
Abstract
The amyloid precursor protein (APP) is part of a larger gene family, which has been found to form homo- or heterotypic complexes with its homologues, whereby the exact molecular mechanism and origin of dimer formation remains elusive. In order to assess the cellular location of dimerization, we have generated a cell culture model system in CHO-K1 cells, stably expressing human APP, harboring dilysine-based organelle sorting motifs [KKAA-endoplasmic reticulum (ER); KKFF-Golgi], accomplishing retention within early secretory compartments. We show that APP exists as disulfide-bonded dimers upon ER retention after it was isolated from cells, and analyzed by SDS-polyacrylamide gel electrophoresis under non-reducing conditions. In contrast, strong denaturing and reducing conditions, or deletion of the E1 domain, resulted in the disappearance of those dimers. Thus we provide first evidence that a fraction of APP can associate via intermolecular disulfide bonds, likely generated between cysteines located in the extracellular E1 domain. We particularly visualize APP dimerization itself and identified the ER as subcellular compartment of its origin using biochemical or split GFP approaches. Interestingly, we also found that minor amounts of SDS-resistant APP dimers were located to the cell surface, revealing that once generated in the oxidative environment of the ER, dimers remained stably associated during transport. In addition, we show that APP isoforms encompassing the Kunitz-type protease inhibitor (KPI) domain exhibit a strongly reduced ability to form cis-directed dimers in the ER, whereas trans-mediated cell aggregation of Drosophila Schneider S2-cells was isoform independent. Thus, suggesting that steric properties of KPI-APP might be the cause for weaker cis-interaction in the ER, compared to APP695. Finally, we provide evidence that APP/APLP1 heterointeractions are likewise initiated in the ER.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The amyloid precursor protein (APP) is a type I transmembrane glycoprotein that is found in all cell types throughout the body, including neurons. Its overall structure resembles a cell surface receptor with a large ectodomain, a single transmembrane domain and a short cytoplasmic tail [1, 2] that may be important for signal transduction and transcription [1, 3]. Proteolytic APP cleavage leads to the production of the β-amyloid peptide (Aβ), which is the major constituent of the amyloid core of senile plaques, found in the brains of patients with Alzheimer’s disease (AD). APP is expressed in three different major isoforms generated by alternative splicing, consisting of 695, 751, or 770 amino acids. The larger two isoforms contain an additional 56 amino acid domain in the middle of the APP sequence, which is structurally and functionally related to Kunitz-type serine protease inhibitors (KPI) [4, 5]. Two paralogues of APP have been identified in mammals, named amyloid precursor like proteins, APLP1 and APLP2 [6]. The redundancy of APP and APLPs is reflected in an analogous protein domain structure and a similar proteolytic processing [7], although the Aβ region is unique to APP [8–10]. On the extracellular side, a cysteine-rich growth factor-like domain (GFLD) with heparin binding properties (HBD) is followed by zinc and copper binding domains (Zn/Cu-BD) referred to as the E1 domain. The E1 domain is then followed by an acidic stretch, rich in aspartic acid and glutamic acid, and the KPI domain, which is not present in APP695 and APLP1. The E2 region consists of another HBD/GFLD and a carbohydrate domain, containing the N-glycosylation sites of the ectodomain [7]. Although APP has been the subject of much study since its identification, its physiological function remains largely undetermined. Recently, a new research direction has begun to emerge, intending to elucidate the mechanisms and importance of APP dimerization. However, there is accumulating evidence from biochemical and structural data that APP can form homodimers [8–10] as well as heterodimers with its paralogues APLP1 and APLP2 [9]. Various experimental approaches have been attempted to visualize dimerization between APP family proteins. The first studies demonstrated the existence of APP–APP interactions at the cell surface, by covalently cross-linking APP at the plasma membrane [8, 9]. Other initial studies showed that a disulfide-bridged loop, encompassing residues 91–111, seemed to be required and necessary for the stabilization of APP homodimers [10, 11], whereas APP interaction was gradually diminished by adding a small peptide mimicking the loop region (loop peptide) [10]. Interestingly, this loop peptide interacted via hydrophobic forces with the APP E1 domain and decreased the generation of sAPPβ as well as Aβ40 and Aβ42 when the synthetic peptide was added to APP-expressing cells, raising the possibility that APP monomer/dimer status somewhat might affect Aβ generation and could play a role in the pathology of AD. In this respect, recent reports have also implicated the APP transmembrane region as a site for cis-dimerization. Introduction of a cysteine mutation in the juxtamembrane/transmembrane (JM/TM) region of APP has been shown to form constitutive dimers by disulfide bridging within the Aβ domain, suggesting close proximity and interaction of the APP TM region [8]. Moreover, the APP transmembrane domain efficiently dimerized in a bacterial assay [12], where APP contains three consecutive glycine-xxx-glycine (GxxxG) motifs [12–15], one embedded within the Aβ sequence. Therefore, this motif was investigated in depth due to its putative impact on Aβ generation. It has been shown that mutations of the glycine residues in the GxxxG motifs to alanine residues attenuate TM dimerization and result in a marked reduction in the processing of APP, and the production of toxic Aβ42 [12, 15]. Similarly, specific gamma-secretase modulators (GSM) were recently demonstrated to lower Aβ levels, by directly interfering with APP TMS dimerization [16]. However, chemically induced dimerization of APP led to decreased APP processing and reduced Aβ levels [17], likewise heterodimerization with its homologues APLP1 and APLP2 was also shown to diminish overall Aβ formation [11]. Other studies have focused more on the extracellular part of APP, and demonstrated by aggregation assays that the APP family members are able to form both homo-and heterotypic complexes in cis-and trans-cellular fashion and that this interaction promoted cell adhesion [9], depending on the E1 domain. Interestingly, the E1 domain was also identified as a major interaction interface for cis-directed dimerization using deletion constructs in co-immunoprecipitations [9], or FRET analysis [11].
Alternatively, it has been proposed by crystallographic and X-ray structure modeling that the E2 region can reversibly dimerize in solution in an antiparallel orientation [18], whereas other conformational studies indicated that the APP ectodomain is monomeric in solution, but forms a complex with extracellular matrix components, such as heparin, which may also regulate dimerization [19].
Despite accumulating data towards APP dimerization, the physiological significance and subcellular localization of its generation are still under debate. Preliminary evidence that APP oligomerization might occur as early as in the ER was obtained when chemically cross-linked [35S]sulfate-labeled APP migrated slower than [35S]methionine-labeled, after being monomerized by reducing agents, suggesting that dimerization was initiated before maturation of N-linked oligosaccharides or tyrosine sulfation [8]. Nevertheless, the precise mode of dimerization remained somewhat elusive, though predominantly hydrophobic interactions between the TM- or ectodomains have been suggested so far [9–12]. Interestingly, few studies have shown that e.g., C99 [14] or myc- and GFP-tagged APP migrates as a dimer on SDS-polyacrylamide gels [8], suggesting that stronger interactions might also play a role in APP dimerization.
To assess the putative dimerization potential of APP in different cellular compartments, we made use of well-described cytosolic dilysine-based motifs, designated as KKAA or KKFF, which are critical for endoplasmic reticulum (ER) localization, exit, or retrieval of type I membrane proteins [20–22]. We have employed a cell culture model system in CHO-K1 cells, stably overexpressing APP chimeric proteins, tagged with respective C-terminal doublelysine retention motifs. Using a variety of biochemical techniques, we could successfully demonstrate in a highly reproducible manner that chimeric APP695, containing the retention signals, was effectively retained within early secretory compartments. Additionally, we obtained high amounts of SDS-stable APP695 homodimers when APP was localized to the ER, but we also recovered minor quantities at the plasma membrane, which dissociate upon strong reducing conditions, suggesting the participation of disulfide bonds. Interestingly, the cis-dimerization capacity of APP within the same cell differed between isoforms, whereas trans-cellular dimerization was isoform independent. Finally, we could show that APP and APLP1 also form SDS-resistant heterodimers, likely initiated in the ER.
Materials and methods
Cell lines and transfections
All Chinese hamster ovary (CHO-K1) cell lines were grown in Alpha Minimum Essential Medium (α-MEM) supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, and 100 U/ml penicillin/streptomycin. All APP constructs were inserted into the retroviral expression vector pLBCX. For generation of stably expressing cells, virus production was induced by double transfection of pLBCX with pVSV-G at a 1:1 ratio into the packaging cell line GP2-293 (Clontech, Mountain View, CA, USA) using the calcium phosphate transfection method. The medium was changed after 48 h and viruses were collected for another 24 h. After infection with recombinant viruses in the presence of 5 μg/ml polybrene for 24 h, CHO-K1 cells were selected with 6 μg/ml blasticidin (Invitrogen, Carlsbad, CA, USA). Stable pools with comparable expression levels were analyzed in further experiments. Co-transfections of stably overexpressing CHO-K1 cells with other plasmid DNAs were performed using LipofectamineTM 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturers’ protocol.
cDNA constructs and cloning
The plasmids pLHCX-APP695wt, pLHCX-APP770wt, and pcDNA3-APP751 [23] were used as templates to subclone APP sequences into the pLBCX vector backbone with a 5′HindIII and 3′ClaI restriction site containing an additional 3′ myc-epitope, using the following fwd primer: 5′-CCCAAGCTTATGCTGCCCGGTTTGGCACTGCTC-3; and rev primer: 5′-CCATCGATTTAGTTCTGCATCTGCTCAAAGAACTT-3′. The same plasmids were used to add the KKAA (APP ER) or KKFF (APP Golgi) retention motifs to the very C-terminus of APP constructs following the myc-tag, using the rev primer 5′-CCATCGATTTAGGCGGCCTTCTTATGGTGATGGTGATGATGACCGGTATGCATATT-3′, for KKAA and 5′CCATCGATTTAGGCGGCTTTTTTATGGTGATGGTGATGATGACCGGTATGCATATT-3′ for KKFF, each containing a ClaI site. To attach an HA epitope to the C-terminus of all three different APP isoforms, the aforementioned cDNAs were used as templates with standard PCR techniques and the following primers: fwd 5′-CCCAAGCTTATGCTGCCCGGTTTGGCACTGCTCCT-3′, introducing a HindIII site, rev 5′- CCATCGATTTAAGCGTAATCTGGAACATCGTATGGGTAGTTCTGCATCTGCTCAAAGAACTTGTAGGTTGGATT-3′. The KKAA motif was subsequently attached to the HA tag at the C-terminus with the following rev primer: 5′- CCATCGATTTAGGCGGCCTTCTTAGCGTAATCTGGAACATCGTATGGGTAG TTCTGCATCTGCTCAAAGAACTTGTAGGTTGGATT-3′. N-terminal truncated APP695∆E1 in pcDNA3 was described elsewhere [11]. The cDNA was also subcloned into pLBCX and amplified by standard PCR techniques with the same primers as designated above, to attach a myc-epitope alone, or followed by an additional KKAA retention motif. All PCR products were digested with HindIII × ClaI and ligated into pLBCX retroviral expression vector. The cDNA of human APLP1 in the peak8 expression vector was kindly provided by Stefan F. Lichtenthaler (Munich, Germany). The coding sequence was amplified by PCR to add HindIII and ClaI restriction sites, using following primers: fwd 5′ CCCAAGCTTTAATGGGGCCCGCCAGCCCCGCTGCTC-3′ and rev 5′-CCATCGATTTAGGGTCGTTCCTCCAGGAAGCGGTAA-3′to subclone into pLBCX. To generate the cysteine mutants APP695 K28C and APP770 K28C, containing a C-terminal myc-epitope, APP695wt pLBCX and APP770wt pLBCX were used as templates and the mutation was introduced by an overlap extension PCR strategy. Two mutagenic primers, containing the desired mismatch K28C, completely complementary to each other were generated as follows: fwd 5′- GTGGGTTCAAACTGTGGTGCAATCATTG-3′ and rev 5′-CAATGATTGCACCACAGTTTGAACCCAC-3′. Each primer was used in a separate reaction with an outer flanking primer (5′HindIII and 3′ClaI, see above), designed to one end of the region of interest, which were used for all other APP constructs so far. In a third PCR, both halves of the construct, which are partly complementary, prime off each other and give the full-length product, comprising the desired mutation. The APP695 ER 98-105ser mutant was generated with the same method as described before, using the following mutagenic primers: fwd 5′-CCAGTGACCATCCAGAACTGGTCCAAGCGGGGCCGCAAGCAGTCCAAGACCCA TCCCCACTTTGT-3′; rev 5′-ACAAAGTGGGGATGGGTCTTGGACTGCTTGCGGCCCCGCTTGGACCAGTTCTGGATGGTCACTGG-3′. All generated constructs were checked by sequencing. Cloning of NT HA APP695 CT Split GFP constructs 1–10 and 11. Generation of plasmid NT HA APP695 CT Split GFP 1–10: for the first PCR, template APP695 NT HA in pcDNA3.1 [9] was amplified with forward primer sAPP-3 (starts at position 1681 in the APP695 ORF) 5′-GAAGTTGAGCCTGTTGATGCC-3′ and reverse primer: 5′-GCTATGACG CGTGTTCTGCATCTGCTC-3′, introducing a MluI restriction site. The resulting PCR product was digested with EcoRI and MluI to obtain a 293-bp fragment. For the second PCR, template GFP 1–10 [24] was amplified with forward primer 5′-GCGATGACGCGTGGTGGTTCGGGTGGTATGTCCAAAGGAGAAG-3′ containing a MluI site and reverse primer 5′-GCCCGCCTCGAGTCATGTTCCTTTTTCATTTGG-3′, including an XhoI site. The resulting PCR product was digested with MluI and XhoI. The two PCR fragments were ligated into vector pcDNA3.1+ containing the 3′ UTR cassette of APP (XhoI/XbaI) [17], using the enzyme sites for EcoRI and XhoI. To add in the 5′ end of the APP fragment, plasmid APP695 NT HA [9] was digested with EcoRI. The resulting 1,900-bp fragment including the HA tag was ligated via EcoRI in the above-mentioned new plasmid. Cloning of NT HA APP695 CT Split GFP 11 was performed in analogy to NT HA APP695 CT Split GFP 1–10. Only the second PCR product was generated differently: Template Split GFP 11 [24] was amplified with forward primer 5′-CTATGACGCGTTCGGGTGGTGGTGGTTCGGGTGGTGGTGGT-3′, including a MluI site and reverse primer 5′-CTATGCTCGAGTTAGGTGATGCCGGGGCGTTC-3′, introducing an XhoI site. The resulting 100-bp PCR product was digested with MluI/XhoI.
Antibodies
The monoclonal antibody 9E10 recognizing the myc-epitope was generated from a mouse hybridoma cell line and used to detect all overexpressed APP constructs in stable CHO-K1 cell lines. Optionally, the polyclonal CT15 [23] antibody was used, raised against the last 15 amino acids of APP. Detection of transiently transfected human APP isoforms, containing a C-terminal HA epitope, was carried out with polyclonal rat HA antibody purchased from Roche. Human APLP1 was detected with C-terminal, polyclonal 171615, purchased from Calbiochem. Monoclonal α-Tubulin antibody was obtained from Sigma-Aldrich. Secondary HRP-conjugated goat antibodies against mouse and rabbit were purchased from Jackson Lab, Maine (USA). NT HA APP695 CT Split GFP 1–10 and NT HA APP695 CT Split GFP 11 constructs were analyzed via Western blotting for APP full-length with monoclonal N-terminal APP antibody 22C11 (Millipore), or APP CTFs with antibody APP CT (clone Y188, rabbit monoclonal, Epitomics). APP-GFP fusions were detected with monoclonal GFP antibody (SC-9996, Santa Cruz).
Western blotting and co-immunoprecipitations
Cells were washed with cold 1× PBS, scraped off culture dishes and lysed in NP-40 lysis buffer (500 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 0.02% sodium azide), plus complete protease inhibitors (Roche). Debris were pelleted by centrifugation at 20,000 × g for 20 min at 4°C in a microcentrifuge. Equal amounts of total protein, determined by BCA protein assay (Pierce Chemicals, Rockford, IL, USA), were used for lysate analysis. Aliquots of CHO-K1 cell lysates, stably overexpressing APP constructs, were either incubated with SDS sample loading buffer (0.625 M Tris-HCl, pH 6.8, 2% w/v SDS, 10% w/v glycerol, 5% β-mercaptoethanol) and directly subjected to SDS-PAGE, or heated at indicated temperatures. Alternatively, samples were incubated with sample buffer without β-mercaptoethanol and heat denatured for 10 min. Proteins were electrophoresed on 4–12% NuPage (Novex®, Invitrogen) gradient gels and transferred onto nitrocellulose membranes (Millipore, Bedford, MA, USA). Non-specific binding to membranes was blocked 1 h with 5% non-fat dry milk in TBS containing 0.01% Tween-20 (Roth, Germany), before incubation with the appropriate primary and secondary antibodies. Proteins were detected using enhanced chemiluminescence (Millipore) by using the LAS-3,000mini (Fujifilm, Duesseldorf, Germany). For co-immunoprecipitation, cells were lysed in NP-40 buffer and equal amounts of total lysates (250 μg) were precipitated using protein A agarose beads (Invitrogen), with either 9E10 antibody for APP, or 171615 for APLP1 (Calbiochem). Beads were collected by low speed centrifugation and washed four times with lysis buffer. Bound proteins were eluted from beads with 30 μl 2 × SDS sample buffer containing BME and analyzed by Western-blot and appropriate primary and secondary antibodies. Immunoreactive bands were visualized using an ECL (enhanced chemiluminescence) system (Millipore), as above.
Cell surface-biotinylation
To examine surface levels of APP, cells were grown in 60-mm dishes to 90% confluency, and rinsed three times with ice-cold PBS. Cell surface proteins were biotinylated with 0.5 mg/ml Sulfo-NHS-LC–LC-Biotin (Pierce) in ice-cold PBS for 40 min at 4°C. The biotin solution was exchanged once after 20 min. Cells were washed four times with ice-cold PBS containing 50 mM NH4Cl to quench unconjugated biotin and lysed in NP-40 buffer. Equal amounts of proteins were incubated with NeutrAvidin Agarose resin (Pierce) at 4°C overnight. Biotinylated proteins were recovered by boiling in 2 × SDS sample buffer for 5 min and separated on 4–12% NuPage (Novex®, Invitrogen).
Metabolic labeling and immunoprecipitation of APP
CHO-K1 cells, stably overexpressing APP695wt or APP695 KKAA were plated in six-well culture dishes. After 24 h, the cultures were pulse-labeled for 15 min at 37°C with 1 ml methionine-free DMEM containing 150 μCi of [35S] methionine/cysteine (EasyTag™ EXPRESS35S Protein Labeling Mix). Cells were lysed immediately after the pulse (time 0), or chased for 15 min–48 h to determine the turnover of APP. Cell pellets were lysed in 500 μl of NP-40 lysis buffer plus protease inhibitors (Roche), and lysates were cleared by centrifugation at 20,000 × g for 20 min. Post-nuclear supernatants were incubated overnight at 4°C with antibody 9E10 (1/20) and protein A-agarose beads. Immunocomplexes were washed three times with NP-40 buffer, one time with PBS, and eluted from the beads by boiling for 10 min in 30 μl of 2 × SDS sample buffer. Proteins were separated on 4–12% NuPage (Novex®, Invitrogen) gradient gels followed by autoradiography on an X-ray film for a minimum of 12 h, at −80°C.
Drug treatments
To inhibit anterograde transport from ER to Golgi compartments, cells were treated with the fungal metabolite brefeldin A (BFA, Sigma; B-7651), ranging from 5 μg/ml to 30 μg/ml. Therefore, medium from cells was replaced 24 h after seeding with fresh medium containing indicated concentrations of BFA or vehicle (DMSO) alone and incubated from 1 to 4 h. Stock solution was prepared as 5 mg/ml in DMSO and stored at −20°C. To inhibit clathrin-mediated endocytosis, cell cultures were treated with 10 μM chlorpromazine (Sigma; C-8138) for 30 min. Chlorpromazine was dissolved in DMSO and stored as stock solutions (50 mM) at −20°C.
Drosophila Schneider (S2) cell aggregation assay
Semi-adherent S2 cells were cultivated at 25°C with growth medium (Schneider’s Medium (Invitrogen); 10% FCS 1% penicillin/streptomycin (10,000 U penicillin, 10 mg streptomycin/ml in 0.9% NaCl). Cells stayed adherent until 70–80% confluency was reached, after which they detached and proliferated in suspension. At this time point, cells were passaged (1/40–1/20) after resuspending to single cell suspension. S2 cells were transiently co-transfected with pMT-Gal4 and the corresponding pUAST constructs containing APP695, APP751, APP770 using Effectene (Qiagen), and subjected to the cell aggregation assay as described previously [9]. Briefly, expression was induced 48 h post-transfection by the addition of CuSO4 (500 μM) for 16 h. In all experiments, comparable transfection efficiencies of about 10% for all analyzed constructs were achieved. For analysis, 4 × 105 cells (single cell suspension) were incubated in a total volume of 1 ml fresh growth medium for 2 h at 90 rpm on a horizontal shaker. Afterwards, cells were transferred to poly-l-lysine coated cover slips using an abscised 1-ml Gilson pipette tip. Cells were allowed to attach to the cover slip for 2–3 h and subjected to immunocytochemical analysis as described before [9], using anti APP antibody (CT-20, Calbiochem) in 5% normal goat serum in PBS (1/1,000) and anti-rabbit AlexaFluor488 (Invitrogen) as secondary antibody. For quantification of aggregated cells, clusters of three or more transfected cells were scored as positive. In total, 900–1,100 transfected cells from at least three independent experiments were counted for each experimental setup in a blinded fashion. Statistical significance was tested using Students’ t test. Representative images of clustered cells were taken on a Leica laser scanning confocal microscope with a 63× water immersion objective.
Immunofluorescence of split GFP constructs
N2a cells were grown on poly-l-lysine (Sigma) coated coverslips (Marienfeld) in 24-well plates (Falcon) and transiently transfected (Lipofectamine 2000, Invitrogen) with NT HA APP695 CT 1–10 and NT HA APP695 CT 11. Fixation of the cells followed 17 h later, for 10 min with 4% paraformaldehyde and 4% sucrose in 1x PBS at 37°C. Permeabilization followed with 0.1% NP40 in 1x PBS. The cells were blocked with 5% (v/v) goat serum (Sigma) in 1× PBS for 1 h and the primary antibody (ER marker Grp78, rabbit polyclonal) (StressGen), or monoclonal mouse HA.11 antibody (Covance) and incubated overnight at 4°C. The secondary antibody, AlexaFlour-594 conjugated goat anti rabbit (Invitrogen), was incubated for 1 h at room temperature. The coverslips were embedded in Mowiol and analyzed by fluorescence microscopy (63× objective, Axio Observer Z.1, Zeiss).
Quantification and statistical analysis
Western blots were quantified by densitometry using ImageJ 1.44.
All graphs and statistical analyses were prepared using GraphPad Prism 4 software (GraphPad, La Jolla, CA, USA). Data were analyzed by one-way analysis of variance (ANOVA) coupled to Newman–Keuls post-test for multiple comparison or t test. p < 0.05 was considered as statistically significant.
Results
Retention of APP in the ER leads to the formation of SDS-stable homodimers
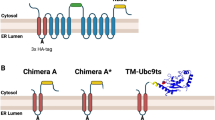
Although many aspects of APP dimerization have been studied, the precise cellular localization of its generation remains unclear. To identify the compartment where APP dimerization is initiated, we first generated chimeric proteins, using APP isoform 695 with a C-terminal myc tag, followed by either a -Lys-Lys-Ala-Ala (KKAA) or -Lys-Lys-Phe-Phe (KKFF) dilysine signal, designated APP ER or APP Golgi as illustrated in Fig. 1a. It has been reported that the sequence context of dilysine signals determines the efficiency of ER retention, shown for CD4-receptor chimeras fused to either retention motif [22]. Specifically, the KKAA chimeras were strictly localized to the ER, unlike KKFF constructs which shuttle between ER-and cis-Golgi compartment, due to the double phenylalanines (-FF) acting as an ER exit determinant [20–22]. We tested the oligomerization potential of APP trafficking mutants, both for KPI and non-KPI isoforms, in stably transfected CHO-K1 mass cultures, expressing comparable levels of the respective APP695 constructs. To determine whether the addition of the KKXX motifs to the cytoplasmic tail of APP would prevent the protein from being transported to the cell surface, we compared retention mutants to APP695 wt, by first employing surface biotinylation experiments (Fig. 1b). Protein expression was analyzed using SDS-PAGE and subsequent Western blotting of total cell lysates (Fig. 1b, left panel). In the biotinylation, no APP full length was detected for any of the retention mutants, indicating a strong intracellular retention mediated by double-lysine motifs (Fig. 1b right panel), whereas APP695 wt appeared in its fully glycosylated form at the plasma membrane (Fig. 1b right panel, APPfl mature). In the lysate controls, APP695 migrated as a set of two bands (~110–130 kDa) representing immature, mostly ER glycosylated APP, and the mature protein that has undergone complex O-glycosylation in the late-Golgi compartment. Surprisingly, for APP ER, two distinct migrating bands appeared in the lysate controls (Fig. 1b, left panel), of which the faster migrating band (~110 kDa) represents immature APP (before cis-Golgi-specific carbohydrate modifications) consistent with permanent ER retention. Additionally, we found an explicit slower migrating band for APP ER, co-migrating at the apparent molecular weight of a putative APP dimer (~220 kDa). This was particularly interesting, because in our experimental conditions we treated all lysate samples with loading buffer, containing SDS and β-mercaptoethanol (BME) prior to electrophoresis, but most importantly, the samples were not heat denatured at the same time (Fig. 1b, left panel, +BME/−95°C). Since the upper band did not disappear upon SDS treatment, we assume that stronger forces, other than hydrophobic associations, must be involved in APP dimer formation, such as intermolecular disulfide bonds. This notion was strengthened by the fact that retention of APP695 in the ER seemed to be sufficient to visualize SDS-stable homodimers, most likely generated via disulfide bonds between cysteine residues located in the E1 ectodomain. However, APP Golgi (containing the “leaky” KKFF motif) did not reveal an upper high-molecular-weight species around ~220 kDa, although it migrated exclusive as immature APP (Fig. 1b, Input) and was absent from the cell surface (Fig. 1b, PD NeutrAvidin, right panel), equivalent to APP ER. We hypothesize that APP bearing the ER exit determinant (-FF) shuttles permanently between the cis-Golgi compartment and the ER, resulting in less APP molecules at steady state in the ER, making it difficult to capture dimerization with our experimental conditions. However, this does not exclude that disulfide-associated APP dimers also exist in other compartments, but under normal cellular conditions this likely represents a sub-population of APP dimers, among others, held together by hydrophobic interactions, normally dissolved by the presence of SDS. Since it was recently proposed that APP can form dimers at the cell surface [9, 11, 19], we intended to include the plasma membrane as a putative source of dimerization. To do so, we transiently co-expressed a dominant negative (D/N) mutant of adaptor protein 180 (AP180) (Fig. 1b, right panel), inhibiting clathrin-mediated endocytosis [25, 26], thereby retaining APP at the plasma membrane and increasing its surface exposure. Indeed, mature APP wt was strongly increased at the cell surface and in the lysates controls upon co-expression of AP180 D/N (Fig. 1b left and right panel, lane four). Nevertheless, we failed to monitor any SDS-stable dimers when retaining APP at the surface, possibly because under normal cellular conditions the amount of covalent APP dimers reaching the surface is below detection limit on Western blots. Nevertheless, we demonstrate that stable APP dimer formation starts early in the secretory pathway, in the ER. Interestingly, when we reversed the experimental conditions and heat denatured the samples, without adding BME, we detected even higher-molecular-weight species, likely representing oligomerized APP (data not shown). These data indicate for the first time that the actual APP dimerization most probably occurs in the ER and that intermolecular disulfide bond(s) might be involved in APP dimerization. Furthermore, this reveals that APP probably exists in a monomer–dimer equilibrium, and that covalent disulfide bonds represent one mechanism of how APP dimerization might be achieved. Additionally, we employed two additional APP antibodies against the C-or N-terminus and confirmed the upper band as specific for APP (data not shown). To further assess the involvement of disulfide bonds in APP dimer formation, we applied stronger denaturing conditions to determine up to which temperatures the dimers remain stable in the presence of the reducing agent BME (Fig. 1c). We established a temperature curve, mixing equal amounts of lysates from APP ER overexpressing CHO-K1 cells with SDS sample buffer (5% BME final conc. in samples), prior to heat samples for 10 min at the indicated temperatures (4–85°C, Fig. 1c). Immunoblotting with 9E10 antibody revealed that dimers persisted up to 50°C, whereas higher temperatures led to a progressive decline, finally resulting in the total disappearance of the upper band at temperatures over 60°C. Quantification of immunoblots clearly showed a corresponding increase of ~50% in monomers vice versa (Fig. 1d). These results indicated that under standard detergent lysis conditions, APP forms disulfide-linked dimers which can only be fully disrupted by extended heating of protein samples in the presence of BME.

ER-retention of APP by addition of the di-lysine motif KKAA induces SDS stable homo-dimers in an oxidizing cellular surrounding. a Schematic illustration of the APP domain structure and the used dilysine retention constructs. APP isoform 695 (APP695) was fused to a C-terminal myc-tag, followed by either a KKAA ER retention motif, or a “leaky” KKFF motif allowing trafficking to the cis-Golgi compartment. The E1 domain, acidic domain, E2 domain, transmembrane domain (TM), and the intracellular domain are indicated. b CHO-K1 cells stably over expressing the respective APP695 wt or APP695 retention constructs were subjected to cell surface biotinylation. To inhibit endocytosis from the cell surface, AP180 D/N (lane four) was transiently co-expressed. At 24-h post-transfection, cell surface proteins were labeled with sulfo NHS-SS-biotin at 4°C, prior to lysis. Biotinylated samples were precipitated with NeutrAvidin agarose beads and analyzed by SDS-PAGE and Western blot (PD: NeutrAvidin; right panel). As input controls, 20 μg of cell lysates were used (input; left panel). APP full length, CTFs and AP180 D/N were probed with 9E10 antibody. Monoclonal α-Tubulin antibody served as a loading control to verify the absence of endomembrane contaminants in the biotinylation. For the input control, all samples were treated with SDS sample loading buffer, containing β-mercaptoethanol (BME) without heat denaturing prior to SDS gel electrophoresis. To elute surface proteins from NeutrAvidin beads, samples were denatured for 5 min at 95°C with BME and separated on 4–12% SDS gels. Molecular weight standards (kDa) are indicated on the left. c Cell lysates (20 μg each lane) of CHO-K1 cells stably overexpressing APP ER (KKAA), were incubated with SDS sample loading buffer (+ BME) and heat denatured for 10 min at the indicated temperatures (4–85°C). APP monomers and SDS stable dimers were detected, using the monoclonal anti myc antibody (9E10), shown on one representative Western blot. d APP dimers and monomers were quantified by densitometric analysis of Western blots, as the one shown in c. In each case, intensities at 4°C were set as 100%. Diagram shows relative intensity (%) of APP monomers and dimers, plotted against different temperatures. Data are (n ≥ 3) ± SEM, one-way ANOVA
ER retention does not affect APP biosynthesis but increases its half-life
We next investigated whether it is possible to visualize the dimerization of unmodified wild-type APP695 (APP695 wt) without adding a C-terminal retention signal. To test this, we applied brefeldin A (BFA), a macrocyclic lactone antibiotic, inhibiting protein secretion at an early step in the secretory pathway [27–30]. It has been shown that BFA leads to rapid redistribution of the Golgi apparatus into the ER, inhibiting the anterograde movement of membrane traffic beyond the mixed ER/Golgi system, thereby retaining secretory and membrane proteins in the ER [27–30]. Cell cultures, overexpressing APP ER or APP wt were treated with 5 μg/ml BFA for different time periods (0–8 h) at 37°C, to block ER export. Subsequently, lysates were separated on a non-reducing SDS-PAGE, followed by immunoblotting with the APP specific polyclonal antibody CT15. Indeed, we found that addition of BFA to cell cultures caused the formation of an SDS-resistant high-molecular-weight band for APP wt, which co-migrated with the upper band of APP ER. A longer exposure of the same blot showed a beginning dimer formation already after 1 h treatment (Fig. 2a, longer exposure and Fig. 2b), with further increase after 8 h. Moreover, the difference between mature and immature APP becomes indistinct, due to the accumulation of particular immature APP in the ER. Interestingly, we also found a stabilizing BFA effect on pre-existing dimers, generated by APP ER (Fig. 2a, c), suggesting that even APP dimerization can be increased when ER exit was further inhibited by the application of BFA. However, the strong increase in monomers for APP ER was due to the employed antibody (CT15), also detecting the fraction of endogenous APP from CHO cells. In contrast, treatment of cell cultures, overexpressing APP ER, with increasing concentrations of BFA (10–30 μg/ml) for 1 h, led to an inverted monomer–dimer equilibrium (Fig. 2d–f) with a 50% decline in monomers, accompanied by a twofold increase in dimers (Fig. 2e, f). Strikingly, this effect was now visible with 9E10 antibody, exclusively detecting overexpressed APP, since endogenous APP from CHO cells obscured this previously (compare Fig. 2a). Nevertheless, this effect was highly reproducible and not dose-dependent. Taken together, these results implicated that not all APP molecules retained in the ER, have necessarily transformed into a dimeric state so far. Thus, extended exposure of APP to an oxidative surrounding had a considerable influence on the monomer–dimer balance, favoring dimerization. To investigate a potential influence of ER-retention on the half-life and biosynthesis of APP, we performed a pulse-chase analysis (Fig. 2g). Following a 15-min pulse with 150 μCi of [35S]-methionine/cysteine, we found no difference in the generation of immature APP between APP695 wt, versus APP695 ER (0 min). At time zero, APP consists predominantly of immature N-glycosylated species in both cell lines. However, fully mature APP appeared after 15 min exclusively in APP695 wt cells, and becomes stabilized after 30 min, thus reflecting traffic through the Golgi compartment. After a 1-h chase period, the APP wt level was dramatically reduced (Fig. 2h, half-life APP wt 45 min) and hardly detectable after 4 h. In contrast, cells overexpressing the AP ER retention mutant did not generate any O-glycosylated, mature APP during the chase period. However, turnover of APP ER is significantly prolonged, and ~50% of total APP can still be detected even after 5.5 h (Fig. 2h). Thus, the half-life of APP ER is more than sevenfold longer than in APP695 wt cells. Note that different chase time points for the APP ER mutant were chosen after an initial pulse-chase experiment already suggesting a prolonged half-life (data not shown). We also confirmed that retention in the ER severely limits APP metabolism, leading to decreased secretion of Aβ and APPsα/β into the culture medium (data not shown), since access to secretases is prevented [31, 32].

Homodimerization of APP wt can be induced by inhibiting protein traffic out of the ER with brefeldin A. a CHO-K1 cells stably overexpressing either APP695 wt or APP695 fused to the ER retention motif KKAA (APP695 ER) were treated with 5 μg/ml brefeldin A (BFA) for the indicated time points (0–8 h). Aliquots of cell lysates were mixed with SDS sample buffer, containing BME without heat denaturing and separated on a 4–12% SDS-PAGE. APP was detected with CT15 antibody, shown on one representative Western blot. The bottom panel shows a longer exposure time, indicating dimer formation of APP695 wt when export of the ER is blocked by BFA. b Diagram showing the ratio of APP wt dimer formation during BFA treatment, plotted against time, with ± SEM of three independent experiments, statistical significance: ***p < 0.001, one-way ANOVA. c Diagram depicting increasing stabilization of APP ER preexisting dimers during BFA treatment, plotted against time. Data represent ± SEM (n ≥ 3), statistical significance: ***p < 0.001, one-way ANOVA. d Cell cultures of CHO-K1 overexpressing APP ER were treated for 1 h with increasing concentrations (10–30 μg) of BFA, or with vehicle (100% ethanol) alone. Samples were treated as under a. APP was detected with monoclonal 9E10 antibody, shown as one representative Western blot. e Diagram showing relative increase of APP ER dimers with higher BFA concentrations, quantified as percentage over vehicle control. Data represent ± SEM as indicated (n ≥ 3). Statistical significance: ***p < 0.001, t test. f Diagram showing simultaneous reduction of APP monomers with increasing BFA concentrations, depicted as percentage of vehicle control. Data represent ± SEM as indicated (n ≥ 3). Statistical significance: ***p < 0.001, t test. g ER retention inhibits APP degradation and increases its half-life. CHO-K1 cell cultures stably overexpressing either APP695 wt or APP695 ER, respectively, were pulse-labeled for 15 min with 150 μCi of [35S] methionine/cysteine. Cells were harvested immediately after the pulse (0 min), or chased for the indicated time points with non-radioactive medium. APP was immunoprecipitated with monoclonal 9E10 antibody and subjected to SDS-PAGE followed by autoradiography on an X-ray film for a minimum of 16 h at −80°C. Note that different time points for APP695 ER were chosen due to its extended half-life. APP mature (APP m) and immature form (APP imm) are indicated. All time points were run in duplicates. For APP ER, samples were run alongside an APP wt control sample to confirm the difference between immature and mature APP. h Half-life graph. Data are mean ± SEM of four independent experiments. *** Statistically significant difference (p < 0.001; t test) between APP wt and APP ER is indicated
APP wild-type homodimers are initially formed in the ER but occur also at the cell surface
We have shown that retention of APP695 in the ER, either by a C-terminal ER retention motif, or blocking its export with BFA, led to the formation of SDS stable homodimers (Fig. 1). To verify that APP dimerization occurs early in the secretory pathway, we attempt to use a different approach. We employed a GFP tagging system for the intracellular detection of APP695, which is based on the auto-assembly capacity of two non-fluorescent portions of GFP, termed GFP 1–10 and GFP 11. When brought together by the association of two interacting partner proteins, individually fused to the fragments, they restore a fully fluorescent GFP, termed bimolecular fluorescence complementation (BiFC) [24]. This enables us to visualize APP–APP interactions and their subcellular localization under physiological conditions [24]. Therefore we fused the two complementary GFP fragments separately to the C-termini of HA-APP695 [9] and co-expressed both constructs in neuroblastoma N2a cells (Fig. 3a). The green fluorescence in cells co-expressing the BiFC constructs clearly indicates an interaction of APP molecules under physiological conditions. We observed a punctate vesicular staining, possibly resembling Golgi or endosomal structures and also an ER staining, as indicated by co-staining with the ER marker protein Grp78 (overlay). These data corroborate, together with our data based on the ER retention signal, that APP dimerization occurs in the endoplasmic reticulum (ER) before its trafficking through the Golgi to the cell surface. Due to its rapid travel along the secretory pathway, we observed the same outcome on SDS-PAGE, only when we mediated APP retention in the ER (Figs. 1, 2). By contrast, no specific GFP fluorescence was detected when single halves of GFP fused to APP (APP-GFP 1–10 and APP-GFP 11) were individually expressed in N2a cells (Fig. 3b). To rule out that the non-detectable GFP signals in the negative controls might result from no expression of transfected plasmids, we performed immunostainings of HA-APP (Fig. 3b, right part), clearly revealing the presence of the APP fusion proteins. Moreover, this confirmed that the split GFP does not perturb the true APP localization in the cell, since expression patterns of negative controls were comparable to those of APP BiFC in the positive controls. Additionally, the expression levels of the APP-GFP fusion proteins in the positive and negative controls were also confirmed by Western blotting (Fig. 3c).These results suggest that APP dimers are initially formed in the ER. However, it cannot be ruled out that dimerization might occur at later stages of the transport. Having established that APP695 wt can form homodimers at an early stage in the secretory pathway, this does not tell us whether these dimers are caused by disulfide bonds or other interactions. We therefore wanted to show that disulfide bridged APP homodimers are in fact generated in the ER, but do also occur naturally in the cell. The APP ER retention construct has proven as a reliable tool to visualize stable dimer formation in the ER, nevertheless since it does not traffic beyond ER, we cannot use this construct to show the existence of disulfide linked APP dimers in other cellular compartments, e.g., at the plasma membrane. We employed a different approach to demonstrate that dimers generated in the ER can indeed reach the cell surface. We first slowed down anterograde ER-to-Golgi transport [27–30] by pre-treating cells overexpressing APP 695 wt with 10 μg/ml BFA for 1 h to allow efficient dimer formation in the ER (Fig. 3d). Next, cells were either directly lysed, or BFA was washed out and cultures were incubated for an additional 30 min at 37°C with medium containing chlorpromazine (CPZ, 10 μM). Chlorpromazine prevents assembly of the adaptor protein-2 (AP-2) on clathrin-coated pits and thus causes a loss of endocytosis [33]. Subsequently, cells were lysed or optionally subjected to a surface biotinylation (Fig. 3e). As expected, upon BFA treatment APP disappeared from the cell surface, verified by biotinylation (Fig. 3e). Furthermore, we were able to trigger APP695 wt dimer formation, as seen previously (compare to Fig. 2a). In contrast, removal of BFA rapidly restored protein secretion from the ER to the Golgi and the cell surface [29, 30], and APP reappeared at the plasma membrane, established by CTF generation and fully glycosylated species, both in lysates and biotinylation experiments (Fig. 3d, e). Strikingly, chlorpromazine (CPZ) treatment led to a marked re-establishment of mature APP at the surface, compared to vehicle control (Fig. 3e), and BFA/CPZ treatment caused the formation of SDS-resistant high-molecular-weight species, migrating as a doublet (Fig. 3d). We assume that the slight size shift of the upper bands represents different glycosylated APP dimers, indicating transport along the secretory route. However, this higher “mature” dimer was absent in APP ER-or BFA-treated APP695 cells, suggesting that dimers were initially generated in the ER, but can be effectively transported through the secretory pathway, reaching the cell surface. This was particularly visible upon blocking endocytosis, thus preventing newly generated dimers from being instantly re-internalized. Note that heating the samples (+95°C) with BME resulted in the disappearance of the upper band(s), supporting our proposed model that APP dimers can associate via covalent intermolecular disulfide bonds.

APP wt homodimers are formed in the ER but also occur at the cell surface. a N2a cells were transiently co-transfected with NT HA APP695 CT Split GFP 1–10 and NT HA APP695 CT Split GFP 11. The cells were fixed with paraformaldehyde and immunostained with ER marker Grp78, followed by incubation with AlexaFlour-594 conjugated secondary antibody. The green fluorescence represents the APP dimer, induced by bimolecular fluorescence complementation (BiFC). Co-staining with Grp78 revealed colocalization of the APP dimer (inset) with the ER. b Single transfections of N2a cells with either NT HA APP695 CT Split GFP 1–10 or NT HA APP695 CT Split GFP 11. APP expression was verified with HA antibody, confirming a non-perturbed distribution (right panel, red). The green channel does not show any specific fluorescence. Scale bar is 10 μm. c N2a cells were transiently transfected with NT HA APP695 CT Split GFP 1–10, NT HA APP695 CT Split GFP 11 or GFP only. Cell lysates were analyzed via Western blotting for APP full length (top part), APP CTFs (middle part) and GFP, or GFP-containing CTFs (bottom part). Note that GFP antibody does not recognize GFP 11 alone. d CHO-K1 cells stably overexpressing APP 695 wt were pretreated for 1 h at 37°C in the presence of 10 μg/ml brefeldin A (BFA), or DMSO (vehicle) for 1 h to block protein secretion from the ER. Subsequently, cells were either directly lysed (BFA), or medium containing BFA was removed, and cells were incubated for additional 30 min at 37°C with 10 μM chlorpromazine (BFA/CPZ), to allow transport to the cell surface, but inhibit further endocytosis. Lysates were analyzed either under non-reducing (+β mercaptoethanol; −95°C), or reducing conditions (+β-mercaptoethanol; +95°C). Note that cells pretreated with BFA, followed by chlorpromazine exposure (BFA/CPZ), display two upper SDS-resistant bands migrating with a slight size shift, indicating different glycosylation patterns (dimer i/m). Note that strong denaturing conditions (+95°C) result in the disappearance of the upper migrating bands, suggesting disulfide bond formation. The separating line indicates different exposure times to visualize dimer formation in treated cells, whereas APP ER overexpressing cells served as size reference for dimers. e Cells were treated as described under d, and subjected to cell surface biotinylation to confirm drug treatments. APP ER is completely absent from the surface, whereas only minor quantities of mature APP reach the surface upon BFA treatment. Removal of BFA, and subsequent incubation with chlorpromazine (BFA/CPZ), strongly increases surface exposed mature APP, compared to vehicle control. Note that no dimers are recovered in biotinylation, because NeutrAvidin beads were boiled in BME containing sample buffer, to release biotin conjugated proteins
APP homodimerization is dependent on the extracellular cysteine-rich E1 domain of the protein
Findings so far, by us and others, strongly argue for a mechanism that drives APP dimerization by its N-terminal cysteine-rich E1-domain [9, 11]. To further dissect the mechanisms by which APP homodimerization is accomplished and to exclude a potential interaction via its cytosolic part, we co-expressed C-terminal adaptor proteins in order to compete for binding, possibly perturbing or diminishing homodimerization (Fig. 4a). The intracellular domain of APP contains one YENPTY motif, which has been shown to interact with several adaptor proteins including FE65, X11, JIP, Shc, Tip60, and Dab1 [34, 35]. Specifically, we overexpressed increasing amounts of either Fe65 or X11α in CHO cells stably transfected with APP ER (Fig. 4a), thus providing a putative sterical hindrance for APP dimers. We observed a slight increase in APP monomers as well as dimers with co-expression of Fe65 and a more severe effect for X11α. This is consistent with previous findings that both adapter proteins stabilize either APP intracellular fragments or increase the half-life of full length APP [36, 37]. Additionally, we confirmed a direct binding of Fe65 to APP wt and APP ER by co-immunoprecipitation experiments and successfully co-purified equal amounts of flag-tagged Fe65 (data not shown). These results showed that Fe65 can interact efficiently with the APP ER mutant via its cytosolic exposed C-terminal part, while the APP N-terminus remains directed into the ER lumen where disulfide-bridged dimerization can occur. We next set out to ascertain that the ~220-kDa band indeed represented an APP homodimer and not any heterodimer of APP with another protein of the same putative molecular mass. Therefore, we generated an additional APP695 construct with a C-terminal HA tag and co-expressed APP695 HA wt or its ER retained counterpart (APP695 HA ER) in CHO cells, stably overexpressing APP695 myc ER (Fig. 4b). The overexpressed APP-myc and APP-HA fusion proteins were probed with mouse anti-myc or rat anti-HA primary antibodies vice versa on different immunoblots. In fact, we confirmed that the high-molecular-weight band represented APP homodimers, since Western blots with either myc-or HA-antibody, independently revealed a band at ~220 kDa in cells co-expressing HA-tagged and myc-tagged APP species. Individually, the APP HA and APP myc proteins migrate as distinct and separable bands under either reducing or non-reducing conditions (Fig. 4b). However, in the presence of reducing and denaturing conditions (+95°C), dimers were converted to monomers, which increased simultaneously (Fig. 4b, upper right blot). Interestingly, co-transfection of APP HA wt into cells already expressing APP myc ER revealed a slightly weaker band around ~220 kDa with HA antibody (Fig. 4b, bottom blot, third lane), in addition to the doublet representing both maturation states of APP. This band most likely represents a dimer composed of APP myc ER and APP HA wt, suggesting that APP695 ER could retain a portion of wt APP within the ER, due to homodimer formation. The most feasible interpretation of our approach is that these high-molecular-weight bands represent APP myc/APP HA “heterodimers”, indicating that APP forms homodimers within the same cellular compartment (cis-dimers). Finally, these findings also minimized the possibility that APP happens to “heterodimerize” with any other unrelated protein of the same molecular weight. Moreover, this is in agreement with our BiFC approach (Fig. 3), already demonstrating a specific interaction of two APP695 molecules, by restoring a functional fluorescent GFP. It has been shown by co-immunoprecipitation experiments and FRET analysis that deletion of the E1 domain impedes APP homodimerization [9, 11] as well as heterointeraction with APLPs [9, 11]. Accordingly, we hypothesized that APP lacking the cysteine-bearing E1 domain is no longer capable of dimer formation in the ER. This region of APP encompasses the growth factor-like domain (GFLD) and a copper-binding domain (CuBD) [38, 39], which contain together 12 cysteine residues, possibly promoting stable covalently linked dimers or higher multimers. We generated CHO-K1 cell lines, overexpressing APP695 deletion constructs, lacking the N-terminal growth factor-like and copper-binding domains (∆E1), thereby omitting all of the cysteines and compared them with cells expressing the corresponding full-length constructs (Fig. 4b). Indeed, the KKAA motif particularly retained APP∆E1 ER in the cell, confirmed by surface biotinylation (Fig. 4c, right panel) and the lack of Golgi-specific carbohydrate modifications (Fig 4 c, left panel). Strikingly, the generated deletion constructs were incapable of forming stable high-molecular-weight species, regardless of ER retention or not (Fig. 4c), indicating that the E1 domain is essential for generating SDS-resistant APP homodimers (compare illustration in Fig. 4e). Note that less mature APP∆E1 was detected at the surface compared to APP695 wt, likely due to less free primary amino groups for biotin attachment, present in the N-terminal truncated APP construct. It has been reported that APP contains a region within the E1 domain, encompassing residues 99–110, with an active heparin binding capacity [38] that might form a loop, stabilized by a disulfide bond between Cys-98 and Cys-105. Based on findings that a synthetic peptide corresponding to the loop residues was able to interfere with APP dimerization [10], we generated an ER-retained APP loop double mutant exchanging Cys-98 and Cys-105 for serine residues (APP ER 98-105ser). We compared cell extracts from CHO-K1 cells transiently transfected with the serine mutant to lysates of our stably expressing APP 695 wt and APP695 ER cells (Fig. 4d). Western-blot analysis clearly demonstrated that the APP ER 98-105ser mutant failed to form homodimers in an oxidizing environment when sulfhydryl groups were exchanged by hydroxyl groups, compared to APP ER, which contains the respective cysteine residues. Thus, we conclude that these cysteines located within the APP heparin-binding site are sufficient to mediate APP homointeraction, and that binding of two N-terminal domains of APP was entirely dependent on the oxidation of the thiol groups. However, at this point we cannot exclude the possibility that cysteine mutants other than those analyzed would have had similar effects on APP dimer formation. Nevertheless, this indicates for the first time that cysteines, located within the E1 domain, might also contribute to cis-dimerization between two adjacent APP molecules, via intermolecular disulfide bonds, as well as stabilizing the APP structure via intramolecular S–S bonds (see illustration in Fig. 4e). We confirmed the necessity of the E1 domain for APP N-terminal interactions, by generating APP truncated at the β-secretase site [40], C99 wt or C99 ER (data not shown). Thereby the entire extracellular part, including the central E2 domain, was absent and Western-blot analysis revealed no SDS-stable dimers for C99 wt or the C99 ER.

APP homodimerization is dependent on cysteine residues in the E1 domain. a CHO-K1 cells stably overexpressing APP695 KKAA (APP ER), were transiently co-transfected with increasing concentrations (1-, 2-, 3 μg) of plasmid DNA encoding C-terminal adaptor proteins Fe65, X11α or empty vector. The upper Western blot shows myc tagged APP ER full length (immature form, lacking Golgi glycosylation pattern) and SDS stable dimers, at molecular weight of ~220 kDa. The lower blots depict transfection controls, using either flag antibody for Fe65, or 9E10 for X11α. b CHO-K1 cells stably overexpressing myc-tagged APP695 ER, were transiently co-transfected with HA-tagged APP constructs, APP695 HA ER or APP 695 HA wt. Western blotting with myc (mouse) or HA (rat) antibody reveals dimers (~220 kDa myc-/HA-APP) under non-reducing conditions, separated on 4–12% acrylamide gradient gels. In the presence of BME and 95°C, APP dimers are converted to monomers, migrating at ~110 kDa. Note that dimers can be detected with both antibodies and myc dimers are increased when APP HA ER is co-expressed (top blot, second lane). Additionally, a weak dimer band occurs when APP HA wt is co-expressed in APP myc ER cells (bottom blot, third lane). This suggests that APP myc ER can retain a portion of APP HA wt in the ER and that dimers are indeed homodimers consisting of APP myc and APP HA. c CHO-K1 cells stably overexpressing APP695 wt, APP695 ER, or the respective constructs, lacking most of the extracellular part (APP∆E1 and APP∆E1ER, compare Fig. 3d), were subjected to surface biotinylation. Biotinylated samples were precipitated with NeutrAvidin agarose beads, followed by SDS-PAGE and Western blot (PD: NeutrAvidin; right panel) and corresponding cell lysates were used as input controls (input; left panel). APP full length, APP∆E1 and CTFs (comigrating ~12 kDa MW) were detected with 9E10 antibody, note that APP lacking the E1 domain migrates faster at ~75 kDa. d Cysteine residues at positions 98 and 105 within the APP extracellular (E1) domain are critical for disulfide bridged dimers, generated in the ER. Cysteines at positions 98 and 105 were substituted by hydrophilic serine residues. Lysates of CHO-K1 cells stably overexpressing APP wt or APP ER, or transiently transfected with APP ER 98-105ser, were analyzed on 4–12% SDS gels and immunoblotted with 9E10 antibody for APP. Note, despite an effective retention, APP homodimerization was impaired in forming homodimers when cysteine residues 98 and 105 are converted to serines, precluding the formation of disulfide bonds between the absent thiol groups. e Schematic illustration of APP695 ER and the corresponding APP∆E1 ER construct. For the APP deletion construct, the signal peptide sequence of APP was introduced in frame with the remaining domains, starting with the acidic region. Deletion of the entire, cysteine-rich E1 domain abolished APP dimerization via disulfide bond formation, as shown under b. Molecular mass standard is indicated on the left side of the blots
Heterointeraction between APP and its homologue APLP1 is initiated and increased in the ER, depending on the APP E1 domain
Various experimental approaches have demonstrated that APP is capable of forming heterodimers with its homologues, APLP1 and APLP2 [9, 11], both intracellular in cis- and intercellular in trans-manner [9]. Since we could define the ER as the cellular compartment providing ideal conditions for triggering APP695 homointeractions via disulfide-bridge formation, we investigated whether this mechanism would also contribute to heterodimerization between APP and APLP1. As a first biochemical approach, we performed a cell surface biotinylation in our CHO-K1 cells stably overexpressing either APP695 wt or APP ER, transiently co-transfected the human APLP1 cDNA (Fig. 5a). Co-expression of APLP1 resulted in a significant reduction of APP695 ER homodimers (Fig. 5a, upper panel, lane 4) representing a decrease of more than 60% (Fig. 5b), accompanied by the simultaneous formation of APP/APLP1 heterocomplexes in the ER (Fig. 5a lower panel, lane 4 and Fig. 5c). At the same time, we monitored an increase of ~50% in APP monomers (Fig. 5b), indicating that a portion of APP homodimers was converted back to monomers, whereas the rest appeared as heterodimers with APLP1. Despite equal expression of APLP1 in both cell lines (Fig. 5a, bottom panel), we found less mature APLP1 at the surface when co-expressed with APP695 ER (Fig. 5a, bottom panel, lane 2 and 4), corresponding to a 50% reduction when compared to APP695 wt (Fig. 5d, bottom panel, lane 2 and 4). This indicates that the interaction of APP and APLP1 occurs already in the ER, and that APP can potentially retain a portion of APLP1 in the ER as a consequence of forming stable heterocomplexes. Interestingly, we found less C-terminal fragments for APLP1 when co-expressed with APP ER, suggesting impaired generation due to ER retention. Note that only fully mature proteins appear at the cell surface whereas secretase derived metabolites (CTFs) are only visible in the lysate controls of APP695 wt and APLP1. To provide further evidence for the interaction between APP and its homologue APLP1, we performed co-immunoprecipitation studies in our stable CHO-K1 cells upon co-expression of human APLP1 (Fig. 6). Lysate controls and co-purified proteins (IP APLP1 or APP) clearly indicate a strong interaction between both proteins in the ER. However, APP homodimerization in the ER is reduced upon APLP1 co-expression (Fig. 6b lysate WB 9E10, lane 4), compared to APP695 ER alone (compare Fig. 6b). At the same time, we observed again the formation of a distinct higher-molecular-weight band for APLP1 in cell lysates, only when co-expressed with APP695 ER, as seen previously (Fig. 5a, bottom blot). In line with the biotinylation approach (Fig. 5a), this band most likely represents APP/APLP1 heterocomplexes, since it co-migrates with the size of APP homodimers (~220 kDa) and both homologues have about the same molecular mass. Furthermore, specific interaction between both proteins was confirmed in APP-and APLP1-directed co-immunoprecipitation experiments (Fig. 6, lower panels). Most interestingly, the amount of co-purified proteins (immature form) was increased when APP was retained in the ER. Taken together, our findings clearly demonstrate a heterocomplex formation between APP and APLP1 as early as in the ER, most likely via disulfide-bond formation between the respective cysteine-bearing E1 domains of both proteins.

Heterodimerization between APP and APLP1 can be initiated and increased in the ER. a CHO-K1 cells stably overexpressing either APP695 wt or APP695 ER were transiently co-transfected with human APLP1 cDNA, or empty vector alone (–). The 24 h post-transfection cells were labeled with sulfo NHS-SS-biotin and biotinylated samples were precipitated with NeutrAvidin agarose beads (PD: NeutrAvidin; right panel). APP full length and CTFs were detected with 9E10 antibody. APLP1 full-length and C-terminal fragments were stained with the polyclonal APLP1 antibody 171615 and α-Tubulin served as a loading control. For detection of surface proteins, NeutrAvidin beads were treated with reducing loading buffer and heated at 95°C, whereas input controls were not heat denatured. All samples were separated on 4–12% SDS gradient gels. Note that heterointeraction of APLP1 and APP695 ER diminishes APP homodimers in the ER and lowers surface levels of mature APLP1. Shown is one representative Western blot. b Diagram showing relative changes in APP dimers and monomers when co-transfected with pcDNA3 or APLP1. Data represent ± SEM as indicated (n ≥ 3). Statistical significance: ***p < 0.001, **p < 0.01, t test. c Graph showing formation of APP/APLP1 heterodimers when co-expressed with APP ER. Data are expressed as percentage increase over APP wt and represent mean ± SEM as indicated (n ≥ 3), statistical significance: ***p < 0.001, t test. d Diagram showing significant reduction in APLP1 surface levels when co-expressed with APP ER, compared to when co-expressed with APP wt, as ± SEM (n ≥ 3) and statistical significance **p < 0.01, t test

CHO-K1 cells stably overexpressing either APP695 wt or APP695 ER were transiently co-transfected with human APLP1 cDNA, or empty vector pLBCX alone (–). At 24-h post-transfection, cells were lysed and equal amounts of total lysates (250 μg) were immunoprecipitated with either 9E10 antibody (for APP), or 171615 antibody (for APLP1). Co-purified proteins were detected with antibodies as indicated (IP: APLP1 WB: 9E10 for APP, lower panel; left) and reverse (IP: 9E10, WB: APLP1, lower part; right). Lysate controls were analyzed with the same antibodies (upper blots) and Tubulin served as loading control. Note that heterointeraction of APP and APLP1 is increased, when APP is retained in the ER
Intracellular cis-dimerization of APP770 in the ER is diminished by the KPI domain, but can be induced by a constitutive cysteine bond at the transmembrane domain
All studies investigating APP dimerization so far have used exclusively the neuronal APP isoform 695 or APLP1 [9, 11, 13, 17]. However, most tissues and glial cells express APP with an additional KPI domain, a 57-amino-acid insert, showing homology to the Kunitz family of serine protease inhibitors [41]. Therefore, we wanted to explore whether APP harboring an additional KPI domain in its extracellular part would display the same dimerization pattern as seen for APP695, since it possesses six further cysteine residues, additional to 12 located within the E1 domain. We generated CHO-K1 cells with stable overexpression of APP770 wt or the respective ER retention mutant (APP770 ER), or we transiently overexpressed APP 751 in CHO-K1 cells to include all different isoforms (Fig. 7a). Surprisingly, when we compared lysates of cells expressing APP695 ER to cells transfected with the longer isoforms APP770 ER and APP751 ER, we did not observe an explicit slower migrating band representing SDS-stable APP-homodimers (Fig. 7a). However, APP770 ER and APP751 ER also appear as single bands representing immature APP, whereas the fully glycosylated form was absent. Moreover, the absence of CTFs confirmed ER retention of all APP isoforms (Fig. 7a, bottom panel) compared to wild-type constructs. Additionally, we generated APP of all three isoforms with a C-terminal HA tag and transiently overexpressed them in a different cell line (HEK 293T) to assure the previous result. Indeed, we found no dimerization for the KPI domain containing isoforms upon ER retention (data not shown). These findings raised the possibility that the KPI domain might hinder cis-directed lateral homodimerization of APP within the cell, possibly due to different intramolecular folding, providing a sterical obstruction. We next wanted to investigate whether it is possible to force lateral cis-dimerization of APP770 in our cellular system. Therefore, we introduced a single cysteine substitution at the APP juxtamembrane region, previously described by Scheuermann and colleagues, to obtain covalently stabilized dimers through an intermolecular disulfide bond [8]. Since substitution of the basic lysine at position 28 of Aβ numbering by a cysteine residue (K28C) was shown to be the most effective mutant in stabilizing APP695 dimers [8], we generated APP K28C mutants of both isoforms. After transfection of native CHO-K1 cells with the cysteine mutants, we detected a slower migrating band at ~220 kDa for both APP695 K28C and APP770 K28C under non-reducing conditions (Fig. 7b), co-migrating at the same molecular weight as dimers from APP695 ER. Most interestingly, this band disappeared after boiling the samples with β-mercaptoethanol (Fig. 7b, +BME, +95°C), as seen for APP695 ER dimers. These findings clearly indicated that dimerization of APP retained in the ER is mediated by disulfide-bond formation, since linking of two thiol groups could only be generated through oxidation in the ER. Furthermore, we demonstrated that it is possible to force lateral cis-dimerization of APP770. The K28C mutation seems to be potent enough to overcome a putative sterical hindrance of the KPI domain, by cross-linking two apposed transmembrane domains of APP770. Next, we questioned whether the artificial disulfide bond, potentially generated in the ER, might affect APP trafficking and processing through the secretory pathway. It has been reported previously by Eggert and colleagues that cysteine substitution at position 28 of Aβ caused a significant decrease in soluble cleavage products such as APPsα/β and Aβ-peptides [17]. We therefore analyzed surface levels of APP695 K28C by surface biotinylation (Fig. 7c). Indeed, we could clearly show that the APP K28C mutant reached the plasma membrane comparable to APP695 wt (Fig. 7c, right panel), whereas trafficking of our ER retention mutant was efficiently inhibited. We therefore conclude that dimerization itself does not necessarily interfere with APP trafficking, nor seems dimerization a prerequisite for plasma membrane localization.

The APP KPI-domain hinders intracellular cis-dimerization in the ER. a Lysates of CHO-K1 cells, stably overexpressing either APP695, APP770, or transiently APP751 (both containing a KPI domain) and the corresponding di-lysine ER retention mutants. SDS-PAGE under non-reducing conditions and Western blot with 9E10 antibody shows no SDS stable cis-homodimers in the ER for both KPI-containing isoforms, compared to APP 695 ER. b CHO-K1 cells were transiently transfected with the K28C mutant (Aβ numbering) of both APP isoforms and run alongside lysates of K1 cells stably overexpressing APP695, APP770 or the respective ER retention constructs (APP ER). Cell lysates were analyzed under non-reducing, versus reducing conditions on 4–12% SDS gels. APP full length, dimers, and CTFs were immunoblotted with 9E10 antibody. K28C mutation led to the formation of SDS stable dimers at ~220 kDa, independent of the used APP isoform. Covalent S–S bonds were dissolved after denaturing the samples with BME at 95°C. c CHO-K1 cells stably overexpressing either APP695 wt or APP695 ER, or transiently overexpressing APP695 K28C, were labeled with sulfo NHS-SS-biotin and analyzed by SDS-PAGE and Western blot (PD: NeutrAvidin; right panel). Lysates were used as input control and APP and its cleavage products (CTFs) were detected with 9E10 antibody. Tubulin verified equal protein loading or cleanness of the biotinylation. Note that the artificial cysteine mutation (K28C) led to efficient dimerization of APP (Input, left panel). The dimer of APP K28C migrated as a doublet, revealing two putative glycosylation patterns (ER and Golgi), because the construct reached the cell surface. Note that in the biotinylation only the mature APP K28C appeared at the cell surface
Trans-dimerization at the cell surface is independent of the APP isoform
As we have shown before, APP770 containing a KPI domain displayed a weakened tendency for intracellular cis-dimerization upon ER retention (Fig. 7a) compared to the neuronal isoform 695. However, in line with previous findings [9, 11], we have verified the E1-domain as the major dimerization interface of two APP molecules (Fig. 4). Consequently, we predicted that the presence of the KPI-domain should not affect intercellular trans-dimerization properties of APP770 (see illustration in Fig. 8d). To compare the homophilic trans-cellular dimerization properties of KPI-containing isoforms versus APP695, we employed a well-established cell aggregation assay using Drosophila Schneider (S2) cells, which lack endogenous expression of the APP homologue APPL [9]. We have previously shown that expression of APP695 and APLPs in different pools of S2 cells caused significant clustering after mixing of the two cell populations, whereas cells expressing only GFP did not co-aggregate with APP/APLP-expressing cells [9]. Thus, cell clustering provides a direct readout for the specific interaction of the two proteins, whereas unspecific interaction of APP/APLPs with endogenous S2 cell proteins was excluded [9]. Therefore, we transfected S2 cells with cDNAs encoding all human APP isoforms (695, 751, 770), or APP695∆E1 and analyzed aggregation via immunocytochemical analysis, as shown in Fig. 8a. Despite a low transfection efficiency (15%), we observed almost equal clustering ratios for cells expressing either APP isoform (Fig. 8b), whereas cells expressing APP695∆E1 did not cluster specifically (Fig 8a, b), similar to untransfected S2 cells. Furthermore, confocal microscopy revealed accumulated APP immunoreactivity at cell–cell contact sites of homotypic clusters (Fig. 8c), in line with previously published data [9]. Together, these results demonstrate that cell–cell interaction by ectodomain-mediated adhesion is independent of the presence of a KPI domain. However, APP dimerization specifically depends on E1 domain associations at both the cell surface and within the cell (compare to Fig. 4). In our studies, only APP695 was able to form lateral intracellular (in the ER) as well as intercellular trans-dimers (at the cell surface). However, the KPI domain of the longer isoforms hindered lateral cis-interaction but not homophilic trans-interaction (Fig. 8d, schematic drawing), because the E1 domains of two molecules can still dimerize.

APP trans-dimerization at the cell surface is not influenced by the KPI domain. a Immunostainings of S2 cells transiently expressing human APP of either isoform 695, 751, 770, or APP695∆E1 were stained by anti APP antibody (CT-20) and secondary anti-rabbit AlexaFluor488. Note that untransfected- or APP695∆E1 expressing cells do not aggregate. b Quantification of clustered cells is shown. Transfected cells were counted and relative amount of aggregated cells is shown (n ≥ 3, ± SD) and statistical significance **p < 0.05, t test. Note that expression of either APP isoform caused equal homotypic clustering of S2 cells. Scale bar equals 20 μm. c A single confocal section homotypic aggregated S2 cells is shown. Scale bar equals 5 μm. d Hypothetical model depicting dimerization properties of APP isoforms within different cellular compartments. APP695 isoform can form homodimers in the ER lumen, favored by oxidizing conditions. However, APP770 failed to dimerize in the same manner, possibly due to the presence of a KPI domain which might adopt an inhibitory, intramolecular structure thereby impeding cis-directed homodimerization. At the surface of Drosophila S2 cells dimerization can occur independent of the particular APP isoform in trans-direction (right panel)
Discussion
In our study, we aimed to elaborate the putative dimerization potential of APP in different cellular locations, and define the precise origin of APP dimer formation. We employed a cell culture model overexpressing APP chimeric proteins with C-terminal double-lysine retention motifs, previously described to retain type I transmembrane proteins in early secretory compartments [20–22]. On SDS-PAGE, KKXX-tagged APP chimeras migrated exclusive as immature species, lacking Golgi-specific carbohydrate modifications, whereas APP wild-type appeared as a set of two bands, equivalent to N-and O-glycosylated forms (Fig. 1b). However, most interestingly we found an explicit upper migrating band for APP695 when it was retained in the ER by the KKAA motif. Strikingly, this band co-migrated at the expected molecular weight of a putative APP dimer (~220 kDa), which remained stable upon SDS treatment, but progressively disappeared under strong reducing conditions (Fig. 1c, d), suggesting that disulfide-linked APP dimers can exist in cells. This was confirmed using various tagged APP species (myc/HA), excluding that the upper SDS-stable band resulted from unspecific aggregation with cellular proteins, other than APP itself (Fig 4b).
Moreover, we detected no SDS-stable dimers with our experimental conditions when APP was shuttled between ER and Golgi compartment due to the KKFF ER-exit-determinant. We therefore believe that there exists an APP monomer-to-dimer equilibrium, which may vary in different cell compartments, or possibly depends on the redox state of a given cell. Moreover, we could demonstrate that once generated in the ER, dimers persisted stable throughout transport, and reached the plasma membrane (Fig. 3d), verifying that they occur normally in the cell. Our hypothesis that APP originally dimerizes in the ER was further strengthened by split GFP experiments. Thus, two non-fluorescent GFP fragments fused individually to the C-termini of APP, assembled into dimers to form a fluorescent signal, when co-localized with a specific ER marker Grp78 (Fig. 3a–c). However, under normal cellular conditions the portion of disulfide linked dimers might be too less to capture them on Western blots. Therefore, our cell culture model exemplified APP dimerization as it might occur under different cellular conditions, particularly visualizing dimerization itself, and thus identifying the ER as the subcellular compartment of its origin, where ideal oxidizing conditions might favor disulfide bond formation between APP molecules [42, 43]. Besides other proposed mechanisms for APP dimerization [10, 12], we believe that intermolecular disulfide linkage, between cysteines located in the E1 domain, represents another mechanism of how at least an APP sub-fraction can dimerize. Indeed, deleting either the E1 domain or the entire N-terminal part (C99, not shown) abolished APP homodimers in our system (Fig. 4c), indicating that this region of the APP ectodomain is indispensable for homointeraction, confirming previously reported results [9, 11]. Interestingly, serine substitution of cysteines at positions 98 and 105, embedded in the conserved loop region within the E1 domain [10, 41], interfered with SDS-stable dimer formation in the ER (Fig. 4d). This indicated that at least one of the respective cysteines might be essential for intermolecular disulfide formation. However, this does not exclude the existence of different dimer species, e.g., driven by hydrophobic interactions between the transmembrane domains of two proximal APP molecules [12–14]. Interestingly, other research efforts have focused on the juxtamembrane (JM) and transmembrane (TM) region, where APP contains three GxxxG motifs [44]. Originally, this motif was shown to mediate close apposition and dimerization of membrane proteins such as glycophorin A (GpA) [45, 46], whereby hydrogen bonds stabilize the helical conformation of transmembrane spanning domains, allowing short interhelical axial distances between two segments in a membrane environment [46]. Recent mutational studies revealed that the GxxxG motifs in the APP TM domain might also participate in C99/βCTF dimerization [12, 14], although their impact on dimerization is still a matter of debate. For example, Munter and colleagues found that the ability of the APP TM sequence to dimerize in a bacterial membrane system (ToxR assays) was diminished, when central glycine residues, Gly29 and Gly33, were substituted by bulkier amino acids, thus modulating Aβ production [12]. Unexpectedly, studies in CHO (mammal) cells indicated that the mutation of the central glycine residues to bulky isoleucine seemed to allow dimerization of the entire C99 protein in cell lines, whereas pairwise replacement with leucines even enhanced the formation of SDS-stable C99 dimers, instead of weakening interaction [14], contradicting previous reported results. Nevertheless, in both studies, mutation of the GxxxG motifs led to a drastic reduction of Aβ42 secretion [12, 14]. In agreement with the existing experimental data [12, 14], molecular dynamics simulation of two APP fragments (Aβ23-55) in a membrane environment, for both the wild-type GxxxG sequence and the leucine mutant, revealed that the γ-site in the mutant was shifted towards the center of the membrane, which would reduce the secretion of Aβ40 and Aβ42 [47]. However, considering that no intermolecular disulfide chains can be involved in C99 oligomerization (no cysteines present), as seen by Kienlen-Campard et al., we speculate that dimerization of APP TM helices is not necessarily taking place in the ER. Thus, probably serving as a prerequisite to accomplish close apposition of two APP full-length molecules, facilitating interactions between E1 domains. Since our studies are primarily based on the APP full-length protein in a cellular context, we do not exclude that other interactions, weaker than disulfide bonds, could mediate dimerization between two proximal TM helices of APP. While we did not identify the formation of stable C99 dimers, irrespective or ER retention or not (data not shown), this might be also cell-specific or dependent on the structure and the composition of the bilayer in which it is anchored [12–14].
To gain further insight into dimerization properties of APP, we investigated whether we might observe a similar mechanism with one of its homologues. It has been recently shown by Kaden and colleagues that APP and APLP1 exhibit strikingly different subcellular localizations, with APP being distributed more intracellularly and APLP1 mainly localized to the cell surface [11]. Due to this different cellular distribution, we were curious whether APP might have a retentive influence over APLP1, likely by forming heterodimers as early as in the ER. Indeed, surface biotinylation experiments revealed that APP was able to retain a portion of APLP1 in the ER, consequently preexisting APP homodimers were diminished whereas an increase in monomers was observed (Fig. 5). Additionally, co-immunoprecipitations confirmed an increased interaction between both homologues, when APP was retained in the ER (Fig. 6). Most interestingly, we detected the formation of SDS-stable APP/APLP1 heterodimers, suggesting an equivalent mechanism for heterodimer formation, likely involving disulfide bond(s). Therefore we provide first evidence that it was possible to visualize a beginning dimer formation between both homologues early in the secretory pathway. This was particularly interesting, due to the fact that hetero-complex formation between APP and APLPs was shown to diminish Aβ production. Therefore, the ER might provide an interesting target possibly favoring dimer equilibrium from homo- to hetero-complexes, thus influencing Aβ generation.
Since all studies so far have predominantly focused on the dimerization properties of APP695 lacking the KPI domain, we analyzed if the longer isoforms interact in the same manner as APP695. Due to the fact that the KPI domain encompasses six further cysteine residues, in addition to the 12 cysteines already present in the E1 domain, one could imagine an increased dimerization potential by disulfide linkage. Surprisingly, when APP770 or APP751 were retained in the ER, no cis-directed homodimer formation was observed on Western blots (Fig. 7a). In contrast, we were able to induce cis-dimerization of APP770 using a previously described [8, 17] constitutive dimerization mutant (APP770 K28C). This led us to the assumption that the KPI might somehow exhibit a sterical obstruction, possibly diverging two E1 domains thus interfering with dimerization. Nevertheless, the close proximity of two APP TM domains might be sufficient to cross-link the full-length proteins by the constitutive K28C mutant (Fig. 7b, c). Interestingly, this cysteine mutant also reached the cell surface (Fig. 7c), yet the disulfide bond can only be formed in the ER via oxidation, confirming our hypothesis that once generated, covalent dimers are trafficked normally in the cell (compare to Fig. 3d, e). Moreover, this suggested close apposition between APP molecules, or else this constitutive disulfide bond could never been generated via oxidation in the ER.
Having demonstrated that dimer formation of KPI-APP seemed less efficient in the ER, but was induced by an engineered disulfide bond, we questioned whether APP isoforms might also display diverse trans-dimerization capacities at the surface, affecting cell adhesion. We employed a well-described cell aggregation assay in Drosophila Schneider (S2)-cells, previously established by Soba et al., where they could demonstrate that APP695 can form homo-as well as hetero-complexes with APLPs at the surface of different cells [9]. However, expression of all three different APP isoforms in S2-cells led to equal cell clustering (Fig. 8), whereas the absence of the extracellular E1 domain abolished specific aggregation of transfected S2 cells (Fig. 8a, b), consistent with observations by Soba and colleagues. Therefore, we speculate that the KPI domain might hinder exclusively cis-dimerization of APP within the same cell, whereas trans-dimers at the surface of two or more cells, were unperturbed (see illustration in Fig. 8d). These findings raised the question: how can the differences in cis/trans interactions between isoforms be explained? Interestingly, it has recently been shown by small-angle X-ray scattering (SAXS), a method to deduce high-resolution, three-dimensional protein structures, that a domain likely corresponding to the KPI domain of APP770 emerges laterally from one end of the molecule, and is freely exposed on the APP surface [48, 49]. Therefore, it is tempting to speculate that the KPI domain might adapt different structural features, possibly depending on the redox state, maintained in a subcellular compartment/microdomain. It seems possible that the six cysteine residues present in the KPI domain [50] could lead to a different folding through the formation of intramolecular disulfide bonds [39] in the ER, but not at the cell surface. Consequently, such conformational changes might potentially hinder dimerization of two adjacent APP molecules only in cis-direction (Fig. 7, 8). In contrast, APP trans-dimerization seemed less sterical obstructed by the presence of the KPI domain, since opposed E1 domains can still interact (see hypothetical illustration in Fig. 8d). While it was possible to force lateral cis-dimerization by introducing an artificial cysteine bond at the TM border of APP (K28C), irrespective of a KPI domain, we speculate that the vicinity of two molecules (or TM helices), is crucial for cis-dimerization [12]. If two APP molecules reside in close proximity within the same membrane, the redox state of the ER might be sufficient to link them between cysteine residues of the E1 domain, or likewise by an engineered disulfide bond. Nevertheless, we assume that trans-dimerization in this aggregation assay is primarily established by hydrophobic interactions, instead of S–S bonds, since cell surface PDI has so far only been identified in few cell types, shown to modify disulfide bonds, e.g., in β-integrins, thus regulating adhesion and migration [51].
In conclusion, our results provide novel observations for the regulation of APP dimerization. To date, the physiological significance of APP–APP interactions has so far remained elusive, but it has been well described that in mammalian cells, the most common mechanism of signal transduction for single-pass transmembrane proteins occurs through a shift in the monomer–dimer equilibrium [52]. Since APP resembles a cell surface receptor, dimerization and dissociation might also regulate various APP functions. One could imagine that cell-bound APP could form dimers in trans-regulating cell–cell adhesion, whereas cis-dimerized APP could act as a growth factor receptor when it is localized to the same cell membrane. In our study, we could demonstrate that APP already dimerizes early in the secretory pathway, within the ER, suggesting that at least a fraction of APP may exist as covalent-associated disulfide dimers, before being transported to the plasma membrane. Therefore this might serve as prerequisite for its correct orientation and exposure to the cell surface, and if APP can be transported once to the cell surface as a dimer, it may achieve the status of a cell surface receptor [53]. Furthermore, we describe a novel mechanism of how APP can form intermolecular dimers. We provide strong evidence that disulfide bridges formed in the ER facilitate cis-dimerization of APP695 within the same cell, whereas KPI-APP seemed to exhibit different dimerization capacities in the ER, but not in trans-direction at the plasma membrane. Moreover, our data might suggest a related mechanism for heterodimer formation of APP, with at least one homologue, APLP1. Nevertheless, further experimental evidence is needed. Taken together, dynamic alterations of APP between monomeric, homodimeric, and possibly heterodimeric status could at least partially explain some of the variety in the functions of APP. However, as our analysis focused on the full-length cellular APP proteins and not on isolated domains, dimerization properties might vary between different cell systems or experimental setups, not excluding the co-existence of diverse dimerization mechanisms in the same cell.
Abbreviations
- Aβ:
-
Amyloid β-peptide
- AD:
-
Alzheimer’s disease
- AICD:
-
APP intracellular domain
- APLP:
-
Amyloid precursor-like protein
- APP:
-
Amyloid precursor protein
- APPs:
-
Soluble APP
- BFA:
-
Brefeldin A
- BME:
-
β-Mercaptoethanol
- CHO:
-
Chinese hamster ovary
- CPZ:
-
Chlorpromazine
- CTF:
-
C-Terminal fragment
- CuBD:
-
Copper binding domain
- D/N:
-
Dominant/negative
- DMSO:
-
Dimethylsulfoxid
- EGF:
-
Epidermal growth factor
- ERGIC:
-
Early Golgi and intermediate compartments
- ER:
-
Endoplasmic reticulum
- FL:
-
Full-length
- FRET:
-
Fluorescence resonance energy transfer
- GFLD:
-
Growth factor-like domain
- kDa:
-
Kilodalton
- KPI:
-
Kunitz type protease inhibitor
- PD:
-
Pull down
- PDI:
-
Protein disulfide isomerase
- SAXS:
-
Small-angle X-ray scattering
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SP:
-
Signal peptide
- TM:
-
Transmembrane domain
- WT:
-
Wild-type
References
Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325:19–25
Lemaire HG, Salbaum JM, Multhaup G, Kang J, Bayney RM, Unterbeck A, Beyreuther K, Müller-Hill B (1989) The PreA4(695) precursor protein of Alzheimer’s disease A4 amyloid is encoded by 16 exons. Nucleic Acids Res 17:517–522
Cao X, Südhof TC (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 29:115–120
Kitaguchi N, Takahashi Y, Tokushima Y, Shiojiri S, Ito H (1988) Novel precursor of Alzheimer’s disease amyloid protein shows protease inhibitory activity. Nature 331:530–532
Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL (1988) Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature 331:528–530
Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F (1992) Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA 89:10758–10762
Coulson EJ, Paliga K, Beyreuther K, Masters CL (2000) What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int 36:175–184
Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G (2001) Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J Biol Chem 276:33923–33929
Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Lower A, Langer A, Merdes G, Paro R, Masters CL, Muller U, Kins S, Beyreuther K (2005) Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J 24:3624–3634
Kaden D, Münter LM, Joshi M, Treiber C, Weise C, Bethge T, Voigt P, Schaefer M, Beyermann M, Reif B, Multhaup G (2008) Homophilic interactions of the amyloid precursor protein (APP) ectodomain are regulated by the loop region and affect beta-secretase cleavage of APP. J Biol Chem 283:7271–7279
Kaden D, Voigt P, Munter LM, Bobowski KD, Schaefer M, Multhaup G (2009) Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J Cell Sci 122:368–377
Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G (2007) GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J 26:1702–1712
Gorman PM, Kim S, Guo M, Melnyk RA, McLaurin J, Fraser PE, Bowie JU, Chakrabartty A (2008) Dimerization of the transmembrane domain of amyloid precursor proteins and familial Alzheimer’s disease mutants. BMC Neurosci 9:17–27
Kienlen-Campard P, Tasiaux B, Van Hees J, Li M, Huysseune S, Sato T, Fei JZ, Aimoto S, Courtoy PJ, Smith SO, Constantinescu SN, Octave JN (2008) Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GxxxG motifs. J Biol Chem 283:7733–7744
Munter LM, Botev A, Richter L, Hildebrand PW, Althoff V, Weise C, Kaden D, Multhaup G (2010) Aberrant amyloid precursor protein (APP) processing in hereditary forms of Alzheimer disease caused by APP familial Alzheimer disease mutations can be rescued by mutations in the APP GxxxG motif. J Biol Chem 285:21636–21643
Richter L, Munter LM, Ness J, Hildebrand PW, Dasari M, Unterreitmeier S, Bulic B, Beyermann M, Gust R, Reif B, Weggen S, Langosch D, Multhaup G (2010) Amyloid beta 42 peptide (Abeta42)-lowering compounds directly bind to Abeta and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc Natl Acad Sci USA 107:14597–14602
Eggert S, Midthune B, Cottrell B, Koo EH (2009) Induced dimerization of the amyloid precursor protein leads to decreased amyloid-beta protein production. J Biol Chem 284:28943–28952
Wang Y, Ha Y (2004) The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell 15:343–353
Gralle M, Oliveira CL, Guerreiro LH, McKinstry WJ, Galatis D, Masters CL, Cappai R, Parker MW, Ramos CH, Torriani I, Ferreira ST (2006) Solution conformation and heparin-induced dimerization of the full-length extracellular domain of the human amyloid precursor protein. J Mol Biol 357:493–508
Pelham HR (1989) Control of protein exit from the endoplasmic reticulum. Annu Rev Cell Biol 5:1–23
Teasdale RD, Jackson MR (1996) Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the Golgi apparatus. Annu Rev Cell Dev Biol 12:27–54
Andersson H, Kappeler F, Hauri HP (1999) Protein targeting to endoplasmic reticulum by dilysine signals involves direct retention in addition to retrieval. J Biol Chem 274:15080–15084
Jager S, Leuchtenberger S, Martin A, Czirr E, Wesselowski J, Dieckmann M, Waldron E, Korth C, Koo EH, Heneka M, Weggen S, Pietrzik CU (2009) Alpha-secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Abeta generation. J Neurochem 111:1369–1382
Feinberg EH, Vanhoven MK, Bendesky A, Wang G, Fetter RD, Shen K, Bargmann CI (2008) GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57:353–363
Zhao X, Greener T, Al-Hasani H, Cushman SW, Eisenberg E, Greene LE (2001) Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. J Cell Sci 114:353–365
Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, Hopkins CR, Evans PR, McMahon HT (2001) Simultaneous binding of PtdIns(4, 5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 291:1051–1055
Pelham HR (1991) Multiple targets for brefeldin A. Cell 67:449–451
Fujiwara T, Oda K, Yokota S, Takatsuki A, Ikehara Y (1988) Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J Biol Chem 263:18545–18552
Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD (1989) Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56:801–813
Miller SG, Carnell L, Moore HH (1992) Post-Golgi membrane traffic: brefeldin A inhibits export from distal Golgi compartments to the cell surface but not recycling. J Cell Biol 118:267–283
Maltese WA, Wilson S, Tan Y, Suomensaari S, Sinha S, Barbour R, McConlogue L (2001) Retention of the Alzheimer’s amyloid precursor fragment C99 in the endoplasmic reticulum prevents formation of amyloid beta-peptide. J Biol Chem 276:20267–20279
Cupers P, Bentahir M, Craessaerts K, Orlans I, Vanderstichele H, Saftig P, De Strooper B, Annaert W (2001) The discrepancy between presenilin subcellular localization and gamma-secretase processing of amyloid precursor protein. J Cell Biol 154:731–740
Wang LH, Rothberg KG, Anderson RG (1993) Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol 123:1107–1117
Jacobsen KT, Iverfeldt K (2009) Amyloid precursor protein and its homologues: a family of proteolysis-dependent receptors. Cell Mol Life Sci 66:2299–2318
Pietrzik CU, Yoon IS, Jaeger S, Busse T, Weggen S, Koo EH (2004) FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J Neurosci 24:4259–4266
Kimberly WT, Zheng JB, Guénette SY, Selkoe DJ (2001) The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem 276:40288–40292
Miller CC, McLoughlin DM, Lau KF, Tennant ME, Rogelj B (2006) The X11 proteins, Abeta production and Alzheimer’s disease. Trends Neurosci 5:280–285 Review
Small DH, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, Masters CL (1994) A heparin-binding domain in the amyloid protein precursor of Alzheimer’s disease is involved in the regulation of neurite outgrowth. J Neurosci 14:2117–2127
Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW (1999) Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol 6:327–333
Lichtenthaler SF, Multhaup G, Masters CL, Beyreuther K (1999) A novel substrate for analyzing Alzheimer’s disease gamma-secretase. FEBS Lett 453:288–292
Oltersdorf T, Fritz LC, Schenk DB, Lieberburg I, Johnson-Wood KL, Beattie EC, Ward PJ, Blacher RW, Dovey HF, Sinha S (1989) The secreted form of the Alzheimer’s amyloid precursor protein with the Kunitz domain is protease nexin-II. Nature 341:144–147
Freedman RB, Hirst TR, Tuite MF (1994) Protein disulphide isomerase: building bridges in protein folding. Trends Biochem Sci 19:331–336
Turano C, Coppari S, Altieri F, Ferraro A (2002) Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol 193:154–163
Khalifa NB, Van Hees J, Tasiaux B, Huysseune S, Smith SO, Constantinescu SN, Octave JN, Kienlen-Campard P (2010) What is the role of amyloid precursor protein dimerization? Cell Adh Migr 4:268–272
Bormann BJ, Knowles WJ, Marchesi VT (1989) Synthetic peptides mimic the assembly of transmembrane glycoproteins. J Biol Chem 264:4033–4037
Senes A, Ubarretxena-Belandia I, Engelman DM (2001) The Cα—HO hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci USA 98:9056–9061
Miyashita N, Straub JE, Thirumalai D, Sugita Y (2009) Transmembrane structures of amyloid precursor protein dimer predicted by replica-exchange molecular dynamics simulations. J Am Chem Soc 131:3438–3439
Gralle M, Botelho MG, Wouters FS (2009) Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J Biol Chem 284:15016–15025
Gralle M, Ferreira ST (2007) Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog Neurobiol 82:11–32
Menendez Gonzalez M, Pérez-Pinera P, Martínez-Rivera M, Calatayud MT, Blázquez Menes B (2005) APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegener Dis 2:277–283
Turano C, Coppari S, Altieri F, Ferraro A (2002) Proteins of the PDI family: Unpredicted non-ER locations and functions. J Cell Physiol 193:154–163
Schlessinger J (2002) Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 110:669–672 Review
Beher D, Hesse L, Masters CL, Multhaup G (1996) Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J Biol Chem 271:1613–1620
Acknowledgments
We thank Michael Plenikowski for the illustrations and Hanne Wesselowski for technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (DFG), as part of FOR1332 grant to SK (819/5-1 and/6-1) and CUP (379/6-1).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Isbert, S., Wagner, K., Eggert, S. et al. APP dimer formation is initiated in the endoplasmic reticulum and differs between APP isoforms. Cell. Mol. Life Sci. 69, 1353–1375 (2012). https://doi.org/10.1007/s00018-011-0882-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0882-4