
Abstract
Throughout the last centuries, the structure and genetic composition of forests have been strongly affected by forest management. Over 30% of European forests are artificially regenerated, very often using translocated forest reproductive material, among these species the Swiss stone pine (Pinus cembra L.). In the late nineteenth and early twentieth centuries, the species was largely used for artificial afforestation in the northern Alps. However, only a few planted trees have survived. Therefore, we aimed to evaluate if the historical afforestation of P. cembra in the northwestern Swiss Alps relied on allochthonous material. We sampled 12 sites, genotyping 11 nuclear microsatellites, to infer the spatial genetic structure of regional populations, to test for genetic differences between natural and planted stands, and to infer potential source regions of planted stands using reference samples covering the entire Alps. Population genetic structure analysis allowed us to distinguish planted from natural stands and to determine that forest reproductive material used for plantations was not of regional origin. We found similar levels of genetic diversity between natural and planted stands. Assignment tests revealed that reproductive material for planting was translocated to the study area from two source regions, i.e., near the border of Switzerland and Austria, and further to the East, between Austria and Italy. Our study shows how genetic tools may inform about historical transfer of forest reproductive material, which still may affect the population genetic make-up of regional occurrences, e.g., because of reduced natural regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
European forests faced extensive deforestation, peaking in the end of the eighteenth and early nineteenth century (Geburek and Myking 2018). To reverse this dramatic development and its respective consequences, such as landslides, flash floods, avalanches, soil erosion, and wood shortage, artificial regeneration by means of planting and sowing of forest reproductive material (FRM) increased considerably toward the end of the nineteenth century (Jansen et al. 2019; Myking et al. 2016). As a consequence, the structure and genetic composition of many forest stands have been strongly affected by forest management (Jansen et al. 2017). Today, more than 30% of European forests are artificially regenerated, very often using translocated FRM (Jansen et al. 2019). There are many reasons why seeds or young plants were transferred from other, sometimes very distant regions, for example lack of local seed sources, intentional search for plant material of desired traits, interest of foresters in exotic species, or simply lower prices and availability (Jansen and Geburek 2016; Jansen et al. 2019). Recently, many countries and international organizations have undertaken reforestation and afforestation projects to mitigate the detrimental effects of global changes, including the loss of habitat and inherent biodiversity (Di Sacco et al. 2021; Naudts et al. 2016; Zheng et al. 2016). Therefore, the debate on seed transfer has gained new attention, mainly to counteract genetic maladaptation and to increase forest stability and efficiency for wood production (Frank et al. 2017).
Conifers, especially Norway spruce (Picea abies) and European larch (Larix decidua), were historically particularly affected by artificial regeneration with non-local FRM, mainly due to their importance in wood production. In the case of P. abies, seed transfer between the Alpine, Carpathian, or Scandinavian regions became a common practice during the last two centuries (Jansen et al. 2017). Similar large-scale translocations of FRM have been carried out for L. decidua, with human-mediated seed transfer between the Alpine and Carpathian regions (Jansen and Geburek 2016; Raffl et al. 2018). However, it remains poorly known how such historic translocations of FRM have altered the native population genetic structure of these species across Europe. Tracing the FRM is complex, mainly since the specific origin of provenances at a given site cannot be identified by phenotypic traits and is usually poorly documented, hence requiring molecular methods (Jansen et al. 2017, 2019). Moreover, the effects of FRM translocations often remain undisclosed in phylogeographic studies because researchers aim at sampling in autochthonous stands to infer the spatial genetic structure imposed by natural dynamics (Jansen et al. 2017; Mátyás et al. 2002).
In Switzerland, the fear of wood penury was widespread in the eighteenth and nineteenth centuries. Many regional authorities began to enact forest laws, allowing the transition to a regulated forest economy (Schuler 2007). The primary aim was to guarantee the long-term forest exploitation and to ensure the conservation and extension of protection forests. In this context, major reforestation and afforestation projects emerged in Switzerland between the nineteenth and early twentieth centuries, mainly in the Northern Alps, including the canton of Fribourg, (Müller 1989; Pfister-Ritter and Pfister-Ritter 1990). In the canton of Fribourg, several conifer species were particularly popular in the afforestation projects, notably Swiss stone pine (Pinus cembra). The objective was not only to restore the forests of the canton of Fribourg for their economic value, but also to avoid problems that arose with the deforestation, such as reducing soil erosion and limiting the negative effects and frequency of avalanches, landslides, and floods (Fankhauser 1898; Rikli 1909). These afforestation projects were ambitious, but not always successful as shown for P. cembra in the canton of Fribourg (Fragnière et al. 2022).
In this study, we focused on P. cembra, a tree species with a geographical range limited to the Alps with patchy occurrences in the Carpathian Mountains (Höhn et al. 2009; Zoller 1991). Pinus cembra is considered an emblematic species of the European tree flora and has attracted the attention of the large public, foresters, and researchers (Caudullo and de Rigo 2016). As a result, there is a large amount of research on various aspects of its biology, ecology, and distribution (Casalegno et al. 2010; Lopez-Saez et al. 2022; Neuschulz et al. 2018; Wieser et al. 2014; Zięba et al. 2020). In a recent study, Fragnière et al. (2022) reported that nearly 450,000 saplings of P. cembra were planted in the canton of Fribourg between 1895 and 1952, of which only 650 individuals could be found on the sites indicated by the cantonal archives during an intensive search in 2020. Some of the planted and natural stands were located close-by, and tracking the status of individual trees was sometimes challenging. Moreover, the origin of the FRM used in these large-scale artificial afforestation efforts remained largely unknown. Generally, there are very few and rather anecdotal studies reporting the historical translocations of P. cembra as they are mainly based on old written sources that are often difficult to access (Zwijacz-Kozica and Żywiec 2007). Recently, genetic analyses suggested the origin of non-native trees and stands of P. cembra (Gugerli et al. 2023; Lendvay et al. 2014), demonstrating the possibility to identify allochthonous origins of forest trees.
Multiple studies have been conducted on the population genetics of P. cembra within its natural range (Dauphin et al. 2020; Dzialuk et al. 2014; Gugerli et al. 2023, 2009, 2001; Höhn et al. 2009; Rellstab et al. 2019; Salzer and Gugerli 2012; Toth et al. 2019). In a range-wide study using nuclear and chloroplast microsatellites, Gugerli et al. (2023) found a distinct East–West structure of five genetic clusters across the Alps and the Carpathian Mountains. This spatial genetic structure may serve as a reference for tracking the origin of allochthonous forest stands. Recently, Dauphin et al. (2021) highlighted the genomic vulnerability of P. cembra in the face of climate warming, showing that juvenile individuals might not be adapted to a warmer and drier climate in the current main elevational range of the species. Hence, knowledge on the genetic composition of P. cembra stands may be informative in view of their sylvicultural management under global warming.
The aim of our study was to assess the origin and legacy of historical artificial afforestation with Swiss stone pine in the northwestern Swiss Alps, using molecular methods. More specifically, we addressed the following questions: (1) Does the spatial genetic composition of P. cembra in the canton of Fribourg allow to distinguish presumably natural and planted stands? (2) Do estimates of genetic diversity differ between natural and planted stands? (3) Are planted populations in the canton of Fribourg genetically similar to the neighboring natural populations occurring in the Western Alps? (4) If not, which source provenances were used for the historical afforestation in the canton of Fribourg? On the basis of our results, we formulate recommendations for future afforestation projects and sylvicultural practices aiming at the sustainable management of the peripheral stands of P. cembra in the Alps, but also for other regions and species.
Materials and methods
Study species
Swiss stone pine (Pinus cembra L.) has a restricted geographical range in the Alps, with disjunct occurrences in the Carpathian Mountains (Höhn et al. 2009; Zoller 1991). The species can cope with the harsh climates of the upper subalpine vegetation zone, with juveniles found at elevations of up to 3000 m a.s.l. in protected sites in the Swiss Alps, even clearly above 3000 m a.s.l. in the southwestern Alps (André et al. 2023). Thus, the species usually forms the upper limit of the tree line within its range (Tranquillini 1979; Ulber et al. 2004). Pinus cembra has wingless seeds, depending primarily on the spotted nutcracker (Nucifraga caryocatactes) for seed dispersal (Mattes 1982; Neuschulz et al. 2015, 2018; Sorensen et al. 2022). In Switzerland, the core distribution of P. cembra is located in the central Alps, where the climate is rather continental, giving it a competitive advantage over P. abies that is otherwise dominating the subalpine vegetation zone of the Alps (Gugerli et al. 2022; Lingua et al. 2008; Ott et al. 1997). In the canton of Fribourg, located in western Switzerland and comprising parts of the northern Alps, P. cembra occurs at the margin of its distribution where climate is more temperate, oceanic than in the core range of the species (Rikli 1909).
Sampling design
Fresh needles of adult P. cembra trees were collected from 12 sites in the canton of Fribourg, in the northwestern Swiss Alps. Six natural and six presumably planted stands were selected to represent the known species’ distribution in the study area (Fragnière et al. 2022). In each stand, 20 adult trees were sampled, and a minimal distance of 30 m between collected trees was maintained to limit relatedness between individuals, except in a few cases where population size was too small (i.e., MOL and SPI). Geographic coordinates of each sampled tree were obtained using an Etrex 32 × GPS (Garmin, Kansas City, USA). Needles of each tree were cut in pieces of 0.5 cm, lyophilized, and conserved in silica gel. In each sampled stand, population census sizes were estimated as described in Fragnière et al. (2022).
DNA extraction and genotyping
Dry needles were grinded using Precellys Evolution tissue homogenizer (Bertin instruments, Montigny-le-Bretonneux, France). Genomic DNA was extracted from approximately 50 mg of grinded tissue using DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The purity and concentration of extracted DNA was estimated with a spectrophotometer (Nanodrop, ThermoFisher, Waltham, USA). The concentration of all samples was normalized to approximately 10 ng/μL.
Samples were genotyped using two multiplex polymerase chain reactions (PCRs) as detailed in Lendvay et al. (2014). The A7 multiplex comprised of six nuclear microsatellite markers (Pc1b, Pc7, Pc18, Pc22, Pc23, Pc35; Salzer et al. 2009). The PCR mix contained 2 μL of extracted DNA, 4.5 μL of Multiplex PCR kit (Qiagen), and 3.5 μL of primer mix. Amplification was performed with the following program on a Veriti 96-well thermocycler (Applied Biosystems, Rotkreuz, Switzerland): (1) An initial denaturation of 15 min at 95 °C, (2) 32 cycles with 30 s at 95 °C, 30 s at 54 °C and 50 s at 72 °C, and (3) a 30 min final extension at 72 °C. The MP1 contained five nuclear microsatellite markers (28Z, BUG, CQG, HJM, YAU; Lendvay et al. 2014). The PCR mix was made with 2 μL of extracted DNA, 4.5 μL of Type-it PCR kit (Qiagen), 1 μL of primer mix, and 2.5 μL of distilled water. The thermo-cycle program for the amplification was the following: (1) An initial denaturation of 5 min at 95 °C, (2) 32 cycles with 30 s at 95 °C, 90 s at 58 °C and 30 s at 72 °C, and (3) a 30 min final extension at 72 °C.
For both multiplex PCRs, amplification products were prepared for the fragment length analysis by diluting the amplified samples 1:3, i.e., adding 20 μL of Merck LiChrosolv water, and 1 μL of this product was mixed with 10 μL of a 1:100 mix of GeneScan Rox400 dye size standard (Applied Biosystems) and Hi-Di formamide. Fragments were analyzed on an ABI 3130 capillary sequencer (Applied Biosystems). Alleles were manually scored with GeneMapper v4.1 (Applied Biosystems). All but one sample in HOC were successfully genotyped (n = 239), and the complete genetic dataset is available as Supplementary Material (ESM2).
Population genetic structure
Genetic structure among populations was investigated using pairwise genetic differentiation, ordination, and Bayesian clustering methods. First, we used the genet.dist function of hierfstat R package v0.04-22 (Goudet 2005) to calculate pairwise genetic differentiation measures (FST) between stands based on Weir and Cockerham’s (1984) equation. We then carried out a hierarchical clustering analysis from pairwise FST estimates to infer phylogenetic relationships and divergence events among natural and planted stands. We used the Euclidean method to compute the distance matrix from pairwise FST values and the Ward D as agglomeration method with the dist and hclust functions of the STATS R package v3.6.1, respectively.
Second, to explore genotypic variation without assumptions of a population model (e.g., Hardy–Weinberg equilibrium, HWE), we carried out a Discriminant Analysis on Principal Components (DAPC) as implemented in the adegenet R package v2.1.2 (Jombart 2008). This method maximizes the variance between genetic clusters while minimizing the variance within them. We performed the unsupervised method that does not rely on a priori defined genetic clusters. We used the K-means clustering analysis to infer the optimal number of genetic clusters (K) based on the lowest Bayesian Information Criterion (BIC) scores across K values, retaining the optimal number of principal components (PCs) given by the alpha score optimization (Jombart et al. 2010).
Third, we evaluated population grouping and compared levels of admixture within and among populations in natural and planted stands using Bayesian clustering analysis as implemented in STRUCTURE v2.3.4 (Pritchard et al. 2000). We ran the analysis with the admixture model and used the LocPrior option (i.e., information on sampling locations; Hubisz et al. 2009) to assist the assignment of individuals to clusters given the restricted spatial scale of sampled genotypes. We ran ten repetitions for each K value ranging from 1 to 8, and each run consisted of a burn-in period of 500,000, followed by 1,000,000 iterations. We inspected the results with STRUCTURE HARVESTER (Earl and vonHoldt 2012) and chose the optimal K value based on the Ln probability distribution of the data. Next, we used the web-based program CLUMPAK (Clustering Markov Packager Across K; Kopelman et al. 2015) to summarize the independent runs and produce graphical representations.
Genetic diversity
The overall number of alleles per locus and per population as well as deviation from HWE, based on expected (Hs) and observed heterozygosity (Ho), were obtained and in the case of HWE tested using the adegenet R package. Measures of allelic richness (Ar), using the rarefaction method per locus and per population, and unbiased inbreeding coefficients (FIS) and other F-statistics were assessed with the hierfstat R package.
Assignment probability of planted trees
To identify the putative origin of planted trees, we used a larger dataset including a representative subset of populations from the entire Alpine distribution presented in Gugerli et al. (2023). The dataset consisted of 845 individuals from 40 Alpine populations, genotyped at the same 11 nuclear microsatellite loci. This Alpine dataset combined with the Fribourg dataset sampled in this study was analyzed with STRUCTURE to assign the possible origin(s) of planted stands from the known genetic pools characterized throughout the species’ Alpine range. We carried out Bayesian clustering analysis with the same parameters and procedure as mentioned above. In addition, GeneClass2 (Piry et al. 2004) was run with the two datasets to assign individuals or groups of individuals from the Fribourg dataset to reference populations of the Alpine dataset. Assignment of the sampled populations to the reference populations was run using the Bayesian method from Rannala and Mountain (1997) with a threshold of 0.05.
Results
Population genetic structure
Pairwise FST values ranged from < 0.001 (BOU and TIS) to 0.123 (SAT and TEY; Table S1). The hierarchical clustering dendrogram based on pairwise FST estimates separated five planted stands (TEY, MOL, SPI, BOU, and TIS; Fig. 1a) from the rest, also indicating a partition into two subgroups (MOL, TEY vs. SPI, BOU, TIS). Likewise, all natural stands formed a single branch, which also included BRE, originally classified as a planted stand, with minor separation of BRE, GAS, and HOC from the other natural stands.

Genetic grouping of planted and natural stands of Pinus cembra from the canton of Fribourg. a Hierarchical clustering analysis of distance matrix based on pairwise FST values. b Individual assignment probabilities from Bayesian clustering at K = 2 (top) and K = 3 (bottom) using STRUCTURE (Pritchard et al. 2000). Stand codes are given in Table 1
The DAPC displayed substantial overlap between stands (discriminant factors DF1 and DF2; Fig. S1). However, five of the six planted stands (TEY, MOL, SPI, BOU, and TIS) formed a distinct cluster along DF1. In these five stands, two subgroups were further separated along DF2, with the stands BOU and TIS on one side and TEY and MOL on the other side, SPI being in-between the two subgroups. As in the hierarchical clustering, BRE grouped with the natural stands.
Bayesian clustering analysis suggested three main genetic clusters (K = 3) to best explain the regional dataset (Fig. S2). At K = 2, we found that natural and planted stands formed two genetic clusters, except for BRE that included individuals with mixed assignment to the two clusters (Fig. 1b). At K = 3, we retrieved a third genetic cluster separating the two planted stands TEY and MOL, and some admixed individuals from SPI (Fig. 1a). This same grouping of populations was also retrieved in the hierarchical clustering based on pairwise FST values (HCPC; Fig. 1a) and in the unsupervised DAPC (Fig. S1).
Genetic diversity assessment
In view of the population genetic structure analysis, we reclassified the BRE stand as being not planted and, therefore, assessed genetic diversity measures with BRE in the group of natural stands. The number of alleles observed ranged from 2 to 22 alleles for Pc35 and Pc23, respectively. Mean estimates of Ho and Hs across loci and populations showed no major deviations from HWE, with values ranging from 0.402 to 0.537 and 0.364 to 0.516, respectively (Table 1). However, we found a significant difference of both Ho and Hs between the two groups of stands (natural vs. planted; p < 0.01; Fig. 2a, b). Correlation between Ho and Hs across loci and among stands showed no major outliers (Fig. S3). Planted stands showed a significantly higher mean allelic richness than natural stands (p < 0.01; Fig. 2c). Inbreeding coefficients (FIS) were relatively low in each population for both the natural and planted stands, ranging from 0.004 to 0.121 (not significantly different from 0; Table 1), and there was no significant difference between population groups (p = 0.461; Fig. 2d). As planted stands had substantially lower population census sizes (p = 0.017; Fig. 2e) with high allelic richness, we found a significant trend of decreasing allelic richness with increasing census sizes for the whole dataset (p < 0.05; Fig. 2f).

Boxplots of genetic diversity estimates and population census size for Pinus cembra from the canton of Fribourg. Observed (Ho) and expected heterozygosity (Hs), mean allelic richness (Ar), and inbreeding coefficient (FIS), grouped according to natural vs. planted stands (a–d). Significant differences were calculated with a t test (a–d) or a Wilcoxon test (e) after inspecting the normal distribution using Shapiro tests. f Correlation between Ar and population census size was significant based on the complete dataset
Assignment probability of planted trees
Based on the Alpine dataset, the analysis of the population genetic structure showed no additional genetic cluster when adding the natural and planted stands sampled from the canton of Fribourg, suggesting that seed provenances originated from the Alps (Fig. 3a). We considered K = 4 as the option that best explains our data (Fig. S4), even though the range-wide structure suggests K = 5 (Gugerli et al. 2023); however, our Alpine dataset ignored populations from the Carpathian Mountains, where the fifth cluster dominates that otherwise only occurs at low probabilities in the easternmost Alpine populations. Moreover, K = 4 depicted a reduced standard deviation among runs compared to higher K values. The natural stands of Fribourg were primarily assigned to the genetic cluster formed by the populations from the western Swiss Alps (CH04, CH05, CH07, and CH08, the latter being a former sample of LAP). For planted stands, individuals of the western group predominantly clustered with populations from the eastern Swiss Alps (CH37 and CH38) and western Austria (AU84 and AU86). Individuals from the eastern group of planted stands were mostly assigned to the cluster comprising populations of the South-Tyrol (IT70 and IT74), Germany and Austria.

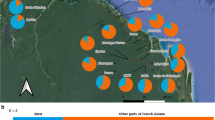
Assignment of natural and planted stands of Pinus cembra in view of the population genetic structure in the Alps. a Bar plot of individual assignment probabilities to K = 4 genetic clusters derived from STRUCTURE analyses. b Population-level assignment probabilities as pie charts across the Alps for K = 4. The inset shows the assignment probabilities for the Fribourg stands only (pie charts: STURUCTRE) and for the assigned populations in the two first ranks of the GeneClass2 analysis (cf., Table S2), with bars in the colors of respective genetic cluster assignments in STRUCTURE (Alpine dataset). GeneClass assignment probabilities (if > 10%) are labeled with potential source population for the planted stands only. Population and stand codes refer to Table 1 (Canton of Fribourg) and (Gugerli et al. 2023) for Alpine reference populations, respectively; bold font refers to populations identified as potential sources in the GeneClass2 analysis
The assignment of natural and planted stands to the known populations from the Alpine dataset using GeneClass2 showed consistent results with those of the Bayesian clustering analysis (Fig. 3b): (i) the natural stands were assigned to the regional populations from the western Swiss Alps, i.e., either CH07, CH05, or CH08 at the first rank with high probabilities (assignment score > 0.63; Table S2). (ii) Of the western group of planted stands, TEY was assigned at first rank to CH14, while MOL was assigned at first rank to CH37. These two populations were both sampled in the canton of Grisons, eastern Switzerland. (iii) Individuals from the eastern group of planted stands were all assigned to populations from western Austria and Italy at first rank: SPI was assigned to AU99 from Austrian South-Tyrol (0.96), BOU to IT70 from Italian South-Tyrol (0.92), and TIS to AU96 from eastern Austria (0.83). iv) BRE was assigned at first rank to CH08 from the Lapé forest (0.94), one of the natural stands (LAP) that had been previously sampled from the study region.
Discussion
Large-scale artificial afforestation to counteract the detrimental forest exploitation in many parts of Europe until the mid-eighteenth century turned out to be unsustainable in some areas for various reasons, including inappropriate species selection, planting practices and, in some cases, the use of non-autochthonous forest reproductive material (FRM). Such FRM transfer is often hard to document, because respective information is commonly lacking in forest archives. However, genetic analyses nowadays allow us to relate the genetic composition of a tree species in a given forest stand to its range-wide spatial genetic structure and for the assignment of FRM to the putative source region. Here, we applied this approach to infer the origin of multiple natural and planted stands of Swiss stone pine in the canton of Fribourg (western Switzerland), where considerable afforestation efforts were made until the middle of the twentieth century (Fragnière et al. 2022). However, only a few planted stands have survived, and this afforestation strategy has to be considered largely unsuccessful. Thus, we were particularly interested to test if the very few remaining planted stands had survived because of the use of regional, potentially locally adapted FRM. Our results clearly demonstrate the non-autochthonous origin of the survived trees in the planted stands, as opposed to the known natural stands. Surprisingly, genetic diversity was higher in the planted than in the natural stands, even though estimates of the population census sizes in the former were lower (Fig. 2e, f), and we detected no signs of inbreeding. Therefore, we assume that FRM used for these plantings was composed of seed collections from genetically diverse stands, from mixed seed sources, and with many seed trees considered that prevented a human-induced bottleneck. While we cannot claim or rule out that adaptive processes contributed to the survival of the remaining planted stands, foreign FRM per se cannot explain why most of the documented planted Swiss stone pine trees did not survive. However, it is likely that ecological factors and benign conditions at the planted stands are part of the reasons why Swiss stone pine still exists at these few remnant sites (Fragnière et al. 2022).
Genetic distinction between natural and planted populations
Neutral genetic differentiation between stands at a regional level may indicate alternative genealogy or demographic history. For Swiss stone pine, a distinct grouping into five genetic clusters is apparent across the natural range (Gugerli et al. 2023), and natural stands in western Switzerland, including the canton of Fribourg, all belong to the same genetic cluster. Hence, genetic differences between stands could inform about divergent demographic histories or possible foreign origin of FRM. Accordingly, genetic relationships among Swiss stone pine stands from the canton of Fribourg included in the present study provided a first support for the non-autochthonous origin(s) of the planted stands. We found a distinct separation of populations into two main genetic groups, using complementary analytical methods (pairwise genetic differentiation, hierarchical clustering, ordination, and Bayesian clustering analyses). All natural stands formed one cluster (Figs. 1 and 3), suggesting close genetic relatedness among them. This finding conforms to the hypothesis that these stands were native (see below). Likewise, five out of the six stands that were considered planted based on information from forest archives (Fragnière et al. 2022) formed a separate cluster. Only the BRE stand did not match with our expectations to be a planted stand, as it clustered together with the natural stands. Accordingly, we consider this stand as largely natural or at least planted using regional FRM, and we, therefore, included it among the natural stands for comparative genetic diversity analyses.
Population genetic structure analysis of the local dataset gave further insights into the hierarchical structuring of the stands from Fribourg. Results of the Bayesian structuring at K = 2 (Fig. 1a) coincided with the HCPC dendrogram (Fig. 1b) in that natural stands were assigned to a different cluster than planted stands (i.e., excluding BRE formerly considered a planted stand). Again, this finding suggests that the FRM used for the plantations did not originate from natural populations from Fribourg but was obtained in genetically differentiated populations. At K = 3, the natural stands remained as one cluster, but planted stands were assigned to two distinct clusters. These results imply that there were at least two FRM sources used for plantations, leading to the observed separation of western (TEY and MOL) and eastern planted stands (SPI, BOU, and TIS). In STRUCTURE results at K = 3, the population from SPI displayed some differences with other planted stands from the eastern part of the study region (BOU and TIS), with proportionally higher assignment probabilities to the cluster with the western planted stands (Table S2). This pattern means that a third source of FRM was used in the afforestation project(s) from which the SPI stand has resulted, or that the FRM used here came from different source individuals or populations. The latter hypothesis is unlikely because all individuals showed admixture, which would only be the case if the genotyped trees were already in the second generation. However, Swiss stone pine requires 40–60 years to reach reproductive age (Ulber et al. 2004), and Fragnière et al. (2022) did not find any regeneration in the planted stands. These circumstances suggest that planted stands still represent the first generation of trees that directly relate to their origin.
Results from both the HCPC dendrogram and STRUCTURE reveal that the samples collected in the BRE stand were a priori misinterpreted as resulting from plantations. This stand clustered together with natural stands in the HCPC dendrogram, and STRUCTURE assigned most individuals to the cluster comprising natural stands from Fribourg. Nonetheless, there were a few individuals that displayed similar assignment probabilities as individuals from eastern planted stands. There is no doubt that an afforestation project was conducted close to this sampling site (Etat de Fribourg 2021). Thus, there are multiple hypotheses that could explain the unexpected results obtained for the BRE stand. First, this could be due to the planting of FRM from a natural, local population. This hypothesis is supported by the fact that the location of this afforestation project is close to natural populations. Second, planted individuals in BRE could be a mix of local and foreign origin, but results from other planted stands do not suggest any mixed origin of FRM. The hypothesis that, in our view, could best explain the results observed in BRE is that most individuals sampled for our analysis were not planted, but rather natural trees. This hypothesis suggests that the same FRM was used for the afforestation project of BRE as for the other eastern planted stands and that either P. cembra trees were planted within an already existing natural population, or seeds from nearby natural populations colonized the site of afforestation thereafter. The latter is unlikely given the slow growth of Swiss stone pine and the respective size of the trees at BRE.
Origin of planted trees from the known Alpine genetic pools
By comparing multilocus genotypes resulting from sampling of native and planted stands in the canton of Fribourg to those found in other Alpine populations, we (i) could confirm the autochthony of the stands considered as natural and (ii) gain insights into the possible origins of the planted stands from Fribourg. Interestingly, natural stands of Fribourg (MOR, PAB, LAP, HOC, SAT, GAS, and likely BRE) shared a similar genetic assignment to one genetic cluster. By analyzing them together with other Alpine populations (K = 4), they were assigned to other populations from the western Swiss Alps (CH04, CH05, CH07, and CH08; Fig. 3a). Furthermore, the results obtained with GeneClass2 (Table S2) were consistent, with all native populations being assigned in the first rank to either CH08, CH07, or CH05 with a high score (> 63%). This finding suggests that the natural colonization of the canton of Fribourg by P. cembra happened from populations from the Western Alps (Gugerli et al. 2023).
The western planted stands (TEY and MOL) consistently clustered together (Figs. 1 and 3), which suggests that their origin, where the FRM was collected for plantations, was in the same region. These two stands are geographically close to each other (Fig. 3), and probably, the same FRM was used for both afforestation initiatives. The STRUCTURE results with the Alpine dataset show that they are genetically similar to populations from the eastern Swiss Alps or western Austria (CH37, CH38, AU84, and AU869; Fig. 3), and GeneClass2 assigned TEY and MOL to populations CH14 and CH37, respectively, which are both located in the Grisons, eastern Switzerland. Taken together, these results point toward an origin of the western planted stands in the eastern-central Alps, close to the border between Switzerland and Austria.
For the eastern planted stands (SPI, BOU and TIS), the STRUCTURE assignments with the Alpine dataset at K = 4 (Fig. 3a) were similar to those of P. cembra populations from Italy (IT70 and IT74). GeneClass2 assigned these population either to IT70, AU96, and AU99 at the two first ranks (Table S2, Fig. 3b). Populations from AU99 and IT70 are both situated in southern Tyrol in Austria and Italy, respectively, and the AU96 population is situated further east, in Styria, an eastern state of Austria. Given that the three populations to which eastern planted stands were assigned to span a large area, it is not possible to give an exact geographical origin. Nonetheless, we can conclude that the seeds collected for raising saplings in the (often local) nursery (Fragnière et al. 2022) were probably coming from an eastern part of the Alps, either in Austria or Italy. Notably, Gugerli et al. (2023) identified a non-autochthonous stand in the Bernese Oberland, nearby the study region in Fribourg, which also related to an origin in the same region of the Alps; information from the forestry archive confirmed that some of the planted material came from South-Tyrol. It is thus likely that Swiss forest services imported FRM from this region for afforestation.
Our main hypothesis explaining the disparate genetic structure between individuals from the BRE stand is that we mainly sampled individuals that were native to the sites and only a few that were planted. GeneClass2 assigned this stand to CH08 in the reference populations, which represents the Lapé forest that had also been genotyped by Gugerli et al. (2023) previously in the Alpine dataset. This confirms that most of the individuals that were collected in BRE were probably native. At the third rank, however, the population from BRE was assigned to CH14, a population from the eastern Swiss Alps to which populations from the western planted group were also assigned. However, STRUCTURE results of both the local (Fig. 1b) and the Alpine dataset (Fig. 3a) indicate that the few presumably non-native individuals in BRE share similarities to populations from the eastern planted group. Thus, it is critical to conclude on the potential origin of the few individuals that were potentially planted, because GeneClass2 and STRUCTURE yielded divergent results. Nevertheless, due to the proximity of this stand to planted populations from the eastern planted group, and because STRUCTURE results show differences at the individual level, it is more likely that FRM involved in the otherwise natural stand BRE had a similar origin as that used in other eastern planted stands.
Conclusions and future perspectives
With our regional study, we showcase how the knowledge on the spatial genetic structure of a species helps to identify potentially allochthonous forest stands even without written documentation about their sylvicultural history. Such information is of great value for forest management that aims at promoting autochthonous stands, e.g., for harvesting seeds. Likewise, applied conservation practice may benefit from such inference on historical translocation beyond tree species. However, this genetic approach requires an a priori knowledge of the main genetic clusters throughout the species’ range, which is more and more becoming available for many taxonomic groups. Our study also highlights the need of using highly variable, ideally bi-parentally inherited molecular markers to capture genetic clusters at high spatial resolution, but also sufficient levels of genetic differentiation necessary for efficient assignment tests. The use of next-generation sequencing data might be powerful to identify local FRM used for afforestation efforts.
While our study does not inform us about the reasons underlying the tremendous afforestation failure in the canton of Fribourg, we assume that it was mainly ecological factors, but also planting procedures, which hampered the success of this vast effort. We postulate that, for minimizing weight, bare-rooted saplings were commonly used for these afforestations at high altitude and in remote places. Such plants have a root system prone to desiccation before and after planting, and the situation was likely accentuated by only shallow soils and lack of shelter by standing canopy. Besides planting practice, further abiotic (climate, topography, and snow) and biotic factors (competition, pathogens, browsing, and mycorrhization) that possibly contributed to the reasons why afforestation largely failed can be found in Fragnière et al. (2022). Moreover, we speculate that the population genetic estimates (i.e., allelic richness and deviation from HWE) retrieved for the still existing planted stands also suggest a genetic component to why most of the other afforested stands did not persist. At least our data demonstrate that the persisting planted stands showed similar levels of genetic diversity and no indication of inbreeding, as did the natural stands, suggesting that FRM used at these sites did not compromise the afforestation success. In turn, one might presume that planted stands that did not survive to the present day may have comprised inbred individuals that made these stands vulnerable to extinction beyond the detrimental effects of ecological factors. Moreover, planted trees may have been maladapted to the local habitat and, therefore, were unable to establish. While it is not possible to test this aspect, because these trees no longer exist, one might evaluate if the still growing planted stands show signs of genomic offset or an increased risk of non-adaptedness (Capblancq et al. 2020; Dauphin et al. 2021; Rellstab et al. 2015) as compared with the natural stands in the region. However, the neutral markers employed in this study preclude any inference on adaptive responses of planted (and naturally occurring) stands.
However, the ultimate reasons for the afforestation failure remain open. We, therefore, advocate that, irrespective of their source of origin, seed collected as FRM should be collected from numerous mother trees to represent a broad genetic composition that will serve as the foundation of future forests.
Data availability
The dataset of this article is available in the Supplementary Information (file2) and also on the WSL environmental data portal EnviDat under https://doi.org/10.16904/envidat.468.
References
André G, Lavergne S, Carcaillet C (2023) Unsuspected prevalence of Pinus cembra in the high-elevation sky islands of the western Alps. Plant Ecol 224:865–873. https://doi.org/10.1007/s11258-023-01341-1
Capblancq T, Fitzpatrick MC, Bay RA, Exposito-Alonso M, Keller SR (2020) Genomic prediction of (mal)adaptation across current and future climatic landscapes. Annu Rev Ecol Evol Syst 51:245–269. https://doi.org/10.1146/annurev-ecolsys-020720-042553
Casalegno S, Amatulli G, Camia A, Nelson A, Pekkarinen A (2010) Vulnerability of Pinus cembra L. in the Alps and the Carpathian mountains under present and future climates. Forest Ecol Manag 259:750–761. https://doi.org/10.1016/j.foreco.2009.10.001
Caudullo G, de Rigo D (2016) Pinus cembra in Europe: distribution, habitat, usage and threats. In: San-Miguel-Ayanz J, de Rigo D, Caudullo G, Houston Dudrrant T, Mauri A (eds) European atlas of forest tree species. Publication Office of the European Union, Luxembourg
Dauphin B, Wüest RO, Brodbeck S, Zoller S, Fischer MC, Holderegger R, Gugerli F, Rellstab C (2020) Disentangling the effects of geographic peripherality and habitat suitability on neutral and adaptive genetic variation in Swiss stone pine. Mol Ecol 29:1972–1989. https://doi.org/10.1111/mec.15467
Dauphin B, Rellstab C, Schmid M, Zoller S, Karger DN, Brodbeck S, Guillaume F, Gugerli F (2021) Genomic vulnerability to rapid climate warming in a tree species with a long generation time. Global Change Biol 27:1181–1195. https://doi.org/10.1111/gcb.15469
Di Sacco A, Hardwick KA, Blakesley D, Brancalion PHS, Breman E, Rebola LC, Chomba S, Koxon K, Elliott S, Ruyonga G, Shaw K, Smith P, Antonelli A (2021) Ten golden rules for reforestation to optimize carbon sequestration, biodiversity recovery and livelihood benefits. Global Change Biol 27:1328–1348. https://doi.org/10.1111/gcb.15498
Dzialuk A, Chybicki I, Gout R, Mączka T, Fleischer P, Konrad H, Curtu AL, Sofletea N, Valadon A (2014) No reduction in genetic diversity of Swiss stone pine (Pinus cembra L.) in Tatra Mountains despite high fragmentation and small population size. Conserv Genet 15:1433–1445. https://doi.org/10.1007/s10592-014-0628-6
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Etat de Fribourg (2021) Archives de l’Etat de Fribourg (AEF)/Saatsarchiv Freiburg (StAF). https://www.fr.ch/cha/aef. Accessed 1 Oct 2020
Fankhauser F (1898) Les inondations e le reboisement de nos montagnes: La Société des forestiers suisses aux autorités fédérales, cantonales et communales
Fragnière Y, Sonnenwyl V, Clément B, Kozlowski G (2022) Large-scale historical afforestation failure with Pinus cembra in the Swiss Prealps. New For 53:533–553. https://doi.org/10.1007/s11056-021-09871-0
Frank A, Howe GT, Sperisen C, Brang P, St Clair JB, Schmatz DR, Heiri C (2017) Risk of genetic maladaptation due to climate change in three major European tree species. Global Change Biol 23:5358–5371. https://doi.org/10.1111/gcb.13802
Geburek T, Myking T (2018) Evolutionary consequences of historic anthropogenic impacts on forest trees in Europe. Forest Ecol Manag 422:23–32. https://doi.org/10.1016/j.foreco.2018.03.055
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Gugerli F, Senn J, Anzidei M, Madaghiele A, Büchler U, Sperisen C, Vendramin GG (2001) Chloroplast microsatellites and mitochondrial nad1 intron 2 sequences indicate congruent phylogeographic relationship of Swiss stone pine (Pinus cembra), Siberian stone pine (P. sibirica), and Siberian dwarf pine (P. pumila). Mol Ecol 10:1489–1497. https://doi.org/10.1046/j.1365-294X.2001.01285.x
Gugerli F, Rüegg M, Vendramin GG (2009) Gradual decline in genetic diversity in Swiss stone pine populations (Pinus cembra) across Switzerland suggests postglacial re-colonization into the Alps from a common eastern glacial refugium. Bot Helv 119:13–22. https://doi.org/10.1007/s00035-009-0052-6
Gugerli F, Brodbeck S, Bebi P, Bollmann K, Dauphin B, Gossner M, Krumm F, Peter M, Queloz V, Reiss G, Rellstab C, Stofer S, von Arx G, Wasem U, Zweifel R (2022) Swiss Stone Pine—Portrait of a Mountain Forest Tree. Eidg. Forschungsanstalt WSL, Birmensdorf, 16 pg
Gugerli F, Brodbeck S, Lendvay B, Dauphin B, Bagnoli F, Tinner W, van der Knaap WO, Höhn M, Vendramin GG, Morales-Molino C, Schwörer C (2023) A range-wide postglacial history of Swiss stone pine based on molecular markers and palaeoecological evidence. J Biogeogr 50:1049–1062. https://doi.org/10.1111/jbi.14586
Höhn M, Gugerli F, Abran P, Bisztray G, Buonamici A, Cseke K, Hufnagel L, Sebastiani F, Quintela-Sabarís C, Vendramin GG (2009) Variation in the chloroplast DNA of Swiss stone pine (Pinus cembra L.) reflects contrasting post-glacial history of populations from the Carpathians and the Alps. J Biogeogr 36:1798–1806. https://doi.org/10.1111/j.1365-2699.2009.02122.x
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Jansen S, Geburek T (2016) Historic translocations of European larch (Larix decidua Mill.) genetic resources across Europe—a review from the 17th until the mid-20th century. For Ecol Manag. https://doi.org/10.1016/j.foreco.2016.08.007
Jansen S, Konrad H, Geburek T (2017) The extent of historic translocation of Norway spruce forest reproductive material in Europe. Ann for Sci 74:56. https://doi.org/10.1007/s13595-017-0644-z
Jansen S, Konrad H, Geburek T (2019) Crossing borders—European forest reproductive material moving in trade. J Environ Manag 233:308–320. https://doi.org/10.1016/j.jenvman.2018.11.079
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genomics 11:94. https://doi.org/10.1186/1471-2156-11-94
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Lendvay B, Höhn M, Brodbeck S, Mindrescu M, Gugerli F (2014) Genetic structure in Pinus cembra from the Carpathian Mountains inferred from nuclear and chloroplast microsatellites confirms postglacial range contraction and identifies introduced individuals. Tree Genet Genomes 10:1419–1433. https://doi.org/10.1007/s11295-014-0770-9
Lingua E, Cherubini P, Motta R, Nola P (2008) Spatial structure along an altitudinal gradient in the Italian central Alps suggests competition and facilitation among coniferous species. J Veg Sci 19:425–436. https://doi.org/10.3170/2008-8-18391
Lopez-Saez J, Corona C, von Arx G, Fonti P, Slamova L, Stoffel M (2022) Tree-ring anatomy of Pinus cembra trees opens new avenues for climate reconstructions in the European Alps. Sci Total Environ 855:158605. https://doi.org/10.1016/j.scitotenv.2022.158605
Mattes H (1982) Die Lebensgemeinschaft von Tannenhäher und Arve. Ber Eidg Forsch.anst Forstl Versuchswes 241:1–74
Mátyás G, Bonfils P, Sperisen C (2002) Autochthon oder allochthon? Ein molekulargenetischer Ansatz am Beispiel der Eiche (Quercus spp.) in der Schweiz. Schweiz Z Forstwesen 153:91–96
Müller U (1989) Schutzwaldaufforstungen des Staates Freiburg im Senseoberland: Forstpolitische Massnahmen des Staates Freiburg seit 1850 am Beispiel der Schutzwaldaufforstungen im Flyschgebiet des Senseoberlandes. Dissertation, ETH
Myking T, Rusanen M, Steffenrem A, Kjaer E, Jansson G (2016) Historic transfer of forest reproductive material in the Nordic region: drivers, scale and implications. Forestry 89:325–337. https://doi.org/10.1093/forestry/cpw020
Naudts K, Chen Y, McGrath MJ, Ryder J, Valade A, Otto J, Luyssaert S (2016) Europe’s forest management did not mitigate climate warming. Science 351:597–600. https://doi.org/10.1126/science.aad727
Neuschulz EL, Mueller T, Bollmann K, Gugerli F, Böhning-Gaese K (2015) Seed perishability determines the caching behaviour of a food-hoarding bird. J Anim Ecol 84:71–78. https://doi.org/10.1111/1365-2656.12283
Neuschulz EL, Merges D, Bollmann K, Gugerli F, Böhning-Gaese K (2018) Biotic interactions and seed deposition rather than abiotic factors determine recruitment at elevational range limits of an alpine tree. J Ecol 106:948–959. https://doi.org/10.1111/1365-2745.12818
Ott E, Frehner M, Frey H-U, Lüscher PU (1997) Gebirgswälder. Haupt, Bern
Pfister-Ritter M, Pfister-Ritter F (1990) Geschichte einer wechselvollen Beziehung… : 100 Jahre Aufforstungen im Sense-Oberland/FR . Forstdepartement des Kantons Freiburg, Direktion des Innern und Landwirtschaft, Fribourg, 106 pg
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A (2004) GeneClass2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raffl H, Konrad H, Curtu LA, Geburek T (2018) Genetic evidence of human mediated, historical seed transfer from the Tyrolean Alps to the Romanian Carpathians in Larix decidua (Mill.) forests. Ann for Sci 75:98. https://doi.org/10.1007/s13595-018-0776-9
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201. https://doi.org/10.1073/pnas.94.17.9197
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R (2015) A practical guide to environmental association analysis in landscape genomics. Mol Ecol 24:4348–4370. https://doi.org/10.1111/mec.13322
Rellstab C, Dauphin B, Zoller S, Brodbeck S, Gugerli F (2019) Using transcriptome sequencing and pooled exome capture to study local adaptation in the giga-genome of Pinus cembra. Mol Ecol Resour 19:536–551. https://doi.org/10.1111/1755-0998.12986
Rikli M (1909) Die Arve in der Schweiz. Neue Denkschr Schweiz Naturforsch Ges 44:1–455
Salzer K, Gugerli F (2012) Reduced fitness at early life stages in peripheral versus core populations of Swiss stone pine (Pinus cembra) is not reflected by levels of inbreeding in seed families. Alp Bot 122:75–85. https://doi.org/10.1007/s00035-012-0106-z
Salzer K, Sebastiani F, Gugerli F, Buonamici A, Vendramin GG (2009) Isolation and characterization of eight polymorphic nuclear microsatellite loci in Pinus cembra L. Mol Ecol Resour 9:858–861. https://doi.org/10.1111/j.1755-0998.2008.02396.x
Schuler A (2007) Lois sur les forêts. https://hls-dhs-dss.ch/fr/articles/013802/2007-08-17/. Accessed 15 May 2023
Sorensen MC, Müller T, Donoso I, Graf V, Merges D, Vanoni M, Fiedler W, Neuschulz EL (2022) Scatter-hoarding birds disperse seeds to sites unfavorable for plant regeneration. Mov Ecol 10:38. https://doi.org/10.1186/s40462-022-00338-1
Toth EG, Tremblay F, Housset JM, Bergeron Y, Carcaillet C (2019) Geographic isolation and climatic variability contribute to genetic differentiation in fragmented populations of the long-lived subalpine conifer Pinus cembra L. in the western Alps. BMC Evol Biol 19:190. https://doi.org/10.1186/s12862-019-1510-4
Tranquillini W (1979) Physiological ecology of the alpine timberline. Tree existence at high altitudes with special reference to the European Alps. Springer, Heidelberg
Ulber M, Gugerli F, Bozic G (2004) EUFORGEN Technical Guidelines for genetic conservation and use for Swiss stone pine (Pinus cembra). International Plant Genetic Resources Institute, Rome, p 6
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wieser G, Gruber A, Oberhuber W (2014) Sap flow characteristics and whole-tree water use of Pinus cembra across the treeline ecotone of the central Tyrolean Alps. Eur J Forest Res 133:287–295
Zheng H, Wang Y, Chen Y, Zhao T (2016) Effects of large-scale afforestation project on the ecosystem water balance in humid areas: an example for southern China. Ecol Eng 89:103–108. https://doi.org/10.1016/j.ecoleng.2016.01.013
Zięba A, Różański W, Szwagrzyk J (2020) Structure of dominance among tree species in relic Swiss stone pine (Pinus cembra L.) forests in Tatra Mountains. Pol J Ecol 68:159–171. https://doi.org/10.3161/15052249PJE2020.68.2.005
Zoller H (1991) Pinus. In: Conert HJ, Hamann U, Schultze-Motel W, Wagenitz G (eds) Gustav Hegi—Illustrierte Flora von Mitteleuropa, 3rd edn. Blackwell, Berlin, pp 77–83
Zwijacz-Kozica T, Żywiec M (2007) Fifty-year changes in a strictly protected stone pine population in the Tatra National Park. Nat Conserv 64:73–82
Acknowledgements
The authors would like to thank Peter Wandeler from the Natural History Museum Fribourg (NHMF) for the financial support of the molecular analyses, the Department of Biology and Botanic Garden of the University of Fribourg for the financial support of the field work, and Dominique Schaller from the Forest and Nature Service of the State of Fribourg (SFN) for the permits. Special thanks go to the following persons for their advice and help during the field work and sample collection: Emanuel Gerber, Gilles Hauser, Henri Descombes and Sophie Giriens (NHMF), Pascal Sonnenwyl (SFN), and Luca Champoud (Botanic Garden of the University of Fribourg).
Funding
Open Access funding provided by Lib4RI – Library for the Research Institutes within the ETH Domain: Eawag, Empa, PSI & WSL.
Author information
Authors and Affiliations
Contributions
VS, YF, and GK conceived the study and performed field sampling, VS, SG, CP, and SB processed the samples in the lab, VS and BD analyzed the data, VS, GK, BD, and FG wrote the manuscript, and all authors provided contributions and agreed to the final text of the article.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no financial and non-financial competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sonnenwyl, V., Dauphin, B., Fragnière, Y. et al. Genetic underpinning of historical afforestation with allochthonous Pinus cembra in the northwestern Swiss Alps. Alp Botany 134, 1–13 (2024). https://doi.org/10.1007/s00035-023-00304-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00035-023-00304-6