
Abstract
Machado–Joseph disease (MJD) is characterized by a pathological expansion of the polyglutamine (polyQ) tract within the ataxin-3 protein. Despite its primarily cytoplasmic localization, polyQ-expanded ataxin-3 accumulates in the nucleus and forms intranuclear aggregates in the affected neurons. Due to these histopathological hallmarks, the nucleocytoplasmic transport machinery has garnered attention as an important disease relevant mechanism. Here, we report on MJD cell model-based analysis of the nuclear transport receptor karyopherin subunit beta-1 (KPNB1) and its implications in the molecular pathogenesis of MJD. Although directly interacting with both wild-type and polyQ-expanded ataxin-3, modulating KPNB1 did not alter the intracellular localization of ataxin-3. Instead, overexpression of KPNB1 reduced ataxin-3 protein levels and the aggregate load, thereby improving cell viability. On the other hand, its knockdown and inhibition resulted in the accumulation of soluble and insoluble ataxin-3. Interestingly, the reduction of ataxin-3 was apparently based on protein fragmentation independent of the classical MJD-associated proteolytic pathways. Label-free quantitative proteomics and knockdown experiments identified mitochondrial protease CLPP as a potential mediator of the ataxin-3-degrading effect induced by KPNB1. We confirmed reduction of KPNB1 protein levels in MJD by analyzing two MJD transgenic mouse models and induced pluripotent stem cells (iPSCs) derived from MJD patients. Our results reveal a yet undescribed regulatory function of KPNB1 in controlling the turnover of ataxin-3, thereby highlighting a new potential target of therapeutic value for MJD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dysfunction in protein homeostasis commonly results in the accumulation of aberrant proteins as toxic aggregates in the cells. Particularly, post-mitotic cells such as neurons are the most affected ones [1]. The formation of intraneuronal proteinaceous inclusions is a histopathological hallmark of polyglutamine (polyQ) diseases comprising Huntington's disease (HD), spinobulbar muscular atrophy (SBMA), dentatorubral–pallidoluysian atrophy (DRPLA), and six types of spinocerebellar ataxias (SCAs). Those inclusions are the consequence of a dynamic expansion of a polyQ tract encoded by a CAG trinucleotide repeat in the respective causative genes [2, 3]. The aggregation propensity of disease proteins depends on the polyQ repeat length, with longer tracts tending to accumulate more intensely [4]. The resulting intracellular aggregates are mainly composed of polyQ-expanded proteins, but recruit molecular chaperones, ubiquitin, proteasome subunits, and transcription factors as well, which may contribute to neuronal dysfunction and death [5,6,7,8]. PolyQ diseases are mainly late-onset, relentlessly progressive, and give rise to the neurological and physical impairments in patients [9].
Machado–Joseph disease (MJD), also known as spinocerebellar ataxia type 3 (SCA3), is the most prevalent type of SCA. It is caused by a CAG repeat expansion in the coding region of the ATXN3/MJD1 gene leading to an elongation of the polyQ tract in the C-terminus of the ataxin-3 protein [10,11,12]. Normal individuals carry 12–44 CAG repeats, while in MJD patients the expanded repeats range between 61 and 87 CAGs [13]. Ataxin-3 is a deubiquitinating enzyme (DUB) proposed to be implicated in the proteasomal degradation of ubiquitinated proteins [14]. It has two structural features including the N-terminal Josephin domain and the C-terminus which contains two or three ubiquitin interacting motifs (UIMs) flanking the polyQ tract [15]. Despite a mass of significant knowledge, the exact physiological mechanism by which polyQ-expanded ataxin-3 elicits neurotoxicity is still far from clear. The expansion of the polyQ tract evokes the formation of protein aggregates, susceptibility to proteolysis, destabilization of functional conformation, leading to abnormal protein–protein interactions, and ultimately neuronal death [16]. Proteolytic processing of ataxin-3 is thought to generate aggregation-prone fragments, which favors cytotoxicity [17, 18]. Despite predominantly cytoplasmic localization of ataxin-3 [19], several lines of evidence point to the nucleus as the preferential site of aggregation, toxicity, and pathology of this polyQ-expanded protein [20, 21]. Indeed, nuclear localization of ataxin-3 was shown to play a pivotal role in the manifestation of symptoms [22].
Nucleocytoplasmic transport involves the recognition of nuclear localization signals (NLSs) or nuclear export signals (NESs) of cargos by nuclear transport receptors referred to as karyopherins. The classical import pathway is mediated by the karyopherin beta family (KPNBs) that binds to the cargos directly or through adaptor proteins, the karyopherin alpha family (KPNAs) [23, 24]. Ataxin-3 possesses a putative NLS and two NESs which might be involved in its nuclear translocation [25, 26]. Nonetheless, the molecular mechanisms implicated in the nucleocytoplasmic transport of ataxin-3 and their contribution to the pathogenesis of MJD are not fully understood. Previously, our group reported that karyopherin subunit alpha-3 (KPNA3) controls the localization of ataxin-3 and is a key player in the pathogenesis and progression of MJD [27]. Importantly, KPNA3 functions as an adaptor protein for karyopherin subunit beta-1 (KPNB1) in the nuclear transport of various cargos, raising the possibility that KPNB1 could be implicated in the localization and modulation of ataxin-3 as well.
Herein, we report that overexpression and knockdown of KPNB1 did not alter the intracellular localization of ataxin-3, but surprisingly influenced its soluble protein levels and aggregate formation by a proteolytic event independent of known MJD-associated proteolytic pathways. Label-free quantitative proteomics-based pathway analysis of cells overexpressing ataxin-3 and KPNB1 suggested an activation of the ATP-dependent Clp protease proteolytic subunit (CLPP) upon KPNB1 overexpression. Consequently, CLPP knockdown counteracted the KPNB1 overexpression-dependent reduction of ataxin-3 levels, while CLPP overexpression enhanced the KPNB1-mediated ataxin-3 lowering. Additionally, we demonstrated decreased KPNB1 protein levels in two different MJD transgenic mouse models and a reduction of both KPNB1 and CLPP protein levels in induced pluripotent stem cells (iPSCs) of MJD patients. Overall, our findings provide evidence that modulation of soluble and aggregated ataxin-3 by KPNB1 is mediated by an activation of the mitochondrial protease CLPP, highlighting a promising new mechanism for targeting polyQ-expanded disease protein levels.
Materials and methods
Mouse housing and tissue sampling
Wild-type mice as well as YAC transgenic mice [28], and CaMKII/MJD77 transgenic mice previously generated in our lab [29], were housed under standard conditions and a 12 h light–dark cycle with 50–55% relative air humidity. Mice were sacrificed using CO2 inhalation and brains were immediately separated on ice, snap-frozen in liquid nitrogen, and stored at −80 °C for further analysis.
Expression constructs and esiRNAs
All plasmids were extracted from DH5α or SURE E. coli using the QIAprep Spin Miniprep Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Overexpression of ataxin-3 for western blot analysis, nucleocytoplasmic fractionation, and filter retardation assays was carried out using the vector pcDNA3.1/Xpress (Thermo Fisher Scientific, Karlsruhe, Germany) containing full-length human ataxin-3 (UniProt ID: P54252-2) with 15, 77, or 148 glutamines. Empty pcDNA3.1/Xpress vector was used as control. Protein stability analysis was performed using the Tet-off system including pTRE responder containing human ataxin-3 with 15 or 77 glutamines and pTET-RCA2 promoter constructs in a 1:1 ratio [30]. pEGFP-N2 constructs (Clontech, Mountain View, CA, US) containing full-length human ataxin-3 with 15 or 148 glutamines were applied for fluorescence microscopy analysis. pGEX-6P-1 constructs (GE Healthcare, Munich, Germany) carrying human ataxin-3 with 15 or 77 glutamines were used to purify GST-ataxin-3, and an empty pGEX-6P-1 vector served as a control. All ataxin-3 constructs contained isoform 3c. pCMV6-XL5 and pCMV6-Entry/myc-DDK vectors (both OriGene Technologies, Rockville, MD, US) were used for overexpressing human KPNB1 and human CLPP, respectively. Endoribonuclease-prepared siRNA (esiRNA) against human KPNB1 (MISSION esiRNA EHU043151, Sigma-Aldrich, St. Louis, MO, US) and against human CLPP (MISSION esiRNA EHU011941, Sigma-Aldrich) were applied to knockdown endogenous KPNB1 and CLPP, respectively, and esiRNA against Renilla luciferase (MISSION esiRNA EHURLUC, Sigma-Aldrich) was used as control.
Antibodies
The following primary and secondary antibodies were applied for immunodetection and immunostaining: anti-ataxin-3 (1:5000; 1H9, MAB5360, Merck, Darmstadt, Germany), anti-ataxin-3/C-terminal (1:2500; SA3637) [20], anti-KPNB1 (1:5000; H-300, sc-11367, Santa Cruz Biotechnology, Heidelberg, Germany), anti-KPNA3 (1:5000; ab137446, Abcam, Cambridge, United Kingdom), anti-CLPP (1:2500; 15,698–1-AP, Proteintech, St. Leon-Rot, Germany), anti-GAPDH (1:5000; 0411, sc-47724, Santa Cruz Biotechnology), anti-β-actin (1:5000; AC-15, A5441, Sigma-Aldrich), anti-vinculin (1:1000; E1E9V, 13,901, Cell Signaling Technologies, Frankfurt am Main, Germany), anti-lamin A/C (1:5000; 346, sc-7293, Santa Cruz Biotechnology), anti-GST (1:2500; B-14, sc-138, Santa Cruz Biotechnology), anti-α-spectrin (1:1000; AA6, Merck), anti-GFP (1:1000; B-2, sc-9996, Santa Cruz Biotechnology), anti-LC3 (1:1000; MBL-PD014, MBL International, Woburn, MA, US), anti-p62 (1:1000; 5114, Cell Signaling Technologies), and IRDye secondary antibodies goat anti-mouse 680LT, goat anti-mouse 800CW, goat anti-rabbit 680RD, goat anti-rabbit 800CW (all 1:10,000; LI-COR Biosciences, Bad Homburg, Germany), and Alexa Fluor 555 (1:500; Thermo Fisher Scientific).
KPNB1 I178D cloning
Mutation on the nucleoporin FxFG binding site of KPNB1 leads to the reduction of both binding and nuclear transport of this protein [31]. I178D mutation was introduced by PCR. Briefly, the mutation (AAT to AGA) was amplified using the following primers: forward, 5’-atgtcacaaaccccaacagc-3′; and reverse, 5’-gtagcagctagcttcacattattactaggctcttctttccttcatcccctggTCTatggcagtcagaatc-3’, and introduced into the pCMV6-XL5 vector containing full-length human KPNB1 using SalI-HF and NheI restriction enzymes (New England Biolabs, Frankfurt am Main, Germany). Afterwards, it was subjected to Sanger sequencing using primers: forward, 5’-accagtggccagaactcatt-3’; and reverse, 5’-agtgcctttcagactctttatca-3’ to confirm the respective mutation and to ensure that no other mutations have been generated.
Cell culture and transfection
Wild-type HEK 293T cells (ATCC: CRL-11268) and ATXN3 KO HEK 293T cells previously generated in our lab [32], were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic (all Gibco, Thermo Fisher Scientific) in a mycoplasma-free incubator at 37 °C and 5% CO2. Transient transfection of HEK 293T cells with constructs and esiRNAs was carried out using TurboFectin 8.0 (OriGene Technologies) and Attractene (QIAGEN) transfection reagents, respectively, according to the manufacturer’s instructions.
Fibroblasts from three MJD patients and three healthy individuals were reprogrammed to iPSCs as described previously [33].
Cell treatments
For KPNB1 inhibition, transfected cells were incubated with 16 µM importazole (IPZ; Sigma-Aldrich) for 48 h. To turn off the expression of Tet-off system, 4.5 µM doxycycline (Sigma-Aldrich) was applied. Activation of endogenous calpains was performed as previously described [18]. Briefly, calpains were activated by incubating transfected cells with 1 µM ionomycin (Sigma-Aldrich) and 5 mM CaCl2 for 1 h. To block major proteolytic pathways, transfected cells were treated with 10 µM calpain inhibitor III (CI-III; Sigma-Aldrich), 10 µM broad-spectrum caspase inhibitor Q-VD-OPh (Sigma-Aldrich), 50 nM autophagy inhibitor bafilomycin A1 (InvivoGen, Toulouse, France), or 10 µM proteasomal inhibitor lactacystin (Enzo Life Sciences, Lausen, Switzerland) for 18 h prior to harvesting.
Cell viability assay
Cell viability was evaluated using the resazurin-based PrestoBlue assay (Thermo Fisher Scientific) according to the manufacturer’s instructions. In brief, 5000 ATXN3 KO HEK 293T cells per well were seeded in a ViewPlate-96 Black (PerkinElmer, Waltham, MA, US) and transfected on the next day. After 72 h, culture medium was aspirated and 100 µl fresh medium containing PrestoBlue reagent in a ratio of 1:10 was added. Cells were incubated at 37 °C and 5% CO2 for 45 min. Afterwards, fluorescence signals were measured at 535 nm (excitation)/620 nm (emission) using an EnVision Multimode Plate Reader and the EnVision Manager software version 1.13.3009.1401 (both PerkinElmer).
Protein lysate preparation
To prepare cell lysates, cells were harvested by removing the media, dissociating the cell layer by pipetting using cold Dulbecco’s phosphate-buffered saline (DPBS; Gibco, Thermo Fisher Scientific), and centrifugation at 300xg for 5 min. RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate) containing cOmplete protease inhibitor EDTA-free (Hoffmann-La Roche, Basel, Switzerland) was added to the cell pellets, incubated on ice for 25 min (vortexing every 5 min), and centrifuged at 16,000xg for 15 min at 4 °C. In the final step, the supernatant was transferred to a fresh reaction tube, supplemented with 10% glycerol, and stored at − 80 °C until further analysis. To obtain mouse brain lysates, brain hemispheres were homogenized in RIPA buffer containing cOmplete protease inhibitor EDTA-free using the ULTRA-TURRAX disperser (VWR International, Ulm, Germany). Subsequently, homogenates were centrifuged at 16,000xg for 30 min at 4 °C and followed as described. Protein concentrations of cell and mouse brain lysates were determined using Bradford protein assay (Bio-Rad Laboratories, Hercules, CA, US).
Western blot analysis
30 μg protein extract were mixed with 4 x LDS sample buffer (1 M Tris-base pH 8.5, 2 mM EDTA, 8% lithium dodecyl sulfate (LDS), 40% glycerol, 0.025% phenol red) supplemented with 100 mM dithiothreitol (DTT) and heat-denatured for 10 min at 70 °C. Protein samples were subjected to electrophoretic separation using 10% Bis–Tris gels and MES SDS (50 mM MES, 50 mM Tris-base pH 7.3, 0.1% SDS, 1 mM EDTA) or MOPS SDS (50 mM MOPS, 50 mM Tris-Base pH 7.7, 0.1% SDS, 1 mM EDTA) running buffer. Afterwards, proteins were transferred onto Amersham Biosciences Protran Premium 0.2 µm nitrocellulose membranes (GE Healthcare) using Bicine/Bis–Tris transfer buffer (25 mM Bicine, 25 mM Bis–Tris pH 7.2, 1 mM EDTA) containing 15% methanol at 80 V for 90 min. After transfer, membranes were blocked using 5% skim milk (Sigma-Aldrich) in Tris-buffered saline (TBS; 10 mM Tris pH 7.5, 150 mM NaCl) for 1 h at room temperature and incubated with primary antibody diluted in TBS-T (TBS containing 0.1% Tween 20) overnight at 4 °C. For visualization, fluorescence-conjugated secondary antibodies were added to the membranes for 1 h at room temperature. Fluorescence signals were detected using the LI-COR ODYSSEY FC system (LI-COR Biosciences) and a ratio of target protein to loading control was quantified using Image Studio 5.2 software (LI-COR Biosciences).
Filter retardation assay
Formation of SDS-insoluble aggregates was analyzed by filter retardation assay as described before [18]. ATXN3 KO HEK 293T cells were harvested 72 h post-transfection and homogenized in lysis buffer (DPBS supplemented with 1% Triton X-100 and cOmplete protease inhibitor EDTA-free) by ultra-sonication for 10 s. Protein concentration of cell homogenates was measured using Bradford protein assay. 10 µg protein extract were diluted in total volume of 100 µl DPBS supplemented with 2% SDS and 50 mM DTT. Samples were boiled for 5 min at 95 °C and cooled down at room temperature to avoid precipitation of SDS. Afterwards, samples were filtered through Amersham Biosciences Protran 0.45 µm nitrocellulose membranes (GE Healthcare) using a Minifold II Slot Blot System (GE Healthcare). Membranes were blocked using 5% skim milk in TBS, and immunodetection was conducted as described in the western blot analysis.
Nucleocytoplasmic fractionation assay
To investigate the subcellular localization of ataxin-3, cytoplasmic and nuclear fractions were separated according to the Rapid, Efficient And Practical (REAP) fractionation method [34] with minor modifications [18]. ATXN3 KO HEK 293T cells were harvested 72 h post-transfection using DPBS and centrifuged at 300xg for 5 min. Cell pellets were triturated in lysis buffer (DPBS supplemented with 0.1% NP-40 and cOmplete protease inhibitor EDTA-free), and an aliquot of cell suspension was taken as the whole cell lysate. The residual volume was centrifuged at 10,000xg for 10 s and the supernatant was collected as the cytoplasmic fraction. Whole cell lysate and cytoplasmic fraction were mixed with 4 x LDS sample buffer and 100 mM DTT. The pellet was rinsed once with lysis buffer and resuspended in 1 x LDS sample buffer and 100 mM DTT, representing the nuclear fraction. All samples were heat-denatured for 10 min at 70 °C followed by ultra-sonication for 10 s. Ultimately, samples were subjected to western blot analysis and immunodetection according to the described protocol.
GST pull-down assay
E. coli BL21 bacteria containing pGEX-6P-1-MJD15Q and pGEX-6P-1-MJD77Q were cultured overnight at 37 °C, and protein expression was induced by adding 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 2 h. Bacteria were centrifuged at 1500xg for 5 min. Pellets were rinsed once and resuspended in GST binding buffer (4.2 mM NaH2PO4, 2 mM KH2PO4, 140 mM NaCl, 10 mM KCl, 1% NP-40) containing cOmplete protease inhibitor EDTA-free. Bacterial lysates were prepared by ultra-sonication for 15 s followed by centrifugation at 16,000xg for 10 min at 4 °C. Eukaryotic protein extracts were obtained by harvesting HEK 293T cells using DPBS and centrifugation at 300xg for 5 min at 4 °C. Pellets were resuspended and lysed in GST binding buffer and centrifuged at 16,000xg for 15 min at 4 °C. Protein concentrations of HEK 293T cells and bacterial lysates were determined using Bradford protein assay. 500 µg prokaryotic protein extract containing GST-15Q or GST-77Q ataxin-3 were immobilized on MagneGST Glutathione Particles (Promega, Walldorf, Germany) for 1 h at 4 °C followed by four times washing with GST binding buffer at 4 °C. Then, MagneGST Glutathione Particles were incubated with 500 µg HEK 293T cells protein extract overnight at 4 °C and washed four times with GST binding buffer. Trapped proteins were eluted from the particles by boiling in 1 x LDS sample buffer containing 100 mM DTT for 5 min at 95 °C and analyzed by western blotting and immunodetection.
Immunofluorescence staining
ATXN3 KO HEK 293T cells were grown on poly-L-lysine-coated glass coverslips and transfected with respective constructs for 72 h. For cell fixation, medium was aspirated and incubation with 4% paraformaldehyde (PFA) in DPBS was performed for 15 min at room temperature. Cells were washed by DPBS briefly, blocked and permeabilized using DPBS (supplemented with 10% BSA and 0.5% Triton X-100) for 1 h at room temperature. Primary antibody diluted in DPBS supplemented with 1% BSA and 0.5% Triton X-100 was added to the fixed cells and incubated overnight at 4 °C. The cells were washed with cold DPBS four times and incubated with fluorescence-tagged secondary antibody (Alexa Fluor 555) for 1 h at room temperature. Subsequently, cells were washed with cold DPBS four times and mounted using VECTASHIELD Antifade Mounting Medium containing DAPI (Vector Laboratories, Peterborough, UK). Fluorescent images were taken with an Axioplan 2 Imaging System, AxioCam MRm (Carl Zeiss Microscopy, Jena, Germany), 400 × magnification, and AxioVision version 4.8 imaging software (Carl Zeiss Microscopy). 20 fields of vision with at least 10 GFP-positive cells were photographed per coverslip under blinded condition. The number of cells with aggregates was counted manually and calculated as percentage of total GFP-positive cells.
Protein in-gel digestion
ATXN3 KO HEK 293T cells were transfected for 72 h in three biological replicates. The whole cell lysates were purified with SDS-PAGE (Thermo Fisher Scientific), and the gel was stained using NOVEX Colloidal Blue Staining Kit (Thermo Fisher Scientific). Stained gel pieces were excised and in-gel digested using trypsin as described previously [35]. Tryptic peptides were desalted using C18-StageTips [36] and subjected to liquid chromatography and tandem mass spectrometry (LC–MS/MS) analysis.
Mass spectrometry and data processing
LC–MS/MS analysis was conducted on an Easy nano-LC (Thermo Fisher Scientific) coupled to an QExactiveHF mass spectrometer (Thermo Fisher Scientific) as described before [37]. Peptides were eluted applying a 60-min segmented gradient with a flow rate of 200 nl/min. The 20 most intensive peaks were selected for fragmentation with HCD.
MS data from all three replicates were processed utilizing MaxQuant software suite version 1.6.7.0 [38]. Database search was carried out using the Andromeda search engine [39], which is integrated in MaxQuant. MS/MS spectra were searched against a target-decoy human UniProt database comprising of 96,817 protein entries and 245 commonly observed contaminants. Cysteine carbamidomethylation was considered as the static modification, while methionine oxidation and acetylation of N-terminal residues were set as variable modifications. Initial mass tolerance was set to 4.5 ppm and 0.5 Da for precursor and fragment ions, respectively. Peptide and modification site identifications were determined at a false discovery rate (FDR) of 0.01, interpreted by the target-decoy approach [40]. Label-free algorithm was enabled, as was the “match between runs” option for samples within one biological replicate [41]. Label-free quantification (LFQ) protein intensities from the MaxQuant data output were employed for relative protein quantification. Bioinformatic analysis (Student’s t-test) was performed using the Perseus software package version 1.6.2.3 [42]. A p-value ≤ 0.05 was considered statistically significant and SO was set to 0 (SO = 0). Volcano plots were generated with FDR ≤ 0.01 and randomization was set to 250. Activation and inhibition of cellular pathways upon KPNB1 overexpression was predicted using the Ingenuity Pathway Analysis (IPA; QIAGEN).
Statistical analysis
Data are presented as mean ± SEM. Statistical analysis was conducted using GraphPad Prism 6.00 software (GraphPad Software Inc., La Jolla, CA, US). Statistical significance of data sets was evaluated using one sample t-test, two-tailed Student’s t-test, or one-way ANOVA with Tukey’s post-test. Significance was considered at p-value ≤ 0.05. Statistical outliers determined by ROUT test (Q = 1%) were excluded from analysis.
Results
Ataxin-3 interacts with karyopherins, but KPNB1 modulation does not affect its intracellular localization
Nuclear shuttling of ataxin-3 is considered as a principal modulator of the MJD pathogenesis [22]. In our previous study, we have shown that KPNA3 has a leading role in the nucleocytoplasmic trafficking of ataxin-3, influencing its aggregation and toxicity [27]. Given that KPNA3 is an adaptor protein for KPNB1 to execute its transport function [24], we sought to study the impact of KPNB1 modulation on ataxin-3. In the first step, we tested whether ataxin-3 interacts directly with KPNB1 as well as KPNA3. For this, we performed a GST pull-down assay using GST-tagged wild-type (15Q) and polyQ-expanded (77Q) ataxin-3 overexpressed in E. coli and lysates of HEK 293T cells, which endogenously express KPNB1 and KPNA3. Western blot analysis of the GST pull-down showed that KPNB1, along with KPNA3, were coprecipitated with both 15Q and 77Q ataxin-3 (Fig. 1a), representing an important indication of a direct functional interaction between ataxin-3 and karyopherins. Next, we investigated the effect of KPNB1 overexpression on subcellular localization of ataxin-3 using fluorescence microscopy and nucleocytoplasmic fractionation. For this purpose, we transfected ATXN3 KO HEK 293T cells with 15Q and 148Q ataxin-3 together with KPNB1 or an empty vector as a control. ATXN3 KO HEK 293T cells were applied for most experiments to avoid the interference of endogenous ataxin-3, otherwise the cell type is mentioned. To our surprise, overexpression of KPNB1 did not show any significant alteration of the intracellular localization of ataxin-3 (Fig. 1b–d). However, KPNB1 overexpression led to an apparent reduction of both nuclear and cytoplasmic 15Q and 148Q ataxin-3 as observed in the western blot analysis of the nucleocytoplasmic fractionation assay (Figs. 1c, d, and S1a, b). Furthermore, the detections revealed the generation of specific ataxin-3 fragments, best visible in the cytoplasmic fraction (Fig. 1c, d). To assess the effect of KPNB1 knockdown on subcellular localization of ataxin-3, cells were cotransfected with either 15Q or 148Q ataxin-3 and endoribonuclease-prepared siRNA (esiRNA) targeting KPNB1 or Renilla luciferase (esiLUC) as a respective control. KPNB1 knockdown did not modulate the localization of ataxin-3, although in contrast to KPNB1 overexpression, an increase of both nuclear and cytoplasmic ataxin-3 was detected in these cells (Fig. S2a, b).

Ataxin-3 interacts with karyopherins, but KPNB1 overexpression does not affect the intracellular localization of ataxin-3. a Interaction of wild-type (15Q) and polyQ-expanded (77Q) ataxin-3 with KPNB1 and its partner KPNA3 was validated by GST pull-down assay. GST-tagged 15Q and 77Q ataxin-3 were purified using glutathione beads followed by incubation with wild-type HEK 293T cell lysates. Input as well as pull-down samples were assessed by western blot analysis. Empty GST was applied as negative control. White Bullets (o) indicate nonspecific bands. b Fluorescence microscopy was performed to visualize the subcellular localization of wild-type and polyQ-expanded ataxin-3 upon KPNB1 overexpression. ATXN3 KO HEK 293T cells co-expressing either EGFP-ataxin-3 15Q or EGFP-ataxin-3 148Q and KPNB1 were analyzed 72 h post-transfection. Microscopy indicated that the intracellular localization of neither wild-type (15Q) nor polyQ-expanded (148Q) ataxin-3 was altered upon KPNB1 overexpression. Blue and green channels show DAPI as nuclear counterstain and GFP signals, respectively. 400 × magnification, scale bar = 20 µm. c, d Nucleocytoplasmic fractionation assay demonstrates a reduction of cytoplasmic as well as nuclear wild-type (15Q) and polyQ-expanded (148Q) ataxin-3 upon KPNB1 overexpression. However, no alteration was observed in the localization of ataxin-3. Moreover, accumulation of mainly cytoplasmic ataxin-3 fragments (black arrowheads) in KPNB1 overexpressing cells was observed. ATXN3 KO HEK 293T cells were cotransfected with either 15Q or 148Q ataxin-3 and KPNB1 or empty vectors. 72 h post-transfection, nucleocytoplasmic fractionation was performed followed by western blot analysis. Blots are shown in low and high exposures. White bullet (o) marks nonspecific bands. Ataxin-3 was detected by 1H9 and C-terminal antibodies. The diagrams illustrate the ratio of nuclear and cytoplasmic ataxin-3 as percentage. GAPDH and lamin A/C were applied as cytoplasmic and nuclear loading controls, respectively. c, n = 4, Cyt/Nuc, unpaired t-test, p = 0.6261; d, n = 4, Cyt/Nuc, unpaired t-test, p = 0.5443. CTRL empty vector, Cyt cytoplasmic fraction, Nuc nuclear fraction, fl full-length, frg fragment, C-term C-terminal, Exp exposure. Values are displayed as means ± SEM. ns not significant
Overall, modulation of KPNB1 protein levels has a potential to influence ataxin-3 through a mechanism that does not appear to implicate the localization of this protein.
KPNB1 overexpression lowers ataxin-3 protein levels independent of its nuclear transport function
After observing potential effects of KPNB1 overexpression on ataxin-3 protein levels in our nucleocytoplasmic fractionation assays, we sought to investigate in detail whether increasing the levels of KPNB1 has any robust effects on ataxin-3 protein levels. First, we transiently transfected cells with either 15Q or 148Q ataxin-3 and KPNB1 constructs. Western blot analysis revealed that overexpression of KPNB1 was accompanied by a significant reduction in the total amount of both 15Q and 148Q ataxin-3 (Fig. 2a, b). In addition to decreased protein levels of ataxin-3, we confirmed an enhanced fragmentation of ataxin-3 upon KPNB1 overexpression. Using two different antibodies recognizing different epitopes, we concluded that these fragments are C-terminal and containing the polyQ tract (Fig. 2a, b). Further analysis demonstrated that the accumulation of ataxin-3 fragments as well as the reduction of full-length ataxin-3 was time-dependent, with the strongest effect being observed after 72 h of KPNB1 overexpression (Fig. S3a, b).

KPNB1 overexpression leads to a reduction of ataxin-3 protein levels, and mutation of the FxFG binding site in KPNB1 does not abolish this effect on ataxin-3. a, b KPNB1 overexpression decreases soluble levels of both wild-type (15Q) and polyQ-expanded (148Q) ataxin-3 and enhances fragmentation of ataxin-3 in comparison to control. ATXN3 KO HEK 293T cells were cotransfected with either 15Q or 148Q ataxin-3 and KPNB1 or empty vectors for 72 h followed by western blot analysis. Detection of ataxin-3 using two different antibodies (1H9 and C-terminal) which recognize different epitopes revealed that ataxin-3 fragments are C-terminal breakdown products (black arrowheads). Western blot analysis confirmed the overexpression of KPNB1. Blots are shown in low and high exposures. GAPDH was applied as loading control. a, 15Q Atx3, n = 6, one sample t-test, p = 0.0008; KPNB1, n = 6, one sample t-test, p < 0.0001; b, 148Q Atx3, n = 6, one sample t-test, p = 0.0006; KPNB1, n = 5, one sample t-test, p = 0.0349. c, d KPNB1 mutation (I178D) in the FxFG binding site does not change the effect of KPNB1 overexpression on either wild-type or polyQ-expanded ataxin-3. ATXN3 KO HEK 293T cells were cotransfected with either 15Q or 148Q ataxin-3 and wild-type KPNB1 or KPNB1 I178D constructs for 72 h. Western blot analysis displays that the ataxin-3-lowering effect of KPNB1 I178D is comparable to wild-type KPNB1, and it leads to a significant reduction of both 15Q and 148Q ataxin-3 protein levels. GAPDH was applied as loading control. c, n = 5, one sample t-test, CTRL vs KPNB1, p = 0.0030; CTRL vs KPNB1 I178D, p = 0.0086; unpaired t-test, KPNB1 vs KPNB1 I178D, p = 0.4784; d, n = 5, one sample t-test, CTRL vs KPNB1, p = 0.0052; CTRL vs KPNB1 I178D, p = 0.0765; unpaired t-test, KPNB1 vs KPNB1 I178D, p = 0.5367. e Western blot analysis of wild-type HEK 293T cells transfected with either KPNB1 or empty vectors. The result displays that KPNB1 overexpression is accompanied by a reduction in protein levels of endogenous ataxin-3. Vinculin was applied as loading control. n = 6, unpaired t-test, p = 0.0221. f Western blot analysis demonstrates that protein levels of GFP do not change upon KPNB1 overexpression. Wild-type HEK 293T cells were cotransfected with GFP and KPNB1 constructs for 72 h. β-actin was applied as loading control. n = 6, unpaired t-test, p = 0.0756. CTRL empty vector, fl full-length, frg fragment, C-term C-terminal, Exp exposure, egAtx3 endogenous ataxin-3, Rel. relative. Values are displayed as means ± SEM. ns not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001
Nuclear translocation of cargos mediated by transport receptors involves the interaction of karyopherin beta with nuclear pore complex proteins (nucleoporins) containing phenylalanine-rich core motifs (FxFG) [43]. It was shown that mutation of isoleucine 178 (I178) in the FxFG binding site of KPNB1 to the less hydrophobic aspartic acid (D), remarkably hampers both binding and nuclear transport of KPNB1 [31]. KPNB1 I178D as well as wild-type KPNB1 were overexpressed in cells transiently expressing either 15Q or 148Q ataxin-3. The effects on full-length ataxin-3 upon KPNB1 I178D overexpression were dissected by western blot analysis. Interestingly, no major difference was identified between the impact of KPNB1 I178D and wild-type KPNB1 on soluble ataxin-3 (Fig. 2c, d), implying that the observed ataxin-3 lowering is a nuclear transport function-independent mechanism.
Next, we wanted to confirm whether KPNB1 overexpression affects the protein levels of endogenous ataxin-3. Moreover, to determine if the impact of KPNB1 on ataxin-3 is substrate-specific, we analyzed the effect of KPNB1 overexpression on a control protein such as GFP. According to its effects on overexpressed ataxin-3, western blot analysis displayed a significant decrease in the protein levels of endogenous ataxin-3 upon KPNB1 overexpression (Fig. 2e), whereas no alteration was observed in the levels of GFP (Fig. 2f). The above results highlight that ataxin-3 protein levels are generally yet specifically lowered by KPNB1 overexpression. These alterations yield KPNB1 as a putative modulator of ataxin-3 protein levels independent of the KPNB1 nuclear transport function.
Knockdown and pharmacological inhibition of KPNB1 increase ataxin-3 protein levels
In the next step, we explored the consequences of lowering KPNB1 levels on wild-type and polyQ-expanded ataxin-3. Cells were transiently transfected with either 15Q or 148Q ataxin-3 together with esiKPNB1 or esiLUC as a respective control and analyzed by western blotting. In contrast to overexpression of KPNB1, knockdown of this protein resulted in an increase in soluble levels of both full-length 15Q and 148Q ataxin-3 (Figs. 3a, b, and S4).

Knockdown and pharmacological inhibition of KPNB1 elevate ataxin-3 protein levels. a, b Western blot analysis reveals that knockdown of KPNB1 is accompanied by a significant increase in the soluble levels of wild-type (15Q) and polyQ-expanded (148Q) ataxin-3. ATXN3 KO HEK 293T cells were cotransfected with either 15Q or 148Q ataxin-3 and esiKPNB1 or esiLUC (control). Cells were harvested 72 h post-transfection, and cell lysates were subjected to western blotting. GAPDH was applied as loading control. a, n = 5, one sample t-test, p = 0.0567; b, n = 5, one sample t-test, p = 0.0315. c, d KPNB1 was inhibited using 16 µM importazole (IPZ) in either wild-type (15Q) or polyQ-expanded (148Q) ataxin-3 expressing HEK 293T cells. 48 h after treatment, cells were harvested and subjected to western blot analysis. Quantification of blots demonstrates that the soluble levels of both 15Q and 148Q ataxin-3 increase upon IPZ treatment compared with DMSO-treated cells (control). GAPDH was applied as loading control. c, n = 6, one sample t-test, p = 0.0162; d, n = 4, one sample t-test, p = 0.0077. e Western blot analysis shows a significant increase of endogenous ataxin-3 protein levels upon IPZ treatment compared with control. Wild-type HEK 293T cells were treated with either 16 µM IPZ or DMSO for 48 h prior to harvesting. GAPDH was applied as loading control. n = 3, unpaired t-test, p = 0.0009. f Protein levels of ataxin-3 can be rescued by IPZ treatment in KPNB1 overexpressing cells. ATXN3 KO HEK 293T cells were cotransfected with 15Q ataxin-3 and KPNB1 constructs followed by 16 µM IPZ or DMSO treatment for 48 h prior to harvesting. Cells were harvested 72 h post-transfection, and ataxin-3 levels were analyzed by western blotting. GAPDH was applied as loading control. n = 4, one sample t-test, CTRL + DMSO vs KPNB1 + DMSO, p = 0.0129; CTRL + DMSO vs KPNB1 + IPZ, p = 0.0615; unpaired t-test, KPNB1 + DMSO vs KPNB1 + IPZ, p = 0.0021. CTRL empty vector, IPZ importazole, egAtx3 endogenous ataxin-3, Rel. relative. Values are displayed as means ± SEM. ns not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001
To further validate our findings, we targeted KPNB1 by pharmacological inhibition. For this, we employed 2,4-diaminoquinazoline (importazole/IPZ) as a specific and reversible inhibitor of KPNB1. The mode of action of IPZ is likely via altering the interaction of KPNB1 with RanGTP [44]. ATXN3 KO HEK 293T cells transiently expressing 15Q or 148Q ataxin-3 as well as wild-type HEK 293T cells were treated with IPZ, and ataxin-3 protein levels were assessed by western blotting. Treatment with IPZ recapitulated the effects of KPNB1 knockdown on ataxin-3 protein levels, demonstrating significantly elevated levels of soluble endogenous or overexpressed 15Q and 148Q ataxin-3 (Fig. 3c–e). To additionally scrutinize the decrease of ataxin-3 protein levels by KPNB1, cells overexpressing 15Q ataxin-3 together with KPNB1 were treated with IPZ. Interestingly, the reduction of ataxin-3 protein levels upon KPNB1 overexpression was restored via IPZ treatment (Fig. 3f). Altogether, both KPNB1 knockdown and its pharmacological inhibition using IPZ led to an increase in soluble levels of wild-type and polyQ-expanded ataxin-3, while the overexpression of KPNB1 had an opposite effect.
KPNB1 overexpression reduces ataxin-3 stability and gives rise to fragments independent of MJD-associated proteolytic pathways
To unravel whether KPNB1 overexpression decreases ataxin-3 stability, we applied the Tet-off system for abrogating the expression of 15Q and 77Q ataxin-3 by administering doxycycline in a time-dependent manner. Western blot analysis indicated that the degradation of both wild-type and polyQ-expanded ataxin-3 accelerated significantly in KPNB1 overexpressing cells as observed 48 h after termination of 15Q ataxin-3, and 24 h and 48 h after termination of 77Q ataxin-3 expression. However, no alteration was observed in the generation of ataxin-3 fragments as they remained stable during the course of the assay (Fig. 4a, b). This result motivated us to investigate the fragmentation of ataxin-3 in KPNB1 overexpressing cells more closely.

KPNB1 overexpression decreases the stability of ataxin-3, and observed ataxin-3 fragments are not mediated by the activation of known MJD-associated proteases or proteolytic pathways. a, b Modulation of ataxin-3 stability was analyzed upon KPNB1 overexpression using the Tet-off system. Western blot analysis revealed a significant reduction in the stability of both wild-type (15Q) and polyQ-expanded (77Q) ataxin-3 in KPNB1 overexpressing cells 48 h after termination of ataxin-3 expression compared with control. ATXN3 KO HEK 293T cells were cotransfected with either pTRE-15Q or pTRE-77Q ataxin-3 and KPNB1 or empty vectors. 24 h post-transfection, ataxin-3 expression was shut off using doxycycline (Dox) at different time intervals (0 h, 6 h, 24 h, and 48 h). GAPDH was applied as loading control. a, n = 3, paired t-test, CTRL vs KPNB1 (6 h), p = 0.6029; CTRL vs KPNB1 (24 h), p = 0.6730; CTRL vs KPNB1 (48 h), p = 0.0132; b, n = 4, paired t-test, CTRL vs KPNB1 (6 h), p = 0.3859; CTRL vs KPNB1 (24 h), p = 0.0202; CTRL vs KPNB1 (48 h), p = 0.0427. c Activation of calpains and caspases upon KPNB1 overexpression was evaluated by detecting cleavage of their common substrate α-spectrin protein in wild-type HEK 293T cells. Western blot analysis demonstrates no alteration in the levels of either full-length (white arrowhead) or breakdown products of α-spectrin (black arrowhead). Moreover, ratio between cleaved and full-length protein remained comparable to control in KPNB1 overexpressing cells. GAPDH was applied as loading control. n = 6, unpaired t-test, p = 0.6296. d Breakdown products of α-spectrin (black arrowhead) are induced in ionomycin (IM) treated cells confirming the activation of calpains compared with control. ATXN3 KO HEK 293T cells were cotransfected with either 15Q or 77Q ataxin-3 followed by KPNB1 overexpression or 1 µM IM treatment for 1 h. β-actin was applied as loading control. e IM administration results in the activation of endogenous calpains and cleavage of ataxin-3. Western blot analysis displays that ataxin-3 fragments induced by KPNB1 overexpression (blue boxes and arrowheads) are not comparable to fragments mediated by calpain cleavage (red boxes and arrowheads). Red and blue channels indicate ataxin-3 detected by 1H9 and C-terminal antibodies, respectively. f Western blot analysis demonstrates that inhibition of calpains and caspases or blocking of autophagy and proteasomal degradation do not prevent ataxin-3 fragmentation in KPNB1 overexpressing cells. ATXN3 KO HEK 293T cells cotransfected with 15Q ataxin-3 and KPNB1 or empty vectors were incubated with 10 µM calpain inhibitor III (CI-III), 10 µM caspase inhibitor (Q-VD-OPh), 50 nM autophagy inhibitor bafilomycin A1 (BafA1), or 10 µM proteasomal inhibitor lactacystin (Lac) for 16 h prior to harvesting. Cells incubated with DMSO were considered as control. The diagram illustrates quantification of ataxin-3 fragments in KPNB1 overexpressing cells. GAPDH was applied as loading control. n = 5, one sample t-test, DMSO vs CI-III, p = 0.3708; DMSO vs Q-VD, p = 0.3136; DMSO vs BafA1, p = 0.4063; DMSO vs Lac, p = 0.2682. CTRL empty vector, fl full-length, frg fragment, Dox Doxycycline, C-term C-terminal, f-Nt fragment-N-terminal, IM ionomycin, CI-III calpain inhibitor III, BafA1 bafilomycin A1, Lac lactacystin, Rel. relative. Values are displayed as means ± SEM. ns not significant; *p ≤ 0.05
Proteolytic degradation and cleavage of polyQ proteins such as ataxin-3, carried out by caspases and calpains or proteolytic pathways such as autophagy and proteasomes, are involved in the pathogenesis of polyQ diseases [45,46,47]. Notably, the activation of caspases and calpains has been identified in numerous MJD models [17, 48]. Therefore, we postulated that the elevated fragmentation of ataxin-3 in KPNB1 overexpressing cells might be mediated by the activation of caspases or calpains. For this purpose, the cleavage of α-spectrin, a natural substrate of caspases and calpains [49] was evaluated in KPNB1 overexpressing wild-type HEK 293T cells. Western blot analysis displayed that neither full-length nor breakdown products of α-spectrin were modulated by KPNB1 overexpression, and the ratio between them was comparable to control (Fig. 4c). To compare the cleavage pattern of ataxin-3 followed by KPNB1 overexpression and calpain activation, a cell-based calpain activation assay was conducted using the ionophore ionomycin (IM). Cells transiently expressing 15Q or 77Q ataxin-3 were subjected to either KPNB1 overexpression or IM treatment to trigger the activation of endogenous calpains, which was confirmed by increased α-spectrin cleavage (Fig. 4d). Using two different antibodies recognizing distinct epitopes, we observed that the cleavage pattern of 15Q or 77Q ataxin-3 followed by activation of calpains differed from ataxin-3 fragments induced by KPNB1 overexpression (Fig. 4e). Moreover, inhibition of calpains and caspases or blocking of autophagic and proteasomal degradation did not prevent the formation of KPNB1 overexpression-associated fragments (Figs. 4f, and S5).
These results provide evidence that the enhancement of ataxin-3 lowering and fragmentation in KPNB1 overexpressing cells does not occur via the activation of known MJD-associated proteases or proteolytic pathways.
KPNB1 overexpression diminishes ataxin-3 aggregation and rescues cell viability
Aggregation of polyQ-expanded ataxin-3 is a hallmark of MJD, and measuring the amount of protein aggregates can provide information on protein toxicity and disease progression [50]. As KPNB1 overexpression strongly reduced soluble levels of both wild-type and polyQ-expanded ataxin-3, we sought to investigate its consequences on the protein aggregation. Thus, aggregation of 148Q ataxin-3 upon KPNB1 overexpression was evaluated by fluorescence microscopy of cells transfected with EGFP-ataxin-3 148Q and KPNB1 constructs (Fig. 5a). In cells overexpressing KPNB1 the percentage of GFP-positive cells with aggregates reduced significantly compared with control (Fig. 5b). This finding was also confirmed by filter retardation assay, where a significant reduction of polyQ-expanded ataxin-3 aggregation was detected in KPNB1 overexpressing cells (Fig. 5c). Moreover, PrestoBlue assay revealed that KPNB1 overexpression rescues the impaired viability of 148Q ataxin-3 expressing cells compared with control (Fig. 5d). In line with the effects on soluble polyQ-expanded ataxin-3, overexpression of mutant KPNB1 (I178D) led to the reduction of ataxin-3 aggregates as well as overexpression of wild-type KPNB1 (Fig. 5e). Conversely, aggregation levels of 148Q ataxin-3 upon KPNB1 knockdown or inhibition with IPZ increased remarkably (Fig. 5f, g), corresponding to the increased soluble levels of polyQ-expanded ataxin-3.

KPNB1 modulation affects ataxin-3 aggregation levels. a, b Fluorescence microscopy was conducted to visualize the alteration of polyQ-expanded ataxin-3 aggregation in KPNB1 overexpressing cells. ATXN3 KO HEK 293T cells co-expressing EGFP ataxin-3 148Q and KPNB1 were fixed 72 h post-transfection, and the number of GFP-positive (EGFP+) cells with and without aggregates was counted manually in 20 fields of vision. The analysis showed a decrease of cells with ataxin-3 aggregates upon KPNB1 overexpression. Blue, green, and red channels show DAPI as nuclear counterstain, GFP, and KPNB1 signals, respectively. White arrowheads mark ataxin-3 aggregates. 400 × magnification, scale bar = 20 µm. The diagram shows the percentage of aggregates in EGFP+ cells. n = 4, unpaired t-test, p = 0.0123. c Filter retardation assay of ATXN3 KO HEK 293T cells co-expressing polyQ-expanded (148Q) ataxin-3 and KPNB1 for 72 h demonstrated a reduction of polyQ-expanded ataxin-3 aggregates compared with control. n = 6, one sample t-test, p = 0.0004. d PrestoBlue assay of ATXN3 KO HEK 293T cells cotransfected with either 15Q or 148Q ataxin-3 and KPNB1 or empty vectors. The viability of cells expressing 148Q ataxin-3 was rescued upon KPNB1 overexpression. Viability was normalized to 15Q ataxin-3 expressing cells. n = 4, one sample t-test, 15Q + CTRL vs 148Q + CTRL, p = 0.0144; 15Q + CTRL vs 148Q + KPNB1, p = 0.0882; paired t-test, 148Q + CTRL vs 148Q + KPNB1, p = 0.0138. e Filter retardation assay of ATXN3 KO HEK 293T cells co-expressing 148Q ataxin-3 and either wild-type or KPNB1 I178D indicates a decrease in the aggregation of 148Q ataxin-3. n = 6, one sample t-test, CTRL vs KPNB1, p = 0.0030; CTRL vs KPNB1 I178D, p = 0.0247; unpaired t-test, KPNB1 vs KPNB1 I178D, p = 0.1883. f Knockdown of KPNB1 using esiRNA shows an increased load of aggregated 148Q ataxin-3 using filter retardation assay. ATXN3 KO HEK 293T cells were cotransfected for 72 h with 148Q ataxin-3 and esiKPNB1 or esiLUC as control. n = 5, one sample t-test, p = 0.0054. g Filter retardation assay shows increased ataxin-3 aggregation in ATXN3 KO HEK 293T cells expressing 148Q ataxin-3 for 72 h and treated with 16 µM IPZ for 48 h. Values were normalized to DMSO-treated cells. n = 5, one sample t-test, p = 0.0279. CTRL empty vector, IPZ importazole, Rel. relative. Values are displayed as means ± SEM. ns not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001
KPNB1 lowers ataxin-3 protein levels via activation of the mitochondrial protease CLPP
Given that KPNB1 overexpression leads to reduction of ataxin-3 protein levels and enhancement of ataxin-3 fragmentation, a question remains concerning which factor directly mediates this effect. To figure out the downstream pathways that might be implicated in this process, unbiased label-free quantitative proteomics was carried out using cells transiently overexpressing either 15Q or 148Q ataxin-3 and KPNB1 or empty vectors (control). Statistical analysis indicated that 23 proteins were upregulated, and 69 proteins downregulated significantly in cells overexpressing 15Q ataxin-3 and KPNB1 compared with control. In addition, 44 upregulated and 127 downregulated proteins were revealed in cells expressing 148Q ataxin-3 upon KPNB1 overexpression, and amongst them, 15 proteins (3 upregulated and 12 downregulated) were shared between 15Q and 148Q ataxin-3 expressing cells which are listed in Table S1 (Fig. 6a–d, and Table S1). To get insight into upstream regulators and identification of cellular processes that have been activated or inhibited, we applied the Ingenuity Pathway Analysis (IPA). Accordingly, activation of mitochondrial protease CLPP was predicted by IPA in both 15Q (z-score = 2, p = 1.43e − 04), and 148Q (z-score = 3.464, p = 7.52e − 14) ataxin-3 expressing cells upon KPNB1 cotransfection, as target proteins of CLPP (Table S2) were modulated.

KPNB1 lowers ataxin-3 levels via activation of the mitochondrial protease CLPP. a–c Label-free quantitative proteomics of cells expressing ataxin-3 reveals activation of mitochondrial protease CLPP upon KPNB1 overexpression. Volcano plots indicating the upregulated and downregulated proteins in ATXN3 KO HEK 293T cells cotransfected with either wild-type (15Q) or polyQ-expanded (148Q) ataxin-3 and KPNB1 or empty vectors. Volcano plots illustrate a direct comparison between the p-value and fold-change of the proteins. Purple circles show proteins with significantly altered levels upon KPNB1 overexpression. The thick dashed line represents the cut-off value at p = 0.05. n = 3, two-sample t-test, p ≤ 0.05. d Venn diagram displays the number of common and unique proteins which have been significantly upregulated or downregulated upon KPNB1 overexpression. e, f Western blot analysis demonstrates that both 15Q and 148Q ataxin-3 protein levels can be rescued partly by knockdown of CLPP in KPNB1 overexpressing cells. ATXN3 KO HEK 293T cells were cotransfected with either 15Q or 148Q ataxin-3, KPNB1, and esiCLPP or esiLUC as control. Cells were harvested 72 h post-transfection and protein levels assessed by western blotting. GAPDH was applied as loading control. e, 15Q Atx3, n = 6, one sample t-test, CTRL + esiLUC vs KPNB1 + esiLUC, p < 0.0001; CTRL + esiLUC vs KPNB1 + esiCLPP, p = 0.0056; unpaired t-test, KPNB1 + esiLUC vs KPNB1 + esiCLPP, p = 0.0264; CLPP, n = 6, one sample t-test, CTRL + esiLUC vs KPNB1 + esiLUC, p = 0.1619; CTRL + esiLUC vs KPNB1 + esiCLPP, p < 0.0001; unpaired t-test, KPNB1 + esiLUC vs KPNB1 + esiCLPP, p < 0.0001; f n = 6, one sample t-test, CTRL + esiLUC vs KPNB1 + esiLUC, p < 0.0001; CTRL + esiLUC vs KPNB1 + esiCLPP, p = 0.0020; unpaired t-test, KPNB1 + esiLUC vs KPNB1 + esiCLPP, p = 0.0219. g Knockdown of CLPP in ATXN3 KO HEK 293T cells cotransfected with 148Q ataxin-3 and KPNB1 for 72 h counteracts the KPNB1-induced lowering of polyQ-expanded ataxin-3 aggregates compared with control. n = 6, one sample t-test, CTRL + esiLUC vs KPNB1 + esiLUC, p = 0.0005; CTRL + esiLUC vs KPNB1 + esiCLPP, p = 0.0107; paired t-test, KPNB1 + esiLUC vs KPNB1 + esiCLPP, p = 0.0187. CTRL empty vector, FC fold-change, Rel. relative. Values are displayed as means ± SEM. ns not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001
As CLPP is a nuclear encoded mitochondrial protease involved in the degradation of unfolded and misfolded proteins [51], we speculated that the reduction of ataxin-3 protein levels in KPNB1 overexpressing cells might be directly associated with the activation of CLPP. Therefore, we validated our hypothesis by knockdown of CLPP using specific esiRNA (esiCLPP) in KPNB1 overexpressing cells. Intriguingly, knockdown of CLPP, as achieved by 47% in esiCLPP-transfected cells, counteracted the reduction of full-length 15Q and 148Q ataxin-3 protein levels (Fig. 6e, f). Filter retardation assay demonstrated an elevated polyQ-expanded ataxin-3 aggregation upon CLPP knockdown in KPNB1 overexpressing cells in comparison to control cells cotransfected with esiLUC (Fig. 6g). To further validate our findings, CLPP was knocked down in 15Q and 148Q ataxin-3 expressing cells, which led to a significant increase in ataxin-3 protein levels (Fig. 7a, b). Conversely, overexpression of CLPP reduced protein levels of both 15Q and 148Q ataxin-3, and this effect was boosted upon additional overexpression of KPNB1 (Fig. 7c, d). These results reinforce the idea that modulation of ataxin-3 by KPNB1 overexpression is likely mediated by the activation of mitochondrial protease CLPP.

CLPP modulation affects ataxin-3 protein levels. a, b Western blot analysis reveals that knockdown of CLPP is accompanied by an increase in protein levels of both wild-type (15Q) and polyQ-expanded (148Q) ataxin-3. ATXN3 KO HEK 293T cells were cotransfected with either 15Q or 148Q ataxin-3 and esiCLPP or esiLUC (control) for 72 h. GAPDH was used as loading control. a, n = 6, one sample t-test, p < 0.0001; b, n = 6, one sample t-test, p = 0.0009. c, d CLPP overexpression reduces both 15Q and 148Q ataxin-3 protein levels and its effect is enhanced by KPNB1 overexpression. ATXN3 KO HEK 293T cells were cotransfected with either 15Q or 148Q ataxin-3 and CLPP or both CLPP and KPNB1 constructs for 72 h. GAPDH was applied as loading control. c, n = 4, one-way ANOVA with Tukey’s post-test, CTRL vs CLPP, p = 0.0089; CTRL vs CLPP + KPNB1, p < 0.0001; CLPP vs CLPP + KPNB1, p = 0.0009; d, n = 4, one-way ANOVA with Tukey’s post-test, CTRL vs CLPP, p = 0.0004; CTRL vs CLPP + KPNB1, p < 0.0001; CLPP vs CLPP + KPNB1, p < 0.0001. CTRL empty vector, C-term C-terminal, Rel. relative. Values are displayed as means ± SEM. **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001
MJD mouse models and iPSCs of MJD patients exhibit reduced KPNB1 protein levels
As we demonstrated KPNB1 as an important modulator of wild-type and polyQ-expanded ataxin-3 protein levels, we sought to investigate its endogenous levels in animal models of MJD. For this purpose, we examined KPNB1 protein levels in the cerebral cortex and whole brain of 5-month-old CaMKII/MJD77 and 15-month-old YAC transgenic mice, respectively. Intriguingly, western blot analysis showed a reduction in KPNB1 protein levels in both MJD mouse models compared with respective controls (Fig. 8a, b). Subsequently, further assessment of KPNB1 and CLPP protein levels was conducted using a patient-derived induced pluripotent stem cell (iPSC) model of MJD. Our findings indicated that both KPNB1 and CLPP protein levels are decreased in MJD patient-derived iPSCs in comparison to healthy controls (Fig. 8c). Investigation of neuronal tissue from two different MJD mouse models and iPSCs of MJD patients laid emphasis on a pathological dysregulation of KPNB1 and CLPP in MJD.

MJD transgenic mouse models and iPSCs of MJD patients exhibit reduced KPNB1 protein levels. a Western blot analysis of whole brain protein extracts of 15-month-old YAC transgenic mice indicates a significant reduction of KPNB1 protein levels compared with controls. β-actin was used as loading control. n = 4–5, unpaired t-test, p = 0.0257. b Western blot analysis of cortical lysates of 5-month-old CaMKII/MJD77 transgenic mice confirms decreased protein levels of KPNB1 in comparison to controls. β-actin was applied as loading control. n = 4–6, unpaired t-test, p = 0.0175. c Western blot analysis of induced pluripotent stem cells (iPSCs) from three MJD patients and three healthy controls demonstrates a reduction of both KPNB1 and CLPP protein levels. β-actin was applied as loading control. n = 3 replicates of 3 patients and 3 controls, unpaired t-test, KPNB1, p = 0.0410; CLPP, p = 0.0325. WT wild-type, iPSCs induced pluripotent stem cells, CTRL healthy control, Rel. relative. Values are displayed as means ± SEM. *p ≤ 0.05
Taken together, we demonstrate that KPNB1 is a modulator of ataxin-3 protein levels, cleavage, and aggregation. Our findings highlight that KPNB1 overexpression lowers ataxin-3 protein levels likely via activation of the mitochondrial protease CLPP (Fig. 9).

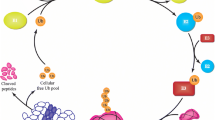
Schematic overview of the proposed ataxin-3 degrading pathway mediated by KPNB1 and CLPP. a KPNB1 overexpression activates mitochondrial protease CLPP, leading to the degradation and reduction of ataxin-3 protein levels. Likewise, overexpression of CLPP has a comparable effect on ataxin-3 protein levels. Cleavage of peptides by the protease component of the Clp complex is an ATP-dependent process [76]. b KPNB1 knockdown and pharmacological inhibition as well as CLPP knockdown prevent ataxin-3 degradation via the proposed pathway. OE overexpression, KD knockdown, Inh. inhibition
Discussion
The appropriate subcellular localization of proteins is essential for their physiological functions and the maintenance of cellular homeostasis [52]. In recent years, there has been a growing appreciation for the idea that dysregulation of the nucleocytoplasmic transport machinery plays a critical role in the pathogenesis of various neurodegenerative diseases. A close relationship between perturbed nuclear pore complexes (NPCs) and pathology of HD and amyotrophic lateral sclerosis (ALS) has been demonstrated [53, 54]. Apart from this, a defective nucleocytoplasmic transport has been indicated in Alzheimer’s disease (AD) [55], Parkinson’s disease (PD) [56], frontotemporal dementia (FTD) [57], and SCA1 [58].
The preferential accumulation of polyQ-expanded proteins in the nucleus of neurons and the formation of intranuclear inclusions are the pathological hallmarks of MJD and other polyQ diseases, leading to neurotoxicity [59]. It has been suggested that the intrinsically low activity of neuronal nuclear ubiquitin–proteasome system (UPS) may account for the nuclear accumulation of polyQ-expanded proteins [60], even though the precise mechanisms remained enigmatic. In this regard, evaluation of the nucleocytoplasmic transport in MJD and other polyQ diseases might unravel its pathological implication and therapeutic targetability.
Previous investigations by our laboratory revealed KPNA3 as a key player in nucleocytoplasmic transport of ataxin-3 and manifestation of symptoms in MJD animal models [27], encouraging us to study the relevance of other karyopherins on ataxin-3 subcellular localization as well. Here, we demonstrated a physical interaction of KPNA3 and KPNB1 with ataxin-3. However, our further analysis of KPNB1 showed no effect of its modulation on the subcellular distribution of either wild-type or polyQ-expanded ataxin-3. Instead, we observed a marked reduction of wild-type and polyQ-expanded ataxin-3 soluble protein levels and enhancement of ataxin-3 cleavage upon KPNB1 overexpression, which is more prominent in wild-type ataxin-3. This effect was not compromised by mutating the FxFG binding site of KPNB1, indicating a nuclear transport function-independent mechanism behind ataxin-3 reduction. Conversely, KPNB1 knockdown and its pharmacological inhibition using importazole resulted in an increase of ataxin-3 protein levels. Given that KPNB1 modulates both wild-type and polyQ-expanded ataxin-3, it seems to be a polyQ-independent and therefore physiological mechanism.
Furthermore, we analyzed the nature of the observed KPNB1-induced ataxin-3 fragments. Ataxin-3 cleavage products have been detected both in the brains of MJD patients and MJD mouse models [18, 61]. Both caspases and calpains have been associated with proteolysis of ataxin-3 in the MJD context [62]. Also, ataxin-3 has been described as a substrate for autophagic and proteasomal degradation [46, 47]. However, we could not substantiate an involvement of either class of proteases in ataxin-3 fragmentation upon KPNB1 overexpression, or the autophagic or proteasomal machineries, indicating the contribution of a yet unknown proteolytic enzyme.
We postulated that modulating KPNB1 might influence the aggregation of polyQ-expanded ataxin-3 as well as its soluble levels. Accordingly, KPNB1 overexpression diminished aggregation and improved the cell viability, whereas KPNB1 knockdown and pharmacological inhibition had the opposite effect on ataxin-3 aggregates. As we observed a modulation of full-length ataxin-3 upon KPNB1 overexpression or knockdown, we concluded that the changes in polyQ-expanded ataxin-3 aggregation were due to its altered soluble levels, as the concentration of polyQ-expanded ataxin-3 has direct consequences for aggregate formation [63].
Interestingly, we observed reduced KPNB1 protein levels in neuronal tissue of two different MJD transgenic mouse models and reduction of both KPNB1 and CLPP protein levels in iPSCs derived from MJD patients, which provides evidence for a pathological dysregulation of KPNB1 and CLPP in MJD.
To further dissect the KPNB1-mediated effects on ataxin-3 levels, we performed label-free quantitative proteomics followed by pathway analysis and discovered a potential activation of the mitochondrial protease CLPP in KPNB1 overexpressing cells. CLPP is a serine protease which plays a pivotal role in the degradation and regulation of synthesis of proteins that are related to cellular metabolic pathways, such as electron transport chain. Nonetheless, little is known about the implication of this mitochondrial protease in cellular pathways and mechanisms [64, 65]. We could corroborate our findings by esiRNA-mediated knockdown of CLPP in KPNB1 overexpressing cells, which led to the rescue of soluble and aggregated forms of ataxin-3. In addition, CLPP overexpression reduced ataxin-3 protein levels, which was further enhanced by KPNB1 cotransfection in these cells. Previous investigations have shown that loss of CLPP is implicated in α-synuclein-associated pathology in induced neurons derived from PD patients. Conversely, overexpression of CLPP inhibits the accumulation of S129-phosphorylated α-synuclein, leading to the promotion of neuronal morphology in these cells [66]. The potential site of the interaction between ataxin-3 and the mitochondrially localized CLPP remains obscure. However, both wild-type and polyQ-expanded ataxin-3 have been shown to be associated with or localized in mitochondria [63, 67]. Some mitochondrial proteins have been identified as interaction partners of wild-type and/or polyQ-expanded ataxin-3, such as cytochrome C oxidase subunit NDUFA4 (NDUFA4), succinate dehydrogenase complex subunits A and B (SDHA and SDHB), and cytochrome C oxidase assembly factor 7 (COA7) [67, 68].
Aside from its role as a nuclear transport protein, KPNB1 has been described as a multifunctional protein involved in cell cycle control, mitosis and replication, promoting endoplasmic-reticulum-associated degradation (ERAD), and also functioning as a chaperone which prevents aggregation of some proteins [69,70,71]. Moreover, dysregulation of KPNB1 has been linked to several types of cancer [72,73,74,75]. The herein described activation of a mitochondrial protease such as CLPP, and thereby the induction of proteolytic turnover would represent a new addition to the functional repertoire of KPNB1.
In sum, our study reveals KPNB1 as a new putative modulator of ataxin-3, which triggers proteolytic degradation via mitochondrial protease CLPP, and consequently, lowers aggregate levels of the polyQ-expanded protein. Further analysis will better assess the therapeutic value of targeting KPNB1 or CLPP not only in MJD but also in other neurodegenerative proteopathies.
Data availability
Not applicable.
References
Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20:421–435
Bauer PO, Nukina N (2009) The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem 110:1737–1765
McLoughlin HS, Moore LR, Paulson HL (2020) Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol Dis 134:104635
Yushchenko T, Deuerling E, Hauser K (2018) Insights into the aggregation mechanism of PolyQ proteins with different glutamine repeat lengths. Biophys J 114:1847–1857
Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME (1998) Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci 21:516–520
Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY (1998) Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19:148–154
Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN (1998) Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol 143:1457–1470
Schmidt T, Lindenberg KS, Krebs A, Schöls L, Laccone F, Herms J, Rechsteiner M, Riess O, Landwehrmeyer GB (2002) Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann Neurol 51:302–310
Rudnicki DD, Margolis RL (2003) Repeat expansion and autosomal dominant neurodegenerative disorders: consensus and controversy. Expert Rev Mol Med 5:1–24
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I et al (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8:221–228
Schöls L, Bauer P, Schmidt T, Schulte T, Riess O (2004) Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 3:291–304
Riess O, Rüb U, Pastore A, Bauer P, Schöls L (2008) SCA3: neurological features, pathogenesis and animal models. Cerebellum 7:125–137
Maciel P, Costa MC, Ferro A, Rousseau M, Santos CS, Gaspar C, Barros J, Rouleau GA, Coutinho P, Sequeiros J (2001) Improvement in the molecular diagnosis of Machado–Joseph disease. Arch Neurol 58:1821–1827
Burnett B, Li F, Pittman RN (2003) The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet 12:3195–3205
Da Silva JD, Teixeira-Castro A, Maciel P (2019) From pathogenesis to novel therapeutics for spinocerebellar ataxia Type 3: evading potholes on the way to translation. Neurotherapeutics 16:1009–1031
Costa Mdo C, Paulson HL (2012) Toward understanding Machado–Joseph disease. Prog Neurobiol 97:239–257
Simões AT, Gonçalves N, Koeppen A, Déglon N, Kügler S, Duarte CB (2012) Pereira de Almeida L: Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado–Joseph disease. Brain 135:2428–2439
Weber JJ, Golla M, Guaitoli G, Wanichawan P, Hayer SN, Hauser S, Krahl AC, Nagel M, Samer S, Aronica E et al (2017) A combinatorial approach to identify calpain cleavage sites in the Machado-Joseph disease protein ataxin-3. Brain 140:1280–1299
Trottier Y, Cancel G, An-Gourfinkel I, Lutz Y, Weber C, Brice A, Hirsch E, Mandel JL (1998) Heterogeneous intracellular localization and expression of ataxin-3. Neurobiol Dis 5:335–347
Schmidt T, Landwehrmeyer GB, Schmitt I, Trottier Y, Auburger G, Laccone F, Klockgether T, Völpel M, Epplen JT, Schöls L, Riess O (1998) An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol 8:669–679
Yang W, Dunlap JR, Andrews RB, Wetzel R (2002) Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet 11:2905–2917
Bichelmeier U, Schmidt T, Hübener J, Boy J, Rüttiger L, Häbig K, Poths S, Bonin M, Knipper M, Schmidt WJ et al (2007) Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J Neurosci 27:7418–7428
Chook YM, Süel KE (2011) Nuclear import by karyopherin-βs: recognition and inhibition. Biochim Biophys Acta 1813:1593–1606
Cautain B, Hill R, de Pedro N, Link W (2015) Components and regulation of nuclear transport processes. Febs j 282:445–462
Antony PM, Mäntele S, Mollenkopf P, Boy J, Kehlenbach RH, Riess O, Schmidt T (2009) Identification and functional dissection of localization signals within ataxin-3. Neurobiol Dis 36:280–292
Breuer P, Haacke A, Evert BO, Wüllner U (2010) Nuclear aggregation of polyglutamine-expanded ataxin-3: fragments escape the cytoplasmic quality control. J Biol Chem 285:6532–6537
Sowa AS, Martin E, Martins IM, Schmidt J, Depping R, Weber JJ, Rother F, Hartmann E, Bader M, Riess O et al (2018) Karyopherin α-3 is a key protein in the pathogenesis of spinocerebellar ataxia type 3 controlling the nuclear localization of ataxin-3. Proc Natl Acad Sci USA 115:E2624-e2633
Cemal CK, Carroll CJ, Lawrence L, Lowrie MB, Ruddle P, Al-Mahdawi S, King RH, Pook MA, Huxley C, Chamberlain S (2002) YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet 11:1075–1094
Schmidt J, Mayer AK, Bakula D, Freude J, Weber JJ, Weiss A, Riess O, Schmidt T (2019) Vulnerability of frontal brain neurons for the toxicity of expanded ataxin-3. Hum Mol Genet 28:1463–1473
Gossen M, Bujard H (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 89:5547–5551
Bayliss R, Littlewood T, Stewart M (2000) Structural basis for the interaction between FxFG nucleoporin repeats and importin-beta in nuclear trafficking. Cell 102:99–108
Weishäupl D, Schneider J, Peixoto Pinheiro B, Ruess C, Dold SM, von Zweydorf F, Gloeckner CJ, Schmidt J, Riess O, Schmidt T (2019) Physiological and pathophysiological characteristics of ataxin-3 isoforms. J Biol Chem 294:644–661
Hayer SN, Schelling Y, Huebener-Schmid J, Weber JJ, Hauser S, Schöls L (2018) Generation of an induced pluripotent stem cell line from a patient with spinocerebellar ataxia type 3 (SCA3): HIHCNi002-A. Stem Cell Res 30:171–174
Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K (2010) REAP: a two minute cell fractionation method. BMC Res Notes 3:294
Borchert N, Dieterich C, Krug K, Schütz W, Jung S, Nordheim A, Sommer RJ, Macek B (2010) Proteogenomics of Pristionchus pacificus reveals distinct proteome structure of nematode models. Genome Res 20:837–846
Rappsilber J, Mann M, Ishihama Y (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2:1896–1906
Franz-Wachtel M, Eisler SA, Krug K, Wahl S, Carpy A, Nordheim A, Pfizenmaier K, Hausser A, Macek B (2012) Global detection of protein kinase D-dependent phosphorylation events in nocodazole-treated human cells. Mol Cell Proteomics 11:160–170
Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372
Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10:1794–1805
Elias JE, Gygi SP (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4:207–214
Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, Akira S, Wiegand M, Hochrein H, O’Keeffe M, Mann M (2010) Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 32:279–289
Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13:731–740
Damelin M, Silver PA (2000) Mapping interactions between nuclear transport factors in living cells reveals pathways through the nuclear pore complex. Mol Cell 5:133–140
Soderholm JF, Bird SL, Kalab P, Sampathkumar Y, Hasegawa K, Uehara-Bingen M, Weis K, Heald R (2011) Importazole, a small molecule inhibitor of the transport receptor importin-β. ACS Chem Biol 6:700–708
Weber JJ, Sowa AS, Binder T, Hübener J (2014) From pathways to targets: understanding the mechanisms behind polyglutamine disease. Biomed Res Int 2014:701758
Pereira Sena P, Weber JJ, Watchon M, Robinson KJ, Wassouf Z, Hauser S, Helm J, Abeditashi M, Schmidt J, Hübener-Schmid J et al (2021) Pathophysiological interplay between O-GlcNAc transferase and the Machado-Joseph disease protein ataxin-3. Proc Natl Acad Sci USA 118(47):e2025810118
Blount JR, Tsou WL, Ristic G, Burr AA, Ouyang M, Galante H, Scaglione KM, Todi SV (2014) Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat Commun 5:4638
Berke SJ, Schmied FA, Brunt ER, Ellerby LM, Paulson HL (2004) Caspase-mediated proteolysis of the polyglutamine disease protein ataxin-3. J Neurochem 89:908–918
Yan XX, Jeromin A, Jeromin A (2012) Spectrin breakdown products (SBDPs) as potential biomarkers for neurodegenerative diseases. Curr Transl Geriatr Exp Gerontol Rep 1:85–93
Schmidt J, Schmidt T, Golla M, Lehmann L, Weber JJ, Hübener-Schmid J, Riess O (2016) In vivo assessment of riluzole as a potential therapeutic drug for spinocerebellar ataxia type 3. J Neurochem 138:150–162
Yu AY, Houry WA (2007) ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett 581:3749–3757
Ding B, Sepehrimanesh M (2021) Nucleocytoplasmic transport: regulatory mechanisms and the implications in neurodegeneration. Int J Mol Sci 22(8):4165
Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, Geater C, Morozko E, Stocksdale J, Glatzer JC et al (2017) Mutant huntingtin disrupts the nuclear pore complex. Neuron 94:93-107.e106
Aizawa H, Yamashita T, Kato H, Kimura T, Kwak S (2019) Impaired nucleoporins are present in sporadic amyotrophic lateral sclerosis motor neurons that exhibit mislocalization of the 43-kDa TAR DNA-binding protein. J Clin Neurol 15:62–67
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ, Dujardin S, Amaral AS et al (2018) Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99:925-940.e927
Chen V, Moncalvo M, Tringali D, Tagliafierro L, Shriskanda A, Ilich E, Dong W, Kantor B, Chiba-Falek O (2020) The mechanistic role of alpha-synuclein in the nucleus: impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum Mol Genet 29:3107–3121
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61
Zhang S, Williamson NA, Duvick L, Lee A, Orr HT, Korlin-Downs A, Yang P, Mok YF, Jans DA, Bogoyevitch MA (2020) The ataxin-1 interactome reveals direct connection with multiple disrupted nuclear transport pathways. Nat Commun 11:3343
Havel LS, Li S, Li XJ (2009) Nuclear accumulation of polyglutamine disease proteins and neuropathology. Mol Brain 2:21
Tydlacka S, Wang CE, Wang X, Li S, Li XJ (2008) Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci 28:13285–13295
Colomer Gould VF, Goti D, Pearce D, Gonzalez GA, Gao H, Bermudez de Leon M, Jenkins NA, Copeland NG, Ross CA, Brown DR (2007) A mutant ataxin-3 fragment results from processing at a site N-terminal to amino acid 190 in brain of Machado-Joseph disease-like transgenic mice. Neurobiol Dis 27:362–369
Matos CA, Almeida LP, Nobrega C (2017) Proteolytic cleavage of polyglutamine disease-causing proteins: revisiting the toxic fragment hypothesis. Curr Pharm Des 23:753–775
Pozzi C, Valtorta M, Tedeschi G, Galbusera E, Pastori V, Bigi A, Nonnis S, Grassi E, Fusi P (2008) Study of subcellular localization and proteolysis of ataxin-3. Neurobiol Dis 30:190–200
Fischer F, Langer JD, Osiewacz HD (2015) Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci Rep 5:18375
Szczepanowska K, Maiti P, Kukat A, Hofsetz E, Nolte H, Senft K, Becker C, Ruzzenente B, Hornig-Do HT, Wibom R et al (2016) CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J 35:2566–2583
Hu D, Sun X, Liao X, Zhang X, Zarabi S, Schimmer A, Hong Y, Ford C, Luo Y, Qi X (2019) Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol 137:939–960
Kristensen LV, Oppermann FS, Rauen MJ, Fog K, Schmidt T, Schmidt J, Harmuth T, Hartmann-Petersen R, Thirstrup K (2018) Mass spectrometry analyses of normal and polyglutamine expanded ataxin-3 reveal novel interaction partners involved in mitochondrial function. Neurochem Int 112:5–17
Laço MN, Oliveira CR, Paulson HL, Rego AC (2012) Compromised mitochondrial complex II in models of Machado-Joseph disease. Biochim Biophys Acta 1822:139–149
Mosammaparast N, Pemberton LF (2004) Karyopherins: from nuclear-transport mediators to nuclear-function regulators. Trends Cell Biol 14:547–556
Zhong Y, Wang Y, Yang H, Ballar P, Lee JG, Ye Y, Monteiro MJ, Fang S (2011) Importin beta interacts with the endoplasmic reticulum-associated degradation machinery and promotes ubiquitination and degradation of mutant alpha1-antitrypsin. J Biol Chem 286:33921–33930
Harel A, Forbes DJ (2004) Importin beta: conducting a much larger cellular symphony. Mol Cell 16:319–330
van der Watt PJ, Maske CP, Hendricks DT, Parker MI, Denny L, Govender D, Birrer MJ, Leaner VD (2009) The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer 124:1829–1840
Kuusisto HV, Jans DA (2015) Hyper-dependence of breast cancer cell types on the nuclear transporter Importin β1. Biochim Biophys Acta 1853:1870–1878
Yang L, Hu B, Zhang Y, Qiang S, Cai J, Huang W, Gong C, Zhang T, Zhang S, Xu P et al (2015) Suppression of the nuclear transporter-KPNβ1 expression inhibits tumor proliferation in hepatocellular carcinoma. Med Oncol 32:128
Kodama M, Kodama T, Newberg JY, Katayama H, Kobayashi M, Hanash SM, Yoshihara K, Wei Z, Tien JC, Rangel R et al (2017) In vivo loss-of-function screens identify KPNB1 as a new druggable oncogene in epithelial ovarian cancer. Proc Natl Acad Sci USA 114:E7301-e7310
Bouchnak I, van Wijk KJ (2021) Structure, function, and substrates of Clp AAA+ protease systems in cyanobacteria, plastids, and apicoplasts: a comparative analysis. J Biol Chem 296:100338
Acknowledgements
We thank Dr. Nicolas Casadei (Institute of Medical Genetics and Applied Genomics, University of Tübingen, Germany) for his contribution to the Ingenuity Pathway Analysis.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was supported by the Federal Ministry of Education and Research (Germany) (BMBF, Project 01DN18020, EuSAge). MA thanks the German Academic Exchange Service (DAAD) for the financial support within the “Research Grants-Doctoral Programmes in Germany” (57129429). OR is a member of the European Reference Network for Rare Neurological Diseases (Project 739510).
Author information
Authors and Affiliations
Contributions
MA, JJW, PPS, OR, and TS designed the study; MA, JJW, PPS, AV, MK, RDIE, and BM performed the experiments and analyzed the data; JS and JH-S provided animal samples; SH provided the iPSCs samples; MA wrote the first draft of the manuscript; all authors commented on the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
All research on human materials was carried out under approval of the Ethics Committee of Tübingen University (598/2011BO1). Mouse tissue was gained and applied for the experiments according to the German Animal Welfare Act and the guidelines of the Federation of European Laboratory Animal Science Associations, based on European Union legislation (Directive 2010/63/EU).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abeditashi, M., Weber, J.J., Pereira Sena, P. et al. KPNB1 modulates the Machado–Joseph disease protein ataxin-3 through activation of the mitochondrial protease CLPP. Cell. Mol. Life Sci. 79, 401 (2022). https://doi.org/10.1007/s00018-022-04372-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04372-5