
Abstract
Even though every cell in a multicellular organism contains the same genes, the differing spatiotemporal expression of these genes determines the eventual phenotype of a cell. This means that each cell type contains a specific epigenetic program that needs to be replicated through cell divisions, along with the genome, in order to maintain cell identity. The stable inheritance of these programs throughout the cell cycle relies on several epigenetic mechanisms. In this review, DNA methylation and histone methylation by specific histone lysine methyltransferases (KMT) and the Polycomb/Trithorax proteins are considered as the primary mediators of epigenetic inheritance. In addition, non-coding RNAs and nuclear organization are implicated in the stable transfer of epigenetic information. Although most epigenetic modifications are reversible in nature, they can be stably maintained by self-recruitment of modifying protein complexes or maintenance of these complexes or structures through the cell cycle.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the recent completion of the human genome, approximately 23,800 genes have been identified. The use of functional genomic approaches has provided much insight into how genes shape a cell or an organism. However, our DNA sequence is not the sole determinant of a phenotype, since each cell of an organism contains the same genetic information. In fact, it is the differential regulation of genes in time (i.e., during development) and space (i.e., tissue) that determines cell fate and eventual phenotype. During every cell cycle, the entire genome needs to be accurately replicated during the S-phase. Similarly, the gene-expression profile needs to be replicated as well; after all, a dividing hepatocyte should give rise to a fully differentiated new hepatocyte even though it shares the same genome with a neuron. The study of “heritable changes in gene function that cannot be explained by changes in the DNA sequence” is referred to as epigenetics [1]. These epigenetic modifications can be stably maintained through mitosis; for example, cells with an epigenetically silenced transgene can be cultured for more than a year without losing the epigenetic information [2]. Additionally, epigenetic information can, in some cases, be transferred through meiosis and therefore affect the next generation [3]. Even though epigenetic information can be inherited through many cell divisions and even generations, it is also reversible. In fact, unlike DNA that, with exceptions, only changes due to mutagenesis or replication errors, epigenetic information is also a product of environmental factors. The reversibility of the system is illustrated by the epigenetic reprogramming of oocytes after fertilization, when widespread demethylation of the genome is observed. After implantation, de novo methylation restores DNA methylation levels in the embryo [4]. Besides physiological reprogramming during development, somatic cells can be reprogrammed into pluripotent stem cells through nuclear transfer or the expression of specific pluripotency-associated transcription factors [4, 5]. In addition, also cancers seem to exploit the reversible nature of the epigenome by silencing tumor suppressors or activating oncogenes [6, 7].
Much research on epigenetic inheritance has focused on DNA methylation, histone variants, and covalent modifications of histone tails [8]. However, throughout the cell cycle, these epigenetic marks face two major caveats; they need to be accurately replicated along with the DNA and, in addition, they need to survive mitosis. During DNA replication this means that any covalent modifications to the DNA need to be copied to the daughter strands. In addition, the passing replication machinery is likely to disrupt many DNA–protein interactions and higher-order chromatin organization and, hence, these need to be either maintained through the S-phase or reorganize after replication. During mitosis, chromatin can be condensed by more than a factor 10,000 [9], again disrupting higher-order chromatin structures and protein–DNA interactions. In this review we describe the molecular mechanisms behind epigenetic inheritance through the cell cycle in mammalian cells. We will focus on DNA methylation, histone modifications and how these are established and maintained by heterochromatin protein 1 (HP1), as well as the PcG/TrxG system. In addition, we consider the influence of small non-coding RNAs (ncRNAs) and nuclear organization of the genome on epigenetic inheritance.
DNA methylation
DNA methylation has been demonstrated to play an important role in epigenetic inheritance.
For example, at the murine Agouti and Axin loci (determining coat color and tail phenotype, respectively [10]) where nearby retrotransposons or intracisternal A-particles (IAP) can affect gene expression. DNA methylation of these IAPs can silence IAP promoter activity and thereby restore wild-type expression. This can result in genetically identical mice that display different coat colors or tail phenotypes. Interestingly, the methylation status of these IAPs can be inherited across generations [11, 12]. Other examples that illustrate the role of DNA methylation in epigenetic inheritance are X-chromosome inactivation and parental imprinting (epigenetic silencing of one allele) which are impaired when DNA methylation machinery is disrupted [13, 14]. In addition, many cancers show local hypermethylation at genomic locations corresponding to tumor suppressors, such as cell cycle inhibitors and genes that mediate the DNA damage response [6].
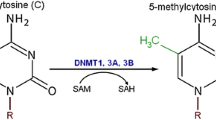
In mammalian cells, DNA methylation occurs on cytosine residues (m5C) and this occurs almost exclusively at cytosine-guanidine dinucleotides (CpG) [15, 16]. These nucleotides are underrepresented in the genome, and approximately 70–80% of all CpGs are methylated [15, 17, 18]. Although most CpG dinucleotides are methylated, the genome also contains CpG islands: short CpG-rich genomic regions which are generally unmethylated (discussed below). Functionally, DNA methylation is associated with a repressed chromatin state and is required for both ‘genome housekeeping’ as well as for gene expression regulation and maintenance of cell identity [19, 20]. The former is illustrated by the widespread methylation of retrotransposons [19]. Abolishing methylation at these sites can result in reactivation of the transposon promoter activity [21]. In addition, methylation might be involved in maintaining chromosomal stability, as noted by chromosomal rearrangements and abnormalities as a result of chemical inhibition of methylation and genetic studies [15, 19]. Global hypomethylation as observed in certain cancers might therefore be a cause of chromosomal instability [22]. Of particular interest when examining the role of DNA methylation in gene regulation are CpG islands [15]. These CpG islands are enriched at genes, since approximately 60% of all genes in the human genome contain a CpG island upstream [15]. Although methylation of CpG islands is infrequent, it does result in a stable repression of genes [23]. The latter is also illustrated by experiments that showed that increased CpG density at a promoter induces more robust silencing of episomes [24]. In addition, although genes with low CpG content at their promoters are readily reactivated by the viral SV40 enhancer, transcriptional silencing of genes with an upstream CpG-dense region is not reversed by the SV40 enhancer [25].
The maintenance of DNA methylation throughout the cell cycle is mainly mediated by three DNA methyltransferases, namely DNMT1, DNMT3a, and DNMT3b [16]. DNMT1 is primarily involved in the propagation of DNA methylation during the cell cycle due to its affinity towards hemimethylated DNA [26]. During the S-phase, the two parental DNA strands are separated for DNA replication, resulting in two hemimethylated strands, and two newly synthesized strands. At this time, DNMT1 associates with proliferating cell nuclear antigen (PCNA), an important member of the replication machinery [27], and NP95, a protein that specifically binds hemimethylated DNA, and subsequently restores methylation on the daughter strands [28]. This process has been shown to be required for both in vitro as in vivo maintenance of DNA methylation [28, 29]. However, even though the accuracy of DNA methylation replication is reported to be approximately 95–99% [30], the maintenance activity of DNMT1 is not sufficient to explain all epigenetic inheritance considering DNA methylation. In addition, both de novo methyltransferases DNMT3a and -b seem to be required for complete epigenetic inheritance. For one, in murine ES cells abrogation of both DNMT3a and -b increases the amount of hemimethylated DNA at repeats by 30% [31]. Furthermore, ES cells deficient for both de novo methylases display reduced levels of methylation over time [32]. Likely, the epigenetic inheritance of DNA methylation requires the maintenance transferase DNMT1 as well as the de novo methyltransferases DNMT3a and -b for perhaps proofreading and methylation of specific DNA sequences [32, 33].
Throughout the cell cycle, the methyltransferases display differential temporal expression and localization. As expected, DNMT1 expression levels peak during the S-phase. During this phase, DNMT1 is responsible for the faithful replication of methylated DNA sequences and, hence, it colocalizes to DNA replication foci [28]. However, the de novo methyltransferases (DNMT3a and -b) show a more heterogeneous profile. DNMT3a seems to be primarily associated with heterochromatin, or inactive chromatin [34]. DNMT3b, however, displays diffuse nuclear localization although DNMT3a and -b colocalize at sites of pericentromeric heterochromatin [34]. The expression levels of the de novo DNMTs also differ during the cell cycle. DNMT3b mRNA levels, like DNMT1, are reduced during the G1/G0 phase of the cell cycle, while they peak during the S-phase [35]. DNMT3a, however, is expressed at approximately constant levels throughout the cell cycle [35]. One possibility is that DNMT3b, in addition to DNMT1, is primarily responsible for DNA methylation maintenance. Abrogation of DNMT3b results in decreased genome-wide methylation levels, while DNMT3a disruption results in a less severe phenotype [36, 37]. The expression peak of DNMT1 and DNMT3b during the S-phase might support the maintenance function DNMT1 and -3b. The abundant DNMT3a expression during the cell cycle indicates a more flexible mechanism for this particular methyltransferase, such as proofreading or DNA methylation of specific loci in response to certain extracellular or intracellular stimuli.
Currently, DNA methylation is the most established system implicated in epigenetic inheritance. The recognition of hemimethylated DNA by DNMT1 provides an efficient method for restoring DNA methylation during the S-phase, supplemented by the de novo methyltransferases DNMT3a and -3b. However, although DNA methylation is generally associated with robust silencing, it is not irreversible. This is illustrated by the demethylation of the paternal and maternal genomes in the zygote after fertilization [38]. In addition, DNA demethylation is required for the reactivation of multiple pluripotency-associated genes during reprogramming of differentiated cells into iPS cells [39]. Nevertheless, the molecular mechanism of DNA demethylation in mammals is disputed. One possibility for cells to demethylate their genome is by passive demethylation. By seizing maintenance methylation activity during DNA replication (i.e., preventing DNMT1 nuclear localization), the genomes of daughter cells become increasingly demethylated [40]. However, this process might not fully explain all observed demethylation events in the mammalian genome. For example, the paternal nucleus in the zygote undergoes rapid genome-wide demethylation after fertilization prior to DNA replication [41]. Furthermore, non-dividing peripheral monocytes undergo DNA demethylation upon differentiation [42], indicating active demethylation, as passive demethylation requires replication.
One candidate for mediating DNA demethylation is activation-induced deaminase (AID). In primordial germ cells, cells that normally undergo genome-wide demethylation, a knockout of this protein results in a significant increase of methylated DNA [43]. Furthermore, the demethylation of pluripotency-associated genes depends on AID [44]. In addition to AID, recent studies also identified several members of the elongator complex that are required for the previously discussed paternal genome demethylation in zygotes [45]. However, although both passive and active DNA demethylation are likely to occur during development, it remains to be determined whether it occurs outside specific developmental events and artificially induced conditions.
All in all, DNA methylation plays an important role in gene expression in mammalian cells. Methylation of CpG islands in promoter regions is often associated with gene silencing and aberrant DNA methylation has been shown to occur in many cancers, leading to silencing of some tumor suppressor genes [15]. Reversal of DNA methylation therefore has emerged as a potential strategy for treatment of cancer [46, 47].
Histones
Although DNA methylation is an established mechanism for stable epigenetic inheritance, organisms that hardly methylate their DNA, such as yeast and Drosophila, can still transmit epigenetic information through the cell cycle, likely by altering chromatin structure [48, 49]. The major determinants of chromatin structure are the histone proteins. DNA coils around histones (complexes referred to as nucleosomes), which eventually contributes to efficient packaging of approximately 2 m of DNA in a single nucleus. Each nucleosome contains an octamere of four core histone proteins, namely H3, H4, H2A, and H2B [50]. However, besides the structural role, histone proteins are crucial in the regulation of gene expression. By altering the local chromatin state, the accessibility of particular DNA sequences to, for example, transcription machinery, can be regulated resulting in activation or repression of genes [51]. In addition, effector proteins can be recruited to mediate transcriptional silencing or activation [52]. Since the local chromatin state contributes to maintaining transcriptional activation or repression, it is not surprising that it needs to be transmittable through the cell cycle, e.g., in order to maintain expression of tissue specific genes. However, this system also needs to allow for dynamic changes, such as required for stress responses or DNA repair. In general, chromatin functionality can be regulated either by the placement of specific histone variants throughout the genome (e.g., the placement of H3.3 at actively transcribed loci, discussed below), or by the covalent modification of histone N-terminal tails [53]. For the case of epigenetic inheritance, a number of histone modifications and histone variants have been identified that might be maintained through the cell cycle or play a role in epigenetic inheritance. Note that the concept of histone modifications as the direct carriers of epigenetic information is disputed as reviewed in [54–56].
Histone modifications and epigenetic inheritance
The nucleosome consists of tightly packed histone proteins, and their N-terminal tails extend from the nucleosome. Numerous residues of these histone tails can be subject to modifications, including the covalent linkage of small molecules (i.e., phosphorylation, acetylation, methylation) as well as macromolecules (e.g., ubiquitination, SUMOylation) [53]. These modifications can directly alter histone or chromatin structure and thereby restrict or facilitate access to transcription factors/machinery, or they can recruit other proteins at the site of interest [57] to facilitate a repressive or accessible chromatin state, such as the heterochromatin-associated protein 1 (HP1) or Polycomb repressive complexes discussed below.
In contrast to other posttranslational modifications, histone methylation is assumed to be a prime candidate for being involved in epigenetic inheritance due to its relatively low turnover [58, 59]. However, the recent identification of demethylases and studies into spatio-temporal histone regulation have also demonstrated the dynamic nature of the histone methylation system [60]. Nonetheless, much recent research has emphasized the role of histone methylation in epigenetic inheritance, with a particular emphasis on lysine methylation on H3 variants [61]. Methylation of H3 at lysine 9 (H3K9) or lysine 27 (H3K27) are known to be repressive marks, while methylation of H3 at lysine 4 (H3K4) is correlated with an open chromatin state and transcription [53]. Each of these residues, can be mono-, di-, and tri-methylated. The active H3K4Me and the repressive H3K27Me are regulated by the TrxG and PcG family, respectively (discussed below). However, a separate system maintains H3K9Me throughout the cell cycle, namely the heterochromatin-associated protein 1 (HP1). In order to be heritable, these methyl marks need to be stably transferred through the S-phase and subsequently mitosis; this has been hypothesized for the H3K9Me3 mark, which will be elaborated on next.
During DNA replication, histones are disassembled in order for the replication machinery to pass and are subsequently rapidly reassembled on the two daughter strands. After disassembly, the parental H2A–H2B dimers and H3–H4 tetramers are redistributed at the daughter strands and complemented with newly synthesized histones [62, 63] in order to provide both strands with a similar number of nucleosomes. Since H3–H4 tetramers can also be split into dimers [64], one potential model is that the parental H3–H4 tetramers along with their modifications are split into dimers and are subsequently evenly distributed at both daughter strands (Fig. 1). In other words, this ‘semi-conservative model’ [65] states that after replication both strands will contain ‘hemi-parental’ nucleosomes, consisting of a parental H3–H4 dimer and a newly synthesized H3–H4 dimer. However, based on experiments with SV40 chromosomes [66, 67], it is assumed that for the bulk of the genome parental H3–H4 are deposited as tetramers and distributed in a random fashion at both daughter strands [8, 62, 64], which means the epigenetic marks on the parental histones are ‘diluted’. However, how can random distribution of parental histones with their epigenetic marks facilitate the faithful replication of local chromatin states? For H3K9 methylation, HP1 might function as a ‘reader’ [8] that detects local H3K9 methylation of parental histones and subsequently induces ‘spreading’ of this mark to the newly synthesized histones (Fig. 1). HP1 is known to be involved in the maintenance of heterochromatin and it binds, through its chromodomain, to di- and trimethylated H3K9 residues [68], an epigenetic mark that is enriched in heterochromatin [69, 70]. Although HP1 does not posses inherent histone lysine methyltransferase (KMT) activity, it can associate with several H3K9 KMTs, including SUV39h1/2 and G9a [71, 72]. Ectopic expression of these KMTs induces heterochromatin abnormalities, illustrating that H3K9 methylation plays a causal role in heterochromatin formation [73]. In addition to its association with KMTs, HP1 also binds to CAF-1, a chaperone that supplies newly synthesized H3–H4 units to the replicated DNA during the S-phase and this interaction is required for heterochromatin formation and S-phase progression [74–76]. Interestingly, CAF-1 is only found in complex with either HP1 or H3. When associated with HP1, CAF-1 also binds SetDB1, a KMT responsible for the monomethylation of H3K9 [77]. This process could serve two purposes, namely to supply HP1 to the site of ongoing replication (i.e., to bind parental H3K9Me3) and to monomethylate the K9 residue of newly synthesized H3 molecules, which serves as a substrate for subsequent trimethylation by SUV39h1/2 [77]. Therefore, HP1 could function in a self-reinforcing mechanism to maintain heterochromatin and its associated H3K9Me3 modification during the S-phase. Upon replication it is recruited to the replication-fork by CAF-1 where it can induce local monomethylation of new H3 molecules. In addition, it can ‘read’ parental H3K9Me3 residues and recruit KMTs that can catalyze the trimethylation of the newly synthesized H3 s, thereby facilitating the spreading of the heterochromatin-associated H3K9Me3 mark [78–80].

Restoration of H3K9 methylation after DNA replication. During the S-phase, nucleosomes are disrupted by the replication machinery and subsequently reassembled at the daughter strands. This requires both parental histones as well as newly synthesized histones. Parental H3–H4 tetramers can be randomly deposited at the daughter strands. Alternatively, in certain instances H3–H4 tetramers can be split into dimers. This would facilitate the semi-conservative model, in which parental H3–H4 dimers pair with newly synthesized H3–H4 dimers in order to form hemi-parental nucleosomes. The propagation of the H3K9Me3 mark is facilitated by HP1 and KMTs, where HP1 binds parental H3K9Me3 marks and induces H3K9 methylation of newly synthesized histones
Although the HP1-reinforcement loop could explain epigenetic inheritance through the S-phase of the cell cycle, the M-phase is also a major caveat due to the intense condensation of higher-order chromatin structure [9]. During mitosis, most HP1 is dissociated from chromatin [81–83] through a ‘methylation/phosphorylation’ (Me/P) switch [84]. Aurora B phosphorylates the neighboring residue of H3K9Me, i.e., serine 10 (H3S10P), which facilitates the release of HP1 from chromatin. In fact, Aurora B depletion results in aberrant HP1 association with chromatin during mitosis [85, 86]. Importantly, H3S10 phosphorylation does not require demethylation of H3K9 [81–83] and, therefore, the epigenetic information is unaltered. After mitosis, the phosphatase PP1 dephosphorylates [85, 87] H3S10 residues, after which HP1 can re-bind methylated H3K9 and resume its functions as heterochromatin regulator. Note that of the three HP1 variants (HP1α, HP1β, HP1γ), HP1α can remain associated with centromeres during mitosis, although this interaction is H3K9Me independent [82]. This indicates the possibility of continued binding throughout the cell cycle as a means for stable epigenetic inheritance.
It has long been assumed that heterochromatin is rather static, since it contains many repeat-rich regions, retrotransposons, and is relatively gene-poor. Recent genome-wide profiling of histone methylations affirmed that H3K9Me3 generally locates to e.g., satellite and long-terminal repeats [88]. In addition, during development, certain PcG/TrxG targets, such as the pluripotency marker Oct4, undergo H3K9 trimethylation which might reflect a more permanent inactivation [89]. However, there is also support for a more dynamic role for heterochromatin during development. When cells exit the cell cycle during G1 and enter a quiescent state (G0), the E2F target genes that mediate G1/S phase progression become H3K9 methylated due to Rb-dependent HP1 and KMT recruitment [90]. These are examples of ‘facultative heterochromatin’ [90]); regions of heterochromatin that can, under certain circumstances, be switched in a euchromatic state that facilitates transcription. Currently, a precise mechanism that distinguishes this more dynamic facultative heterochromatin from the more static (e.g., pericentric) heterochromatin remains elusive.
Histone variants and epigenetic memory
Besides histone modifications, different histone variants could also mediate epigenetic inheritance. For H3, five variants have been identified, including H3.1 and H3.2 that are only incorporated during the S-phase to complement parental histones on the two daughter strands, CENP-A, a H3 variant that marks centromeres, and H3.3, a histone variant that can be incorporated in every phase of the cell cycle [91]. Especially the latter variant is implicated in epigenetic inheritance. H3.3 can replace the H3.1 that is deposited during the S-phase and is deposited by a dedicated histone chaperone HIRA independent of DNA replication [92]. In general, H3.3 is enriched in actively transcribed chromatin [93]. In addition, H3.3 is enriched for H3K4 trimethylation, a modification associated with transcriptional activation [94]. Nuclear transfer experiments in Xenopus demonstrated that H3.3 mediates inherited transcription of MyoD. The epigenetic mark H3K4Me3 was shown to be required for this epigenetic memory, and, additionally, the overexpression of H3.3 increased the inheritance of the transcriptionally active state [95]. However, how are these H3.3 variants inherited through the cell cycle?
Upon gene transcription, local chromatin gains active marks (i.e., H3K4Me) [96] and H3.3 is deposited [97]. During replication, newly synthesized H3.1 and H3.2 variants complement the parental histones in a random fashion, thereby diluting the local H3.3 concentration approximately twofold. However, the local H3.3 concentration is still likely to be elevated as compared to a random genomic position, and thereby can still reflect an accessible chromatin structure that can facilitate further transcription [98]. This continued transcription, in turn, might induce more H3.3 deposition throughout the cell cycle and thereby fully restore the active chromatin state. Alternatively, recognition of a “H3.3 environment”, perhaps through the H3K4Me3 mark, might induce increased H3.3 deposition [95, 98].
In summary, both specific covalent modifications of histones as well as certain histone variants are likely to be inherited through the cell cycle. Since during the S-phase parental H3–H4 dimers or tetramers are complemented with newly synthesized H3–H4 units, the epigenetic information becomes diluted. For the H3K9Me3 mark, binding of HP1 to the parental modifications and interactions with KMTs and newly synthesized histones, results in faithful restoration of H3K9 trimethylation after replication. For the H3 variant H3.3, its ability to be incorporated throughout the cell cycle at actively transcribed regions is likely to ensure its propagation. Besides the inheritance of H3K9Me3, other H3 methylation marks, such as the repressive H3K27Me3 and the active H3K4Me3 are likely to be inherited, which is mediated by the Polycomb and Trithorax complexes.
Polycomb/Trithorax
The Polycomb and Trithorax (PcG/TrxG) family of proteins was first identified in Drosophila where it was shown to be essential for the stable inheritance of a repressed or active gene state, respectively [99]. During Drosophila development, specific homeotic genes (Hox) are activated in a patterned fashion throughout the embryo [99, 100], and although the PcG/TrxG proteins are not required for the initiation of Hox gene transcription, they are essential for the maintenance of these genetic programs through cell divisions [99]. More recent genome-wide analyses have identified hundreds genes besides Hox genes that can be silenced or activated by the PcG/TrxG proteins, which exert their effects at Polycomb regulatory elements (PRE) throughout the genome [101, 102]. In mammalian cells, PREs have not been identified on a large scale [103], although Woo et al. [104] recently proposed the existence of a mammalian PRE. However, PcG/TrxG proteins are known to play crucial roles in mammalian development, cell fate determination, cancer [7, 105], and even immunity [106]. On a molecular level, the PcG and TrxG proteins function in distinct multiprotein complexes and act as chromatin modifiers by both inducing the covalent modification of histones as well as through other mechanisms. Before examining the potential mechanisms by which PcG and TrxG complexes can confer epigenetic inheritance, first a more in-depth look at their specific methods of silencing is provided.
Polycomb-mediated silencing
The Polycomb system mediates silencing through two distinct complexes, namely the Polycomb repressive complex 1 (PRC1) and PRC2. Although there seems to be some functional redundancy between the two complexes [107], their mechanisms of action are rather distinct. PRC2 contains the histone lysine methyltransferase EZH2, which trimethylates the H3 lysine 27 residue. Although H3K27 mono- and dimethylation occurs at approximately 50% of the nucleosomes in mammalian genomes [108], H3K27 trimethylation is more restricted and correlates with PcG silencing [109]. However, H3K27Me3 itself is not likely to be responsible for silencing by directly altering chromatin state [109]. Instead, H3K27Me3 might serve as a recruitment site for PRC1, which acts as a silencing complex [110, 111]. PRC1 recruitment is facilitated by the Polycomb protein (PC), which is one of the members of PRC1, through its chromodomain. Unlike the chromodomain of HP1 which predominantly binds H3K9Me2/3, PC preferentially binds H3K27Me3 [68, 112]. Although there is evidence for PRC1 binding in an H3K27Me3-independent fashion [109, 113], recent experiments in mammalian cells do support a causal role for H3K27Me3 in PRC1 recruitment. For one, abrogation of the H3K27 demethylase (discussed below) UTX increases H3K27Me3 levels at Hox target genes, which results in increased PRC1 deposition at these sites [114]. Moreover, knockdown of the PRC2 KMT EZH2 results in decreased H3K27 trimethylation and reduced PRC1 binding at Hox genes, which can be rescued by the expression of the viral H3K27 KMT vSET [115].
Although the exact mechanism of silencing by PRC1 is not known, several plausible theories have been suggested (Fig. 2). For one, H2AK119 ubiquitination [116] has been reported to be essential for PRC1-mediated silencing [117, 118]. Two constituents of PRC1, namely RING and Bmi1, seem to be essential for this function [118]. Although H2AK119Ubi does not inhibit the association of RNA Polymerase II, it seems to mediate silencing by preventing elongation of the transcript [117]. In addition to H2A ubiquitination, PRC1 might induce silencing through chromatin compaction [119], although this is disputed [99]. Furthermore, there is evidence for PRC1 silencing through the formation of higher-order chromatin structures or ‘looping’ [99, 120].

Mechanisms of PcG/TrxG-mediated silencing and activation. PRC2 exerts its function by methylating H3K27 residues. H3K27Me3, in turn, recruits PRC2 in a positive feedback loop which propagates these marks through the S-phase. In addition, H3K27Me3 recruits PRC1 which induces transcriptional repression through several mechanisms: 1 chromatin compaction, 2 looping, 3 H2K119 ubiquitination. The TrxG proteins mediate transcriptional activation by H3K4 trimethylation. In addition, TrxG proteins might mediate the deposition of H3.3. Interestingly, both TrxG as PcG members have been shown to associate with KDMs that remove the H3K27Me and H3K4Me, respectively
Trithorax-mediated activation
Unlike the PcG proteins that mediate the epigenetic repression of genes, the TrxG proteins function in an antagonistic fashion and are implicated in transcriptional activation. In Drosophila, TrxG proteins were identified to be required for maintaining Hox gene expression in appropriate segments during development, like PcG proteins are required for repression [121]. However, TrxG proteins regulate more than Hox genes in both Drosophila and mammalian cells. Interestingly, several members of the TrxG complex are histone lysine methyltransferases that trimethylate the H3 lysine 4 residue [99, 121–124]. Unlike the repressive H3K27 methylation, the methylation of H3K4 is a known mark of transcriptionally active heterochromatin and H3K4Me3 can even replace the silencing PcG and HP1 proteins from chromatin [122] in Drosophila. In general, it is assumed that rather than being a (co)activator of transcription, TrxG is likely involved in stimulating or facilitating transcriptional elongation [7, 99, 120]. In addition to H3K4 methylation, TrxG is also implicated in H3K27 acetylation [125]. Unlike methylation of H3K27, which is a known repressive mark, acetylation of this residue is related to gene activation and competes with H3K27 methylation [125, 126]. Lastly, in Drosophila, the trithorax member GAGA-factor mediates H3.3 replacement by direct association with HIRA, a H3.3 chaperone [127].
Interestingly, both PcG as TrxG complexes have been shown to associate with histone demethylases (KDM) (Fig. 2). More specifically, PcG complexes associate with Jarid1 and LSD1, two H3K4 KDMs, while TrxG complexes associate with UTX and Jmjd3, both H3K27 KDMs [106, 114]. This indicates that besides the previously discussed silencing and activating functions, the two systems can antagonize each other by directly removing the repressive and activating trimethylation of histones. As expected from their association with PcG/TrxG, these KDMs also play a role in epigenetic cell fate determination. For example, fully differentiated macrophages can, under circumstances, transdifferentiate; a process that requires Jmjd3 to remove the trimethylation of H3K27 by PRC2 at specific genes, such as Bmp-2 [106]. Whether deubiquinating enzymes (DUB) associate with TrxG in order to counteract PRC1 silencing by removing the H2AK119Ubi is not known, although H3K4 methylation and H2A deubiquitination have been shown to act in a reciprocal fashion [128].
PcG/TrxG during the cell cycle
As previously discussed, the major caveats for epigenetic inheritance is that this information needs to be maintained throughout DNA replication as well as mitosis. The former induces the dilution of epigenetic marks at histones, since both daughter strands receive a mixture of parental histones (carrying epigenetic information) as well as newly synthesized histones. In addition, mitosis involves the severe condensation of chromatin and the dissociation of many chromatin-binding proteins in order to facilitate this [9, 129]. The transmission of transcriptionally silent and active states by the PcG/TrxG system through cell divisions implies that either these proteins need to remain associated with their chromatin targets, or that chromatin ought to be somehow marked for re-association during DNA replication and mitosis. Both of these conditions have been reported for the Polycomb proteins.
PRC2 seems to adhere to the latter possibility, both during DNA replication as well as mitosis. During the S-phase, PRC2 localizes to the replication fork and colocalizes with PCNA [130], a crucial constituent of the replication machinery. In addition, it can directly bind to trimethylated H3K27; its own catalytic target. This might suggest that during replication, when epigenetic information gets diluted due to the addition of newly synthesized histones, PRC2 binds the parental lysine 27 trimethylated nucleosomes and spreads this mark to the neighboring new nucleosomes, thereby fully restoring the epigenetic silencing of the transcriptional state (Fig. 2). In accordance with their theory, Hansen et al. [130] observed stable PRC2-dependent silencing after the establishment of H3K27Me3 at a reporter gene that was integrated in the genome. In other words, once established the H3K27Me3 can be perpetuated by PRC2 during replication. In addition to PRC2 being closely associated with the replication fork, PRC2 also remains associated with chromatin during mitosis [130, 131]. This might suggest the ‘simplest’ form of epigenetic inheritance for PRC2: it can remain at target sites and thereby maintains epigenetic information throughout the entire cell cycle.
The transmission of PRC1 is more complicated. In vitro, PRC1 can remain associated with DNA through DNA replication [132]. This process is H3K27Me3-independent. However, in vivo studies suggest that PRC1 dissociates from chromatin during the S-phase, G2 and mitosis, and that PRC1-mediated silencing is restored during the G1-phase [132, 133]. The restoration of PRC1 foci in G1 is dependent on H3K27Me3, since PRC2-component knockdown results in disrupted PRC1 foci formation, and, in addition, delays entry into the S-phase [131]. These latter findings support the previously discussed model that PRC2-mediated H3K27Me3 precedes PRC1 recruitment and subsequent silencing. However, the findings that PRC1 can remain associated with chromatin during DNA replication in addition to other findings that suggest PRC2-independent functions of PRC1 (previously discussed) might suggest a similar model as for HP1-mediated heterochromatin maintenance. There might be a ‘pool’ of PRC1 that dissociates from chromatin during S-phase and mitosis and re-associates in G1, and there might be pool that remains bound to specific genomic locations during the cell cycle. Interestingly, similar as discussed for the dissociation/re-association of HP1, the presence of a serine next to the H3K27 residue might suggest a Me/P-switch [84], during which the phosphorylation of H3S28 could displace the Polycomb complexes. Later, in G1, the dephosphorylation of H3S28 again facilitates the binding of PcG complexes.
Ringrose and Paro [134], however, dispute that the histone modifications themselves mediate epigenetic inheritance. Instead, they propose that histone modifications are not the carriers of epigenetic inheritance, but only reflect the active/inactive state of the gene. Instead, they propose that Polycomb responsive elements (PREs) might induce a standard state of transcriptional silence [134], as supported by PRE deletion resulting in a loss of silencing [135, 136]. Actively transcribed regions of the genome that are continuously transcribed should be marked as such by non-coding RNAs (ncRNAs), specific DNA organization in the nucleus (discussed below), specific chromatin-binding proteins, or by H3.3 replacement [134]. These can subsequently be transferred through cell divisions and perpetuate the transcriptional activity. However, the lack of in-depth knowledge of mammalian PREs and the differences between mammalian and Drosophila epigenetics makes it difficult to confirm/refute either theory.
Stability versus flexibility
Interestingly, although PcG/TrxG complexes are known to mediate stable epigenetic inheritance, they are also implicated in regulating dynamic processes, such as cell cycle regulation and immunity. In addition, they can be regulated by signal transduction pathways, like hedgehog signaling [137] and are known to associate with the Retinoblastoma (Rb) gene [138]. However, how can some genes remain stably silenced or activated while others are dynamically regulated by the same system? One possible explanation for this apparent paradox comes from embryonic stem (ES) cells [89, 139]. In ES cells, many PcG/TrxG targets show ‘bivalency’, meaning that these genes contain both the PcG-mediated repressive H3K27Me3 mark as well as the TrxG-mediated H3K4Me3 mark [89, 139]. However, during differentiation and cell fate determination, more and more of these bivalent genes will acquire monovalent histone modifications (either H3K27Me3 or H3K4Me3), although a small fraction will remain bivalent. These bivalent genes are prone to rapid activation, while the monovalent genes become stably repressed or activated. Fluctuation of PcG or TrxG activity due to extracellular signaling is more likely to affect bivalent genes that are already ‘halfway there’, than monovalent genes. Therefore, even though bivalent domains can also be inherited [89], the monovalent genes are likely to be more stably heritable over time.
One example of such a bivalent locus is the INK4A locus which encodes p16INK4A and p14Arf, two genes that mediate cell cycle arrest and senescence in response to oncogenic stress and aging [140]. In cycling cells, PcG complexes regulate the silencing of these tumor suppressors. However, abrogation of Bmi1, a constituent of PRC1, results in premature senescence, while overexpression of Bmi1 results in the bypass of senescence, even in the presence of oncogenic stress [89, 141, 142]. In addition, the replicative-senescence of MEFs involves TrxG-mediated H3K4 methylation at the INK4A locus, H3K27 demethylation by Jmjd3, and PcG displacement [140]. In accordance, cells from aging organisms are difficult to reprogram and this inefficiency is dependent on the increased expression INK4A locus [143], which supports the notion that the increasing monovalency of H3K4Me3 or H3K27Me3 at a genomic location can increase the robustness of silencing. However, whether the activated/repressed status of all PcG/TrxG targets can be inherited is not clear.
Non-coding RNAs
In addition to the mechanisms of epigenetic inheritance mentioned above, RNA might also mediate the stable transmission of epigenetic states, in particular small interfering RNAs (siRNAs) and long non-coding RNAs (ncRNA). An established example of this is Xist, a long ncRNA that mediates the inactivation of one of two X-chromosomes in females. Xist only associates with the inactive X-chromosome and subsequently recruits epigenetic silencing machinery, such as DNA methylases and PcG complexes [144]. Interestingly, the translocation of the Xist gene to autosomes results in subsequent silencing of the specific autosome [145]. Besides X-inactivation, there is also ample evidence for the involvement of ncRNAs in the previously discussed epigenetic inheritance mechanisms, especially heterochromatin formation by HP1, and the PcG/TrxG system. Since much of the knowledge on the role of ncRNAs in epigenetic inheritance is based on model organisms such as yeast (S. pombe), plants, and Drosophila, the exact involvement and specific mechanisms of mammalian ncRNA-mediated silencing is still uncertain.
RNA-mediated epigenetic inheritance is implicated in the formation and maintenance of heterochromatin [70, 146–148]. In S. pombe, the disruption of the RNAi machinery leads to heterochromatin defects, including H3K9 methylation defects [149]. One potential molecular model for RNAi-mediated heterochromatin silencing [70, 148, 150] involves the transcription of (peri) centromeric repeats [151, 152]. These transcripts are required for RNAi production; the resulting small interfering RNAs (siRNAs) together with Ago1, Tas3 and Chp1 form the RITS (RNA-induced transcriptional silencing) complex, which in turn recruits other heterochromatin-modifying factors, such as Swi6 (yeast homologue of HP1) and Clr4 (yeast homologue of Suv39h1/2). The recruitment of RITS, which in itself requires H3K9Me3 [150], results in subsequent heterochromatin formation and H3K9 methylation. Interestingly, the transcription of these repeats occurs mostly in the S-phase [153, 154], and therefore provides an interesting candidate for epigenetic inheritance of heterochromatin during DNA replication; transcription of repeats during the S-phase results in a positive feedback loop resulting in heterochromatin formation and maintenance [70, 148]. However, whether similar mechanisms occur in mammalian cells is somewhat disputed. There is evidence for the involvement of RNAi in mammalian heterochromatin formation [155–157]. For example, the formation of higher-order heterochromatin structures and HP1 localization to these requires an RNA component [157]. However, one study reports that Dicer-knockout ES cells display little pericentric heterochromatin defects [158].
Besides heterochromatin formation, siRNAs have also been implicated in mediating DNA methylation. Using short-hairpin RNAs (shRNA) against RASSF1A, Castanotto et al. [159] showed increased promoter methylation in HeLa cells. In plants, RNAi-mediated DNA methylation is readily observed, although in mammals this might be limited to the rare non-CpG DNA methylation as observed mostly early in development [160]. Currently, more research in mammalian cell systems and organisms are required for an established role of RNAi in DNA methylation.
RNAs are also likely involved in mediating some epigenetic functions of the PcG/TrxG system, although this seems mostly based on long ncRNAs rather than RNAi [161]. In Drosophila, many PREs are transcribed and produce ncRNAs, and although mammalian PREs are currently disputed, there is also evidence for the production of ncRNAs near mammalian PcG/TrxG targets [134, 161]. Furthermore, PcG/TrxG proteins have been shown to bind RNA, although whether this is sequence-dependent is disputed [161]. Interestingly, in Drosophila the mere transcription of PREs might promote transcriptional activity of PcG-targeted genes and thereby counteract PcG silencing [134]. For example, using a transgene containing a PRE followed by a reporter gene, Schmitt et al. [162] showed that mere transcription through the PRE is sufficient to activate the reporter gene. In addition, the transcribed ncRNA of a PRE results in TrxG recruitment, resulting in gene activation [163]. These findings support the previously discussed model of ‘standard silencing’, which states that active transcriptional sites should be marked by e.g., H3.3 replacement or RNA components, in order to transmit a heritable active chromatin state. In mammalian cells, the transcriptional activation of HoxA genes has also been linked to transcription of intergenic ncRNAs at the HoxA cluster [164]. However, several findings seem to contradict the standard silencing theory. Importantly, several mammalian ncRNAs have been detected that upon transcription induce PcG-mediated silencing, rather than to alleviate it [161]. For example, the Kcnq1ot1 ncRNA [165] is required for the silencing of the Kcnq1 locus. This ncRNA associates with PcG proteins in a tissue specific manner. In addition, the abrogation of the HOTAIR ncRNA results in upregulation of the HoxD cluster [166]. Interestingly, in mammalian cells most non-transcribed genes still produce short ncRNA transcripts [167]. Many of these ncRNAs can bind PRC2 [168, 169] and ncRNA production at activating genes declines. One model, as proposed by Guenther and Young [170], is that short transcribed ncRNAs recruit PRC2 and results in repression. However, transcription initiation as promoted by transcription factors recruits TrxG proteins and results in activation.
Although the exact mechanism and causality of the interaction between ncRNAs and the PcG/TrxG remains elusive, the involvement of an RNA component is highly likely. Similarly, an RNA component is likely to function in mammalian heterochromatin formation. However, how these ncRNAs might be regulated throughout the cell cycle is currently unclear.
DNA organization
The current view of the nucleus is a dynamic one; rather than being just a place of storage of genetic information, the nucleus contains specific functional domains, such as the nucleolus, the inactive heterochromatin, and the active euchromatin [171–173]. These latter two are even replicated in a spatio-temporal distinct manner during the S-phase. In recent years, much research has focused on long-range DNA interactions. These interactions can occur in cis, so in neighboring sequences on the same chromosome, as well as in trans, interactions with distant regions on the same chromosome or even interactions with sequences at different chromosomes. An example of each is the regulation of β-globin genes through looping to a cis-regulatory ‘master enhancer’ that also controls other globin genes, and the brief interaction of the X-inactivation centers (Xic) of two X-chromosomes in female cells just before the random X-inactivation of one of the copies [174]. These higher-order chromatin rearrangements can also be induced. For example HoxB genes can loop out of their ‘chromosome territory’ upon retinoic acid-induced expression, and steroid-induced activation of nuclear receptors results in the formation of interchromosomal contacts required for enhanced transcription of the induced genes [175, 176]. Interestingly, nuclear organization and long-range DNA structures might potentially function in epigenetic inheritance. For example, there is some evidence for mitotic transmissibility of chromosome organization in the nucleus, at least through several cell divisions [177, 178]. These findings, however, are somewhat disputed. Other studies confirm that chromosome positions in the nucleus are fixed throughout the cell cycle, although they change after mitosis. Subsequently, in G1 the chromosomes are unpacked into a different nuclear organization [179–181]. Regardless of the heritability of chromosome organization in the nucleus, considerable evidence has accumulated for long-range DNA interactions in heterochromatin formation and the PcG/TrxG system. However, are these DNA structures themselves involved in epigenetic inheritance? How could they survive DNA replication and mitosis? These questions will be addressed below.
Evidence for long-range DNA contacts during heterochromatin formation comes from studies in Drosophila, where the insertion of a heterochromatic sequence in the brown gene (bw), results in dominant repression and localization of both the mutant allele, as well as the wild type allele to heterochromatic regions, a process termed position-effect variegation (PEV) [182]. In mammalian cells there is also evidence for a role of DNA organization in heterochromatin formation. It was reported that localization of the β-globin locus away from heterochromatin influences its histone modification (acetylation) and transcriptional state [183]. Although localization of genes to heterochromatin at the nuclear periphery correlates with their transcriptional inactivity [184], the maintenance of these structures throughout the cell cycle is unclear. In Drosophila, it was reported that at least the interaction of the dominant repressive bw mutant with heterochromatin is lost during DNA replication and is re-established in G1 [185].
DNA organization is also likely involved in the PcG/TrxG system [173, 186]. For one, PcG proteins are known to localize to PcG bodies [187, 188] that might cluster Polycomb targets. In Drosophila, the introduction of extra copies of the PRE Fab-7 induces clustering with each other and the endogenous Fab-7 locus [189] in a PcG-dependent manner. Interestingly, the loss of the endogenous locus results in the loss of PcG silencing at the site of the inserted artificial Fab-7 fragment. This loss of silencing can be stably passed onto the next generation, even after restoring the endogenous Fab-7 locus. This indicates a potential role for nuclear organization in epigenetic inheritance of PcG silencing [186, 189]. Although it was previously discussed that a direct role for RNAi in PcG silencing is currently disputed [161], the nuclear localization of the Fab-7 fragments depends on the RNAi machinery [190, 191].
Insulator proteins, and the insulator sequences that they bind, may play a role in mediating epigenetic inheritance. Although several functions have been published, the most established function of insulator proteins is to insulate promoters from enhancer elements and thereby affecting transcription. In Drosophila the expression of a dominant-negative form of the insulator protein BEAF (boundary element-associated factors) results in enhanced PEV through the disruption of chromatin organization [192]. In mammalian cells, CTCF, an insulator protein, is an interesting candidate for mediating epigenetic inheritance by altering nuclear organization [193–195]. CTCF binds to insulator sequences throughout the mammalian genome where it performs its function by looping DNA and hence altering nuclear organization of genes [193, 195]. One example of CTCF function is the expression of the paternally imprinted Igf2 gene [193, 194] on the murine chromosome 7. Genomic imprinting of genes is the monoallelic expression of certain genes transferred either paternally or maternally [196]. Under normal circumstances, the maternal imprinting control region (ICR) is unmethylated leading to CTCF binding, subsequent looping, and silencing of the maternal Igf2 gene. However, the paternal ICR is methylated which prevents CTCF binding and therefore facilitates transcription. In addition to disrupting Igf2, the abrogation of CTCF function at the ICR can also disrupt the transcription at specific genes located at different chromosomes [197].
Interestingly, there are several indications that CTCF might be stably transmitted through replication and mitosis. For one, some insulator sequences and CTCF-binding sites have been detected within introns of genes. In experiments in Drosophila, insertion of an insulator binding sequence in an intron retains its function [198]. Also in mammalian cells, CTCF-binding sites have been detected in introns [199]. Although this does not directly implicate maintenance through DNA replication, at least local double-strand disruption by transcription machinery does not hamper insulator function. In addition, several studies have shown continued CTCF binding at mitotic chromosomes, and even continued Igf2–ICR interaction through mitosis [200]. Note that CTCF has also been shown to interact with PRC2 and is a known regulator of heterochromatin spreading, which also might indicate a role for CTCF in epigenetic inheritance through these systems.
Similarities and differences between epigenetic systems
As discussed, cells depend on the inheritance of epigenetic information throughout the cell cycle to maintain their state of differentiation. So far, DNA methylation is the most established epigenetic mark that can stably confer heritable phenotypes. However, more and more evidence is accumulating for the epigenetic inheritance of chromatin states and even higher-order chromatin structures. The H3K9Me3-HP1 system and the PcG/TrxG systems are the obvious candidates for the transfer of chromatin states through cell divisions. Interestingly, although HP1-mediated heterochromatin and PcG-silenced areas generally do not colocalize, DNA methylation seems to be involved in both systems.
Heterochromatin is, in general, fairly gene-poor, repeat-rich, and relatively CpG-poor, although most CpG dinucleotides that are present are methylated [70, 201]. The interaction between HP1, H3K9 KMTs and the DNA methylating enzymes have been shown in multiple studies [202, 203]. However, the causality of the interaction is still disputed and evidence exists for both the recruitment of the HP1-KMT machinery to methylated DNA, as well as the recruitment of DNMTs to sites of H3K9 methylation [204]. This could indicate a self-reinforcing mechanism of both DNA methylation and H3K9 methylation as proposed by Fuks [204].
Interestingly, although the methylation of H3K9 does occur at promoters with relatively low CpG content, it does not occur at promoters containing CpG islands [88, 201, 205]. As previously discussed, CpG islands are generally unmethylated CpG-dense areas found upstream of ~60% of human genes and the methylation of such islands results in robust silencing. The PcG/TrxG system and hence H3K27Me3 and H3K4Me3, however, does associate with CpG-islands. In fact, in ES cells more than 97% of PRC2 target sites contain CpG-islands or CpG-rich areas [205]. One option is that DNA methylation might reinforce PcG-mediated silencing [89, 137, 206]. In agreement with this, there is evidence that PcG target genes become methylated during development [207]. Both PRC2 as PRC1 might be responsible for this methylation by direct recruitment of DNMTs [208, 209]. Intriguingly, H3K4Me3 does not colocalize with DNA methylation [201]. In fact, DNA methylation can not occur at sequences that contain methylated H3K4, and conversely, DNA demethylation is correlated with H3K4 methylation [42]. Since the TrxG proteins depend on H3K4 trimethylation in order to maintain an open chromatin conformation, it is possible that DNA methylation at CpG islands can only occur at monovalent PcG sites that do not contain TrxG.
All in all, it is tempting to suggest a model as depicted in Fig. 3, where the DNMTs can interact with both PcG/TrxG and HP1-KMT system to alter the local chromatin state.

Interactions of the PcG/TrxG system and the HP1-KMT system with DNA methyltransferases. Both the repressive PcG proteins as well as HP1 are known to interact with DNMTs. For the former, both PRC1 as PRC2 directly interact with DNMTs, and methylation of PcG sites might reflect a state of stable transcriptional silencing. HP1 also interacts with DNMTs although the causality of the interaction is not yet resolved; there is evidence for both the recruitment of DNMTs by H3K9Me and HP1, as well as recruitment of HP1-KMT to methylated DNA. The TrxG proteins, however, are likely to oppose DNMT action, as H3K4Me3 and DNA methylation are mutually exclusive. The fact that both repressive chromatin-modifying systems (PcG and HP1) interact with DNMTs, while the activating TrxG antagonizes DNMTs could support the notion that DNA methylation might be, in certain genomic regions, an additional ‘layer’ of silencing
Hierarchical model of epigenetic stability
In general, DNA methylation seems to be the most stable epigenetic modification displayed in Fig. 4. Although DNA demethylation is likely to occur during specific developmental phases, under normal circumstances cycling cells will not likely undergo genome-wide demethylation of the DNA. HP1-KMT mediated heterochromatin is also likely to be a stable system, due to its role in retrotansposon and repeat silencing. In addition, certain early developmental genes become H3K9 trimethylated during development which might reflect a stable silenced state, although facultative heterochromatin might be more dynamically regulated. For the PcG/TrxG system, genomic regions that contain only monovalent modifications (either H3K27Me3 or H3K4Me3) reflect a relatively stable state, while bivalent regions remain ‘poised’ for transcription upon environmental stimuli. As noted by Kaufman and Rando [56], most hypothesized heritable chromatin modifications are involved in repression of genes instead of activation, with the TrxG proteins and the histone variant H3.3 being an exception. How the stability and heritability of monovalent TrxG-regulated genes compares the repressive PcG-regulated genes is as of yet unclear and, hence, not included in Fig. 4.

Proposed stability of the discussed epigenetic modifications. DNA methylation, especially when it occurs at CpG islands or CpG-rich areas, reflects a robust silenced state that is difficult to reverse. Heterochromatin is also likely to be relatively stably, due to its role in silencing repeats, retrotransposons, and other gene-poor regions. However, facultative heterochromatin can be dynamically regulated, e.g., during the cell cycle. The PcG/TrxG system features both bivalently marked target genes which are ‘poised’ for transcription and can react to dynamic stimuli, as well as monovalent sites which are less susceptible to fluctuation of PcG/TrxG protein activity
As previously discussed, both ncRNAs as DNA organization are likely to play a role in epigenetic inheritance. However, how these emerging topics fit into a hypothesized hierarchical model as shown in Fig. 4, remains elusive. In addition, the causality of these concepts in epigenetics warrants further investigation; e.g., can a change in DNA organization confer heritability of gene expression/repression as such, or is the change in DNA organization induced through a different mechanism? In addition, can DNA organization and ncRNAs confer heritability autonomously or do they function as a part of different systems (e.g., such as the discussed siRNA role in H3K9Me3-HP1 heterochromatin formation or long ncRNAs in PcG-mediated repression)?
Concluding remarks
All in all, we have reviewed the major epigenetic systems that are implicated in the stable maintenance of cell fate. In order to confer a similar phenotype after cell division, the epigenetic systems need to be faithfully replicated at their genomic locations during the S-phase and survive mitosis, or at least be re-recruited after the M-phase. From the reviewed literature two general mechanisms become apparent for the transmission of epigenetic information, namely self-recruitment and maintained binding. In the case of DNA methylation, hemimethylated DNA recruits DNMT1 in order to copy the majority of methylated CpGs. For the propagation of methylated H3K27 and H3K9 through the S-phase, cells could rely on PRC2 and HP1, respectively. The former binds H3K27Me3 directly and possesses intrinsic KMT activity for that specific residue, while the latter binds H3K9Me3 and can associate with known H3K9 KMTs as well as histone chaperones, In addition to self-recruitment, there is also evidence for the continued binding of certain epigenetic modifiers through phases of the cell cycle. PRC1, for example, can remain bound to DNA during replication, and PRC2 was shown can be detected on chromosomes during mitosis. Even CTCF can remain bound at imprinted loci in order to maintain DNA organization during mitosis at specific loci.
Nonetheless, many questions remain on the exact mechanisms of epigenetic inheritance. For one, although the previously discussed positive-feedback mechanisms suggest transmission of histone methylation, it remains a question whether the histone modifications themselves are the primary determinants of epigenetic inheritance or whether other mechanisms (e.g., ncRNA transcription or genome organization) underlie this inheritance. Furthermore, although much research has focused on epigenetic repression, the stability and heritability of the activating TrxG and the histone variant H3.3 requires additional research in mammalian cells, as well as how these interact. Overall, an in-depth understanding of epigenetic inheritance can also aid in our understanding of human disease, as already DNA methylation and the PcG/TrxG proteins have been implicated in the pathogenesis of numerous diseases, such as cancers and infectious diseases.
References
Haig D (2004) The (dual) origin of epigenetics. Cold Spring Harb Symp Quant Biol 69:67–70
Feng YQ, Desprat R, Fu H, Olivier E, Lin CM, Lobell A, Gowda SN, Aladjem MI, Bouhassira EE (2006) DNA methylation supports intrinsic epigenetic memory in mammalian cells. PLoS Genet 2:e65. doi:10.1371/journal.pgen.0020065
Silva AJ, White R (1988) Inheritance of allelic blueprints for methylation patterns. Cell 54:145–152
Li E (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3:662–673
Yamanaka S (2009) Elite and stochastic models for induced pluripotent stem cell generation. Nature 460:49–52
Das PM, Singal R (2004) DNA methylation and cancer. J Clin Oncol 22:4632–4642
Lund AH, van Lohuizen M (2004) Epigenetics and cancer. Genes Dev 18:2315–2335
Probst AV, Dunleavy E, Almouzni G (2009) Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol 10:192–206
Li G, Sudlow G, Belmont AS (1998) Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: precise choreography of condensation/decondensation and nuclear positioning. J Cell Biol 140:975–989
Rakyan VK, Preis J, Morgan HD, Whitelaw E (2001) The marks, mechanisms and memory of epigenetic states in mammals. Biochem J 356:1–10
Morgan HD, Sutherland HG, Martin DI, Whitelaw E (1999) Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 23:314–318
Rakyan V, Whitelaw E (2003) Transgenerational epigenetic inheritance. Curr Biol 13:R6
Li E, Beard C, Jaenisch R (1993) Role for DNA methylation in genomic imprinting. Nature 366:362–365
Mohandas T, Sparkes RS, Shapiro LJ (1981) Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science 211:393–396
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Jones PA, Liang G (2009) Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 10:805–811
Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10:2709–2721
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33:245–254
Weber M, Schubeler D (2007) Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr Opin Cell Biol 19:273–280
Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9:465–476
Walsh CP, Chaillet JR, Bestor TH (1998) Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet 20:116–117
Eden A, Gaudet F, Waghmare A, Jaenisch R (2003) Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300:455
Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D (2007) Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39:457–466
Hsieh CL (1994) Dependence of transcriptional repression on CpG methylation density. Mol Cell Biol 14:5487–5494
Boyes J, Bird A (1992) Repression of genes by DNA methylation depends on CpG density and promoter strength: evidence for involvement of a methyl-CpG binding protein. EMBO J 11:327–333
Hermann A, Goyal R, Jeltsch A (2004) The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem 279:48350–48359
Moldovan GL, Pfander B, Jentsch S (2007) PCNA, the maestro of the replication fork. Cell 129:665–679
Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, Tajima S, Mitsuya K, Okano M, Koseki H (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450:908–912
Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317:1760–1764
Richards EJ (2006) Inherited epigenetic variation—revisiting soft inheritance. Nat Rev Genet 7:395–401
Liang G, Chan MF, Tomigahara Y, Tsai YC, Gonzales FA, Li E, Laird PW, Jones PA (2002) Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol 22:480–491
Chen T, Ueda Y, Dodge JE, Wang Z, Li E (2003) Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol 23:5594–5605
Hattori N, Abe T, Suzuki M, Matsuyama T, Yoshida S, Li E, Shiota K (2004) Preference of DNA methyltransferases for CpG islands in mouse embryonic stem cells. Genome Res 14:1733–1740
Bachman KE, Rountree MR, Baylin SB (2001) Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem 276:32282–32287
Robertson KD, Keyomarsi K, Gonzales FA, Velicescu M, Jones PA (2000) Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res 28:2108–2113
Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzle KW, Baylin SB, Vogelstein B (2002) DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416:552–556
Dodge JE, Okano M, Dick F, Tsujimoto N, Chen T, Wang S, Ueda Y, Dyson N, Li E (2005) Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J Biol Chem 280:17986–17991
Haaf T (2006) Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr Top Microbiol 310:13–22
Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A (2008) Dissecting direct reprogramming through integrative genomic analysis. Nature 454:49–55
Morgan HD, Santos F, Green K, Dean W, Reik W (2005) Epigenetic reprogramming in mammals. Hum Mol Genet 14:R47–R58
Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J (2000) Active demethylation of the paternal genome in the mouse zygote. Curr Biol 10:475–478
Klug M, Heinz S, Gebhard C, Schwarzfischer L, Kraus SW, Andreesen R, Rehli M (2010) Active DNA demethylation in human postmitotic cells correlates with activating histone modifications, but not transcription levels. Genome Biol 11:R63. doi:10.1186/gb-2010-11-6-r63
Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W (2010) Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463:1101–1105. doi:10.1038/nature08829
Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM (2010) Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature 463:1042–1047
Okada Y, Yamagata K, Hong H, Wakayama T, Zhang Y (2010) A role for the elongator complex in zygotic paternal genome demethylation. Nature 463:554–558
Ellis L, Atadja PW, Johnstone RW (2009) Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther 8:1409–1420
Ying Y, Tao Q (2009) Epigenetic disruption of the WNT/β-catenin signaling pathway in human cancers. Epigenetica 4:307–312
Bernstein BE, Meissner A, Lander ES (2007) The mammalian epigenome. Cell 128:669–681
Henikoff S, Furuyama T, Ahmad K (2004) Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet 20:320–326
Talbert PB, Henikoff S (2010) Histone variants—ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 11:264–275
Orphanides G, Reinberg D (2000) RNA polymerase II elongation through chromatin. Nature 407:471–475
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128:707–719
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705
Ptashne M (2007) On the usage of the word ‘epigenetic’. Curr Biol 17:r233–r236
Campos EI, Reinberg D (2009) Histones: annotating chromatin. Annu Rev Genet 43:559–599
Kaufman PD, Rando OJ (2010) Chromatin as a potential carrier of heritable information. Curr Opin Cell Biol. doi:10.1016/j.ceb.2010.02.002
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45
Rice JC, Allis CD (2001) Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol 13:263–273
Byvoet P, Shepherd GR, Hardin JM, Noland BJ (1972) The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch Biochem Biophys 148:558–567
Volkel P, Angrand PO (2007) The control of histone lysine methylation in epigenetic regulation. Biochimie 89:1–20
Loyola A, Almouzni G (2007) Marking histone H3 variants: how, when and why? Trends Biochem Sci 32:425–433
Ehrenhofer-Murray AE (2004) Chromatin dynamics at DNA replication, transcription and repair. Eur J Biochem 271:2335–2349
Groth A, Rocha W, Verreault A, Almouzni G (2007) Chromatin challenges during DNA replication and repair. Cell 128:721–733
Xu M, Long C, Chen X, Huang C, Chen S, Zhu B (2010) Partitioning of histone H3–H4 tetramers during DNA replication-dependent chromatin assembly. Science 328:94–98
Nakatani Y, Tagami H, Shestakova E (2006) How is epigenetic information on chromatin inherited after DNA replication? Ernst Schering Res Found Workshop, pp 89–96
Cusick ME, DePamphilis ML, Wassarman PM (1984) Dispersive segregation of nucleosomes during replication of simian virus 40 chromosomes. J Mol Biol 178:249–271
Sogo JM, Stahl H, Koller T, Knippers R (1986) Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J Mol Biol 189:189–204
Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17:1870–1881
Eissenberg JC, Elgin SC (2000) The HP1 protein family: getting a grip on chromatin. Curr Opin Genet Dev 10:204–210
Grewal SI, Jia S (2007) Heterochromatin revisited. Nat Rev Genet 8:35–46
Aagaard L, Laible G, Selenko P, Schmid M, Dorn R, Schotta G, Kuhfittig S, Wolf A, Lebersorger A, Singh PB, Reuter G, Jenuwein T (1999) Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3–9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J 18:1923–1938
Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, Harel-Bellan A, Ameyar-Zazoua M, Ait-Si-Ali S (2010) A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell 37:46–56
Melcher M, Schmid M, Aagaard L, Selenko P, Laible G, Jenuwein T (2000) Structure-function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Mol Cell Biol 20:3728–3741
Quivy JP, Roche D, Kirschner D, Tagami H, Nakatani Y, Almouzni G (2004) A CAF-1 dependent pool of HP1 during heterochromatin duplication. EMBO J 23:3516–3526
Quivy JP, Gérard A, Cook AJL, Roche D, Almouzni G (2008) The HP1-p150/CAF-1 interaction is required for pericentric heterochromatin replication and S-phase progression in mouse cells. Nat Struct Mol Biol 15:792–797
Kwon SH, Workman JL (2008) The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol Cell 26:217–227
Loyola A, Tagami H, Bonaldi T, Roche D, Quivy JP, Imhof A, Nakatani Y, Dent SYR, Almouzni G (2009) The HP1–CAF1–SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep 10:769–775
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120–124
Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292:110–113
Talbert PB, Henikoff S (2006) Spreading of silent chromatin: inaction at a distance. Nat Rev Genet 7:793–803
Minc E, Allory Y, Worman HJ, Courvalin JC, Buendia B (1999) Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 108:220–234
Hayakawa T, Haraguchi T, Masumoto H, Hiraoka Y (2003) Cell cycle behavior of human HP1 subtypes: distinct molecular domains of HP1 are required for their centromeric localization during interphase and metaphase. J Cell Sci 116:3327–3338
Schmiedeberg L, Weisshart K, Diekmann S, Meyer Zu Hoerste G, Hemmerich P (2004) High- and low-mobility populations of HP1 in heterochromatin of mammalian cells. Mol Biol Cell 15:2819–2833
Fischle W, Wang Y, Allis CD (2003) Binary switches and modification cassettes in histone biology and beyond. Nature 425:475–479
Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD (2005) Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438:1116–1122
Hirota T, Lipp JJ, Toh BH, Peters JM (2005) Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438:1176–1180
Prigent C, Dimitrov S (2003) Phosphorylation of serine 10 in histone H3, what for? J Cell Sci 116:3677–3685
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560
Pietersen AM, van Lohuizen M (2008) Stem cell regulation by Polycomb repressors: postponing commitment. Curr Opin Cell Biol 20:201–207
Trojer P, Reinberg D (2007) Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell 28:1–13
Hake SB, Allis CD (2006) Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc Natl Acad Sci USA 103:6428–6435
Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y (2004) Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116:51–61
Mito Y, Henikoff JG, Henikoff S (2005) Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet 37:1090–1097
McKittrick E, Gafken PR, Ahmad K, Henikoff S (2004) Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci USA 101:1525–1530
Ng RK, Gurdon JB (2008) Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol 10:102–109
Berger SL (2007) The complex language of chromatin regulation during transcription. Nature 447:407–412
Wirbelauer C, Bell O, Schubeler D (2005) Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev 19:1761–1766
Ng RK, Gurdon JB (2008) Epigenetic inheritance of cell differentiation status. Cell Cycle 7:1173–1177
Schwartz YB, Pirrotta V (2007) Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 8:9–22
Ringrose L, Paro R (2004) Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 38:413–443
Schwartz YB, Kahn TG, Nix DA, Li XY, Bourgon R, Biggin M, Pirrotta V (2006) Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat Genet 38:700–705
Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K (2006) Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 20:1123–1136
Schuettengruber B, Cavalli G (2009) Recruitment of Polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 136:3531–3542
Woo CJ, Kharchenko PV, Daheron L, Park PJ, Kingston RE (2010) A region of the human HOXD cluster that confers Polycomb-group responsiveness. Cell 140:99–110
Lund AH, van Lohuizen M (2004) Polycomb complexes and silencing mechanisms. Curr Opin Cell Biol 16:239–246
De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G (2007) The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of Polycomb-mediated gene silencing. Cell 130:1083–1094
Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A (2010) Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev 24:265–276
Peters AH, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12:1577–1589
Simon JA, Kingston RE (2009) Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10:697–708
Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441:349–353
Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS (2004) Hierarchical recruitment of Polycomb group silencing complexes. Mol Cell 14:637–646
Cao R, Zhang Y (2004) The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev 14:155–164
Bantignies F, Cavalli G (2006) Cellular memory and dynamic regulation of Polycomb group proteins. Curr Opin Cell Biol 18:275–283
Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R (2007) Demethylation of H3K27 regulates Polycomb recruitment and H2A ubiquitination. Science 318:447–450
Mujtaba S, Manzur KL, Gurnon JR, Kang M, Van Etten JL, Zhou MM (2008) Epigenetic transcriptional repression of cellular genes by a viral SET protein. Nat Cell Biol 10:1114–1122
Weake VM, Workman JL (2008) Histone ubiquitination: triggering gene activity. Mol Cell 29:653–663
Stock JK, Giadrossi S, Casanova M, Brookes E, Vidal M, Koseki H, Brockdorff N, Fisher AG, Pombo A (2007) Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol 9:1428–1435
Cao R, Tsukada Y, Zhang Y (2005) Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell 20:845–854
Francis NJ, Kingston RE, Woodcock CL (2004) Chromatin compaction by a Polycomb group protein complex. Science 306:1574–1577
Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G (2007) Genome regulation by Polycomb and trithorax proteins. Cell 128:735–745
Grimaud C, Nègre N, Cavalli G (2006) From genetics to epigenetics: the tale of Polycomb group and trithorax group genes. Chromosome Res 14:363–375
Beisel C, Imhof A, Greene J, Kremmer E, Sauer F (2002) Histone methylation by the Drosophila epigenetic transcriptional regulator Ash1. Nature 419:857–862
Byrd NB, Shearn A (2003) ASH1, a Drosophila trithorax group protein, is required for methylation of lysine 4 residues on histone H3. Proc Natl Acad Sci USA 10:11535–11540
Gregory GD, Vakoc CR, Rozovskaia T, Zheng X, Patel S, Nakamura T, Canaani E, Blobel GA (2007) Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol Cell Biol 27:8466–8479
Schwartz YB, Kahn TG, Stenberg P, Ohno K, Bourgon R, Pirrotta V (2010) Alternative epigenetic chromatin states of Polycomb target genes. PLoS Genet 6:e1000805. doi:10.1371/journal.pgen.1000805
Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ (2009) CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136:3131–3141
Nakayama T, Nishioka K, Dong YX, Shimojima T, Hirose S (2007) Drosophila GAGA factor directs histone H3.3 replacement that prevents the heterochromatin spreading. Genes Dev 21:552–561
Nakagawa T, Kajitani T, Togo S, Masuko N, Ohdan H, Hishikawa Y, Koji T, Matsuyama T, Ikura T, Muramatsu M, Ito T (2008) Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev 22:37–49
Martinez-Balbas MA, Dey A, Rabindran SK, Ozato K, Wu C (1995) Displacement of sequence-specific transcription factors from mitotic chromatin. Cell 83:29–38
Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10:1291–1300
Aoto T, Saitoh N, Sakamoto Y, Watanabe S, Nakao M (2008) Polycomb group protein-associated chromatin is reproduced in post-mitotic G1 phase and is required for S phase progression. J Biol Chem 283:18905–18915
Francis NJ, Follmer NE, Simon MD, Aghia G, Butler JD (2009) Polycomb proteins remain bound to chromatin and DNA during DNA replication in vitro. Cell 137:110–122
Voncken JW, Schweizer D, Aagaard L, Sattler L, Jantsch MF, van Lohuizen M (1999) Chromatin-association of the Polycomb group protein BMI1 is cell cycle regulated and correlates with its phosphorylation status. J Cell Sci 112:4627–4639
Ringrose L, Paro R (2007) Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134:223–232
Busturia A, Wightman CD, Sakonju S (1997) A silencer is required for maintenance of transcriptional repression throughout Drosophila development. Development 124:4343–4350
Sengupta AK, Kuhrs A, Muller J (2004) General transcriptional silencing by a Polycomb response element in Drosophila. Development 131:1959–1965
Sparmann A, van Lohuizen M (2006) Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 6:846–856
Dahiya A, Wong S, Gonzalo S, Gavin M, Dean DC (2001) Linking the Rb and Polycomb pathways. Mol Cell 8:557–569
Ringrose L (2006) Polycomb, trithorax and the decision to differentiate. Bioessays 28:330–334
Agherbi H, Gaussmann-Wenger A, Verthuy C, Chasson L, Serrano M, Djabali M (2009) Polycomb mediated epigenetic silencing and replication timing at the INK4a/ARF locus during senescence. PLoS One 4:e5622. doi:10.1371/journal.pone.0005622
Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Monch K, Minucci S, Porse BT, Marine JC, Hansen KH, Helin K (2007) The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 21:525–530
Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M (1999) The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397:164–168
Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, Blasco MA, Serrano M (2009) The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460:1136–1139
Wutz A (2007) Xist function: bridging chromatin and stem cells. Trends Genet 23:457–464
Popova BC, Tada T, Takagi N, Brockdorff N, Nesterova TB (2006) Attenuated spread of X-inactivation in an X; autosome translocation. Proc Natl Acad Sci USA 103:7706–7711
Lippman Z, Martienssen R (2004) The role of RNA interference in heterochromatic silencing. Nature 431:364–370
Grewal SI (2010) RNAi-dependent formation of heterochromatin and its diverse functions. Curr Opin Genet Dev 20:134–141
Kloc A, Martienssen R (2008) RNAi, heterochromatin and the cell cycle. Trends Genet 24:511–517
Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297:1833–1837
Buhler M, Moazed D (2007) Transcription and RNAi in heterochromatic gene silencing. Nat Struct Mol Biol 14:1041–1048
Djupedal I, Portoso M, Spahr H, Bonilla C, Gustafsson CM, Allshire RC, Ekwall K (2005) RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev 19:2301–2306
Kato H, Goto DB, Martienssen RA, Urano T, Furukawa K, Murakami Y (2005) RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science 309:467–469
Kloc A, Zaratiegui M, Nora E, Martienssen R (2008) RNA interference guides histone modification during the S phase of chromosomal replication. Curr Biol 18:490–495
Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI (2008) Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 451:734–737
Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K (2005) Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19:489–501
Fukagawa T, Nogami M, Yoshikawa M, Ikeno M, Okazaki T, Takami Y, Nakayama T, Oshimura M (2004) Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat Cell Biol 6:784–791
Maison C, Bailly D, Peters AH, Quivy JP, Roche D, Taddei A, Lachner M, Jenuwein T, Almouzni G (2002) Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet 30:329–334
Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ (2005) Characterization of Dicer-deficient murine embryonic stem cells. Proc Natl Acad Sci USA 102:12135–12140
Castanotto D, Tommasi S, Li M, Li H, Yanow S, Pfeifer GP, Rossi JJ (2005) Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol Ther 12:179–183
Wassenegger M (2005) The role of the RNAi machinery in heterochromatin formation. Cell 122:13–16
Hekimoglu B, Ringrose L (2009) Non-coding RNAs in Polycomb/trithorax regulation. RNA Biol 6:129–137
Schmitt S, Prestel M, Paro R (2005) Intergenic transcription through a Polycomb group response element counteracts silencing. Genes Dev 19:697–708
Sanchez-Elsner T, Gou D, Kremmer E, Sauer F (2006) Noncoding RNAs of trithorax response elements recruit Drosophila Ash1 to Ultrabithorax. Science 311:1118–1123
Sessa L, Breiling A, Lavorgna G, Silvestri L, Casari G, Orlando V (2007) Noncoding RNA synthesis and loss of Polycomb group repression accompanies the colinear activation of the human HOXA cluster. RNA 13:223–239
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32:232–246
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323
Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA (2007) A Chromatin landmark and transcription initiation at most promoters in human cells. Cell 130:77–88
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA 106:11667–11672
Kanhere A, Viiri K, Araújo CC, Rasaiyaah J, Bouwman RD, Whyte WA, Pereira F, Brookes E, Walker K, Bell GW, Pombo A, Fisher AG, Young RA, Jenner RG (2010) Short RNAs are transcribed from repressed Polycomb target genes and interact with Polycomb Repressive Complex-2. Mol Cell 38:675–688
Guenther MG, Young RA (2010) Repressive transcription. Science 329:150–151
Cremer T, Cremer C (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2:292–301
Lamond AI, Earnshaw WC (1998) Structure and function in the nucleus. Science 280:547–553
Lanctot C, Cheutin T, Cremer M, Cavalli G, Cremer T (2007) Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet 8:104–115
de Laat W, Grosveld F (2007) Inter-chromosomal gene regulation in the mammalian cell nucleus. Curr Opin Genet Dev 17:456–464
Chambeyron S, Bickmore WA (2004) Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev 18:1119–1130
Hu Q, Kwon YS, Nunez E, Cardamone MD, Hutt KR, Ohgi KA, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, Fu XD (2008) Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci USA 105:19199–19204
Essers J, van Cappellen WA, Theil AF, van Drunen E, Jaspers NG, Hoeijmakers JH, Wyman C, Vermeulen W, Kanaar R (2005) Dynamics of relative chromosome position during the cell cycle. Mol Biol Cell 16:769–775
Gerlich D, Beaudouin J, Kalbfuss B, Daigle N, Eils R, Ellenberg J (2003) Global chromosome positions are transmitted through mitosis in mammalian cells. Cell 112:751–764
Abney JR, Cutler B, Fillbach ML, Axelrod D, Scalettar BA (1997) Chromatin dynamics in interphase nuclei and its implications for nuclear structure. J Cell Biol 137:1459–1468
Chubb JR, Boyle S, Perry P, Bickmore WA (2002) Chromatin motion is constrained by association with nuclear compartments in human cells. Curr Biol 12:439–445
Walter J, Schermelleh L, Cremer M, Tashiro S, Cremer T (2003) Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J Cell Biol 160:685–697
Csink AK, Bounoutas A, Griffith ML, Sabl JF, Sage BT (2002) Differential gene silencing by trans-heterochromatin in Drosophila melanogaster. Genetics 160:257–269
Schubeler D, Francastel C, Cimbora DM, Reik A, Martin DI, Groudine M (2000) Nuclear localization and histone acetylation: a pathway for chromatin opening and transcriptional activation of the human beta-globin locus. Genes Dev 14:940–950
Harmon B, Sedat J (2005) Cell-by-cell dissection of gene expression and chromosomal interactions reveals consequences of nuclear reorganization. PLoS Biol 3:e67. doi:10.1371/journal.pbio.0030067
Csink AK, Henikoff S (1998) Large-scale chromosomal movements during interphase progression in Drosophila. J Cell Biol 143:13–22
Cavalli G (2007) Chromosome kissing. Curr Opin Genet Dev 17:443–450
Saurin AJ, Shiels C, Williamson J, Satijn DP, Otte AP, Sheer D, Freemont PS (1998) The human Polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J Cell Biol 142:887–898
Buchenau P, Hodgson J, Strutt H, Arndt-Jovin DJ (1998) The distribution of Polycomb-group proteins during cell division and development in Drosophila embryos: impact on models for silencing. J Cell Biol 141:469–481
Bantignies F, Grimaud C, Lavrov S, Gabut M, Cavalli G (2003) Inheritance of Polycomb-dependent chromosomal interactions in Drosophila. Genes Dev 17:2406–2420
Grimaud C, Bantignies F, Pal-Bhadra M, Ghana P, Bhadra U, Cavalli G (2006) RNAi components are required for nuclear clustering of Polycomb group response elements. Cell 124:957–971
Lei EP, Corces VG (2006) A long-distance relationship between RNAi and Polycomb. Cell 124:886–888
Gilbert MK, Tan YY, Hart CM (2006) The Drosophila boundary element-associated factors BEAF-32A and BEAF-32B affect chromatin structure. Genetics 173:1365–1375
Phillips JE, Corces VG (2009) CTCF: master weaver of the genome. Cell 137:1194–1211
Bushey AM, Dorman ER, Corces VG (2008) Chromatin insulators: regulatory mechanisms and epigenetic inheritance. Mol Cell 32:1–9
Bartkuhn M, Renkawitz R (2008) Long range chromatin interactions involved in gene regulation. Biochim Biophys Acta 1783:2161–2166
Ideraabdullah FY, Vigneau S, Bartolomei MS (2008) Genomic imprinting mechanisms in mammals. Mutat Res 647:77–85
Ling JQ, Li T, Hu JF, Vu TH, Chen HL, Qiu XW, Cherry AM, Hoffman AR (2006) CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science 312:269–272
Geyer PK, Corces VG (1992) DNA position-specific repression of transcription by a Drosophila zinc finger protein. Genes Dev 6:1865–1873
Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B (2007) Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128:1231–1245
Burke LJ, Zhang R, Bartkuhn M, Tiwari VK, Tavoosidana G, Kurukuti S, Weth C, Leers J, Galjart N, Ohlsson R, Renkawitz R (2005) CTCF binding and higher order chromatin structure of the H19 locus are maintained in mitotic chromatin. EMBO J 24:3291–3300
Mohn F, Schubeler D (2009) Genetics and epigenetics: stability and plasticity during cellular differentiation. Trends Genet 25:129–136
Fuks F, Hurd PJ, Deplus R, Kouzarides T (2003) The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res 31:2305–2312
Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH (2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13:1192–1200
Fuks F (2005) DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev 15:490–495
Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE (2008) Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4:e1000242. doi:10.1371/journal.pgen.1000242
Cedar H, Bergman Y (2009) Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10:295–304
Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schubeler D (2008) Lineage-specific Polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30:755–766
Negishi M, Saraya A, Miyagi S, Nagao K, Inagaki Y, Nishikawa M, Tajima S, Koseki H, Tsuda H, Takasaki Y, Nakauchi H, Iwama A (2007) Bmi1 cooperates with Dnmt1-associated protein 1 in gene silencing. Biochem Biophys Res Commun 353:992–998
Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439:871–874
Acknowledgments
We would like to thank Dr. F.A.C. Wiegant for critical reading of the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Blomen, V.A., Boonstra, J. Stable transmission of reversible modifications: maintenance of epigenetic information through the cell cycle. Cell. Mol. Life Sci. 68, 27–44 (2011). https://doi.org/10.1007/s00018-010-0505-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0505-5