
Abstract
Activation of telomerase is a critical step in the development of about 85 % of human cancers. Levels of Tert, which encodes the reverse transcriptase subunit of telomerase, are limiting in normal somatic cells. Tert is subjected to transcriptional, post-transcriptional and epigenetic regulation, but the precise mechanism of how telomerase is re-activated in cancer cells is poorly understood. Reactivation of the Tert promoter involves multiple changes which evolve during cancer progression including mutations and chromosomal re-arrangements. Newly described non-coding mutations in the Tert promoter region of many cancer cells (19 %) in two key positions, C250T and C228T, have added another layer of complexity to telomerase reactivation. These mutations create novel consensus sequences for transcription factors which can enhance Tert expression. In this review, we will discuss gene structure and function of Tert and provide insights into the mechanisms of Tert reactivation in cancers, highlighting the contribution of recently identified Tert promoter mutations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Telomere and telomerase holoenzyme
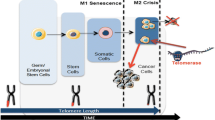
Telomeres are conserved, repetitive sequences located at the ends of eukaryotic chromosomes which protect the integrity of genomic DNA [1, 2]. DNA polymerase is unable to replicate the 5′ ends of chromosomes, hence, cells require a RNA dependent DNA polymerase called telomerase to synthesize DNA on the lagging strand [3–5]. Telomerase activity is tightly regulated and seen mainly in germ cells, stem cells and some immune cell types which have high proliferative needs. In contrast, somatic cells do not display detectable telomerase activity [6]. As a result, the chromosomes of normal somatic cells shorten 50–200 bp each replication at the telomeres due to the problem of end replication. Thus, somatic cells are eventually burdened with DNA damage, replication crisis, cellular senescence or apoptosis and can divide only limited number of times [7], whereas cells that have active telomerase possess unlimited proliferative potential.
Telomerase was discovered by Carol Greider and Elizabeth Blackburn in 1984 from ciliate Tetrahaymana [5]. Telomerase holoenzyme is comprised of a catalytic subunit, hTERT (human telomerase reverse transcriptase) that has reverse transcriptase activity and an RNA component, hTR (human telomerase RNA component) which primes DNA synthesis from telomere repeats. The three dimensional structure of human telomerase is yet to be fully understood. To date, it remains a challenge to purify and crystallize the entire telomerase complex due to its low abundance (estimated to be approximately 20–50 molecules of telomerase per HEK-293 cells [8], and estimated ~250 molecules even in the highly telomerase active cancer cell lines [9]), insolubility problems, and also the requirement of substantial enrichment. Biochemical characterization, however, suggests that hTERT and hTR are sufficient to recapitulate telomerase activity in vitro; however, other cellular factors may be required for basal in vivo activity. Mass spectrometric studies and reconstitution approaches suggest that telomerase has a minimum molecular weight of 650–670 kD and each component appears as a dimer in the core complex including hTERT 127kd, hTR 156kd, DKC1 (dyskerin) 57 kD and Nop10 9 kD [10]. Cohen et al. also demonstrated that telomerase can occur as a dimer [8], but in a recent study Wu et al. showed that telomerase can also function as a monomer and is able to add telomeric ends [11]. Furthermore, proteins like Pontin, Reptin, Gar1, Nhp2, and Tcab1 were shown to be transiently associated with the telomerase core complex and are thought to be required for proper telomerase assembly and recruitment to chromosomes [12, 13]. Current knowledge proposes that the limiting factor for telomerase activity is the level of TERT which is kept under tight transcriptional control. Expression of Tert mRNA in somatic cells is sufficient to reconstitute telomerase activity [14–16] and expression levels of Tert always show strong correlation with telomerase activity [17, 18]. Although the level of RNA component of the telomerase holoenzyme hTR is high in all tissues regardless of telomerase activity [19] it is generally expressed at a lower level in normal somatic cells as compared to cancer cells [20]. Furthermore, half-life experiments for hTR showed a higher turnover rate (~5 days) in somatic cells, whereas the half-life is proposed to be between 3 and 4 weeks in cancer and highly proliferative stem cells [20]. Most cancer cells have more than 10,000 hTR molecules per cell whereas quantification studies by competitive RT-PCR assays show that hTert mRNA range between 1 and 30 molecules in human cancer cells [21]. Other quantification studies determined that TERT protein numbers range between 600 and 700 molecules per cell in HeLa cells [22]. It was also reported that the half-life of hTert mRNA is only 2–3 h [23], while the half-life of active telomerase complex appears to be approximately 24 h according to activity assays [24]. These observations indicate that hTR is essential for the activity of telomerase; however, the limiting factor for telomerase activity is strictly dependent on transcription of hTert mRNA with confounding effects on the stability of the complex by yet unidentified factors. On the other hand, it is noteworthy that hTR can be the limiting factor for telomerase activity in some cases like in the fibrosarcoma-derived HT1080 cells [25]. In general, these results underlie the complexity of telomere maintenance but overall any defect in telomere maintenance failure results in general genomic instability. In particular, DNA damage, increased cellular senescence and organ degeneration are key features of aging phenomena due to telomere shortening [26–28].
Telomerase reactivation in cancer cells
Tert expression is reactivated in ~85 % of all cancers [29]. Recently numerous reports have indicated oncogenic effects of TERT independent of its role in telomere elongation [30–32]. Telomere length in mice (20–50 kb) is greater than in humans (5–10 kb) [33–35]. However, Tert expression is upregulated in murine breast [36] and skin [37] tumors despite their long telomeres. These results suggested that TERT could be playing other roles in cancer. Moreover, overexpression of Tert resulted in increased cell proliferation in mammary carcinomas [38] and epidermal tumors [39] in mice. Similarly, increased expression of Tert initiated T cell lymphomas in mouse thymocytes [40] without significant changes in telomere length, thereby supporting its telomere independent role [32]. Moreover, in human cancer cell lines, knocking down Tert resulted in rapid decreases in cell proliferation and growth [41]. Mechanistically, TERT was shown to indirectly associate with promoters of NFκB target genes interleukin (IL)-6, tumor necrosis factor (TNFα) and IL-8, which are critical for inflammation and cancer progression, to increase expression [42]. Expression of catalytically inactive Tert in mouse model led to the activation of hair follicle stem cells and induction of hair growth [43]. Although the major function of telomerase is thought to be telomere elongation, accumulating evidence has suggested that it can modulate expression of various genes including target of Wnt/β-catenin [44] and NFκB signaling [42] which affect cancer progression and tumorigenesis [45]. High throughput genomic and transcriptomic analysis has revealed that TERT can regulate expression of about 300 genes involved in cell cycle regulation, cellular signaling, and cell proliferation [46]. Non-canonical roles of telomerase have been discussed previously [31, 32], hence in this review we will be focusing on Tert re-activation via regulation of Tert transcription.
Structure of Tert gene
The hTert gene is 42-kb long and located on chromosome 5 with 16 exons. The reverse transcriptase domain is coded by exons 5–6–7–8–9. It has been suggested that the telomerase transcript containing 16 exons can be spliced into 22 isoforms [47] but only full length Tert transcript possesses reverse transcriptase activity essential for elongating telomeres [48, 49]. The most commonly studied isoforms in cancer cell lines involve exon 6–9 that partially encode the reverse transcriptase domain [49]. Alternative spliced isoforms within this region produce isoforms named minus alpha, minus beta and minus alpha/beta. Minus alpha isoform is spliced from a 36-bp acceptor site into exon 6 that creates dominant-negative protein without reverse transcriptase activity [49]. Overexpression of the minus alpha form resulted in inhibition of telomerase which leads to either cell death or senescence [50]. The minus beta isoform has a stop codon in exon 10 due to a frameshift after skipping exons 7 and 8. This isoform is the most common form among cancer cell lines. Although minus beta Tert is subjected to non-sense mediated mRNA decay due to a premature stop codon, it has been shown that its transcripts can be translated into protein [51]. Listerman et al. have reported that overexpression of the minus beta form competed for hTR with endogenous telomerase activity. In their study, Listerman and colleagues reported that overexpressed minus beta form conferred a growth advantage in breast cancer cells by protecting cells from cisplatin-induced apoptosis [52]. However, it remains to be determined why cancer cells preferentially express the catalytically inactive minus beta spliced variant of Tert. Tert isoforms can be expressed in a given cell simultaneously; however, ratios may differ among different cell types according to external stimuli [51]. Deletions/insertions distant from the RT domain also result in truncated forms of TERT protein, the functions of which are not fully understood and need careful characterization.
The Tert promoter has complex regulation dynamics whereby multiple transcriptional regulatory elements play functional roles in different contexts either individually or interactively. Although the Tert promoter does not have typical transcription regulatory elements like TATA and CAAT boxes [53], it contains recognition sequences for multiple important transcription factors such as p53, p21, SP1, ETS, E2F, AP1, HIF1 and c-myc (Fig. 1) [54–62]. Increasing evidence shows that the level of telomerase activity is primarily regulated through levels of Tert which are primarily controlled by transcription of Tert gene. Although the transcription factors stated above may alter the ability to regulate Tert transcription under specific cell type and physiological conditions, none of them are sufficient on their own to promote immortalization of somatic cells [63].

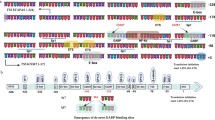
Schematic of Tert promoter region with regulatory protein binding sites and de-novo mutations. The +1 and +78 (ATG) indicates transcription start site (TSS) and first codon for TERT protein, respectively. Dark blue regions correspond to CpG islands. Highly recurrent mutations C228T and C250T are shown at −46 and −68 positions from TSS. The rs2853669 polymorphism is shown in ETS2 binding site (−167 from TSS). The sites on the promoter are not precisely scaled
The Tert promoter region contains E-boxes and GC-boxes which are the consensus binding sites for transcription factors c-myc and SP1, respectively. These factors are known to regulate many cellular events like cell growth, apoptosis, and chromatin remodeling. Sp1 and c-myc cooperatively regulate Tert expression in a cell type specific manner [58]. One of the prominent characteristics of Tert promoter is its high GC-rich content without a typical TATA box or a CCAAT box. There are five GC-boxes in the proximal Tert promoter which serve as SP1 binding port and two E-boxes (CACGTG) located in the −165 and +44 regions which are targetable by c-myc and\or max proteins through their helix–loop–helix leucine zipper domains [58]. Luciferase reporter assays reveal that deletion of the E-box at the −165 position results in 60 % reduction in transcriptional activity in C33A and ME180 cells but not in SiHa cells. Interestingly, mutating the other E-box domain at position +44 led to a 60 % reduction only in ME180 cells. Expression of estrogen, a sex hormone, exhibits a strong concordance with Tert transcription. Mutations in the estrogen receptor (ER) binding sites of Tert promoter dramatically reduced Tert transcription. Moreover, c-myc is a known target of ER and some other growth factors. Kyo et al. demonstrated that the effect of estrogen was completely abrogated upon mutating E-boxes in proximal Tert promoter [57]. Furthermore, mutating the GC-box, located −32 from TSS, reduced transcription by 20–40 % in all cell lines and a 90 % reduction was detected when all SP1 binding sites were mutated [58]. We can thus conclude that Sp1 binding sites (GC-boxes) are essential for Tert transcriptional activity and E-boxes can accelerate transcription in a cell-type specific manner. E2F transcription factors are involved in cell cycle regulation and DNA synthesis [64, 65]. Ectopic expression of E2F1 repressed Tert promoter activity through inhibition of SP1 binding to DNA in cancer cells. In contrast, overexpression of E2F1 in normal human fibroblasts increased Tert promoter activity through a non-canonical E2F1 site which is located –51 to –88 [66]. All these data suggest that E2F1 has a dual role for regulating Tert expression that requires further investigation.
The tumor suppressor gene p53 has two binding sites −1240 and −1877 upstream of the transcription start site of the Tert promoter (Fig. 1). Overexpression of p53 together with SP1 leads to suppression of Tert expression while siRNA silencing of p53 delays senescence but is insufficient to drive cells to an immortal state [67, 68]. Overexpression of Tert is not sufficient to drive cells to the immortal state as well. Tert expression and telomerase activity could be enhanced by suppression of p53; however, both actions are required for immortalization of primary human ovarian surface epithelial cells [67, 68].
NFκB is a key regulator of innate and adaptive immunity which is essential for host defense [69, 70]. Activated NFκB activates the expression of target genes that are responsible for cell survival, proliferation, differentiation of cells and for mounting appropriate immune responses for host defense [71]. NFκB was also reported to be associated with tumorigenesis and chemoresistance [71, 72]. The Tert promoter contains a NFκB binding site upstream of its translation start site. A significant increase in Tert expression was observed through NFκB activation [73]. Moreover, NFκB can mediate TERT translocation to the nucleus from the cytoplasm whilst IKK2 inhibitors can reverse this effect in MM.1S cells [74].
The PI3K-AKT pathway regulates various cellular events such as proliferation and differentiation which are important in tumorigenesis [75]. It has been reported that Tert expression was increased by receptor tyrosine kinases and VEGF which are controlled by PI3K in ovarian cancer cells [76]. Moreover, Tert expression is increased depending on two VEGF isoforms, VEGF165 and VEGF189, which are also positively regulated upon Tert expression regardless of its telomeric function in HeLa cells [77]. Under physiological conditions, all of the molecules listed above are responsible for specific cellular processes and almost all somatic cells can achieve these processes without reactivating their Tert gene. However, multiple changes and impairments evolve during cancer progression that cause chromosomal re-arrangements, point mutations, deletions/insertions, translocations and copy number changes.
The Tert gene has a GC rich promoter which contains three CpG islands [53, 78]. Methylation of gene promoters is generally known to repress transcription; however, several studies revealed complex methylation patterns for the active/inactive Tert promoter. The Tert promoter region has a CpG island (positions −1100 to +150), that is mostly hypermethylated through specific DNA methyltransferases (DNMTs) in cancer cells [79]. The Tert promoter between −150 and +150 represents a usual pattern of gene expression. Absence of methylation causes constitutive expression particularly in this region, hypermethylation of 5′ Tert promoter prevents binding of methylation sensitive CTCF repressor to the first exon; however, partial hypomethylation of core promoter is required for Tert transcription. Thus, Tert promoter methylation represents a unique model for transcription in which hypermethylation of cytosine islands causes inhibition of Tert expression and this differs among different cell types [79, 80]. Hypermethylation of the Tert core promoter results in low telomerase activity and better survival rate in B cell chronic lymphocytic leukemia [81]. On the contrary, several studies reported that hypermethylation in telomerase active cancer cells and hypomethylation of normal tissues may inhibit binding of repressor elements to the Tert promoter region and enhance transcriptional activity [78, 80, 82–84]. A comprehensive study by Renaud et al. demonstrated that the Tert promoter in most cancer cell lines is heavily methylated between −500 and −600 bp upstream of TSS; however, they tend to be partially methylated at TSS region [79]. The Myc/Max proteins are responsible for recruiting Histone Acetyl transferases (HATs) to the promoter regions where they can bind through their consensus sequences [85]. Increased acetylation of H3 and H4 histones creates more open chromatin structure, thereby enhancing the accessibility of other transcription regulatory elements. However, Mad proteins could bind to the same E-boxes and heterodimerize with Max proteins that act antagonistically to inhibit transcription [86].
In conclusion, Tert promoter specific methylation analysis revealed a complex/unusual methylation pattern for the promoter region, indicating that distal and proximal promoter regions have different methylation patterns that can be partially modified due to activation or inactivation of the Tert gene. These results highlight importance of chromatin modifiers, especially the roles of histone methyl transferases and demethylases in transcription dynamics.
Tert promoter mutations in cancer
Point mutations in the Tert promoter are highly recurrent in cancer cases including glioblastoma, melanoma, urothelial, bladder, hepatocellular carcinoma, and thyroid cancers [87–91]. These mutations generally create new consensus motifs for transcriptional regulators like ETS/TCF factors and are associated with increased Tert mRNA (Fig. 2). Therefore, point mutations in the Tert promoter could unravel a novel mechanism for Tert reactivation in cancer cells. Recurrent mutations have been identified in Chr.5:1,295,228(C>T) or (CC>TT), Chr.5:1,295,250 C>T, Chr.5:1,295,242_1,295,243CC>TT mutations in 19 % of cancers [90].

Highly recurrent mutations C228T and C250T create novel ETS binding sites in Tert promoter. Tert promoter with wild type, C250T and C228T are shown with their reported binding partners. Red and green rectangular boxes indicate ETS and p52 consensus sites on Tert promoter, respectively. ETS1/2 and p52 binds Tert promoter harboring C250T mutation while GABP binds to Tert promoter bearing either C250T or C228T mutation, resulting in increased Tert mRNA expression
Alternative lengthening of telomeres (ALT) is a recombination-based mechanism that is activated in the absence of telomerase activity. ALT recombination is stimulated by the signals of double-strand DNA break and meiotic HOP2-MND1 heterodimer induces RAD51 and DMC1 mediated recombination [92]. ALT is observed frequently in sarcomas (25–60 %) [93], brain tumors (10–25 %) [93], and pancreatic neuroendocrine tumors (40 %) [92], but is rare in colon, breast, lung, prostate and pancreas cancers. Generally, cancers with ALT show poor prognosis as compared to telomerase activity. However, glioblastomas with ALT represent two- to threefold longer survival [92, 94]. Recently it was also shown that mutations in ATRX or DAXX genes cause increase in non-coding RNA TERRA expression which is known to inhibit telomerase activity [95–98]. Furthermore, TERRA can induce ALT through R-loop formation in telomeres [92]. ATRX and DAXX alter telomeric chromatin dynamics that function as a chromatin remodeler and histone chaperone, respectively. Therefore, cells having these mutations depend on ALT for telomere maintenance. Interestingly, ATRX/DAXX genes and Tert promoter mutations are mutually exclusive [87]. Besides, there are subpopulations that display both ALT and telomerase mechanisms for telomere maintenance [99] which could be a temporary/transient stage before final commitment to ALT and/or telomerase as the major telomerase mechanism depending on the cellular context and other signaling milieu. However, there is very little knowledge regarding the mechanism by which cells can utilize both mechanisms or how they choose between ALT or telomerase as a preferred mechanism to maintain their telomeres.
Hepatocellular carcinomas (HCCs) are the third leading cause of cancer deaths and are mostly associated with hepatitis B or C virus infection while a minority is associated with alcoholic cirrhosis, obesity and rare genetic diseases [100, 101]. Recently, somatic mutations in the Tert promoter region have been reported in HCC. Killela et al. reported that 44 % of HCC tumor samples have Tert promoter mutations (27/61) and the mutations tend to occur relatively early during tumorigenesis [87]. Other teams have also identified these mutations and, according to recent reports, Tert promoter mutations are less frequent in Eastern countries (29 to 31 % of HCC), whereas 54–60 % of HCC are mutated in Western countries [87, 102–104]. The most frequent Tert promoter mutations in HCC (93 %) were observed at the first hot spot and consist of G to A (−124G>A) mutations. While mutations in the second hot spot −146 bp G to A substitutions (−146G>A) were significantly less frequent than those found in melanoma (46 %) [105]. Interestingly, Tert promoter mutations are more frequent in older patients and significantly associated with activating mutations of the beta catenin pathway [103, 105]. These data suggest that aberrant activation of the β-catenin pathway associated with Tert promoter mutations can be one of the mechanisms of telomerase reactivation in HCC. Taken together, these data suggest that somatic mutations in the Tert promoter region are one of the most frequently observed genetic alterations in human HCC. Although Tert amplification and insertion of HBV in the Tert promoter has been considered as the alternative mechanism leading to telomerase reactivation in HCC [106, 107], identification of Tert promoter mutations in HCCs has opened new insights into telomerase reactivation and telomere maintenance in liver carcinogenesis.
Bladder cancers represent the most common urinary tract cancer worldwide [108, 109] and recent studies have showed that Tert promoter mutations are the most common mutations in all stages and grades of bladder cancer with an even distribution [110–112]. In addition, the frequency of Tert promoter mutations detected in these studies was higher than any earlier reported genetic alteration in any gene in bladder cancer [112, 113]. Tert promoter mutations were detected in both low-grade and high-grade tumors of bladder cancer [90] and, in particular, the −124C>T mutation was detected as the most frequent alteration in 175 (53.5 %) tumor samples [112]. In contrast, the prevalence of ALT in bladder cancer is as low as 1 % in 188 samples [114]. Intriguingly, Tert promoter mutations in conjunction with the identified common polymorphism have effects on both survival and recurrence in bladder cancer. The common polymorphism rs2853669 from −245 bp ATG start site in the Tert promoter acted as a modifier of survival and recurrence in bladder cancer patients. Bladder cancer patients who do not harbor a variant allele of rs2853669 showed almost twofold reduction in survival and increased disease recurrence. Mechanistically, the variant allele of polymorphism disrupts a pre-existing non-canonical ETS2 binding site in the proximal region of the Tert promoter, adjacent to an E-box (Fig. 1). This mechanism is in contrast to the two highly recurrent Tert mutations which generate an ETS/TCF binding site [112]. Disruption of the ETS2 binding site in Tert promoter or silencing of ETS2 in breast cancer cells has been shown to result in decreased Tert expression and cell proliferation due to the disabling of c-Myc binding to the E-box in Tert promoter [62]. Moreover, the variant allele of the SNP has been previously shown to affect telomerase expression and telomere length maintenance in non-small lung cancers [115]. Tert promoter mutations in combination with the polymorphism can have the potential to serve as clinical biomarkers for prediction of survival and recurrence in bladder cancer patients [112].
Another cancer type that harbors Tert promoter mutations is urothelial cancers (UC). Borah et al. reported that when compared to −146C>T mutation, −124C>T mutation is a frequently altered lesion in 23 UC cell lines derived from different stages and grades, including muscle invasive and non-invasive tumors. These promoter mutations provide increased Tert mRNA levels with an 18-fold increase in median value according to qRT-PCR analyses. Although most of these transcripts were found to be alternatively spliced variants of Tert which lack functional reverse transcriptase activity, both TERT protein and telomerase enzymatic activity were higher in tumor samples harboring promoter mutations. However, compared to the 18-fold increase in Tert mRNA level, they reported much more modest 2-fold increases in the TERT protein and telomerase activity that may indicate higher expression of inactive Tert isoforms [91]. These results revealed that Tert promoter mutations are an effective strategy to boost the level of telomerase activity in UC which do not employ ALT [114]. It remains to be determined why cancer cells express catalytically inactive TERT isoforms and whether expression of specific isoforms would change in cancers harboring promoter mutations.
Thyroid cancer is an endocrine malignancy that can be classified into two common groups: papillary (~85 %) and follicular (~10 %) thyroid cancers. C228T mutation was observed in 30 of 257 (11.7 %) papillary thyroid cancers (PTC) and 9 of 79 (11.4 %) follicular thyroid cancers (FTC) while interestingly, no mutations were detected in benign thyroid tumors (0 of 85). C250T mutation was uncommon in all groups of thyroid tumors, but is mutually exclusive with C228T mutation. BRAF (v-Raf murine sarcoma viral oncogene homolog B) mutation (V600E), which activates the MAPK pathway, is commonly observed in thyroid cancers [116]. It is also known that cancers harboring BRAF mutation are more aggressive than WT counterparts [117]. Tert C228T mutation was correlated with BRAF mutation and it was reported that 19 of 104 cases (18.3 %) coexist in PTC while 11 of 153 (7.2 %) BRAF negative tumors exclusively harbor C228T mutation. In a separate study, BRAF mutation was found to exist in 19 of 30 (63.3 %) C228T mutant tumors, and 85 of 227 (37.4 %) C228T negative tumors [118]. Similarly, Vinagre et al. investigated Tert promoter mutations in large thyroid cancers categorized as normal, benign and malignant lesions. Tert promoter mutations were only detected in malignant lesions in 11 % of PTCs, 14 % of FTCs, 21 % of poorly differentiated thyroid carcinomas and 13 % of anaplastic carcinomas. The majority of these mutations were C228T. Correlation analysis revealed that Tert promoter mutations were highly associated with larger tumor size, older patients, lymph node metastasis and BRAF (V600E) mutation. Tert mRNA was also significantly higher in patients with BRAF and Tert promoter mutation [90]. These results indicate that Tert promoter mutation is prevalent in more aggressive tumors.
Gliomas are the most common cancer of the central nervous system (CNS), with four major subtypes including primary glioblastoma, astrocytoma, oligodendriogliomas and oligoastrocytomas [119]. Primary glioblastomas are the most common glioma among adults with very poor survival rates. It was shown that 83 % of the 78 tumors analyzed harbored Tert promoter mutations which were correlated with high Tert expression levels and poorer patient survival [87]. Remarkably, EGFR amplification, a common phenomenon in GBM tumors, was observed to be mutually exclusive with Tert mutations [87]. Astrocytomas are another frequently occurring glioma subtype. This group displays rare Tert promoter mutations (10 %); however, IDH1 and IDH2 mutations (75 %) as well as ATXR mutations (70 %) are common, which is also correlated with the high prevalence of ALT (63 %) in these gliomas. In contrast to astrocytomas, oligodendrogliomas frequently (78 %) harbor Tert promoter mutations. Interestingly, more than 90 % of this glioma subtype seems to strictly depend on mutations either in ATRX gene or Tert promoter for telomere maintenance, indicating the requirement of these genetic alterations for tumorigenesis of this subtype. Oligoastrocytomas have features of both astrocytomas and oligodendriogliomas and have a 25 % frequency of Tert mutations [87].
Recently it was reported that 15 % of grade II and III glioblastoma patients had Tert promoter mutations in their genome; however, the mutation rate increases dramatically in grade IV patients to 76 % [90]. Patients with Tert promoter mutations in combination with common gene alterations found in gliomas including IDH mutations and 1p19q deletion showed poorer survival rate as compared to patients without Tert mutations [120]. This is a clear indication that these promoter mutations increase the malignancy of gliomas through Tert activation.
Although a majority of neuroblastoma tumors possess Tert promoter mutations, alternative genetic alterations like copy number increase and Tert gene re-arrangements leading activation of Tert have been observed in cancer patients. Genome wide sequencing analysis of neuroblastoma patients revealed recurrent Tert gene re-arrangements occurring at 5p15.33 region leading to a juxtaposition of the Tert promoter with strong enhancers. Tert re-arrangement was observed in 13 % (28 of 217) of high-risk neuroblastoma tumors. Patients with these re-arrangements had increased Tert mRNA expression and poor survival rate. Moreover, multiple active enhancer clusters were detected in the translocated regions adjacent to Tert rearrangements indicating that translocation of cis-acting regulatory elements can cause up-regulation of Tert expression [121]. These results suggest that genomic rearrangements rather than Tert copy number changes could be the major cause of aberrant Tert expression in neuroblastoma in addition to Tert promoter mutations.
Similar observations have been shown in melanoma patients and cell lines. Melanomas frequently harbor mutations in oncogenes like BRAF, NRAS (neuroblastoma RAS viral oncogene homolog), KIT (v-kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog) and tumor suppressors CDKN2A and PTEN. These mutations occur according to cancer stage. Recently a high frequency of Tert promoter mutations was reported in melanoma patients and cell lines by several groups [90, 122–124]. Horn et al., observed Tert promoter mutations in a melanoma-prone family where all members developed melanomas at early ages. Furthermore, the same group reported that 125 of 168 (74 %) metastatic melanoma cell lines carry Tert promoter mutations which are located 124 (C>T), (CC>TT), 138 and 146 bp upstream from the ATG start codon with frequencies of 27.4, 4.2, 4.8 and 38.1 %, respectively [122]. Whole genome sequencing analyses revealed that Tert promoter mutations are the most frequent mutations after BRAF and NRAS genes in melanomas [123]. Both groups observed two- to fourfold increases in Tert expression by Luciferase reporter assays due to novel ETS site generation upon mutations [122, 123]. It is noteworthy that Tert mutations are more common in patients with BRAF and/or NRAS mutations [124] and Tert mRNA levels are higher when Tert and BRAF mutations coexist [90].
Recently two independent groups have studied binding of ETS factors on mutated sites. Bell et al. identified several ETS factors such as ELF1, ETS1 and ETV4 which can bind to the mutation site significantly; however, GABP was enriched more in mutation regions together with PolII (Fig. 2). In addition, chromatin immunoprecipitation experiments for GABP did not show enrichment in K562, A549, HeLa, MCF-7 cells which do not harbor the Tert promoter mutation. GABP knockdown also led to a rapid decrease in Tert transcription [125]. Li et al. identified binding partner p52 for ETS factor—ETS1/2 to drive Tert transcription in cells containing C250T Tert mutation (Fig. 2). They showed that non-canonical NFκB signaling is necessary to drive Tert transcription particularly in C250T mutant Tert promoter by direct interaction with ETS factor. Consistent with their biochemical data, knock down of p52 during non-canonical NFκB activation abolished tumorigenesis in C250T-mutant glioblastoma cells transplanted mouse models. C250T mutation creates a half site NFκB consensus sequence (5′-GGGGG-3′ or 5′-GGAA-3′) and increased p52 binding was observed in the mutant promoter as compared to WT Tert sequence in electrophoretic mobility shift assays (EMSA). The group further demonstrated that Tert expression increased upon binding of p52 to the novel half site in cells harboring C250T but not C228 mutation. Moreover, ETS1/2 heterodimerized with p52 at C250T region and cooperatively activated Tert gene expression, thereby demonstrating the non-canonical role of NFκB in telomerase reactivation in cancer cells harboring Tert promoter mutations [126].
Conclusion
Reactivation of telomerase has been considered as a strategy for telomere maintenance and is a major hallmark of cancer. Telomerase reactivation mostly depends on the amount of TERT in the cell since there are sufficient amounts of other telomerase complex molecules as summarized above. Therefore, increases in TERT could be substituted by increasing gene copy number as in HeLa cells (five copies), overexpression of oncogenes that can bind its promoter for transcription like c-Myc or using alternative splicing to form catalytically active/inactive proteins.
More importantly, Tert promoter mutations which create new consensus sequence for ETS and NFκB binding result in increased transcription of Tert mRNA. It is also noteworthy that these mutations could lead to chromatin conformation changes by novel short, middle and long distance interactions, which could, in turn, directly regulate the Tert gene and possibly other genes simultaneously. Since these mutations are highly recurrent in many cancer types and occur at high frequency, the mechanisms underlying regulation of Tert expression at its promoter need to be deciphered. We can speculate that the presence of these mutations could modulate core transcriptional machinery by recruiting additional factors to the Tert promoter to regulate expression of specific isoforms of Tert transcript preferentially in cancers. Furthermore, it would be interesting to determine why mutations occur persistently in the −146 and −124 positions in a wide range of various cancer types. Recently, Chiba et al. revealed that introducing any of the three most frequent Tert promoter mutations using CRISPR/Cas9 genome editing in human embryonic stem cells did not increase Tert expression, activity or telomere length. However, when these engineered stem cells were differentiated to either fibroblast or nerve cells, all of the Tert promoter mutations, without any additional oncogenic mutations, prevented silencing of the Tert promoter and resulted in enhanced Tert expression, telomerase activity, telomere length and growth comparable to cancer cell lines [127]. These results indicate that Tert promoter mutations do not affect already active promoters but prevent proper silencing of the Tert gene in differentiated cells. Recently, it was shown that cancer cell lines harboring Tert promoter mutations represent display histone marks [128]. It would be intriguing to speculate that the presence of Tert promoter mutations may affect recruitment of epigenetic modulators and enhancers to the Tert promoter to drive Tert expression.
Since Tert promoter mutations are not present in stem and healthy proliferating cells, it will be crucial to decipher the mechanistic pathways which regulate Tert gene silencing and understand how they are affected by these promoter mutations. It will also be a wise strategy to identify specific therapeutic approaches targeting only Tert promoter mutations so that only tumor cells but not telomerase positive stem cells are eliminated in cancer patients.
References
Moyzis RK et al (1988) A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA 85:6622–6626
Blackburn EH (2000) The end of the (DNA) line. Nat Struct Biol 7:847–850. doi:10.1038/79594
Watson JD (1972) Origin of concatemeric T7 DNA. Nature New Biol 239:197–201
Olovnikov AM (1973) A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol 41:181–190
Greider CW, Blackburn EH (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43:405–413
Greider CW (2012) Molecular biology. Wnt regulates TERT—putting the horse before the cart. Science 336:1519–1520. doi:10.1126/science.1223785
Huffman KE, Levene SD, Tesmer VM, Shay JW, Wright WE (2000) Telomere shortening is proportional to the size of the G-rich telomeric 3′-overhang. J Biol Chem 275:19719–19722. doi:10.1074/jbc.M002843200
Cohen SB et al (2007) Protein composition of catalytically active human telomerase from immortal cells. Science 315:1850–1853. doi:10.1126/science.1138596
Xi L, Cech TR (2014) Inventory of telomerase components in human cells reveals multiple subpopulations of hTR and hTERT. Nucleic Acids Res 42:8565–8577. doi:10.1093/nar/gku560
Sauerwald A et al (2013) Structure of active dimeric human telomerase. Nat Struct Mol Biol 20:454–460. doi:10.1038/nsmb.2530
Wu RA, Dagdas YS, Yilmaz ST, Yildiz A, Collins K (2015) Single-molecule imaging of telomerase reverse transcriptase in human telomerase holoenzyme and minimal RNP complexes. Elife 4:e08363. doi:10.7554/eLife.08363
Venteicher AS, Meng Z, Mason PJ, Veenstra TD, Artandi SE (2008) Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132:945–957. doi:10.1016/j.cell.2008.01.019
Vulliamy T et al (2008) Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci USA 105:8073–8078. doi:10.1073/pnas.0800042105
Bodnar AG et al (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279:349–352
Counter CM et al (1998) Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene 16:1217–1222. doi:10.1038/sj.onc.1201882
Vaziri H, Benchimol S (1998) Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Current Biol CB 8:279–282
Kilian A et al (1997) Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet 6:2011–2019
Nakamura TM et al (1997) Telomerase catalytic subunit homologs from fission yeast and human. Science 277:955–959
Avilion AA et al (1996) Human telomerase RNA and telomerase activity in immortal cell lines and tumor tissues. Cancer Res 56:645–650
Yi X, Tesmer VM, Savre-Train I, Shay JW, Wright WE (1999) Both transcriptional and posttranscriptional mechanisms regulate human telomerase template RNA levels. Mol Cell Biol 19:3989–3997
Yi X, Shay JW, Wright WE (2001) Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res 29:4818–4825
Akincilar SC et al (2015) Quantitative assessment of telomerase components in cancer cell lines. FEBS Lett 589:974–984. doi:10.1016/j.febslet.2015.02.035
Xu D, Gruber A, Bjorkholm M, Peterson C, Pisa P (1999) Suppression of telomerase reverse transcriptase (hTERT) expression in differentiated HL-60 cells: regulatory mechanisms. Br J Cancer 80:1156–1161. doi:10.1038/sj.bjc.6690480
Holt SE, Aisner DL, Shay JW, Wright WE (1997) Lack of cell cycle regulation of telomerase activity in human cells. Proc Natl Acad Sci USA 94:10687–10692
Cristofari G, Lingner J (2006) Telomere length homeostasis requires that telomerase levels are limiting. EMBO J 25:565–574. doi:10.1038/sj.emboj.7600952
De Lange T (2005) Telomere-related genome instability in cancer. Cold Spring Harb Symp Quant Biol 70:197–204. doi:10.1101/sqb.2005.70.032
Jaskelioff M et al (2011) Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469:102–106. doi:10.1038/nature09603
Sahin E, Depinho RA (2010) Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464:520–528. doi:10.1038/nature08982
Shay JW, Wright WE (2005) Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis 26:867–874. doi:10.1093/carcin/bgh296
Koh CM et al (2015) Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest 125:2109–2122. doi:10.1172/JCI79134
Li Y, Tergaonkar V (2014) Noncanonical functions of telomerase: implications in telomerase-targeted cancer therapies. Cancer Res 74:1639–1644. doi:10.1158/0008-5472.CAN-13-3568
Low KC, Tergaonkar V (2013) Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem Sci 38:426–434. doi:10.1016/j.tibs.2013.07.001
Kipling D, Cooke HJ (1990) Hypervariable ultra-long telomeres in mice. Nature 347:400–402. doi:10.1038/347400a0
Zijlmans JM et al (1997) Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci USA 94:7423–7428
de Lange T et al (1990) Structure and variability of human chromosome ends. Mol Cell Biol 10:518–527
Broccoli D, Godley LA, Donehower LA, Varmus HE, de Lange T (1996) Telomerase activation in mouse mammary tumors: lack of detectable telomere shortening and evidence for regulation of telomerase RNA with cell proliferation. Mol Cell Biol 16:3765–3772
Bednarek A, Budunova I, Slaga TJ, Aldaz CM (1995) Increased telomerase activity in mouse skin premalignant progression. Cancer Res 55:4566–4569
Artandi SE et al (2002) Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc Natl Acad Sci USA 99:8191–8196. doi:10.1073/pnas.112515399
Gonzalez-Suarez E et al (2001) Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J 20:2619–2630. doi:10.1093/emboj/20.11.2619
Canela A, Martin-Caballero J, Flores JM, Blasco MA (2004) Constitutive expression of tert in thymocytes leads to increased incidence and dissemination of T-cell lymphoma in Lck-Tert mice. Mol Cell Biol 24:4275–4293
Li S et al (2004) Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and anti-telomerase short-interfering RNA. Cancer Res 64:4833–4840. doi:10.1158/0008-5472.CAN-04-0953
Ghosh A et al (2012) Telomerase directly regulates NF-kappaB-dependent transcription. Nat Cell Biol 14:1270–1281. doi:10.1038/ncb2621
Choi J et al (2008) TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet 4:e10. doi:10.1371/journal.pgen.0040010
Park JI et al (2009) Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460:66–72. doi:10.1038/nature08137
Shin EM et al (2014) DEAD-box helicase DP103 defines metastatic potential of human breast cancers. J Clin Invest 124:3807–3824. doi:10.1172/JCI73451
Perrault SD, Hornsby PJ, Betts DH (2005) Global gene expression response to telomerase in bovine adrenocortical cells. Biochem Biophys Res Commun 335:925–936. doi:10.1016/j.bbrc.2005.07.156
Hrdlickova R, Nehyba J, Bose HR Jr (2012) Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation. Mol Cell Biol 32:4283–4296. doi:10.1128/MCB.00550-12
Saeboe-Larssen S, Fossberg E, Gaudernack G (2006) Characterization of novel alternative splicing sites in human telomerase reverse transcriptase (hTERT): analysis of expression and mutual correlation in mRNA isoforms from normal and tumour tissues. BMC Mol Biol 7:26. doi:10.1186/1471-2199-7-26
Yi X et al (2000) An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia 2:433–440
Colgin LM et al (2000) The hTERTalpha splice variant is a dominant negative inhibitor of telomerase activity. Neoplasia 2:426–432
Wong MS et al (2013) Regulation of telomerase alternative splicing: a target for chemotherapy. Cell Rep 3:1028–1035. doi:10.1016/j.celrep.2013.03.011
Listerman I, Sun J, Gazzaniga FS, Lukas JL, Blackburn EH (2013) The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res 73:2817–2828. doi:10.1158/0008-5472.CAN-12-3082
Cong YS, Wen J, Bacchetti S (1999) The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet 8:137–142
Horikawa I, Cable PL, Afshari C, Barrett JC (1999) Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res 59:826–830
Takakura M et al (1999) Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res 59:551–557
Wu KJ et al (1999) Direct activation of TERT transcription by c-MYC. Nat Genet 21:220–224. doi:10.1038/6010
Kyo S et al (1999) Estrogen activates telomerase. Cancer Res 59:5917–5921
Kyo S et al (2000) Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res 28:669–677
Goueli BS, Janknecht R (2003) Regulation of telomerase reverse transcriptase gene activity by upstream stimulatory factor. Oncogene 22:8042–8047. doi:10.1038/sj.onc.1206847
Takakura M, Kyo S, Inoue M, Wright WE, Shay JW (2005) Function of AP-1 in transcription of the telomerase reverse transcriptase gene (TERT) in human and mouse cells. Mol Cell Biol 25:8037–8043. doi:10.1128/MCB.25.18.8037-8043.2005
Anderson CJ, Hoare SF, Ashcroft M, Bilsland AE, Keith WN (2006) Hypoxic regulation of telomerase gene expression by transcriptional and post-transcriptional mechanisms. Oncogene 25:61–69. doi:10.1038/sj.onc.1209011
Xu D, Dwyer J, Li H, Duan W, Liu JP (2008) Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. J Biol Chem 283:23567–23580. doi:10.1074/jbc.M800790200
Zhu J, Zhao Y, Wang S (2010) Chromatin and epigenetic regulation of the telomerase reverse transcriptase gene. Protein Cell 1:22–32. doi:10.1007/s13238-010-0014-1
Nevins JR (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ 9:585–593
Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12:2245–2262
Won J, Yim J, Kim TK (2002) Opposing regulatory roles of E2F in human telomerase reverse transcriptase (hTERT) gene expression in human tumor and normal somatic cells. Faseb J 16:1943–1945. doi:10.1096/fj.02-0311fje
Kanaya T et al (2000) Adenoviral expression of p53 represses telomerase activity through down-regulation of human telomerase reverse transcriptase transcription. Clin Cancer Res Off J Am Assoc Cancer Res 6:1239–1247
Yang G et al (2007) Knockdown of p53 combined with expression of the catalytic subunit of telomerase is sufficient to immortalize primary human ovarian surface epithelial cells. Carcinogenesis 28:174–182. doi:10.1093/carcin/bgl115
Li Q, Withoff S, Verma IM (2005) Inflammation-associated cancer: NF-kappaB is the lynchpin. Trends Immunol 26:318–325. doi:10.1016/j.it.2005.04.003
Tong L, Tergaonkar V (2014) Rho protein GTPases and their interactions with NFkappaB: crossroads of inflammation and matrix biology. Biosci Rep 34:e00115. doi:10.1042/BSR20140021
Correa RG et al (2005) Zebrafish IkappaB kinase 1 negatively regulates NF-kappaB activity. Curr Biol CB 15:1291–1295. doi:10.1016/j.cub.2005.06.023
Perkins ND (2012) The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer 12:121–132. doi:10.1038/nrc3204
Kote-Jarai Z et al (2013) Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression. Hum Mol Genet 22:2520–2528. doi:10.1093/hmg/ddt086
Akiyama M et al (2003) Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res 63:18–21
Dey A et al (2008) Hexamethylene bisacetamide (HMBA) simultaneously targets AKT and MAPK pathway and represses NF kappaB activity: implications for cancer therapy. Cell Cycle 7:3759–3767
Bermudez Y et al (2007) VEGF- and LPA-induced telomerase in human ovarian cancer cells is Sp1-dependent. Gynecol Oncol 106:526–537. doi:10.1016/j.ygyno.2007.05.005
Zhou L, Zheng D, Wang M, Cong YS (2009) Telomerase reverse transcriptase activates the expression of vascular endothelial growth factor independent of telomerase activity. Biochem Biophys Res Commun 386:739–743. doi:10.1016/j.bbrc.2009.06.116
Renaud S, Bosman FT, Benhattar J (2003) Implication of the exon region in the regulation of the human telomerase reverse transcriptase gene promoter. Biochem Biophys Res Commun 300:47–54
Renaud S et al (2007) Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res 35:1245–1256. doi:10.1093/nar/gkl1125
Devereux TR et al (1999) DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res 59:6087–6090
Bechter OE, Eisterer W, Dlaska M, Kuhr T, Thaler J (2002) CpG island methylation of the hTERT promoter is associated with lower telomerase activity in B-cell lymphocytic leukemia. Exp Hematol 30:26–33
Dessain SK, Yu H, Reddel RR, Beijersbergen RL, Weinberg RA (2000) Methylation of the human telomerase gene CpG island. Cancer Res 60:537–541
Nomoto K et al (2002) Methylation status and expression of human telomerase reverse transcriptase mRNA in relation to hypermethylation of the p16 gene in colorectal cancers as analyzed by bisulfite PCR-SSCP. Jpn J Clin Oncol 32:3–8
Renaud S, Loukinov D, Bosman FT, Lobanenkov V, Benhattar J (2005) CTCF binds the proximal exonic region of hTERT and inhibits its transcription. Nucleic Acids Res 33:6850–6860. doi:10.1093/nar/gki989
McMahon SB, Wood MA, Cole MD (2000) The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol 20:556–562
Ge Z et al (2010) Chromatin remodeling: recruitment of histone demethylase RBP2 by Mad1 for transcriptional repression of a Myc target gene, telomerase reverse transcriptase. Faseb J 24:579–586. doi:10.1096/fj.09-140087
Killela PJ et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA 110:6021–6026. doi:10.1073/pnas.1303607110
Griewank KG et al (2013) TERT promoter mutations in ocular melanoma distinguish between conjunctival and uveal tumours. Br J Cancer 109:497–501. doi:10.1038/bjc.2013.312
Landa I et al (2013) Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab 98:E1562–E1566. doi:10.1210/jc.2013-2383
Vinagre J et al (2013) Frequency of TERT promoter mutations in human cancers. Nat Commun 4:2185. doi:10.1038/ncomms3185
Borah S et al (2015) Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 347:1006–1010. doi:10.1126/science.1260200
Dilley RL, Greenberg RA (2015) ALTernative telomere maintenance and cancer. Trends Cancer 1:145–156. doi:10.1016/j.trecan.2015.07.007
Henson JD, Reddel RR (2010) Assaying and investigating alternative lengthening of telomeres activity in human cells and cancers. FEBS Lett 584:3800–3811. doi:10.1016/j.febslet.2010.06.009
Hakin-Smith V et al (2003) Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 361:836–838
Goldberg AD et al (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140:678–691. doi:10.1016/j.cell.2010.01.003
Flynn RL et al (2015) Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347:273–277. doi:10.1126/science.1257216
Episkopou H et al (2014) Alternative lengthening of telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res 42:4391–4405. doi:10.1093/nar/gku114
Redon S, Reichenbach P, Lingner J (2010) The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res 38:5797–5806. doi:10.1093/nar/gkq296
Reddel RR (2014) Telomere maintenance mechanisms in cancer: clinical implications. Curr Pharm Des 20:6361–6374
Forner A, Llovet JM, Bruix J (2012) Hepatocellular carcinoma. Lancet 379:1245–1255. doi:10.1016/S0140-6736(11)61347-0
El-Serag HB (2011) Hepatocellular carcinoma. N Engl J Med 365:1118–1127. doi:10.1056/NEJMra1001683
Schulze K et al (2015) Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 47:505–511. doi:10.1038/ng.3252
Totoki Y et al (2014) Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 46:1267–1273. doi:10.1038/ng.3126
Huang DS et al (2015) Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur J Cancer 51:969–976. doi:10.1016/j.ejca.2015.03.010
Nault JC et al (2013) High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 4:2218. doi:10.1038/ncomms3218
Paterlini-Brechot P et al (2003) Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 22:3911–3916. doi:10.1038/sj.onc.1206492
Sung WK et al (2012) Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet 44:765–769. doi:10.1038/ng.2295
Burger M et al (2013) Epidemiology and risk factors of urothelial bladder cancer. Eur Urol 63:234–241. doi:10.1016/j.eururo.2012.07.033
Cohen SM, Shirai T, Steineck G (2000) Epidemiology and etiology of premalignant and malignant urothelial changes. Scand J Urol Nephrol Suppl 34(203):105–115. doi:10.1080/00365590050509869
Hurst CD, Platt FM, Knowles MA (2014) Comprehensive mutation analysis of the TERT promoter in bladder cancer and detection of mutations in voided urine. Eur Urol 65:367–369. doi:10.1016/j.eururo.2013.08.057
Allory Y et al (2014) Telomerase reverse transcriptase promoter mutations in bladder cancer: high frequency across stages, detection in urine, and lack of association with outcome. Eur Urol 65:360–366. doi:10.1016/j.eururo.2013.08.052
Rachakonda PS et al (2013) TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci USA 110:17426–17431. doi:10.1073/pnas.1310522110
Dulak AM et al (2013) Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 45:478–486. doi:10.1038/ng.2591
Heaphy CM et al (2011) Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 179:1608–1615. doi:10.1016/j.ajpath.2011.06.018
Hsu CP, Hsu NY, Lee LW, Ko JL (2006) Ets2 binding site single nucleotide polymorphism at the hTERT gene promoter—effect on telomerase expression and telomere length maintenance in non-small cell lung cancer. Eur J Cancer 42:1466–1474. doi:10.1016/j.ejca.2006.02.014
Davies H et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. doi:10.1038/nature00766
Xing M et al (2013) Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA 309:1493–1501. doi:10.1001/jama.2013.3190
Liu X et al (2013) Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer 20:603–610. doi:10.1530/ERC-13-0210
Jansen M, Yip S, Louis DN (2010) Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet Neurol 9:717–726. doi:10.1016/S1474-4422(10)70105-8
Eckel-Passow JE et al (2015) Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med 372:2499–2508. doi:10.1056/NEJMoa1407279
Peifer M et al (2015) Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. doi:10.1038/nature14980
Horn S et al (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339:959–961. doi:10.1126/science.1230062
Huang FW et al (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339:957–959. doi:10.1126/science.1229259
Griewank KG et al (2014) TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J Natl Cancer Inst 106:dju246. doi:10.1093/jnci/dju246
Bell RJ et al (2015) Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 348:1036–1039. doi:10.1126/science.aab0015
Li Y et al (2015) Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol 17:1327–1338. doi:10.1038/ncb3240
Chiba K et al (2015) Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife 4:e07918. doi:10.7554/eLife.07918
Stern JL, Theodorescu D, Vogelstein B, Papadopoulos N, Cech TR (2015) Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev 29:2219–2224. doi:10.1101/gad.269498.115
Acknowledgments
We are grateful to Prof. Anne Frary for proof-reading and suggestions. We also thank Dr. Ekta Khattar and Yinghui Li for critical reading. We thank the Agency for Science Technology and Research, Singapore (A*STAR) for funding and support to the V.T. laboratory.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Akincilar, S.C., Unal, B. & Tergaonkar, V. Reactivation of telomerase in cancer. Cell. Mol. Life Sci. 73, 1659–1670 (2016). https://doi.org/10.1007/s00018-016-2146-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2146-9