), 4.83 (s, 2H), 3.91 (s, 3H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 382.1108, found 382.1113.
4-(N-Benzyl-N-phenylsulfamoyl)benzoic acid (6)
Compound 5 (63 mg, 0.17 mmol) was suspended in methanol (10 mL). After addition of aqueous 10% KOH solution (5 mL), the mixture was stirred overnight at room temperature. To the obtained solution, aqueous HCl solution (10%) was added until the product precipitated. The precipitate was filtered off by suction, washed with water and dried in vacuo to give 6 as a solid (42 mg, 69%). C20H17NO4S (367.4); mp 254 °C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 13.53 (br, 1H), 8.13 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 8.7 Hz, 2H), 7.31–7.18 (m, 8H), 7.07–7.02 (m, 2H), 4.83 (s, 2H); 13C-NMR (101 MHz, DMSO-d6) δ (ppm) 166.2, 141.3, 138.2, 136.0, 134.1, 130.2, 128.9, 128.5, 128.3, 128.1, 127.9, 127.6, 127.5, 53.6; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 368.0951, found 368.0938.
Methyl 4-(N-phenylsulfamoyl)benzoate (12)
A solution of aniline (117 μL, 1.28 mmol) in a mixture of dry dichloromethane/dry THF/pyridine (4:1:1, 6 mL) was portionwise treated with methyl 4-(chlorosulfonyl)benzoate (250 mg, 1.07 mmol). The resulting mixture was stirred at room temperature overnight. After dilution with aqueous HCl (1 M), the organic phase was separated. The aqueous phase was extracted exhaustively with dichloromethane. The combined organic phases were washed with water and brine, dried over anhydrous Na2SO4 and concentrated in vacuo to yield 12 as a solid (243 mg, 78%). C14H13NO4S (291.3); mp 129–130 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 10.41 (br, 1H), 8.06 (d, J = 8.7 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.23–7.17 (m, 2H), 7.08–6.99 (m, 3H), 3.83 (s, 3H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 292.0638, found 292.0623.
Methyl 4-[N-(naphthalen-1-ylmethyl)-N-phenylsulfamoyl]benzoate (13)
A solution of 12 (100 mg, 0.34 mmol) in dry DMF (6 mL) was treated with K2CO3 (94 mg, 0.68 mmol) and KI (57 mg, 0.34 mmol). After stirring for 30 min at room temperature, 1-(chloromethyl)naphthalene (103 μL, 0.68 mmol) was added dropwise. The mixture was heated at 85 oC for 6 h, diluted with water and exhaustively extracted with ethyl acetate. The combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 8:2) to give 13 as a solid (109 mg, 74%). C25H21NO4S (431.5); mp 196 °C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 8.43 (d, J = 8.1 Hz, 1H), 8.19 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 7.5 Hz, 1H), 7.85 (d, J = 8.2 Hz, 2H), 7.75 (dd, J = 8.2 and 2.5 Hz, 1H), 7.63 (ddd, J = 8.4, 6.8 and 1.4 Hz, 1H), 7.54 (ddd, J = 8.0, 6.8 and 1.2 Hz, 1H), 7.31–7.22 (m, 2H), 7.19–7.11 (m, 3H), 6.96–6.88 (m, 2H), 5.27 (s, 2H), 3.93 (s, 3H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 432.1264, found 432.1300.
4-[N-(Naphthalen-1-ylmethyl)-N-phenylsulfamoyl]benzoic acid (14)
Compound 13 (60 mg, 0.14 mmol) was suspended in methanol (10 mL). After addition of aqueous KOH solution (5 M, 5 mL), the mixture was stirred at 60 °C for 4 h. The cooled reaction mixture was treated with aqueous HCl solution (10%) until the product precipitated. The precipitate was filtered off by suction, washed with water and dried in vacuo to give 14 as a solid (37 mg, 64%). C25H21NO4S (417.5); mp 219–220 °C; 1H-NMR (600 MHz, DMSO-d6) δ (ppm) 13.56 (br, 1H), 8.45 (d, J = 8.5 Hz, 1H), 8.17 (d, J = 8.5 Hz, 2H), 7.89 (dd, J = 8.2 and 1.3 Hz, 1H), 7.81 (d, J = 8.5 Hz, 2H), 7.75 (dt, J = 7.7 and 1.2 Hz, 1H), 7.63 (ddd, J = 8.4, 6.8 and 1.3 Hz, 1H), 7.54 (ddd, J = 8.2, 6.8 and 1.1 Hz, 1H), 7.30–7.27 (m, 1H), 7.25–7.21 (m, 1H), 7.16– 7.14 (m, 3H), 6.95–6.92 (m, 2H), 5.27 (s, 2H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.2, 140.6, 137.8, 134.9, 133.2, 131.3, 130.6, 130.2, 128.7, 128.6, 128.5, 128.5, 128.3, 127.9, 127.8, 126.4, 125.9, 125.0, 123.6, 51.9; HR-MS (ESI) m/z [M–H]–: calc. 416.0962, found 416.0958.
Methyl 4-{N-[2-(indol-3-yl)ethyl]sulfamoyl}benzoate (51)
A solution of tryptamine (410 mg, 2.56 mmol) in dry dichloromethane (10 mL) was treated with triethylamine (1.5 mL). After portionwise addition of methyl 4-(chlorosulfonyl)benzoate (500 mg, 2.13 mmol), the mixture was stirred for 16 h at room temperature, diluted with ethyl acetate and washed with aqueous HCl solution (1 M) followed by water. The organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure to yield 51 as a solid (735 mg, 96%). C18H18N2O4S (358.4); mp 139 °C.; 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 10.79 (s, 1H), 8.09 (d, J = 8.7 Hz, 2H,), 7.95 (t, J = 5.8 Hz, 1H), 7.89 (d, J = 8.7 Hz, 2H), 7.36 (dd, J = 7.9 and 0.7 Hz, 1H), 7.29 (dt, J = 8.1 and 0.9 Hz, 1H), 7.10 (d, J = 2.3 Hz, 1H), 7.03 (ddd, J = 8.2, 7.0 and 1.2 Hz, 1H), 6.93 (ddd, J = 8.0, 7.0 and 1.1 Hz, 1H), 3.89 (s, 3H), 3.09–3.02 (m, 2H), 2.83–2.75 (m, 2H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 359.1060, found 359.0987.
Methyl 4-{N-[2-(indol-3-yl)ethyl]-N-(naphthalen-2-ylmethyl)sulfamoyl}benzoate (52)
To a mixture of K2CO3 (77 mg, 0.56 mmol), 51 (100 mg, 0.28 mmol) and dry DMF (5 mL) was added 2-(chloromethyl)naphthalene (99 mg, 0.56 mmol). The resulting mixture was stirred at 80 °C for 6 h, cooled, diluted with ethyl acetate and washed three times with water. The organic phase was dried over anhydrous Na2SO4 and concentrated. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 7:3) to yield 52 as an oil (83 mg, 60%). C29H26N2O4S (498.6); 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 10.74 (d, J = 2.5 Hz, 1H), 8.10 (d, J = 8.5 Hz, 2H), 7.98 (d, J = 8.5 Hz, 2H), 7.93–7.88 (m, 2H), 7.85–7.82 (m, 1H), 7.81 (s, 1H), 7.53–7.46 (m, 3H), 7.23 (d, J = 8.1 Hz, 1H), 7.03 (d, J = 7.9 Hz, 1H,), 6.98 – 6.93 (m, 2H), 6.73 (ddd, J = 8.0, 6.9 and 1.0 Hz, 1H), 4.59 (s, 2H), 3.35–3.30 (m, 2H,), 2.70–2.65 (m, 2H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 499.1686, found 499.1681.
4-{N-[2-(Indol-3-yl)ethyl]-N-(naphthalen-2-ylmethyl)sulfamoyl}benzoic acid (53)
Compound 52 (75 mg, 0.15 mmol) was suspended in methanol (8 mL). After addition of aqueous KOH solution (20%, 4 mL), the mixture was stirred at 70 °C for 4 h. The cooled reaction mixture was treated with aqueous HCl solution (3 M) until the product precipitated. The precipitate was filtered off by suction, washed with water and dried in vacuo to give 53 as a solid (43 mg, 59%). C28H24N2O4S (484.6); mp 245 °C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 13.46 (s, 1H), 10.76 (s, 1H), 8.12 (d, J = 8.6 Hz, 2H), 8.01 (d, J = 8.6 Hz, 2H), 7.96–7.90 (m, 2H), 7.88–7.79 (m, 2H), 7.55–7.47 (m, 3H), 7.25 (d, J = 8.1 Hz, 1H), 7.05 (d, J = 7.9 Hz, 1H), 7.00–6.95 (m, 2H), 6.76 (ddd, J = 8.0, 7.0 and 1.0 Hz,), 4.61 (s, 2H), 3.40–3.35 (m, 2H), 2.74–2.66 (m, 2H); 13C-NMR (101 MHz, DMSO-d6) δ (ppm) 166.2, 143.2, 136.1, 134.4, 134.2, 132.8, 132.5, 130.2, 128.3, 127.6, 127.6, 127.2, 127.1, 126.6, 126.4, 126.1, 126.1, 123.1, 121.0, 118.3, 117.9, 111.4, 110.3, 51.7, 48.5, 24.5; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 485.1530, found 485.1525.
4-(N-{2-[1-(2,4-Dichlorobenzyl)indol-3-yl]ethyl}-N-(naphthalen-2-ylmethyl)sulfamoyl)benzoic acid (54)
To a solution of 52 (190 mg, 0.38 mmol) in dry DMF (6 mL) was added sodium hydride (60% dispersion in mineral oil) (29 mg, 0.73 mmol) under ice cooling in a nitrogen atmosphere. The mixture was stirred at room temperature for 30 min. After addition of 2,4-dichlorobenzyl bromide (137 mg, 0.57 mmol) in portions, stirring was continued for 10 min. The reaction mixture was carefully quenched with water and stirred for another 15 min. The suspension was diluted with ethyl acetate and washed three times with water. The organic phase was dried over anhydrous Na2SO4 and the solvent was removed in vacuo. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate/formic acid, 8:2:0.1) to yield 54 as a solid (65 mg, 27%). C35H28Cl2N2O4S (643.6); mp 185 °C; 1H-NMR (600 MHz), DMSO-d6, δ (ppm) 13.54 (s, 1H), 8.10 (d, J = 8.5 Hz, 2H), 7.98 (d, J = 8.5 Hz, 2H), 7.94–7.89 (m, 2H), 7.84–7.81 (m, 2H), 7.63 (d, J = 2.1 Hz, 1H), 7.53–7.51 (m, 2H), 7.49 (dd, J = 8.5 and 1.8 Hz, 1H), 7.25–7.21 (m, 2H), 7.12 (d, J = 8.0 Hz, 1H), 7.04–7.00 (m, 2H), 6.82 (ddd, J = 7.9, 6.9 and 0.9 Hz, 1H), 6.53 (d, J = 8.4 Hz, 1H), 5.30 (s, 2H), 4.61 (s, 2H), 3.39– 3.34 (m, 2H), 2.74–2.69 (m, 2H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.2, 143.0, 136.0, 134.6, 134.2, 132.8, 132.7, 132.6, 132.5, 130.3, 129.6, 128.9, 128.3, 128.1, 127.6, 127.6, 127.2, 127.2, 127.1, 126.8, 126.4, 126.2, 126.1, 121.6, 118.9, 118.4, 110.9, 109.9, 51.6, 48.4, 46.1, 24.4; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 643.1220, found 643.1252.
4-{N-[2-(1-Benzhydrylindol-3-yl)ethyl]-N-(naphthalen-2-ylmethyl)sulfamoyl}benzoic acid (55)
Compound 52 (230 mg, 0.46 mmol) was reacted with benzhydryl bromide (171 mg, 0.69 mmol) following the protocol described for the synthesis of 54 to yield 55 as a solid (120 mg, 40%). C41H34N2O4S (650.8); mp 174 °C; 1H-NMR (600 MHz, CDCl3) δ (ppm) 8.18 (d, J = 8.5 Hz, 2H), 7.94 (d, J = 8.5 Hz, 2H), 7.84–7.81 (m, 1H), 7.79 (d, J = 8.5 Hz, 1H,), 7.73–7.70 (m, 1H), 7.58 (s, 1H), 7.50–7.48 (m, 2H), 7.43 (dd, J = 8.5 and 1.8 Hz, 1H), 7.32–7.29 (m, 6H), 7.16–7.12 (m, 2H), 7.05 (ddd, J = 8.4, 7.1 and 1.2 Hz, 1H), 7.03 (ddd, J = 5.9, 3.2 and 1.4 Hz), 6.88 (ddd, J = 8.0, 7.0 and 1.0 Hz, 1H), 6.71 (s, 1H), 6.47 (s, 1H), 4.54 (s, 2H), 3.46 – 3.40 (m, 2H), 2.83–2.77 (m, 2H); 13C-NMR (151 MHz, CDCl3) δ (ppm) 169.6, 145.1, 139.8, 136.9, 133.4, 133.3, 133.2, 132.9, 131.0, 128.9, 128.8, 128.4, 128.1, 128.0, 127.9, 127.9, 127.7, 127.3, 126.6, 126.4, 126.2, 124.9, 121.9, 119.5, 118.9, 111.2, 110.4, 63.6, 52.8, 48.8, 25.4; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 651.2312, found 651.2326.
tert-Butyl-{2-[1-(2,4-dichlorobenzyl)indol-3-yl]ethyl}carbamate (56)
To a solution of tert-butyl [2-(indol-3-yl)ethyl]carbamate [21] (300 mg, 1.15 mmol) in dry DMF (6 mL) was added sodium hydride (60% in mineral oil) (88 mg, 2.20 mmol) under ice cooling in a nitrogen atmosphere. The mixture was stirred at room temperature for 30 min. Then 2,4-dichlorobenzyl bromide (415 mg, 1.73 mmol) was added in portions and stirring was continued for 10 min. The reaction mixture was quenched with water and stirred for an additional 15 min. Ethyl acetate was added and the organic solution washed three times with water, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 8:2) to give 56 as an oil (305 mg, 63%). C22H24Cl2N2O2 (419.3); 1H-NMR (400 MHz, CDCl3) δ (ppm) 7.63 (d, J = 7.8 Hz, 1H,), 7.43 (d, J = 2.1 Hz, 1H), 7.21–7.18 (m, 2H), 7.17–7.11 (m, 1H), 7.07 (dd, J = 8.3 and 2.1 Hz, 1H), 6.95 (s, 1H), 6.53 (d, J = 8.4 Hz, 1H), 5.33 (s, 2H), 4.59 (s, 1H), 3.50–3.43 (m, 2H), 3.02–2.94 (m, 2H), 1.43 (s, 9H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 419.1288, found 419.1298.
2-[1-(2,4-Dichlorobenzyl)indol-3-yl]ethan-1-amine (57)
A solution of 56 (300 mg, 0.72 mmol) in dry dichloromethane was treated with trifluoroacetic acid (2 mL) and stirred at room temperature for 3 h. The reaction mixture was made weakly alkaline with aqueous NaOH (1 M) and extracted exhaustively with ethyl acetate. The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to yield 57 as an oil (228 mg, quantitative). C17H16Cl2N2 (319.2); 1H-NMR (400 MHz, CDCl3) δ (ppm) 7.64 (d, J = 7.7 Hz, 1H), 7.42 (d, J = 2.1 Hz, 1H), 7.20–7.17 (m, 2H), 7.16–7.10 (m, 1H), 7.06 (dd, J = 8.3 and 2.1 Hz, 1H), 6.97 (s, 1H), 6.53 (d, J = 8.3 Hz, 1H), 5.32 (s, 2H), 3.11–3.02 (m, 2H), 3.00–2.91 (m, 2H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 319.0673, found 319.0742.
Methyl 4-(N-{2-[1-(2,4-dichlorobenzyl)indol-3-yl]ethyl}sulfamoyl)benzoate (58)
A solution of 57 (290 mg, 0.91 mmol) in dry dichloromethane (5 mL) was treated with pyridine (1 mL) and stirred at room temperature for 15 min. Then methyl 4-(chlorosulfonyl)benzoate (178 mg, 0.76 mmol) was added in portions and the mixture was stirred at room temperature for 16 h. The reaction mixture was exhaustively extracted with ethyl acetate and the combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 8:2) to yield 58 as an oil (261 mg, 66%). C25H22Cl2N2O4 (517.4); 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.05 (d, J = 8.7 Hz, 2H), 7.79 (d, J = 8.7 Hz, 2H), 7.44–7.41 (m, 2H), 7.19–7.16 (m, 2H), 7.10–7.04 (m, 2H), 6.86 (s, 1H), 6.53 (d, J = 8.3 Hz, 1H), 5.29 (s, 2H), 4.48 (t, J = 6.0 Hz, 1H), 3.96 (s, 3H), 3.38–3.28 (m, 2H), 2.98–2.91 (m, 2H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 517.0750, found 517.0747.
4-(N-{2-[1-(2,4-Dichlorobenzyl)indol-3-yl]ethyl}sulfamoyl)benzoic acid (59)
A suspension of compound 58 (35 mg, 0.068 mmol) in a mixture of methanol (6 mL) and aqueous KOH solution (20%) (3 mL) was stirred at 70 °C for 5 h. The cooled reaction mixture was treated with aqueous HCl solution (3 M) until the product precipitated, and then extracted exhaustively with ethyl acetate. The combined organic phases were dried over anhydrous Na2SO4 and concentrated to give 59 as a solid (21 mg, 62%). C24H20Cl2N2O4S (503.4); mp 178 °C; 1H-NMR (600 MHz, DMSO-d6) δ (ppm) 13.39 (s, 1H), 8.06 (d, J = 8.5 Hz, 2H), 7.95 (t, J = 5.8 Hz, 1H), 7.87 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 2.2 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H,), 7.30–7.27 (m, 2H), 7.20 (s, 1H), 7.08 (ddd, J = 8.2, 7.0 and 1.2 Hz, 1H), 7.01 (ddd, J = 7.9, 7.0 and 1.0 Hz, 1H), 6.60 (d, J = 8.4 Hz, 1H), 5.39 (s, 2H), 3.10–3.05 (m, 2H), 2.81 (t, J = 7.5 Hz, 2H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.2, 144.3, 136.0, 134.7, 134.0, 132.7, 132.6, 130.0, 129.6, 128.9, 127.7, 127.5, 126.9, 126.7, 121.6, 119.0, 118.6, 111.4, 109.9, 46.2, 43.3, 25.3; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 503.0594, found 503.0594.
Methyl 4-(N-{2-[(tert-butoxycarbonyl)amino]ethyl}-N-{2-[1-(2,4-dichlorobenzyl)indol-3-yl]ethyl}sulfamoyl)benzoate (60)
A solution of 58 (390 mg, 0.75 mmol) in dry dimethylacetamide (8 mL) was successively treated with Cs2CO3 (368 mg, 1.13 mmol) and 2-(Boc-amino)ethyl bromide (203 mg, 0.91 mmol). The obtained suspension was stirred at 60 °C for 16 h. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate and washed three times with water. The organic phase was dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel (cylcohexane to cyclohexane/ethyl acetate, 7:3) to yield 60 as a solid (151 mg, 30%). C32H35Cl2N3O6S (660.6); mp 69 °C; 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.10 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.61 (d, J = 2.1 Hz, 1H), 7.43 (d, J = 7.7 Hz, 1H), 7.20–7.11 (m, 3H), 7.06 (dd, J = 8.3 and 2.1 Hz, 1H), 6.91 (s, 1H), 6.50 (d, J = 8.4 Hz, 1H), 5.29 (s, 2H), 4.81 (s, 1H), 3.95 (s, 3H), 3.50 – 3.44 (m, 2H), 3.33–3.27 (m, 4H), 3.09–3.03 (m, 2H), 1.43 (s, 9H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 660.1696, found 660.1763.
Methyl 4-(N-(2-aminoethyl)-N-{2-[1-(2,4-dichlorobenzyl)indol-3-yl]ethyl}sulfamoyl)benzoate (61)
To a solution of 60 (150 mg, 0.23 mmol) in dry dichloromethane (5 mL) was added trifluoroacetic acid (0.5 mL). The solution was stirred for 4 h at room temperature. A saturated aqueous NaHCO3 solution was added in portions until the mixture was slightly alkaline. After exhaustive extraction with ethyl acetate, the combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give 61 as an oil (127 mg, quantitative). C27H27Cl2N3O4S (560.5); 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 8.08 (d, J = 8.7 Hz, 2H), 7.91 (d, J = 8.7 Hz, 2H,), 7.67 (d, J = 2.2 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.34–7.28 (m, 1H), 7.22 (s, 1H), 7.11 (ddd, J = 8.2, 7.0 and 1.3 Hz, 1H), 7.04 (ddd, J = 8.0, 7.0 and 1.1 Hz, 1H), 6.63 (d, J = 8.4 Hz, 1H), 5.41 (s, 2H), 3.89 (s, 3H), 3.45 – 3.38 (m, 2H), 3.19 (t, J = 7.1 Hz, 2H), 2.96–2.91 (m, 2H), 2.72–2.63 (m, 2H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 560.1172, found 560.1186.
Methyl 4-(N-{2-[1-(2,4-dichlorobenzyl)indol-3-yl]ethyl}-N-[2-(phenylsulfonamido)ethyl]sulfamoyl)benzoate (62)
To a solution of 61 (120 mg, 0.21 mmol) in dry dichloromethane (5 mL) was added triethylamine (0.5 mL) followed by benzenesulfonyl chloride (55 mg, 0.31 mmol). After stirring at room temperature for 16 h, the mixture was diluted with ethyl acetate and washed with aqueous HCl solution (1 M) and water. The organic phase was dried over anhydrous Na2SO4 and the solvent was removed in vacuo. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 6:4) to yield 62 as an oil (75 mg, 50%). C33H31Cl2N3O6S2 (700.6); 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.10 (d, J = 8.5 Hz, 2H), 7.83–7.76 (m, 4H), 7.59–7.52 (m, 2H), 7.50–7.45 (m, 2H), 7.42 (d, J = 2.1 Hz, 1H), 7.21–7.11 (m, 3H), 7.04 (ddd, J = 8.3, 7.1 and 2.1 Hz, 1H), 6.90 (s, 1H), 6.50 (d, J = 8.3 Hz, 1H), 5.28 (s, 2H,), 4.92 (t, J = 6.0 Hz, 1H), 3.96 (s, 3H), 3.46 – 3.39 (m, 2H), 3.26 (t, J = 6.2 Hz, 2H), 3.12 (q, J = 6.1 Hz, 2H), 3.04–2.92 (m, 2H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 700.1104, found 700.1068.
4-(N-{2-[1-(2,4-Dichlorobenzyl)indol-3-yl]ethyl}-N-[2-(phenylsulfonamido)ethyl]sulfamoyl)benzoic acid (63)
Compound 62 (75 mg, 0.11 mmol) was saponified according to the procedure described for the synthesis of 59 to give 63 as a solid (71 mg, 97%). C32H29Cl2N3O6S2 (686.6); mp 204 °C; 1H-NMR (600 MHz, DMSO-d6) δ (ppm) 13.47 (s, 1H), 8.06 (d, J = 8.5 Hz, 2H,), 7.84–7.80 (m, 3H), 7.75 (dd, J = 8.4 and 1.3 Hz, 2H), 7.67 (d, J = 2.2 Hz, 1H), 7.65–7.62 (m, 1H), 7.59–7.55 (m, 2H), 7.52 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.3 Hz, 1H), 7.27 (dd, J = 8.4 and 2.2 Hz, 1H), 7.20 (s, 1H), 7.11 (ddd, J = 8.2, 7.0 and 1.2 Hz, 1H), 7.05 (ddd, J = 7.9, 7.0 and 1.0 Hz, 1H), 6.60 (d, J = 8.4 Hz, 1H), 5.41 (s, 2H), 3.41–3.37 (m, 2H), 3.24 (t, J = 7.3 Hz, 2H), 2.92–2.85 (m, 4H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.1, 142.6, 140.1, 136.0, 134.7, 134.5, 132.8, 132.6, 132.5, 130.2, 129.6, 129.3, 128.9, 127.6, 127.5, 127.0, 126.9, 126.4, 121.7, 119.1, 118.6, 111.0, 109.9, 49.1, 47.4, 46.2, 41.8, 24.4; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 686.0948, found 686.0951.
tert-Butyl [2-(5-chloro-2-methylindol-3-yl)ethyl]carbamate (76)
A solution of 2-(5-chloro-2-methylindol-3-yl)ethan-1-amine [22, 23] (400 mg, 1.92 mmol) in dry dioxane (20 mL) was treated with triethylamine (1.34 mL, 9.61 mmol). After stirring at room temperature for 15 min, di-tert-butyl dicarbonate (503 g, 2.30 mmol) was added and the mixture was stirred at room temperature for a further 4 h. The solvent was then removed in vacuo and the residue chromatographed on silica gel (cyclohexane/ethyl acetate, 7:3) to afford 76 [24] as a waxy substance (195 mg, 33%). C16H21ClN2O2 (308.8); 1H-NMR (400 MHz, CDCl3) δ (ppm) 7.86 (s, 1H), 7.44 (d, J = 2.0 Hz, 1H), 7.19–7.15 (m, 1H), 7.06 (dd, J = 8.5 and 2.0 Hz, 1H), 4.57–4.52 (m, 1H), 3.38 – 3.26 (m, 2H), 2.84 (t, J = 6.7 Hz, 2H), 2.37 (s, 3H), 1.44 (s, 9H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 309.1364, found 309.1333.
tert-Butyl [2-(1-benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]carbamate (77)
To a solution of 76 (150 mg, 0.49 mmol) in dry DMF (5 mL) was added sodium hydride (60% dispersion in mineral oil) (39 mg, 0.98 mmol) in portions under ice cooling in a nitrogen atmosphere. The mixture was stirred for 30 min at room temperature and then benzhydryl bromide (192 mg, 0.78 mmol) was added. After stirring for another 10 min, the reaction mixture was carefully quenched with water, stirred for further 15 min, diluted with ethyl acetate, and washed three times with water. The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 9:1) to yield 77 as an oil (135 mg, 59%). C29H31ClN2O2 (475.0); 1H-NMR (400 MHz, CDCl3) δ (ppm) 7.45 (d, J = 2.0 Hz, 1H), 7.41–7.27 (m, 6H), 7.13–7.07 (m, 4H), 6.87 (s, 1H), 6.80 (dd, J = 8.8 and 2.1 Hz, 1H), 6.55 (d, J = 8.8 Hz, 1H), 4.57–4.53 (m, 1H), 3.36–3.28 (m, 2H), 2.88 (t, J = 6.9 Hz, 2H), 2.25 (s, 3H), 1.43 (s, 9H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 475.2147, found 475.2147.
Methyl 4-{N-[2-(1-benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]sulfamoyl}benzoate (78)
A solution of 77 (150 mg, 0.32 mmol) in a solution of HCl in dioxane (4 M, 10 mL) was stirred for 4 h at room temperature. The mixture was then concentrated under reduced pressure, diluted with ethyl acetate, washed three times with aqueous NaOH solution (1 M) and dried over anhydrous Na2SO4. The solvent was removed in vacuo to yield 2-(1-benzhydryl-5-chloro-2-methylindol-3-yl)ethan-1-amine as an oil (120 mg). This compound was dissolved in a mixture of dry dichloromethane/dry THF (6 mL, 3:2) and triethylamine (1 mL). After stirring in an ice bath for 15 min, methyl 4-(chlorosulfonyl)benzoate (86 mg, 0.37 mmol) was added portionwise and the mixture was stirred at room temperature for 16 h. The reaction mixture was washed with an aqueous HCl solution (1 M), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 7:3) to yield 78 as a waxy substance (128 mg, 71%). C32H29ClN2O4S (573.1); 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.07 (d, J = 8.7 Hz, 2H), 7.80 (d, J = 8.8 Hz, 2H), 7.38 – 7.30 (m, 6H), 7.18 (dd, J = 2.1 and 0.5 Hz, 1H), 7.11–7.05 (m, 4H), 6.85 (s, 1H), 6.78 (dd, J = 8.8 and 2.1 Hz, 1H), 6.53 (dd, J = 8.8 and 0.5 Hz, 1H), 4.35 (t, J = 6.1 Hz, 1H), 3.95 (s, 3H), 3.29–3.18 (m, 2H), 2.89 (t, J = 6.8 Hz, 2H), 2.25 (s, 3H); HR-MS (ESI) m/z [M–H]–: calc. 571.1464, found 571.1467.
4-{N-[2-(1-Benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]sulfamoyl}benzoic acid (79)
Compound 77 (100 mg, 0.17 mmol) was saponified according to the procedure described for the synthesis of 59. The product was additionally purified by chromatography on silica gel (cyclohexane to cyclohexane/ethyl acetate/formic acid, 7:3:0.1) to give 79 as a solid (65 mg, 67%). C31H27ClN2O4S (559.1); mp 244 °C; 1H-NMR (600 MHz, DMSO-d6) δ (ppm) 13.41 (s, 1H), 8.09 (d, J = 8.5 Hz, 2H), 7.92–7.88 (m, 3H), 7.38 – 7.30 (m, 7H), 7.13 (s, 1H), 7.06 (dd, J = 7.8 and 1.4 Hz, 4H), 6.77 (dd, J = 8.9 and 2.1 Hz, 1H), 6.63 (d, J = 8.9 Hz, 1H), 2.94–2.90 (m, 2H), 2.77 (t, J = 7.5 Hz, 2H), 2.19 (s, 3H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.2, 144.3 139.1, 136.7, 134.3, 134.2, 130.1, 129.3, 128.6, 128.0, 127.7, 126.7, 123.5, 120.0, 116.7, 112.7, 108.2, 61.5, 43.2, 24.7, 11.2; HR-MS (ESI) m/z [M–H]– calc. 557.1307, found 557.1314.
Methyl 4-{N-[2-(5-chloro-2-methylindol-3-yl)ethyl]-N-methylsulfamoyl}benzoate (80)
To a solution of 76 (241 mg, 0.78 mmol) in dry THF (5 mL) was added dropwise, under a nitrogen atmosphere and cooling on ice, a LiAlH4 solution (1 M in dry THF) (3.12 mL). The mixture was stirred under reflux for 2 h. After cooling to room temperature, the reaction mixture was carefully quenched with water and then treated with aqueous NaOH solution (1 M). The resulting precipitate was filtered off. The filtrate was dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford 2-(5-chloro-2-methylindol-3-yl)-N-methylethan-1-amine as an oil (138 mg, 79%). An aliquot of this compound (130 mg, 0.58 mmol) was dissolved in a mixture of dry dichloromethane/dry THF (8 mL, 4:1) and triethylamine (1 mL). After addition of methyl 4-(chlorosulfonyl)benzoate (151 mg, 0.64 mmol), the mixture was stirred at room temperature for 16 h. The reaction mixture was diluted with ethyl acetate, washed with an aqueous HCl solution (1 M), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 7:3) to yield 80 as an oil (70 mg, 28%). C20H21ClN2O4S (420.9); 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.11 (d, J = 8.7 Hz, 2H), 7.88–7.61 (m, 3H), 7.33 (d, J = 1.9 Hz, 1H), 7.16 (dd, J = 8.5 and 0.5 Hz, 1H), 7.04 (dd, J = 8.5 and 2.0 Hz, 1H), 3.95 (s, 3H), 3.29–3.19 (m, 2H), 2.96–2.90 (m, 2H), 2.82 (s, 3H), 2.39 (s, 3H); HR-MS (APCI, direct probe) m/z [M + H]+: ber. 559.1453, gef. 559.1393.
4-{N-[2-(1-Benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]-N-methylsulfamoyl}benzoic acid (81)
Compound 80 (65 mg, 0.15 mmol) was reacted with benzhydryl bromide (61 mg, 0.25 mmol) in presence of sodium hydride (60% dispersion in mineral oil) (12 mg, 0.30 mmol) following the protocol described for the synthesis of 54 to yield 81 as a solid (35 mg, 40%). C32H29ClN2O4S (573.1); mp 227 °C; 1H-NMR (600 MHz, DMSO-d6) δ (ppm) 13.45 (s, 1H), 8.10 (d, J = 8.5 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 2.2 Hz, 1H), 7.38–7.31 (m, 6H), 7.17 (s, 1H), 7.09–7.06 (m, 4H), 6.81 (dd, J = 8.8 and 2.2 Hz, 1H), 6.66 (d, J = 8.9 Hz, 1H), 3.17–3.13 (m, 2H), 2.91 (m, 2H), 2.75 (s, 3H), 2.25 (s, 3H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.2, 141.2, 139.1, 136.8, 134.5, 134.4, 130.3, 129.3, 128.6, 128.0, 127.7, 127.2, 123.6, 120.1, 116.9, 112.7, 108.1, 61.5, 50.2, 35.1, 23.2, 11.3; HR-MS (ESI) m/z [M–H]–: ber. 571.1464, gef. 571.1440.
Methyl 4-{N-(2-acetoxyethyl)-N-[2-(1-benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]sulfamoyl}benzoate (82)
A mixture of 78 (105 mg, 0.18 mmol), K2CO3 (50 mg, 0.36 mmol) and dry DMF (5 mL) was treated with 2-bromoethyl acetate (37 mg, 0.22 mmol) and stirred at room temperature for 4 h. After dilution with ethyl acetate, the reaction mixture was washed three times with water, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 7:3) to yield 82 as a waxy substance (56 mg, 46%). C36H35ClN2O6S (659.2); 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 8.12 (d, J = 8.6 Hz, 2H), 7.96 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 2.1 Hz, 1H), 7.41 – 7.30 (m, 6H), 7.15 (s, 1H), 7.09 – 7.05 (m, 4H), 6.80 (dd, J = 8.8 and 2.1 Hz, 1H), 6.63 (d, J = 8.8 Hz, 1H), 4.05 (t, J = 5.6 Hz, 2H), 3.88 (s, 3H), 3.49 (t, J = 5.7 Hz, 2H), 3.32–3.27 (m, 2H), 2.95–2.89 (m, 2H), 2.26 (s, 3H), 1.91 (s, 3H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 659.1977, found 659.1995.
4-{N-[2-(1-Benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]-N-(2-hydroxyethyl)sulfamoyl}benzoic acid (83)
To a solution of 82 (50 mg, 0.076 mmol) in a mixture of methanol and THF (3:1, 8 ml) was added aqueous KOH solution (10%) (4 mL). After stirring at room temperature overnight, the mixture was treated with aqueous HCl solution (3 M) until the product precipitated, and then extracted exhaustively with ethyl acetate. The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate/formic acid, 6:4:0.1) to yield 83 as a solid (27 mg, 59%). C33H31ClN2O5S (603.1); mp 235 °C (decomp.); 1H-NMR (600 MHz, DMSO-d6) δ (ppm) 13.46 (s, 1H), 8.10 (d, J = 8.5 Hz, 2H), 7.93 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 2.1 Hz, 1H), 7.39–7.30 (m, 6H), 7.15 (s, 1H), 7.07 (d, J = 7.3 Hz, 4H), 6.80 (dd, J = 8.9 and 2.1 Hz, 1H), 6.63 (d, J = 8.8 Hz, 1H), 4.86 (s, 1H), 3.51–3.45 (m, 2H), 3.30 – 3.25 (m, 4H), 2.97–2.92 (m, 2H), 2.25 (s, 3H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.2, 143.2, 139.1, 136.7, 134.5, 134.3, 130.3, 129.2, 128.6, 128.0, 127.7, 127.0, 123.6, 120.1, 116.8, 112.8, 108.0, 61.5, 60.0, 50.5, 49.3, 24.4, 11.1; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 603.1715, found 603.1722.
Methyl 4-{N-(5-acetoxypentyl)-N-[2-(1-benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]sulfamoyl}benzoate (84)
Compound 84 was prepared from 78 (135 mg, 0.24 mmol) and 5-bromopentyl acetate (59 mg, 0.28 mmol) according to the procedure described for the preparation of 82. The reaction time was 16 h. The product was obtained as an oil (103 mg, 62%). C39H41ClN2O6S (701.3); 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.15 (d, J = 8.8 Hz, 2H), 7.89 (d, J = 8.7 Hz, 2H), 7.36 (dd, J = 2.1 and 0.5 Hz, 1H), 7.33–7.29 (m, 6H), 7.12–7.03 (m, 4H), 6.85 (s, 1H), 6.79 (dd, J = 8.8 and 2.1 Hz, 1H), 6.53 (dd, J = 8.8 and 0.5 Hz, 1H), 4.01 (t, J = 6.5 Hz, 2H), 3.95 (s, 3H) 3.30 – 3.23 (m, 2H), 3.24–3.16 (m, 2H), 3.00 – 2.92 (m, 2H), 2.29 (s, 3H), 2.03 (s, 3H), 1.63–1.51 (m, 4H), 1.38–1.29 (m, 2H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 701.2447, found 701.2429.
4-{N-[2-(1-Benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]-N-(5-hydroxypentyl)sulfamoyl}benzoic acid (85)
A solution of 84 (95 mg, 0.14 mmol) in a mixture of methanol and THF (2:1, 6 ml) was treated with aqueous KOH solution (10%) (2 mL) and stirred at room temperature overnight. Then aqueous HCl solution (3 M) was added until the product precipitated. After storage in a refrigerator at 5 oC overnight, the precipitate was filtered off by suction, washed with water and dried in vacuo to give 85 as a solid (79 mg, 90%). C36H37ClN2O5S (645.2); mp 123 °C; 1H-NMR (600 MHz, DMSO-d6) δ (ppm) 8.11 (d, J = 8.5 Hz, 2H), 7.92 (d, J = 8.5 Hz, 2H), 7.43 (d, J = 2.2 Hz, 1H), 7.38–7.29 (m, 6H), 7.14 (s, 1H), 7.07 (dd, J = 7.8 and 1.5 Hz, 4H), 6.80 (dd, J = 8.8 and 2.1 Hz, 1H), 6.64 (d, J = 8.9 Hz, 1H), 4.36 (s, 1H), 3.32 (t, J = 6.5 Hz, 2H), 3.26–3.20 (m, 2H), 3.15 – 3.10 (m, 2H), 2.94–2.88 (m, 2H), 2.25 (s, 3H), 1.44–1.36 (m, 2H,), 1.32 (dt, J = 8.4 and 6.6 Hz, 2H), 1.24–1.15 (m, 2H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.2, 143.2, 139.2, 136.8, 134.5, 134.3, 130.4, 129.3, 128.7, 128.0, 127.8, 127.1, 123.7, 120.2, 116.8, 112.8, 108.1, 61.6, 60.5, 48.5, 48.2, 32.0, 28.0, 24.6, 22.6, 11.2; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 645.2184, found 645.2175.
Methyl 4-[N-[2-(1-benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]-N-(2-{2-[(tert-butyldiphenylsilyl)oxy]ethoxy}ethyl)sulfamoyl]benzoate (86)
A stirred mixture of 78 (120 mg, 0.21 mmol), K2CO3 (58 mg, 0.42 mmol) and dry DMF (5 mL) was treated with tert-butyl[2-(2-chloroethoxy)ethoxy]diphenylsilane [25] (38 mg, 0.10 mmol) and heated at 100 °C for 2 h. After cooling to room temperature, the addition of tert-butyl[2-(2-chloroethoxy)ethoxy]diphenylsilane was repeated three times (38 mg, 0.10 mmol each) over a period of 6 h. Then the reaction mixture was diluted with ethyl acetate and washed three times with water. The organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel (cyclohexane to cyclohexane/ethyl acetate, 8:2) to yield 86 as an oil (85 mg, 45%). C40H35ClN2O8S2 (899.6); 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.12 (d, J = 8.7 Hz, 2H), 7.90 (d, J = 8.7 Hz, 2H), 7.66–7.61 (m, 4H), 7.44 – 7.27 (m, 13H), 7.11–7.04 (m, 4H), 6.83 (s, 1H), 6.79 (dd, J = 8.8 and 2.1 Hz, 1H), 6.52 (dd, J = 8.8 and 0.5 Hz, 1H), 3.94 (s, 3H), 3.76 (t, J = 5.8 Hz, 2H), 3.60 (t, J = 5.5 Hz, 2H), 3.49 (t, J = 5.7, 2H), 3.46–3.41 (m, 2H), 3.39–3.33 (m, 2H), 3.06–2.99 (m, 2H), 2.25 (s, 3H), 1.44 (s, 9H); HR-MS (APCI, direct probe) m/z [M + H]+: calc. 899.3311, found 899.3131.
Methyl 4-{N-[2-(1-benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]-N-[2-(2-hydroxyethoxy)ethyl]sulfamoyl}benzoate (87)
To a solution of 86 (80 mg, 0.089 mmol) in dry THF (5 mL) was added dropwise a solution of tetrabutylammonium fluoride in dry THF (1 M) (180 µL) under ice cooling. After stirring the mixture at room temperature overnight, the solvent was distilled off in vacuo and the residue purified by chromatography on silica gel (cyclohexane/ethyl acetate, 2:8) to give 87 as an oil (29 mg, 49%). C36H37ClN2O6S (661.2); 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.14 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 8.7 Hz, 2H), 7.36 (dd, J = 2.1 and 0.5 Hz, 1H), 7.33–7.29 (m, 6H), 7.12–7.03 (m, 4H), 6.84 (s, 1H), 6.78 (dd, J = 8.8 and 2.1 Hz, 1H), 6.52 (dd, J = 8.8 and 0.5 Hz, 1H), 3.95 (s, 3H), 3.70 – 3.66 (m, 2H), 3.60 (t, J = 5.5 Hz, 2H), 3.51–3.44 (m, 4H), 3.39–3.33 (m, 2H), 3.04–2.95 (m, 2H,), 2.28; HR-MS (APCI, direct probe) m/z [M + H]+: calc. 661.2134, found 661.2200.
4-{N-[2-(1-Benzhydryl-5-chloro-2-methylindol-3-yl)ethyl]-N-[2-(2-hydroxyethoxy)ethyl]sulfamoyl}benzoic acid (88)
A solution of 87 (25 mg, 0.038 mmol) in a mixture of methanol and THF (2:1, 3 ml) was treated with aqueous KOH solution (10%) (1.5 mL). After stirring at room temperature overnight, the mixture was treated with aqueous HCl solution (3 M) until the product precipitated, and then extracted exhaustively with ethyl acetate. The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane to cyclohexane/ethyl acetate, 1:1) to yield 88 as a wax-like substance (21 mg, 86%). C40H38ClN3O6S2 (647.2); 1H-NMR (600 MHz, DMSO-d6) δ (ppm) 13.47 (s, 1H), 8.10 (d, J = 8.5 Hz, 2H), 7.94 (d, J = 8.5 Hz, 2H), 7.46 (d, J = 2.1 Hz, 1H), 7.38–7.29 (m, 6H), 7.15 (s, 1H), 7.08 (dd, J = 7.0 and 0.8 Hz, 4H), 6.80 (dd, J = 8.9 and 2.2 Hz, 1H), 6.64 (d, J = 8.8 Hz, 1H), 4.56 (s, 1H), 3.48 (t, J = 5.7 Hz, 2H), 3.43 (t, J = 5.3 Hz, 2H), 3.39 (t, J = 5.8 Hz, 2H,), 3.34 (t, J = 5.2 Hz, 2H), 3.31–3.27 (m, 2H), 2.98–2.91 (m, 2H), 2.26 (s, 3H); 13C-NMR (151 MHz, DMSO-d6) δ (ppm) 166.2, 143.2, 139.1, 136.7, 134.6, 134.3, 130.2, 129.2, 128.6, 128.0, 127.7, 127.1, 123.6, 120.1, 116.8, 112.8, 108.0, 72.2, 69.2, 61.6, 60.1, 49.1, 47.9, 24.3, 11.1; HR-MS (ESI) m/z [M–H]–: calc. 645.1832, found 645.1825.
Biochemical and physicochemical analysis
Inhibition of cytosolic phospholipase A2α (cPLA2α)
Inhibition of cPLA2α was determined according to a published procedure [20]. Briefly, cPLA2α isolated from porcine platelets was incubated with co-vesicles consisting of the substrate 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (200 µM) and 1,2-dioleoyl-sn-glycerol (100 µM). The enzyme reactions were terminated after 60 min, and cPLA2α activity was measured by quantifying the arachidonic acid released by the enzyme in the presence and the absence of a test compound using reversed-phase HPLC and single quad MS-detection. In parallel, blank incubations were performed in the absence of the enzyme. Inhibition of cPLA2α was calculated from the amount of arachidonic acid liberated by the enzyme in the presence and the absence of a test compound (corrected for blank). IC50 values were determined by probit transformation [26].
Determination of log P values
The partition coefficients (log P) were determined by reversed-phase HPLC according to a published OECD method as recently described [27].
References
Shimizu T, Wolfe LS. Arachidonic acid cascade and signal transduction. J Neurochem. 1990;55:1–15. https://doi.org/10.1111/j.1471-4159.1990.tb08813.x.
Tsuge K, Inazumi T, Shimamoto A, Sugimoto Y. Molecular mechanisms underlying prostaglandin E2-exacerbated inflammation and immune diseases. Int Immunol. 2019;3:597–606. https://doi.org/10.1093/intimm/dxz021.
Di Gennaro A, Haeggström JZ. The leukotrienes: immune-modulating lipid mediators of disease. Adv Immunol. 2012;116:51–92. https://doi.org/10.1016/B978-0-12-394300-2.00002-8.
Soubhye J, van Antwerpen P, Dufrasne F. Targeting cytosolic phospholipase A2α for novel anti-inflammatory agents. Curr Med Chem. 2018;25:2418–47. https://doi.org/10.2174/0929867325666180117103919. PMID: 29345571.
Lehr M. Inhibitors of cytosolic phospholipase A2α as anti-inflammatory drugs. In: Levin JI, Laufer S, editors. Anti-Inflammatory Drug Discovery (RSC Drug Discovery Series No. 26). Cambridge: The Royal Society of Chemistry; 2012. p. 35-57.
Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, et al. Pyrrolidine inhibitors of human cytosolic phospholipase A2. J Med Chem. 2000;43:1041–4. https://doi.org/10.1021/jm9905155.
Shimizu H, Ito A, Sakurada K, Nakamura J, Tanaka K, Komatsu M, et al. AK106-001616, a potent and selective inhibitor of cytosolic phospholipase A : In vivo efficacy for inflammation, neuropathic pain, and pulmonary fibrosis. J Pharm Exp Ther. 2019;369:511–22. https://doi.org/10.1124/jpet.118.255034.
Yamanishi K, Ellis B, Kudo K, Kayanoki T, Dews I, Ostor A, et al. The novel cPLA2 inhibitor AK106-001616 is a new category of anti-inflammatory/analgesic drug demonstrating efficacy and favorable tolerability in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2013;72(Suppl. 3):240 https://doi.org/10.1136/annrheumdis-2013-eular.751.
McKew JC, Foley MA, Thakker P, Behnke ML, Lovering FE, Sum FW, et al. Inhibition of cytosolic phospholipase A2α: hit to lead optimization. J Med Chem. 2006;49:135–58. https://doi.org/10.1021/jm0507882.
McKew JC, Lee KL, Shen MW, Thakker P, Foley MA, Behnke ML, et al. Indole cytosolic phospholipase A2 α inhibitors: discovery and in vitro and in vivo characterization of 4-{3-[5-chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(diphenylmethyl)-1H-indol-3-yl]propyl}benzoic acid, efipladib. J Med Chem. 2008;51:3388–413. https://doi.org/10.1021/jm701467e.
Chen L, Wang W, Lee KL, Shen MW, Murphy EA, Zhang W, et al. Reactions of functionalized sulfonamides: application to lowering the lipophilicity of cytosolic phospholipase A2α inhibitors. J Med Chem. 2009;52:1156–71. https://doi.org/10.1021/jm8009876.
Connolly S, Bennion C, Botterell S, Croshaw PJ, Hallam C, Hardy K, et al. Design and synthesis of a novel and potent series of inhibitors of cytosolic phospholipase A2 based on a 1,3-disubstituted propan-2-one skeleton. J Med Chem. 2002;45:1348–62. https://doi.org/10.1021/jm011050x.
Ludwig J, Bovens S, Brauch C, Elfringhoff AS, Lehr M. Design and synthesis of 1-indol-1-yl-propan-2-ones as inhibitors of human cytosolic phospholipase A2α. J Med Chem. 2006;49:2611–20. https://doi.org/10.1021/jm051243a.
Drews A, Bovens S, Roebrock K, Sunderkötter C, Reinhardt D, Schäfers M, et al. 1-(5-Carboxyindol-1-yl)propan-2-one inhibitors of human cytosolic phospholipase A2α with reduced lipophilicity: synthesis, biological activity, metabolic stability, solubility, bioavailability, and topical in vivo activity. J Med Chem. 2010;53:5165–78. https://doi.org/10.1021/jm1001088.
Garznisky D. Cytosolic phospholipase A2α and fatty acid amide hydrolase as potential drug targets: synthesis and testing of inhibitors and identification of new lead structures by computer-assisted drug design. Dissertation Universtity of Münster, 2018. https://d-nb.info/1156953685.
Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, et al. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol. 2009;5:220–6. https://doi.org/10.1038/nchembio.150.
Gianella-Borradori M, Christou I, Bataille CJ, Cross RL, Wynne GM, Greaves DR, et al. Ligand-based virtual screening identifies a family of selective cannabinoid receptor 2 agonists. Bioorg Med Chem. 2015;23:241–63. https://doi.org/10.1016/j.bmc.2014.11.002.
Hawkins PC, Skillman AG, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J Med Chem 2007;50:74–82. https://doi.org/10.1021/jm0603365.
Branum ST, Colburn, RW, Dax SL, Flores CM, Jetter MC, Liu Y, et al. N-Benzothienyl sulfonamides as TRPM8 modulators and their preparation, pharmaceutical compositions and use in the treatment of diseases. WO2009012430, 2009.
Barth M, Kampschulze J, Rudolph S, Meyer zu Vilsendorf I, Hanekamp W, Mulac D, et al. Hexafluoroisopropyl carbamates as selective MAGL and dual MAGL/FAAH inhibitors: Biochemical and physicochemical properties. Chem Med Chem 2022;e202100757. https://doi.org/10.1002/cmdc.202100757.
Roy S, Haque S, Gribble GW. Synthesis of novel oxazolyl-indoles. Synthesis. 2006;23:3948–54. https://doi.org/10.1055/s-2006-950318.
Di Cesare MA, Minetti P, Tarzia G, Spadoni G. 5-Halo-tryptamine derivatives used as ligands of the 5-HT6 and/or 5-HT7 serotonin receptors. WO2003000252, 2003.
Cheng YZ, Zhao QR, Zhang X, You SL. Asymmetric dearomatization of indole derivatives with N-hydroxycarbamates enabled by photoredox catalysis. Angew Chem Int Ed Engl. 2019;58:18069–74. https://doi.org/10.1002/anie.201911144.
Rene O, Fauber BP. Syntheses of 4-, 5-, 6-, and 7-substituted tryptamine derivatives and the use of a bromine atom as a protecting group. Tetrahedron Lett. 2014;55:830–3. https://doi.org/10.1016/j.tetlet.2013.12.025.
Christensen CA, Batsanov AS, Bryce MR, Howard JA. Molecular saddles. 7. New 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene cyclophanes: synthesis, redox properties, and x-ray crystal structures of neutral species and a dication salt. J Org Chem. 2001;66:3313–20. https://doi.org/10.1021/jo001524k.
Finney DJ, Probit Analysis: A Statistical Treatment of the Sigmoid Response Curve, Cambridge University Press, 1952.
Rudolph S, Dahlhaus H, Hanekamp W, Albers C, Barth M, Michels G, et al. Aryl N-[ω-(6-Fluoroindol-1-yl) alkyl]carbamates as inhibitors of fatty acid amide hydrolase, monoacylglycerol lipase, and butyrylcholinesterase: structure-activity relationships and hydrolytic stability. ACS Omega. 2021;6:13466–83. https://doi.org/10.1021/acsomega.1c01699.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borecki, D., Lehr, M. N-Substituted 4-sulfamoylbenzoic acid derivatives as inhibitors of cytosolic phospholipase A2α. Med Chem Res 31, 975–992 (2022). https://doi.org/10.1007/s00044-022-02895-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-022-02895-x