
Abstract
Fibrosis is a typical aging-related pathological process involving almost all organs, including the heart, kidney, liver, lung, and skin. Fibrogenesis is a highly orchestrated process defined by sequences of cellular response and molecular signals mechanisms underlying the disease. In pathophysiologic conditions associated with organ fibrosis, a variety of injurious stimuli such as metabolic disorders, epigenetic changes, and aging may induce the progression of fibrosis. Sirtuins protein is a kind of deacetylase which can regulate cell metabolism and participate in a variety of cell physiological functions. In this review, we outline our current understanding of common principles of fibrogenic mechanisms and the functional role of SIRT3/6 in aging-related fibrosis. In addition, sequences of novel protective strategies have been identified directly or indirectly according to these mechanisms. Here, we highlight the role and biological function of SIRT3/6 focus on aging fibrosis, as well as their inhibitors and activators as novel preventative or therapeutic interventions for aging-related tissue fibrosis.
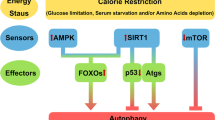
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fibrosis is considered to be the outcome of a chronic healing response marked by excessive deposition and formation of extracellular matrix (ECM), chronic inflammation, and loss of parenchymal cells. Activated pro-fibroblasts play a prominent role in this process [37, 55, 68, 114].
Many factors such as age, genetic factors, gender, and obesity can contribute to the development of fibrosis [30]. Fibrosis eventually affects all tissues and organs, contributing to increased morbidity and mortality from age-related diseases [111]. A complex cellular cascade induced by organ damage may underlie the coagulation disorder between ECM synthesis and ECM degradation [32]. Although the organ damage may be triggered by organ-specific ways, the fibrosis process and the involved cascades are conserved across organs [68]. The organ fibrosis is regulated by different cascades and signaling pathways including Wingless/Int (Wnt) [32] and transforming growth factor-β (TGF-β) [80, 116]. There is increasing evidence that fibrosis is the main cause of the deterioration of various human organ functions during aging.
Cell senescence refers to a permanent arrest of the cell cycle, resulting in a steady loss of the cell function to proliferate, with the continuously decreased metabolic states such as autophagy function, energy metabolism regulation and anti-stress, despite the remaining cell viability and metabolic activity [126]. Aging is accompanied by the gradually declining structural integrity of an organism [19], during which the risk of death increases [5, 22, 26, 27, 61].
Sirtuins as the niacinamide adenine dinucleotides (NAD)+-dependent protein deacetylases are considered the primary regulators of cell function. As a key player in regulating metabolism and preventing oxidative stress, sirtuin 3 (SIRT3) is commonly found in organs and tissues with high metabolic rate capacity, including the cardiac. Metabolic diseases and cardiac disorders related to its activity have been proposed as therapeutic targets. Maintaining genomic stability and maintaining telomere function may be accomplished by SIRT6 deacetylating histone H3 at lysine 9 and lysine 56 [99].
An important step in tissue fibrosis during chronic disease and aging is the TGF-β1-mediated fibroblasts into myofibroblasts, cells capable of synthesizing ECM. Studies have demonstrated that inhibiting pro-fibrotic TGF-β1 signaling by SIRT3 inhibits fibroblast to myofibroblast differentiation, suggesting SIRT3 is important for controlling age-related tissue fibrosis [91]. Moreover, SIRT6 functions as a key anti-aging molecule by controlling multiple cellular processes associated with aging and preventing age-induced cardiac hypertrophy and fibrosis [76]. This paper summarizes the biological structure and function of SIRT3 and SIRT6, and describes the role of SIRT3 and SIRT6 in fibrosis and aging.
SIRT family
A sirtuin is a NAD+-dependent class III histone deacetylase that deacetylates histones with NAD+. SIRTs can exert a regulatory role in a wide range of physiological and pathological processes, including energy production, oxidative stress, mitochondrial homeostasis, cell aging and apoptosis, DNA damage, playing a prominent role in the pathogenesis progression [7, 44, 70, 77, 78].
To date, seven SIRTs subtypes, SIRT1 7, have been identified in mammalian cells [110]. SIRT1, SIRT6, and SIRT7 are located in the nucleus, SIRT2 are located in the cytoplasm, and SIRT3, SIRT4, and SIRT5 are located in the mitochondria. A translocation of SIRT1 to the cytoplasm and a relocalization of SIRT3 between mitochondria and nuclei can occur under certain conditions [85].
Different SIRTs subtypes exert different functions. SIRT1, SIRT2, SIRT3, and SIRT-7 mainly exhibit the properties of NADH-dependent deacetylases. SIRT4 and SIRT6 may function as deacetylases and ADP-ribosomal transferases. SIRT5 has weak deacetylation activity, yet it has strong desuccinylation and depropanediylation activity [106]. As mitochondrial SIRTs, SIRT3/4/5 play an important role in mitochondrial biogenesis and the regulation of oxidative stress. SIRT6, a nuclear deacetylase, plays a role in ADP-ribosyltransferase activity, inflammation and metabolism [25]. Intriguingly, SIRT6 and SIRT3 maintain each other’s levels, SIRT3 inhibits oxidative stress, and SIRT6 activates SIRT3 transcription by upregulating nuclear factor erythroid 2 (NF-E2) -related factor 2 (Nrf2) -dependent transcription [42].
Structure and function of SIRT3
The SIRT3 gene is located on chromosome 11p15.5, a chromosome region associated with longevity [2]. Two functional domains are present on the human SIRT3 protein: a large Rossmann folding and NAD+ binding site, and a small helix complex and zinc binding site [38]. There is a crack between the two domains where the acetylated substrate is inserted. While being transported into the mitochondria, SIRT3 is cleaved by mitochondrial matrix processing peptidase (MPP), which results in a short, more active form 28 kDa.
Among the mitochondrial SIRTs, SIRT3 is the only one with strong NAD+-dependent deacetylase activity [46]. According to initial reports, SIRT3 is primarily responsible for regulating acetyl bodies in mitochondria. In the mitochondrial electron transport chain (ETC), SIRT3 directly binds to succinate dehydrogenase A and ATP synthase (Complex V) of complex I and II and regulates them, thereby vigorously raising ATP levels [60]. In energy-demanding cells, SIRT3 plays an important role in mitochondrial function and cell metabolism, such as fatty acid oxidation, tricarboxylic acid cycle (TCA), and ETC [67, 123]).
SIRT3 regulates core mitochondrial processes, but its function may differ in tissues contributing to fuel production and fuel utilization, depending on the metabolic pathways involved [18]. Thus, SIRT3 may play distinct roles at the tissue level as well as at the cellular level. In previous studies, SIRT3 deficiency caused mitochondrial respiration impairment and elevated reactive oxygen species (ROS) production in myoblasts and cancer cells [6]. SIRT3 has been speculated to be associated with human longevity, and studies have demonstrated the reduced SIRT3 expression in sedentary older adults compared to younger adults [82].
Structure and biological function of SIRT6
The human SIRT6 gene is located at 13.3 of the short arm of chromosome 19 and consists of 8 exons, with exon 8 being the longest at 838 bases. On the other hand, exon 4 is the shortest with only 60 bases. The SIRT6 protein molecule comprises 355 amino acid residues and has a molecular weight of 39.1 kDa [63]. Structurally, SIRT6 consists of an N-terminal extension (NTE), a C-terminal extension (CTE), and a structurally conservative central domain. The NTE is associated with its catalytic activity, which is crucial for chromatin binding and the deacetylation of lysine 9 and 56 (H3K9 and H3K56) of the intrinsic histone H3. The CTE is essential for nuclear localization and recognition of nucleosome DNA. Both extensions play an important role in nucleosome binding [97]. Unlike other Sirtuin proteins, SIRT6 lacks the classical zinc finger binding sequence, Rossmann folding structure, and highly flexible NAD+ binding ring in its central domain. However, SIRT6 possesses a unique unfolded zinc finger binding domain and a highly stable single helix structure. Because of this unique characteristic, SIRT6 binds NAD+ with high affinity, even in the absence of a deacetylation substrate. This feature may explain why SIRT6 effectively promotes the ADP-ribosyltransferase reaction [24, 73].
SIRT6 exhibits both ADP-ribosyltransferase and NAD+-dependent histone deacetylase activities. It can utilize NAD+ as a substrate for intramolecular ADP-ribosylation. The currently identified glycosylation substrates of SIRT6 mainly include the K521 site of PARP1 and the nuclear helper inhibitor KAP1. As for histone deacetylation, three substrates have been identified: lysine 9, 18, and 56 of histone H3 (H3K9, H3K18, and H3K56). SIRT6 can be actively recruited to target gene promoters to inhibit the transcriptional activity of these genes through deacetylation of H3K9, H3K18, or H3K56 sites. This process helps maintain genomic stability, telomere integrity, promotes DNA repair, and prevents aging [9]. In addition, SIRT6 can deacetylate forkhead box O1 (FoxO1), associated factor histone acetyltransferase 5 (general control non-derepressible-5, GCN5), and several non-histone substrates such as C-terminal binding protein interacting proteins (CTIP). These deacetylation events further contribute to the modulation of glucose homeostasis, among other processes [66, 96].
The regulation of SIRT6 abundance is complex and precise in vivo. At the transcriptional level, SIRT6 expression is significantly activated by pharmacological inhibition of poly ADP-ribose polymerase 1 (PARP1) [108]. C-FOS and SIRT1-FOXO3a-NRF1 (SFN) complexes can also upregulate SIRT6 expression by binding to SIRT6 promoters [29]. Endogenous microRNAs (miRNAs), including miR-33a/b, miR-122, miR-330-5p, and miR-495, regulate SIRT6 translation by binding to its 3'-untranslated region (UTR) [87]. SIRT6 protein stability is largely controlled by proteasome-dependent degradation pathways.
Fibrosis
The aging process predisposes people to fibrosis and can affect many tissues (Fig. 1) [8]. SIRT3 and SIRT6 have been found to play important roles in tissue fibrosis (Table 1).

The role of SIRT3 and SIRT6 in several tissue fibrosis diseases. Mechanically, tissue fibrosis diseases include liver fibrosis, kidney fibrosis, lung fibrosis, and cardiac fibrosis
Hepatic fibrosis
Hepatic fibrosis occurs when stressed or damaged liver cells and activated macrophages (Kupffer cells) stimulate the activation of hepatic stellate cells, which then secrete excessive ECM components, including type I and type III collagen, leading to the formation of liver fibrosis [84]. Hepatic fibrosis is a common feature of various chronic liver diseases and can progress to cirrhosis, liver failure, and hepatocellular carcinoma [16, 17].
SIRT3, which is predominantly located in the mitochondria, has been strongly associated with oxidative stress and liver-related diseases [62]. It has been shown that adenosine 5′-monophosphate-activated protein kinase (AMPK), a protein involved in the pathophysiology of liver fibrosis [41], can reduce liver fibrosis [33]. SIRT3 has been identified as a downstream effector of AMPK in several disease models, and activation of the AMPK-SIRT3 signaling pathway helps improve mitochondrial function, thereby alleviating disease progression. The anti-fibrotic effect of celastrol, which is attributed to its anti-inflammatory properties, depends on the activation of AMPK-SIRT3 signaling. Celastrol acts as an anti-fibrotic agent by suppressing inflammation, and its effects are believed to be mediated by the activation of AMPK-SIRT3 signaling. Depletion of AMPK or SIRT3 compromises the anti-inflammatory effects of celastrol [107].
Studies in mice fed a high-fat, high-fructose diet for 16 weeks and in liver samples from patients with non-alcoholic steatohepatitis (NASH) have shown that SIRT6 plays a regulatory role in the progression of NASH to liver fibrosis. Hepatocyte-specific knockout of SIRT6 in mice aggravated liver fibrosis, and reduced SIRT6 expression levels were observed in the livers of NASH patients as the disease progressed to fibrosis [39, 125]. It has been demonstrated that SIRT6 inhibits Smad3 activation, a member of the Smad family involved in TGF-β signaling, through H3K9 deacetylation, thereby suppressing the expression of key genes related to liver fibrosis and contributing to fibrosis regression [64]. Mechanistic studies have revealed that SIRT6 inhibits TGF-β-induced activation of hepatic stellate cells by deacetylating specific sites (K333 and K378) on Smad3, resulting in the downregulation of liver fibrosis-related genes and the inhibition of fibrosis progression [125].
Renal fibrosis
An important regulator of mitochondrial function, SIRT3 participates in the injury and repair processes of acute kidney injury (AKI). It has been suggested that SIRT3 may play a significant role in the early stages of fibrosis following ischemia–reperfusion injury (IR-AKI) by regulating mitochondrial dynamics. Furthermore, deficiency of SIRT3 can potentially worsen renal insufficiency and promote renal fibrosis [13]. Proximal renal tubular epithelial cells (TECs), which are rich in mitochondria and heavily rely on mitochondrial oxidative phosphorylation as their primary energy source, are particularly affected by the dysregulation of SIRT3. In the initial stages of renal fibrosis, decreased expression of SIRT3 is accompanied by increased acetylation of mitochondria isolated from TECs. Studies using SIRT3 knockout mice have shown that these mice are more susceptible to renal fibrosis, particularly characterized by high levels of acetylated mitochondrial proteins [13]. Interestingly, the administration of honokiol has been found to activate SIRT3, leading to improved acetylation and prevention of renal fibrosis. Furthermore, in the context of unilateral ureteral obstruction, a condition associated with renal fibrosis, it has been observed that most renal proteins, accounting for 26.76% of mitochondrial proteins, undergo hyperacetylation. These hyperacetylated proteins are localized within a wide range of mitochondrial pathways. Notably, pyruvate dehydrogenase E1α (PDHE1α), a crucial link between glycolysis and the TCA cycle, undergoes hyperacetylation at lysine 1 in TECs following stimulation with TGF-β. Importantly, this process is regulated by SIRT3 [123].
In the context of AKI, studies have demonstrated that overexpression of SIRT6 can prevent its occurrence [54]. In the context of renal tubular epithelial cells, depletion of SIRT6 leads to aggravated epithelial-mesenchymal transition (EMT), accompanied by upregulation of homeodomain-interacting protein kinase 2 (HIPK2). A protein kinase called HIPK2 is involved in multiple molecular pathways that lead to cell death and development. It is noteworthy that this protein kinase can regulate a variety of pro-fibrotic pathways, such as Wnt/β-catenin, TGF-β and Notch, which are involved in kidney, lung, liver, and heart fibrosis [23]. Interestingly,, SIRT6 has the ability to down-regulate HIPK2 at the post-transcriptional level [52].
Pulmonary fibrosis
Pulmonary fibrosis is a severe and chronic interstitial lung disease for which effective treatments are limited. Apoptosis and mitochondrial dysfunction in alveolar epithelium cells are key factors in idiopathic pulmonary fibrosis and asbestosis. Through deacetylation of manganese superoxide dismutase (MnSOD) and mitochondrial 8-oxygen guanine DNA glycosylase, SIRT3 plays a partial role in mitochondrial reactive oxygen species removal. By inhibiting acetylation of OGG1 at K338/341, SIRT3 counteracts mtDNA damage and apoptosis induced by reductive oxidant expression. Conversely, silencing of SIRT3 promotes these detrimental effects, ultimately leading to pulmonary fibrosis. Deficiency of SIRT3 contributes to increased mitochondrial DNA damage and apoptosis in alveolar epithelial cells, exacerbating the progression of pulmonary fibrosis [35]. Induction of SIRT3 expression by bayicalein reduces lung fibroblast senescence and fibrosis induced by bleomycin [36].
SIRT6, on the other hand, has demonstrated its ability to reduce fibrosis in various organs. In the context of pulmonary fibrosis, SIRT6 has been found to inhibit bleomycin-induced injury in alveolar epithelial cells both in vitro and in mice. High-throughput sequencing studies have revealed that SIRT6-overexpressing lung tissue exhibits enhanced lipid catabolism. SIRT6 mitigates bleomycin-induced ectopic lipid toxicity by promoting lipid degradation, thereby increasing energy supply and reducing lipid peroxide levels. Notably, peroxisome proliferator-activated receptor α (PPARα) has been identified as a critical mediator of SIRT6’s effects on lipid catabolism, anti-inflammatory response, and anti-fibrotic signaling. These findings suggest that targeting the SIRT6-PPARα-mediated lipid catabolic pathway holds promise as a potential therapeutic strategy for pulmonary fibrosis and related disorders [31]. In addition, other studies have demonstrated that SIRT6 inhibits NF-κB signaling pathway and blocks TGF-β1-induced lung myofibroblast differentiation [95, 124].
Cardiac fibrosis
Cardiac fibrosis is a common patho-physiological remodeling process, which greatly affects the structure and function of the heart and further causes heart failure. Abnormal proliferation, differentiation and migration of cardiac fibroblasts are responsible for excessive deposition of ECM in cardiac muscle [57]. Acetylation plays an important role in the development of cardiac fibrosis by regulating various pathogenic conditions, including oxidative stress, mitochondrial dysfunction and energy metabolism disorders [57].
SIRT3 is expressed at high levels in the heart and improves heart health by regulating cardiac energy [67]. Studies have demonstrated that SIRT3 inhibits inflammation and fibrosis in cardiomyocytes by promoter specific deacetylation of histone H3 lysine K27 to inhibit FOS transcription and reduce activator protein-1 (AP-1) DNA binding activity [71]. Su et al. found that cardiac fibrosis was partly achieved by the mechanism of SIRT3 inducing ferroptosis in myofibroblasts through p53 acetylation [89]. Pericytes are the progenitors of myofibroblasts and fibroblasts and contribute to the deposition of ECM [21]. In another study by Su et al., SIRT3 knockdown was found to promote angiotensin II (Ang-II)-induced NADPH oxidase-derived ROS formation and increase the expression of TGF-β1. This suggests that Ang-II-induced myocardial fibrosis may involve both SIRT3-mediated transformation of pericyte into myofibroblast/fibroblasts and ROS-TGF-1 activity [90]. Cardiovascular remodeling due to obesity involves structural and functional disorders, in which cardiac inflammation and fibrosis play a critical role. Studies have found that SIRT3 upregulates monocyte chemoattractant protein-1 (MCP-1) by activating and regulating ROS-NF-κB, thereby inhibiting cardiac inflammation and fibrosis [28]. Daidzein (DAI), an isoflavone found in soy foods, has antioxidant and anti-inflammatory properties. DAI not only affects cardiac energy metabolism by regulating SIRT3 but also plays an antioxidant role through the SIRT3/FOXO3a pathway [48]. Therapeutic hypothermia intervention may inhibit inflammation and fibrosis by modulating SIRT3/NLRP3 signaling pathway [119]. Through regulation of the SIRT3/MnSOD pathway, LCZ696 ameliorates pathological cardiac remodeling caused by oxygen stress and pressure overload [75].
SIRT6 is an important regulator of cardiovascular function in health and disease. Zhang et al. demonstrated that SIRT6 negatively regulates pathological remodeling, fibrosis, and myocardial injury by activating AMPK-angiotensin-converting enzyme 2 (ACE2) signaling and inhibiting the connective tissue growth factor (CTGF)-pro-inflammatory chemokine fractalkine (FKN) pathway [124]. In addition, Sundaresan et al. found that SIRT6 knockdown can enhance histone H3 lysine 9 (H3K9) acetylation and c-Jun promoter transcriptional activity, resulting in the over-activation of a variety of IGF signaling related genes. These results indicate that SIRT6 can negatively regulate IGF-Akt signaling pathway and reduce myocardial hypertrophy and myocardial fibrosis [58]. The differentiation of cardiac fibroblasts into myofibroblasts represents a key event in cardiac fibrosis and contributes to pathological cardiac remodeling. SIRT6 deletion has been reported to induce the transcriptional activity and DNA binding activity of nuclear factor κB, further exacerbating Ang-II-induced myofibroblast differentiation [100, 112]. In addition, SIRT6 has also been shown to enhance mitochondrial biogenesis and mitophagy by deacetylation and inhibition of Sgk1, capable of ameliorating the cardiotoxicity induced by the anthracycline doxorubicin [74].
SIRT3/6 and aging
Ageing is a natural process characterized by the gradual decline in structural integrity of an organism over time, resulting in decreased functioning and an increased risk of biological death [61]. Cellular senescence refers to a permanent state of cell cycle arrest, where cells lose their proliferative capacity while maintaining viability and metabolic activity [126]. Ageing has emerged as a major risk factor for various human diseases, including diabetes, cancer, cardiovascular disease, and neurodegenerative disorders. Cellular senescence can be categorized into two types: replicative senescence and stress-induced premature senescence (SIPS). Replicative senescence occurs due to the cessation of cell division, which is a consequence of telomere depletion [15]. On the other hand, stressors such as oxidative stress and DNA damage can induce SIPS, leading to growth arrest within a few days. It is worth noting that SIPS does not necessarily involve telomere shortening [69]. During cellular ageing, functions like autophagy, energy metabolism regulation, stress resistance, and metabolic status gradually decline.
SIRT3 and aging
SIRT3 is one of the first genes identified to extend lifespan [40]. As early as 2003, SIRT3 was reported to be associated with longevity in humans [82]. Scientists have demonstrated that mice without SIRT3 have significantly shorter lifespans and spontaneously develop cancer, metabolic syndrome, cardiovascular disease, and neurodegenerative diseases [3]. Clinical studies have indicated that the decline in SIRT3 activity in the elderly is mainly attributed to the reduction in NAD levels, which can be partially offset by appropriate activities [14]. Furthermore, SIRT3, as a major mitochondrial deacetylase, has been found to increase the acetylation of mitochondrial proteins in various tissues of knockout mice [18].
Multiple studies have posited that the advantageous impacts of SIRT3 on the processes of aging and disease primarily occur through its facilitation of ROS clearance [86]. SIRT3 is involved in multiple antioxidant pathways. SIRT3 actively participates in numerous antioxidant pathways. The ETC serves as a significant generator of ROS, and SIRT3 can indirectly diminish ROS production by regulating the efficiency of the ETC. Furthermore, SIRT3 directly modulates the activity of various superoxide scavengers through deacetylation, thereby mitigating superoxide production and averting oxidative stress (van [103]. In addition, the transcription coactivator PGC-1α and the transcription factor FOXO3a are involved in the regulation of antioxidant enzymes expression by SIRT3 [92].
SIRT6 and aging
SIRT6 is a nucleolar chromatin-associated protein that plays a crucial role in stabilizing the genome and telomeres, thereby preventing premature cell aging [66]. Animal studies have demonstrated that male mice with ineffective SIRT6 exhibit a phenotype of premature aging, while mice overexpressing SIRT6 show an extended lifespan [41, 66]. The deacetylation activity of SIRT6 is essential for maintaining genomic stability. As a result of SIRT6 deacetylation of H3K9ac, WRN is stabilized in telomere chromatin, preventing replication-related telomere defects, fusion of end-to-end chromosomes, and premature cell aging [66]. SIRT6 has been found to engage in interactions with the RELA subunit of NF-κB, resulting in the deacetylation of H3K9ac at the promoter region of NF-κB target genes [9]. This process effectively hinders cell senescence. In addition, SIRT6 recruits the chromatin remodeler SNF2H to the DNA cleavage site, leading to the deacetylation of histone H3K56ac. This mechanism serves to prevent genomic instability and facilitates the repair of damaged sites through chromatin remodeling [104].
In human cells, SIRT6 is indispensable for the maintenance of telomere position effect silencing and plays a crucial role in preserving the structure of silenced telomere chromatin [98]. SIRT6 promotes the deacetylation of H3K18ac, which leads to the silencing of peripheral centromeric heterochromatin and prevents abnormal accumulation of peripheral centromeric transcripts [96].
Furthermore, SIRT6 regulates glucose homeostasis and NAD metabolic balance, contributing to the slowing down of the aging process. As a result of increased lipolysis and elevated precursor levels, SIRT6 maintains the youthfulness of both the gluconeogenesis and tricarboxylic acid (TCA) cycles [81, 109].
Pathological mechanism of SIRT3/6 in fibrosis and aging
The pathological process of fibrosis is characterized by inflammation, oxidative stress, and apoptosis and energy metabolism (Fig. 2).

Schematic representation of the pathological mechanism of SIRT3/6 in fibrosis and aging. The pathological process of fibrosis is characterized by inflammation, oxidative stress, and apoptosis and energy metabolism. ETC electron transport chain; MnSOD manganese superoxide dismutase; PI3K phosphatidylinositol 3-kinase
Fibrosis is characterized by the aberrant accumulation of ECM proteins within the interstitial space, representing a fundamental pathological reaction to persistent inflammation. SIRT6 was recruited to the promoter regions of NF-κB target genes, leading to the suppression of gene expression. This inhibition occurs through the deacetylation of histone H3K9 at the target gene’s promoter by SIRT6 [43]. Sirt6 knockout mice exhibited obesity-related insulin resistance and increased inflammation in adipose tissue [45]. The loss of SIRT6 in macrophages resulted in the activation of NF-κB, leading to the production of IL-6 in the endothelium. This, in turn, activated the positive feedback loop involving STAT3 and NF-κB [47]. The interaction between SIRT6 and NF-κB activation enhances the pro-inflammatory M1 polarization of macrophages in the bone marrow and augments the migratory capacity of macrophages toward adipose tissue.
SIRT3 plays a critical role in mitigating mitochondrial oxidative stress through direct regulation of MnSOD [79, 94]. Furthermore, SIRT3 and SIRT4 collaborate to maintain mitochondrial NAD levels and safeguard against cell death following induced stress [117]. SIRT6 has been identified as a crucial element in the process of aging and age-related illnesses. SIRT6 interacts with NRF2, a transcription factor that regulates the expression of antioxidant genes, including heme oxygenase 1 in human mesenchymal stem cells [72]. Therefore, SIRT6-mediated activation of NRF2 protects cells from decay by protecting them from oxidative stress. Phosphatidylinositol 3-kinase (PI3K), as one of the central regulators of aging, plays a key role in the regulation of aging-related diseases [59]. Through PI3K activation, miR-34a becomes upregulated in epithelial cells in response to oxidative stress. Oxidative stress triggers the activation of miR-34a, leading to a decrease in the expression of SIRT6 and its abilities to combat aging. During periods of hyperosmotic stress, the cell utilizes aldose reductase (AR) to control its stress reactions [101, 102]. It is interesting to note that the increased AR expression under hyperosmotic stress is the result of SIRT6-mediated regulation. By overexpressing SIRT6, serum levels of IGF1 were reduced, IGF-binding protein 1 was increased, and major components of the IGF1 signaling pathway were phosphorylated [41].
Through autophagy, damaged cells are eliminated from the body, preventing cellular senescence [83]. IGF-Akt signaling through mTOR exerts a negative regulation on the process. It is reported that SIRT6 triggers autophagy in human bronchial epithelial cells by attenuating IGF-Akt-mTOR signaling [93]. Induction of autophagy prevents cellular damage and the aging process, further supporting SIRT6’s role in aging.
SIRT3, known as longevity promoting sirtuin, is also known as the “guardian of mitochondria” [65]. SIRT3 regulates mitochondrial DNA integrity, mitochondrial structural dynamics, and functional homeostasis by affecting metabolism. SIRT3 exhibits deacetylase activity against hundreds of mitochondrial proteins and is able to regulate stress pathways and energy homeostasis [1]. The energetic regulation of SIRT3 is further enhanced by its role in the TCA, as it activates the functions of AceCS2 and JNK2 [12].
Therapeutic strategies for SIRT3/6 in aging and organ fibrosis
SIRT3/6 has been shown by more and more studies to alleviate the progression of multi-organ fibrosis and is a promising target (Table 2).
Zhang et al. found that probucol, as a cholesterol-lowering drug with strong antioxidant properties, improved EMT and pulmonary fibrosis by restoring SIRT3 expression [118].In addition, a recent study found that baicalein attenuates bleomycin-induced lung fibroblast senescence and pulmonary fibrosis by restoring SIRT3 expression and inhibiting TGF-β1/Smad signaling pathway [36]. Renal interstitial fibrosis is a common pathway for the progressive development of chronic renal diseases (CKD) with different etiology, and is the main pathological basis leading to end-stage renal disease. Poricoic acid A is an anti-fibrotic drug isolated from Poria cocos. It was shown to attenuate renal fibroblast activation and interstitial fibrosis by upregulating SIRT3 and inducing β-catenin K49 deacetylation [11]. Uncoupling protein 1 is a nuclear encoded protein located in the inner mitochondrial membrane, which has been shown to inhibit the occurrence of oxidative stress by stabilizing SIRT3, thereby reducing EMT and ECM accumulation, and ultimately alleviating renal interstitial fibrosis [115]. Liver fibrosis, a chronic inflammatory healing reaction, progresses to hepatocirrhosis without effective intervention. Physion 8-O-β-glucopyranoside (PSG), an anthraquinone derived from Rumex japonicus Houtt, was shown to be able to increase the enzymatic activity and promoter activity of SIRT3 in fibrotic liver and activated hematopoietic stem cells. In addition, PSG significantly increased SIRT3 mRNA and protein expression. In brief, PSG can reduce inflammatory response by regulating SIRT3-mediated NF-κB P65 expression in liver fibrosis, which is an effective anti-fibrotic effect [10]. Hesperetin derivative (HD-16), a monomer compound extracted from hesperetin, was proved by Li et al. to reduce ccl4-induced hepatitis and liver fibrosis by activating AMPK/SIRT3 pathway [49]. Studies have found that γ-man is a strong candidate for the treatment of oxidative stress-induced liver fibrosis. γ-man can induce SIRT3 to inhibit NAD(P)H oxidase activity, thereby reducing oxidative stress in cells. In addition, γ-man enhanced SIRT3 expression and decreased HMGB1 expression, resulting in reduced accumulation of type I collagen and α-SMA in the liver [105]. Zhang and colleagues demonstrated that therapeutic hypothermia inhibits inflammation and fibrosis through SIRT3/NLRP3 signaling pathway to protect myocardial ischemia—reperfusion injury in an isolated rat heart model [119]. Liu et al. explored the role of hydrogen sulfide through the detection of SIRT3 myocardial fibrosis. They found that NaHS enhanced the activity of the SIRT3 promoter and increased SIRT3 mRNA expression. Altogether, NaHS attenuated myocardial fibrosis through oxidative stress inhibition via a SIRT3-dependent manner [56].
SIRT6 regulates DNA repair, glucose and lipid metabolism, cellular senescence, and inflammation. Studies have found that Yishen Tongluo formula significantly improves renal fibrosis by regulating SIRT6/TGF-β1/Smad2/3 signaling pathway, promoting TGF-β1 degradation, and then inhibiting the expression of type I collagen, α-smooth muscle actin, type IV collagen and fibronectin [122]. It has been found that calorie restriction can delay age-dependent renal degeneration and replicative senescence of human fibroblast WI38 by enhancing SIRT6 expression. In addition to this, SIRT6 interacts with NF-κB to regulate inflammation and apoptosis [120]. Li and colleagues demonstrated that exogenous H2S could activate cystathionine-lyase and autophagy through SIRT6/AMPK signaling pathway, inhibit cardiomyocyte senescence and improve diabetic myocardial fibrosis [53]. In addition, SIRT6 is also a potentially favorable therapeutic target for diabetic cardiomyopathy. SIRT6-specific inhibitor OSS-12816 can increase the levels of inflammatory factors and ROS in vitro and in vivo, and aggravate the apoptosis and fibrosis of cardiomyocytes induced by diabetes in mice [34].
Perspective
Interstitial fibrosis is a prevalent pathological characteristic observed in various tissues during the process of aging, resulting in the gradual decline of organ functionality. Among the organs commonly affected by age-related ailments, the kidneys are particularly susceptible, rendering older individuals more prone to chronic kidney disease.
Mammalian sirtuins have emerged as a group of metabolic regulators that facilitate the connection between protein acetylation and energy metabolism. While the comprehension of the roles of distinct sirtuins across different organ levels is still in its nascent phase, there has been some advancement in identifying sirtuin targets that have a wide range of effects on cellular protection and regeneration mechanisms. Consequently, a logical progression from these discoveries is the exploration for compounds that activate sirtuins [20, 88]. Resveratrol has the ability to function as an allosteric modulator, inducing structural alterations in the substrate, consequently enhancing its affinity for sirtuins [4]. Encouraging results have been achieved in the areas of diabetes, cardiovascular disease, and neuropathy. Regarding SIRT3, magnolol has demonstrated the ability to selectively activate SIRT3, thereby exhibiting anti-inflammatory and antioxidant effects in both chronic and acute kidney disease models [50, 51].
Ageing is a major risk factor for chronic diseases and is highly associated with cardiovascular disease, cancer, metabolic disorders, and decline in organ function over time [113]. It is important to note that most fibrotic diseases become more common with age. In an increasingly aging population, the use of effective anti-fibrosis treatments is essential to extend healthy life. In recent years, the important physiological functions of SIRTs in the pathophysiology of organ fibrosis have attracted extensive attention. SIRT regulates a variety of biological functions in different processes. In addition, SIRT is considered a therapeutic target for age-related diseases. As two different subtypes of SIRTs, SIRT3 and SIRT6 play an irreplaceable role in the prevention and treatment of aging and fibrosis. Based on the description of aging and fibrosis, the function of SIRT3/6 in aging and organ fibrosis was summarized, and some prevention and treatment strategies were provided for aging and organ fibrosis.
This review also has some limitations, fibrosis and aging are complex processes and mutual influence. Insights into the regulatory mechanisms of SIRT have been extrapolated from in vitro studies, lacking more data from clinical trials. In addition, other members of the SIRT family also play an important role in fibrosis and aging, which requires further study and comparison of the interaction and interaction between SIRT family members.
The advancements made in the targeting of SIRT3/6 have shown promising potential for the development of novel therapies aimed at addressing tissue injury and fibrosis associated with aging. In addition, these investigations have elucidated the underlying mechanisms of SIRT3/6-mediated signaling pathways involved in oxidative stress, inflammation, and apoptosis across various tissues. In conclusion, the findings from previous research serve as a valuable foundation for future studies focused on the prevention and mitigation of age-related tissue fibrosis.
Availability of data and material
N/A.
References
Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 105:14447–14452
Bellizzi D, Dato S, Cavalcante P, Covello G, Di Cianni F, Passarino G, Rose G, De Benedictis G (2007) Characterization of a bidirectional promoter shared between two human genes related to aging: SIRT3 and PSMD13. Genomics 89:143–150
Benigni A, Cassis P, Conti S, Perico L, Corna D, Cerullo D, Zentilin L, Zoja C, Perna A, Lionetti V, Giacca M, Trionfini P, Tomasoni S, Remuzzi G (2019) Sirt3 deficiency shortens life span and impairs cardiac mitochondrial function rescued by Opa1 gene transfer. Antioxid Redox Signal 31:1255–1271
Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17:679–690
Borgoni S, Kudryashova KS, Burka K, de Magalhães JP (2021) Targeting immune dysfunction in aging. Ageing Res Rev 70:101410
Brillo V, Chieregato L, Leanza L, Muccioli S, Costa R (2021) Mitochondrial dynamics, ROS, and cell signaling: a blended overview. Life 11:332
Brouwers FP, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, Hillege HL, van Veldhuisen DJ, van Gilst WH (2013) Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J 34:1424–1431
Cardoso AL, Fernandes A, Aguilar-Pimentel JA, de Angelis MH, Guedes JR, Brito MA, Ortolano S, Pani G, Athanasopoulou S, Gonos ES, Schosserer M, Grillari J, Peterson P, Tuna BG, Dogan S, Meyer A, van Os R, Trendelenburg AU (2018) Towards frailty biomarkers: Candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res Rev 47:214–277
Chang AR, Ferrer CM, Mostoslavsky R (2020) SIRT6, a mammalian deacylase with multitasking abilities. Physiol Rev 100:145–169
Chen C, Gu J, Wang J, Wu Y, Yang A, Chen T, Zhou T, Liu Z (2021) Physcion 8-O-β-glucopyranoside ameliorates liver fibrosis through inflammation inhibition by regulating SIRT3-mediated NF-κB P65 nuclear expression. Int Immunopharmacol 90:107206
Chen D-Q, Chen L, Guo Y, Wu X-Q, Zhao T-T, Zhao H-L, Zhang H-J, Yan M-H, Zhang G-Q, Li P (2023) Poricoic acid A suppresses renal fibroblast activation and interstitial fibrosis in UUO rats via upregulating Sirt3 and promoting β-catenin K49 deacetylation. Acta Pharmacol Sin 44:1038–1050
Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y, Huang J (2014) Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis 5:e1047
Cheng L, Yang X, Jian Y, Liu J, Ke X, Chen S, Yang D, Yang D (2022) SIRT3 deficiency exacerbates early-stage fibrosis after ischaemia-reperfusion-induced AKI. Cell Signal 93:110284
Chini CCS, Tarragó MG, Chini EN (2017) NAD and the aging process: role in life, death and everything in between. Mol Cell Endocrinol 455:62–74
Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130:223–233
Dawood RM, El-Meguid MA, Salum GM, El Awady MK (2020) Key players of hepatic fibrosis. J Interferon Cytokine Res 40:472–489
Dhar D, Baglieri J, Kisseleva T, Brenner DA (2020) Mechanisms of liver fibrosis and its role in liver cancer. Exp Biol Med 245:96–108
Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, FotuhiSiahpirani A, Kemmerer ZA, Prolla TA, Roy S, Coon JJ, Denu JM (2015) SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab 21:637–646
Dodig S, Čepelak I, Pavić I (2019) Hallmarks of senescence and aging. Biochemia medica 29:483–497
Farghali H, KutinováCanová N, Lekić N (2013) Resveratrol and related compounds as antioxidants with an allosteric mechanism of action in epigenetic drug targets. Physiol Res 62:1–13
Frangogiannis NG (2019) Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med 65:70–99
Gao X, Tian X, Huang Y, Fang R, Wang G, Li D, Zhang J, Li T, Yuan R (2022) Role of circular RNA in myocardial ischemia and ageing-related diseases. Cytokine Growth Factor Rev 65:1–11
Garufi A, Pistritto G, D’Orazi G (2023) HIPK2 as a novel regulator of fibrosis. Cancers 15:1059
Gertler AA, Cohen HY (2013) SIRT6, a protein with many faces. Biogerontology 14:629–639
Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR (2021) SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res 128:474–491
Gu C, Li T, Jiang S, Yang Z, Lv J, Yi W, Yang Y, Fang M (2018) AMP-activated protein kinase sparks the fire of cardioprotection against myocardial ischemia and cardiac ageing. Ageing Res Rev 47:168–175
Guo J, Huang X, Dou L, Yan M, Shen T, Tang W, Li J (2022) Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduct Target Ther 7:391
Guo X, Yan F, Li J, Zhang C, Su H, Bu P (2020) SIRT3 ablation deteriorates obesity-related cardiac remodeling by modulating ROS-NF-κB-MCP-1 signaling pathway. J Cardiovasc Pharmacol 76:296–304
Guo Z, Li P, Ge J, Li H (2022) SIRT6 in aging, metabolism, inflammation and cardiovascular diseases. Aging Dis 13:1787
Han X, Ding C, Sang X, Peng M, Yang Q, Ning Y, Lv Q, Shan Q, Hao M, Wang K, Wu X, Zhang H, Cao G (2022) Targeting Sirtuin1 to treat aging-related tissue fibrosis: from prevention to therapy. Pharmacol Ther 229:107983
He J, Yu C, Shen Y, Huang J, Zhou Y, Gu J, Cao Y, Zheng Q (2023) Sirtuin 6 ameliorates bleomycin-induced pulmonary fibrosis via activation of lipid catabolism. J Cell Physiol. https://doi.org/10.1002/jcp.31027
Hu HH, Cao G, Wu XQ, Vaziri ND, Zhao YY (2020) Wnt signaling pathway in aging-related tissue fibrosis and therapies. Ageing Res Rev 60:101063
Hu YB, Ye XT, Zhou QQ, Fu RQ (2018) Sestrin 2 attenuates rat hepatic stellate cell (HSC) activation and liver fibrosis via an mTOR/AMPK-dependent mechanism. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 51:2111–2122
Huang Y, Zhang J, Xu D, Peng Y, Jin Y, Zhang L (2021) SIRT6-specific inhibitor OSS-128167 exacerbates diabetic cardiomyopathy by aggravating inflammation and oxidative stress. Mol Med Rep. https://doi.org/10.3892/mmr.2021.12006
Jablonski RP, Kim SJ, Cheresh P, Williams DB, Morales-Nebreda L, Cheng Y, Yeldandi A, Bhorade S, Pardo A, Selman M, Ridge K, Gius D, Budinger GRS, Kamp DW (2017) SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J Off Publ Fed Am Soc Exp Biol 31:2520–2532
Ji-Hong Y, Yu M, Ling-Hong Y, Jing-Jing G, Ling-Li X, Lv W, Yong-Mei J (2023) Baicalein attenuates bleomycin-induced lung fibroblast senescence and lung fibrosis through restoration of Sirt3 expression. Pharm Biol 61:288–297
Jiang S, Li T, Yang Z, Yi W, Di S, Sun Y, Wang D, Yang Y (2017) AMPK orchestrates an elaborate cascade protecting tissue from fibrosis and aging. Ageing Res Rev 38:18–27
Jin L, Wei W, Jiang Y, Peng H, Cai J, Mao C, Dai H, Choy W, Bemis JE, Jirousek MR, Milne JC, Westphal CH, Perni RB (2009) Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J Biol Chem 284:24394–24405
Ka SO, Bang IH, Bae EJ, Park BH (2017) Hepatocyte-specific sirtuin 6 deletion predisposes to nonalcoholic steatohepatitis by up-regulation of Bach1, an Nrf2 repressor. FASEB J Off Publ Fed Am Soc Exp Biol 31:3999–4010
Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13:2570–2580
Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY (2012) The sirtuin SIRT6 regulates lifespan in male mice. Nature 483:218–221
Kanwal A, Pillai VB, Samant S, Gupta M, Gupta MP (2019) The nuclear and mitochondrial sirtuins, Sirt6 and Sirt3, regulate each other’s activity and protect the heart from developing obesity-mediated diabetic cardiomyopathy. FASEB J Off Publ Fed Am Soc Exp Biol 33:10872–10888
Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, Chua KF (2009) SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136:62–74
Kitada M, Ogura Y, Monno I, Koya D (2019) Sirtuins and type 2 diabetes: role in inflammation, oxidative stress, and mitochondrial function. Front Endocrinol 10:187
Kuang J, Zhang Y, Liu Q, Shen J, Pu S, Cheng S, Chen L, Li H, Wu T, Li R, Li Y, Zou M, Zhang Z, Jiang W, Xu G, Qu A, Xie W, He J (2017) Fat-specific Sirt6 ablation sensitizes mice to high-fat diet-induced obesity and insulin resistance by inhibiting lipolysis. Diabetes 66:1159–1171
Kumar A, Dvir-Ginzberg M (2021) Sirtuins as NAD+-dependent deacetylases and their potential in medical therapy. Medical epigenetics. Elsevier, Amsterdam, pp 633–664
Lee Y, Ka SO, Cha HN, Chae YN, Kim MK, Park SY, Bae EJ, Park BH (2017) Myeloid Sirtuin 6 deficiency causes insulin resistance in high-fat diet-fed mice by eliciting macrophage polarization toward an M1 phenotype. Diabetes 66:2659–2668
Li H, Zhang M, Wang Y, Gong K, Yan T, Wang D, Meng X, Yang X, Chen Y, Han J (2022) Daidzein alleviates doxorubicin-induced heart failure via the SIRT3/FOXO3a signaling pathway. Food Funct 13:9576–9588
Li J-J, Jiang H-C, Wang A, Bu F-T, Jia P-C, Zhu S, Zhu L, Huang C, Li J (2022) Hesperetin derivative-16 attenuates CCl4-induced inflammation and liver fibrosis by activating AMPK/SIRT3 pathway. Eur J Pharmacol 915:174530
Li N, Xie H, Li L, Wang J, Fang M, Yang N, Lin H (2014) Effects of honokiol on sepsis-induced acute kidney injury in an experimental model of sepsis in rats. Inflammation 37:1191–1199
Li N, Zhang J, Yan X, Zhang C, Liu H, Shan X, Li J, Yang Y, Huang C, Zhang P, Zhang Y, Bu P (2017) SIRT3-KLF15 signaling ameliorates kidney injury induced by hypertension. Oncotarget 8:39592–39604
Li X, Li W, Zhang Z, Wang W, Huang H (2022) SIRT6 overexpression retards renal interstitial fibrosis through targeting HIPK2 in chronic kidney disease. Front Pharmacol 13:1007168
Li Y, Liu M, Song X, Zheng X, Yi J, Liu D, Wang S, Chu C, Yang J (2020) Exogenous hydrogen sulfide ameliorates diabetic myocardial fibrosis by inhibiting cell aging through SIRT6/AMPK autophagy. Front Pharmacol 11:1150
Li Z, Xu K, Zhang N, Amador G, Wang Y, Zhao S, Li L, Qiu Y, Wang Z (2018) Overexpressed SIRT6 attenuates cisplatin-induced acute kidney injury by inhibiting ERK1/2 signaling. Kidney Int 93:881–892
Liang Z, Li T, Jiang S, Xu J, Di W, Yang Z, Hu W, Yang Y (2017) AMPK: a novel target for treating hepatic fibrosis. Oncotarget 8:62780–62792
Liu L, Gong W, Zhang S, Shen J, Wang Y, Chen Y, Meng G (2021) Hydrogen sulfide attenuates angiotensin II-induced cardiac fibroblast proliferation and transverse aortic constriction-induced myocardial fibrosis through oxidative stress inhibition via Sirtuin 3. Oxid Med Cell Longev 2021:9925771
Liu W, Yuan Q, Cao S, Wang G, Liu X, Xia Y, Bian Y, Xu F, Chen Y (2023) Acetylation mechanisms and targeted therapies in cardiac fibrosis. Pharmacol Res 193:106815
Liu Y-P, Wen R, Liu C-F, Zhang T-N, Yang N (2023) Cellular and molecular biology of sirtuins in cardiovascular disease. Biomed Pharmacother 164:114931
Liu Y, Liu Q, Zhang Z, Yang Y, Zhou Y, Yan H, Wang X, Li X, Zhao J, Hu J, Yang S, Tian Y, Yao Y, Qiu Z, Song Y, Yang Y (2023) The regulatory role of PI3K in ageing-related diseases. Ageing Res Rev 88:101963
Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV Jr, Weissman S, Verdin E, Schwer B (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27:8807–8814
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217
Ma Y, Chai H, Ding Q, Qian Q, Yan Z, Ding B, Dou X, Li S (2019) Hepatic SIRT3 upregulation in response to chronic alcohol consumption contributes to alcoholic liver disease in mice. Front Physiol 10:1042
Mahlknecht U, Ho AD, Voelter-Mahlknecht S (2006) Chromosomal organization and fluorescence in situ hybridization of the human Sirtuin 6 gene. Int J Oncol 28:447–456
Maity S, Muhamed J, Sarikhani M, Kumar S, Ahamed F, Spurthi KM, Ravi V, Jain A, Khan D, Arathi BP, Desingu PA, Sundaresan NR (2020) Sirtuin 6 deficiency transcriptionally up-regulates TGF-β signaling and induces fibrosis in mice. J Biol Chem 295:415–434
Mazumder S, Barman M, Bandyopadhyay U, Bindu S (2020) Sirtuins as endogenous regulators of lung fibrosis: A current perspective. Life Sci 258:118201
Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, Chang HY, Bohr VA, Ried T, Gozani O, Chua KF (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452:492–496
Murugasamy K, Munjal A, Sundaresan NR (2022) Emerging roles of SIRT3 in cardiac metabolism. Front Cardiovasc Med 9:850340
Mutsaers HA, Olinga P (2016) Editorial: organ fibrosis: triggers, pathways, and cellular plasticity. Front Med 3:55
Naka K, Tachibana A, Ikeda K, Motoyama N (2004) Stress-induced premature senescence in hTERT-expressing ataxia telangiectasia fibroblasts. J Biol Chem 279:2030–2037
Ogura Y, Kitada M, Koya D (2021) Sirtuins and renal oxidative stress. Antioxidants 10:1198
Palomer X, Román-Azcona MS, Pizarro-Delgado J, Planavila A, Villarroya F, Valenzuela-Alcaraz B, Crispi F, Sepúlveda-Martínez Á, Miguel-Escalada I, Ferrer J, Nistal JF, García R, Davidson MM, Barroso E, Vázquez-Carrera M (2020) SIRT3-mediated inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal Transduct Target Ther 5:14
Pan H, Guan D, Liu X, Li J, Wang L, Wu J, Zhou J, Zhang W, Ren R, Zhang W, Li Y, Yang J, Hao Y, Yuan T, Yuan G, Wang H, Ju Z, Mao Z, Li J, Qu J, Tang F, Liu GH (2016) SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res 26:190–205
Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM (2011) Structure and biochemical functions of SIRT6. J Biol Chem 286:14575–14587
Peng K, Zeng C, Gao Y, Liu B, Li L, Xu K, Yin Y, Qiu Y, Zhang M, Ma F (2023) Overexpressed SIRT6 ameliorates doxorubicin-induced cardiotoxicity and potentiates the therapeutic efficacy through metabolic remodeling. Acta Pharm Sin B 13(6):2680–2700
Peng S, Lu X-F, Qi Y-D, Li J, Xu J, Yuan T-Y, Wu X-Y, Ding Y, Li W-H, Zhou G-Q, Wei Y, Li J, Chen S-W, Liu S-W (2020) LCZ696 ameliorates oxidative stress and pressure overload-induced pathological cardiac remodeling by regulating the Sirt3/MnSOD pathway. Oxid Med Cell Longev 2020:9815039
Pillai VB, Samant S, Hund S, Gupta M, Gupta MP (2021) The nuclear sirtuin SIRT6 protects the heart from developing aging-associated myocyte senescence and cardiac hypertrophy. Aging 13:12334–12358
Pillai VB, Sundaresan NR, Jeevanandam V, Gupta MP (2010) Mitochondrial SIRT3 and heart disease. Cardiovasc Res 88:250–256
Pole A, Dimri M, Dimri GP (2016) Oxidative stress, cellular senescence and ageing. AIMS Mol Sci 3:300–324
Qiu X, Brown K, Hirschey MD, Verdin E, Chen D (2010) Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 12:662–667
Ren L-L, Li X-J, Duan T-T, Li Z-H, Yang J-Z, Zhang Y-M, Zou L, Miao H, Zhao Y-Y (2022) Transforming growth factor-β signaling: from tissue fibrosis to therapeutic opportunities. Chem Biol Interact 369:110289
Roichman A, Elhanati S, Aon MA, Abramovich I, Di Francesco A, Shahar Y, Avivi MY, Shurgi M, Rubinstein A, Wiesner Y, Shuchami A, Petrover Z, Lebenthal-Loinger I, Yaron O, Lyashkov A, Ubaida-Mohien C, Kanfi Y, Lerrer B, Fernández-Marcos PJ, Serrano M, Gottlieb E, de Cabo R, Cohen HY (2021) Restoration of energy homeostasis by SIRT6 extends healthy lifespan. Nat Commun 12:3208
Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V, Passarino G, Feraco E, Mari V, Barbi C, BonaFe M, Franceschi C, Tan Q, Boiko S, Yashin AI, De Benedictis G (2003) Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol 38:1065–1070
Rubinsztein DC, Mariño G, Kroemer G (2011) Autophagy and aging. Cell 146:682–695
Seki E, Brenner DA, Karin M (2012) A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 143:307–320
Sharma A, Mahur P, Muthukumaran J, Singh AK, Jain M (2023) Shedding light on structure, function and regulation of human sirtuins: a comprehensive review. 3 Biotech 13:29
Shen Y, Wu Q, Shi J, Zhou S (2020) Regulation of SIRT3 on mitochondrial functions and oxidative stress in Parkinson’s disease. Biomed Pharmacother 132:110928
Shi MY, Bang IH, Han CY, Lee DH, Park BH, Bae EJ (2020) Statin suppresses sirtuin 6 through miR-495, increasing FoxO1-dependent hepatic gluconeogenesis. Theranostics 10:11416–11427
Sinclair DA, Guarente L (2014) Small-molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol 54:363–380
Su H, Cantrell AC, Chen J-X, Gu W, Zeng H (2023) SIRT3 deficiency enhances ferroptosis and promotes cardiac fibrosis via p53 acetylation. Cells 12:1428
Su H, Zeng H, Liu B, Chen J-X (2020) Sirtuin 3 is essential for hypertension-induced cardiac fibrosis via mediating pericyte transition. J Cell Mol Med 24:8057–8068
Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY, Gupta M, Nagalingam RS, Wolfgeher D, Verdin E, Gupta MP (2015) SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3β. Mol Cell Biol 36:678–692
Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP (2009) Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Investig 119:2758–2771
Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y, Wakui H, Yoshii Y, Yumino Y, Fujii S, Minagawa S, Tsurushige C, Kojima J, Numata T, Shimizu K, Kawaishi M, Kaneko Y, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Nakayama K, Kuwano K (2014) Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol (Baltimore, MD: 1950) 192:958–968
Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40:893–904
Tao Z, Jin Z, Wu J, Cai G, Yu X (2023) Sirtuin family in autoimmune diseases. Front Immunol. https://doi.org/10.3389/fimmu.2023.1186231
Tasselli L, Xi Y, Zheng W, Tennen RI, Odrowaz Z, Simeoni F, Li W, Chua KF (2016) SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat Struct Mol Biol 23:434–440
Tennen RI, Berber E, Chua KF (2010) Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech Ageing Dev 131:185–192
Tennen RI, Bua DJ, Wright WE, Chua KF (2011) SIRT6 is required for maintenance of telomere position effect in human cells. Nat Commun 2:433
Tennen RI, Chua KF (2011) Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci 36:39–46
Tian K, Liu Z, Wang J, Xu S, You T, Liu P (2015) Sirtuin-6 inhibits cardiac fibroblasts differentiation into myofibroblasts via inactivation of nuclear factor κB signaling. Transl Res 165:374–386
Timucin AC, Basaga H (2016) SIRT6 is a positive regulator of aldose reductase expression in U937 and HeLa cells under osmotic stress: in vitro and in silico insights. PLoS ONE 11:e0161494
Ungurianu A, Zanfirescu A, Margină D (2023) Sirtuins, resveratrol and the intertwining cellular pathways connecting them. Ageing Res Rev 88:101936
van de Ven RAH, Santos D, Haigis MC (2017) Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol Med 23:320–331
Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, Bredbenner K, Park R, Sinclair DA, Bohr VA, Gorbunova V, Seluanov A (2016) JNK phosphorylates SIRT6 to stimulate DNA double-strand break repair in response to oxidative stress by recruiting PARP1 to DNA breaks. Cell Rep 16:2641–2650
Wang A, Zhou F, Li D, Lu J-J, Wang Y, Lin L (2019) γ-Mangostin alleviates liver fibrosis through Sirtuin 3-superoxide-high mobility group box 1 signaling axis. Toxicol Appl Pharmacol 363:142–153
Wang Y, He J, Liao M, Hu M, Li W, Ouyang H, Wang X, Ye T, Zhang Y, Ouyang L (2019) An overview of Sirtuins as potential therapeutic target: structure, function and modulators. Eur J Med Chem 161:48–77
Wang Y, Li C, Gu J, Chen C, Duanmu J, Miao J, Yao W, Tao J, Tu M, Xiong B, Zhao L, Liu Z (2020) Celastrol exerts anti-inflammatory effect in liver fibrosis via activation of AMPK-SIRT3 signalling. J Cell Mol Med 24:941–953
Wencel PL, Lukiw WJ, Strosznajder JB, Strosznajder RP (2018) Inhibition of poly(ADP-ribose) polymerase-1 enhances gene expression of selected sirtuins and APP cleaving enzymes in amyloid beta cytotoxicity. Mol Neurobiol 55:4612–4623
Wiley CD, Campisi J (2021) The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab 3:1290–1301
Winnik S, Auwerx J, Sinclair DA, Matter CM (2015) Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J 36:3404–3412
Thaiss WM, Sannwald L, Kloth C, Ekert K, Hepp T, Bösmüller H, Klag T, Nikolaou K, Horger M, Kaufmann S (2019) Quantification of hemodynamic changes in chronic liver disease: correlation of perfusion-CT data with histopathologic staging of fibrosis. Acad Radiol 26:1174–1180
Wu K, Wang Y, Liu R, Wang H, Rui T (2023) The role of mammalian Sirtuin 6 in cardiovascular diseases and diabetes mellitus. Front Physiol 14:1207133
Wu X, Qian L, Zhao H, Lei W, Liu Y, Xu X, Li J, Yang Z, Wang D, Zhang Y, Zhang Y, Tang R, Yang Y, Tian Y (2023) CXCL12/CXCR4: an amazing challenge and opportunity in the fight against fibrosis. Ageing Res Rev 83:101809
Xin Z, Ma Z, Hu W, Jiang S, Yang Z, Li T, Chen F, Jia G, Yang Y (2018) FOXO1/3: potential suppressors of fibrosis. Ageing Res Rev 41:42–52
Xiong W, Xiong Z, Song A, Lei C, Ye C, Su H, Zhang C (2023) UCP1 alleviates renal interstitial fibrosis progression through oxidative stress pathway mediated by SIRT3 protein stability. J Transl Med 21:521
Xu X, Hong P, Wang Z, Tang Z, Li K (2021) MicroRNAs in transforming growth factor-beta signaling pathway associated with fibrosis involving different systems of the human body. Front Mol Biosci 8:707461
Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130:1095–1107
Zhang H-X, Li Y-N, Wang X-L, Ye C-L, Zhu X-Y, Li H-P, Yang T, Liu Y-J (2019) Probucol ameliorates EMT and lung fibrosis through restoration of SIRT3 expression. Pulm Pharmacol Ther 57:101803
Zhang J, Lu Y, Yu P, Li Z, Liu Y, Zhang J, Tang X, Yu S (2022) Therapeutic hypothermia alleviates myocardial ischaemia-reperfusion injury by inhibiting inflammation and fibrosis via the mediation of the SIRT3/NLRP3 signalling pathway. J Cell Mol Med 26:4995–5007
Zhang N, Li Z, Mu W, Li L, Liang Y, Lu M, Wang Z, Qiu Y, Wang Z (2016) Calorie restriction-induced SIRT6 activation delays aging by suppressing NF-κB signaling. Cell Cycle 15:1009–1018
Zhang Q, Tu W, Tian K, Han L, Wang Q, Chen P, Zhou X (2019) Sirtuin 6 inhibits myofibroblast differentiation via inactivating transforming growth factor-β1/Smad2 and nuclear factor-κB signaling pathways in human fetal lung fibroblasts. J Cell Biochem 120:93–104
Zhang X, Zhao L, Xiang S, Sun Y, Wang P, Chen JJ, Teo BS-X, Xie Z, Zhang Z, Xu J (2023) Yishen Tongluo formula alleviates diabetic kidney disease through regulating Sirt6/TGF-β1/Smad2/3 pathway and promoting degradation of TGF-β1. J Ethnopharmacol 307:116243
Zhang Y, Wen P, Luo J, Ding H, Cao H, He W, Zen K, Zhou Y, Yang J, Jiang L (2021) Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis 12:847
Zhang Z-Z, Cheng Y-W, Jin H-Y, Chang Q, Shang Q-H, Xu Y-L, Chen L-X, Xu R, Song B, Zhong J-C (2017) The sirtuin 6 prevents angiotensin II-mediated myocardial fibrosis and injury by targeting AMPK-ACE2 signaling. Oncotarget 8:72302–72314
Zhong X, Huang M, Kim HG, Zhang Y, Chowdhury K, Cai W, Saxena R, Schwabe RF, Liangpunsakul S, Dong XC (2020) SIRT6 protects against liver fibrosis by deacetylation and suppression of SMAD3 in hepatic stellate cells. Cell Mol Gastroenterol Hepatol 10:341–364
Zurgil U, Ben-Ari A, Atias K, Isakov N, Apte R, Livneh E (2014) PKCη promotes senescence induced by oxidative stress and chemotherapy. Cell Death Dis 5:e1531
Acknowledgements
We thanked Home for Researcher for language editing service and Figdraw (www.figdraw.com) for expert assistance in the pattern drawing.
Funding
None.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval
N/A.
Consent to participate
N/A.
Consent for publication
N/A.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, W., Li, T., Chen, J. et al. SIRT3/6: an amazing challenge and opportunity in the fight against fibrosis and aging. Cell. Mol. Life Sci. 81, 69 (2024). https://doi.org/10.1007/s00018-023-05093-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-05093-z