
Abstract
The objective of this study was to map the quantitative trait loci (QTLs) for chip color after harvest (AH), cold storage (CS) and after reconditioning (RC) in diploid potato and compare them with QTLs for starch-corrected chip color. Chip color traits AH, CS, and RC significantly correlated with tuber starch content (TSC). To limit the effect of starch content, the chip color was corrected for TSC. The QTLs for chip color (AH, CS, and RC) and the starch-corrected chip color determined with the starch content after harvest (SCAH), after cold storage (SCCS) and after reconditioning (SCRC) were compared to assess the extent of the effect of starch and the location of genetic factors underlying this effect on chip color. We detected QTLs for the AH, CS, RC and starch-corrected traits on ten potato chromosomes, confirming the polygenic nature of the traits. The QTLs with the strongest effects were detected on chromosomes I (AH, 0 cM, 11.5% of variance explained), IV (CS, 43.9 cM, 12.7%) and I (RC, 49.7 cM, 14.1%). When starch correction was applied, the QTLs with the strongest effects were revealed on chromosomes VIII (SCAH, 39.3 cM, 10.8% of variance explained), XI (SCCS, 79.5 cM, 10.9%) and IV (SCRC, 43.9 cM, 10.8%). Applying the starch correction changed the landscape of QTLs for chip color, as some QTLs became statistically insignificant, shifted or were refined, and new QTLs were detected for SCAH. The QTLs on chromosomes I and IV were significant for all traits with and without starch correction.
Similar content being viewed by others

Introduction
Starch is the major storage carbohydrate in plants. It is also a key molecule in mediating plant responses to abiotic stresses (Thalmann and Santelia 2017). Environmental stress can induce changes in the source–sink relationship (Dong and Beckles 2019). Starch plays a double role in carbon allocation: it can act as a sugar source when carbon is needed or as a sugar sink when sugars are in excess (MacNeill et al. 2017). The released sugars can function as osmoprotectants to mitigate the negative effect of the stress (Thalmann and Santelia 2017). In potato, tuber starch content (TSC) varies between 10 and 25% of fresh weight depending on the intended use of different potato cultivars (Ellis et al. 1998; Verma et al. 1975). During tuber cold storage (CS), at approximately 4 °C, starch is reversibly broken down into the reducing sugars glucose and fructose. This phenomenon is referred to as cold-induced sweetening (Isherwood 1973). TSC and reducing sugar content determine the suitability of potato tubers for the market and processing industry. High reducing sugar content is not desirable for thermal treatment because it causes a dark coloration in final potato products, such as fries and chips (Kumar et al. 2004). Pearson’s correlation coefficient between chip color and reducing sugar content r = − 0.94 (Fuller and Huges 1984). The average content of reducing sugars in chipping cultivars should not exceed 0.33% (Duplessis et al. 1996). However, during cold-induced sweetening, sugar content can reach 2% fresh weight (Isherwood 1973). Therefore, chipping quality is typically evaluated as the color of a potato chip following CS. Chipping quality also depends on starch properties (Werij et al. 2012). To improve potato quality after storage, tubers can be subjected to controlled warming, known as a reconditioning (RC) process, during which higher temperatures (approximately 20 °C) initiate the conversion of reducing sugars into the starch (Isherwood 1973).
During maturation, potato tubers accumulate nearly 100% of their carbohydrate as starch and only small amount of starch is broken down to soluble sugars (Schreiber et al. 2014). Starch biosynthesis and degradation in potato tubers are pivotal pathways affecting tuber quality. These two pathways are spatially and temporarily separated from each other. The chipping quality of potato tubers is an outcome of biochemical processes occurring at each stage of tuber formation, from starch biosynthesis, granule formation and degradation pathways to the sucrose release and reducing sugars formation in vacuole. There is evidence of significant correlations between chip color and starch content after harvest (AH) (Pearson’s correlation coefficient: r = 0.57) and after CS (r = 0.68) (Li et al. 2013) and between starch properties such as starch gelatinization temperature of fresh tissues, described as thermal transition peak temperature (r = 0.54, O’Donoghue et al. 1996). The correlation between starch content and chipping quality is a result of the activity of various enzymes that can modify starch structure (crystalline or amorphous) due to the extent to which starch is broken down into reducing sugars (Barichello et al. 1990, 1991). The crystalline regions of starch granules determine their stability and make them less sensitive to enzymatic degradation. High amylose content makes the starch granules of potato tubers less susceptible to cold-induced sweetening (Jansky and Fajardo 2014).
Mapping of quantitative trait loci (QTLs) has been performed in a number of experimental populations of diploid potatoes (Douches and Freyre 1994; Menendez et al. 2002, Werij et al. 2012; Sołtys-Kalina et al. 2015; Śliwka et al. 2016; Braun et al. 2017; Xiao et al. 2018) in addition to a few association studies (Li et al. 2008, 2010; Schreiber et al. 2014). The genes involved in starch synthesis and degradation, the Calvin cycle and photorespiration, the glycolysis, oxidative pentose phosphate pathway, and the Krebs cycle are distributed across all twelve potato chromosomes (Chen et al. 2001). Notably, certain gene alleles play dual roles in starch synthesis and hydrolysis pathways. For example, the α-glucan phosphorylase (StPho) is an enzyme directly involved in the formation and size of starch granules and affects gelling properties (Werij et al. 2012), but the allele of the gene encoding StPho, StPho1a, is also considered a candidate gene for chipping quality (Werij et al. 2012; Van Harsselaar et al. 2017). There is also evidence of the involvement in starch and reducing sugar formation, of genes such as transcription factors (Van Harsselaar et al. 2017) and genes responsible for plant hormone signaling (auxin-regulated protein, AuxRP) or heat-shock proteins (Hsp90) (Sołtys-Kalina et al. 2015).
In our recent paper, we used 12-3 population, an F1 progeny from a cross of high-starch maternal and low-starch paternal components where starch content in potato tubers strongly segregated with normal distribution. We described 12 QTLs for starch content located on seven potato chromosomes in the diploid population 12-3 (Śliwka et al. 2016). The most important QTL spanned a wide region of chromosome I. In the present study, we used the same population 12-3 to explore the genetic basis of starch–sugar interconversion in non-stressed and cold-stressed potato tubers. Reciprocal relationships between starch biosynthesis and degradation pathways at genetic and biochemical levels have significant influence on starch and reducing sugar content. Many of the genes significantly associated with both traits may be located on the map near each other and their allele effects overlap within the same locus (Schönhals et al. 2017). To differentiate between QTL affecting chip color only, from QTL affecting starch content and in consequence chip color also, we used an approach that was applied earlier for other traits: maturity-corrected resistance to late blight (Bormann et al. 2004) and for tuber starch-corrected blackspot bruise susceptibility (Hara-Skrzypiec et al. 2018). To diminish the effect of starch-related genes as well as the influence of starch content and chip color correlation, we introduced starch correction on chip color. The goal of the investigation was to compare the location of the genetic factors underlying chip color and starch-corrected chip color.
QTL analyses for chip color AH, after CS and after RC, and starch-corrected analysis of chip color were performed. For the first time, the QTLs for chip color observed in three conditions (AH, CS, and RC) and starch-corrected chip color (SCAH, SCCS, and SCRC) were compared to assess the extent of the effect of starch on chip color.
Materials and methods
Plant material and phenotype analysis
The plant material consisted of the potato diploid population 12-3 (F1 progeny, N = 183) from a cross of the seed parent DG 00-683 and pollen parent DG 08-28/13 (Śliwka et al. 2016). Population 12-3 was grown in 2012–2014 in pots under plant tents. In 2012, F1 progeny were propagated from true seeds, and in 2013–2014 (first and second tuber generation), each individual was planted in three independent replications. The plants were watered regularly, fertilized, and protected against insects and pathogens.
For chip color analysis, six undamaged tubers from each diploid clone were collected. After harvesting tubers each year, four slices from two tubers were taken and fried (first set AH), and then four tubers were transferred to controlled storage lockers set at 4 °C. The storage temperature was held constant for 70 days, and a second set was fried after CS. Subsequently, two tubers were removed for RC at 20 °C for an additional 40 days, and a third frying set was evaluated. Chip color was visually assessed using a nine-point color chart (score 1 = black chip color).
TSC was estimated using the underwater weight method according to Lunden (1956), as described by Śliwka et al. (2016).
Statistical analysis
The phenotypic data were tested for fitness to normal distribution with the Shapiro–Wilk test. The reproducibility of the analyzed traits between years was estimated by calculating linear Pearson’s correlation coefficients. The coefficients of determination (R2) for chip color and TSC were estimated using analysis of variance (two-way ANOVA). The broad-sense heritability (H2) was estimated as in Guo et al. (2015), according to the following formula:
Using previously calculated variation components by fitting a linear mixed-effects model to the phenotyping data via a restricted maximum likelihood (REML) approach, the correction of the chip color results by starch content after harvest (SCAH), starch content after cold storage (SCCS) and starch content after reconditioning (SCRC) was carried out in a proportional method based on regression analysis. After determination of the regression function describing the relationship between the starch content and chip color, corrective values were calculated based on the determined regression coefficients. The corrective values calculated in this way (individual for each case/observation) were used to adjust the observed chip color values.
All statistical analyses were performed using STATISTICA for Windows (Stat Soft, Inc. and StatSoft Polska Ltd., Kraków, Polska). Because tuber starch and chip color evaluation was performed in 2012 in only one replication, the analysis of variance and broad-sense heritability were calculated on data sets from 2013 to 2014.
QTL analysis
A previously published genetic map of the diploid potato population 12-3 was used (Śliwka et al. 2016). The map consisted of 1597 markers, including 1584 diversity array technology (DArT) markers, 11 cleaved amplified polymorphic sequence (CAPS) markers, one sequence characterized amplified region (SCAR) marker, and one phenotypic (purple flower color) marker. QTL analysis was performed using interval mapping with the MapQTL 6 software (Van Ooijen 2009). QTLs were detected using a logarithm of odds (LOD) ≥ 3.0 as the threshold of significance.
Results
Phenotyping
The mean values of the 3-year evaluation of chip color and starch-corrected chip color of mapping population 12-3 are presented in Table 1. The data distribution of the traits was evaluated for fitness to a normal curve by the Shapiro–Wilk test. A normal distribution of chip color was observed AH, and in the case of other traits, distributions approached normal (Table 1). The distribution of TSC was normal (Śliwka et al. 2016). The widest range of variation was identified for chip color after RC (2.4–9.0), and the range was narrowest for chip color AH (3.3–8.2). Analysis of variance in the mapping population demonstrated a significant effect of genotype and year on chip color and starch-corrected chip color (Table 2). Genotype explained 41.4 (SCAH) to 56.8% of the variance (RC) observed in particular datasets. The variance explained by years of experiments ranged from 0.2% (RC) to 5.9% (SCAH). Interaction genotype × year was significant only for RC and explained 10.8% of the variance.
The genotype and interaction genotype × year significantly influenced the TSC phenotype and explained 58.2% and 12.7% of the variance, respectively (Table 2).
Chip color and TSC were significantly correlated for all tested combinations, and Pearson’s correlation coefficients ranged from 0.596 (TSC × AH) to 0.630 (TSC × RC) (Table 3). Pearson’s correlation coefficients between the results of assessments from particular years of all tested traits are presented in Supplementary Table 1.
The H2 value for chip color ranged from 72.4% (SCRC) to 81.9% (CS and RC) and was 66.7% for TSC (Table 2).
QTL analysis
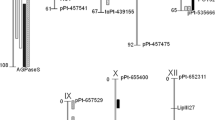
The QTLs for AH, CS and RC were localized on nine potato chromosomes: I, II, III, IV, VI, VIII, IX, X and XI (Fig. 1, Table 4).

Overview of QTL for chip color detected in the diploid potato mapping population 12-3. Interval QTL mapping was performed using MapQTL® 6 (Van Ooijen 2009) and 3-year datasets (mean chip colour 2012–2014) obtained in different conditions and starch-corrected as listed in the figure. Black bars symbolize chromosomes, only the chromosomes harboring QTL are shown. Color-coded bars represent QTL on the right while the locations of candidate gene markers are shown on the left of chromosomes (ADP/ATPtr ADP, ATP carrier protein; AGPaseS-a1334 ADP-glucose pyrophosphorylase S; AOX1a, AOXb alternative oxidases; β-amyl β-amylase; NL-AAM α-amylase; PPCK1a phosphoenolpyruvate carboxylase kinase 1a; pk1 pyruvate kinase; SnRK1 sucrose non-fermenting-1-related protein kinase; Sus4 sucrose synthase 4). For details and single-year QTL analyses, see Supplementary Table 2
For AH, the most important QTLs were found on chromosomes I and III, explaining 11.5% (LOD = 4.79, 0.0 cM) and 11.4% (LOD = 4.72, 98.1 cM) of the variance, respectively (Table 4). The QTLs for AH were also detected on chromosomes II, IV and VIII.
The most prominent QTLs for CS were detected on chromosome IV, and they were significant in mean trait value and datasets from particular testing years (2012–2014) (Supplementary Table 2). Two QTLs for CS between 10.8 and 24.4 cM (peak at 17.6 cM, LOD = 4.22) and between 35.5 and 61.1 cM (peak at 43.9 cM, LOD = 5.32) on chromosome IV (Table 4) explained 10.2% and 12.7% of the variance, respectively. For CS, the QTLs were detected also on chromosomes I, VI, VIII, IX, X, and XI (Table 4, Fig. 1).
The most significant QTL for RC, which had a peak at 49.7 cM and explained 14.1% (LOD = 5.96) of the variance, was detected on chromosome I (Table 4). Partially overlapping QTL regions on chromosome IV between 13.7 and 22.6 cM (significant in mean trait value) and 33.5 and 60.2 cM (significant in mean trait value and datasets from particular years 2012–2014) were also detected for RC (Supplementary Table 2). Notably, analogous QTL peaks for RC and CS were detected, and the peak for RC at 17.6 cM explained 9.3% (LOD = 3.83) of the variance, while the peak at 43.9 cM explained 12.4% (LOD = 5.19) of the variance (Table 4). The QTLs for RC were also detected on chromosomes I, II, VIII, and X (Table 4, Fig. 1).
The QTLs for SCAH, SCCS and SCRC were detected on nine chromosomes: I, III, IV, VI, VIII, IX, X, XI and XII (Fig. 1, Table 4).
The most significant QTLs for SCAH were localized on chromosomes III and VIII explaining 10.0% (LOD = 4.11) and 10.8% (LOD = 4.48) of the variance, respectively (Table 4). The QTLs for SCAH were also detected on chromosomes IX and XII. For SCCS, the most prominent QTLs were localized on chromosomes VI and XI at 48.2 cM and 79.5 cM, explaining 10.5% (LOD = 4.33) and 10.9% (LOD = XI) of the variance, respectively. The QTLs for SCCS were also detected on chromosomes VIII, IX, and X (Table 4, Supplementary Table 2).
Location of QTLs for starch-corrected chip color on chromosomes I and IV were similar but shifted in comparison to those for AH, CS and RC (Fig. 1, Table 4). The position of the QTL peak at 56.6 cM for SCAH on chromosome IV, explaining 9.3% of the variance (LOD = 3.80), overlapped with the peak for AH. For SCAH, the QTL on chromosome I was similar to this AH at 0.0–14.3 cM; however, QTL peak was shifted to 12.3 cM and explained 9.8% of the variance (LOD = 4.05).
Two QTL peaks for SCCS were localized at 17.6 and 43.9 cM on chromosome IV and explained 9.8% (LOD = 4.03) and 8.9% (LOD = 3.64) of the variance, respectively (Table 4).
For this trait SCCS, the QTL peak on chromosome I was localized at 11.3 cM and explained 7.9% (LOD = 3.23) of the variance.
For SCRC, the most prominent QTL (34.5–60.2 cM) was localized on chromosome IV, the QTL peak at 43.9 cM explained 10.8% (LOD = 4.48) of the variance and overlapped with the peak for RC. The second QTL (12.7–22.6 cM) on chromosome IV was similar to this localized for RC; however, QTL peak was shifted and detected at 16.6 cM, explaining 9.7% (LOD = 3.98) of the variance. On chromosome I, the most significant QTL for SCRC was detected at 13.3 cM, explaining 7.7% (LOD = 3.14) of the variance. The QTLs for SCRC were also detected on chromosome IX.
Of 11 CAPS markers, six were associated with chip color: α-amylase (NL-AAM), phosphoenolpyruvate carboxylase kinase 1a (PPCK1a), pyruvate kinase (pk1), alternative oxidase (AOX1a, AOXb) and β-amylase (β-amyl) (Table 5, Supplementary Table 3). Most significant linkage between chip color and the candidate gene marker was found for CS and AOXb; this marker explained 11.4% of variance (LOD = 7.77). After starch correction, only one marker PPCK1a was significant, for SCAH and SCRC explaining 7.9% of variance (LOD = 3.20) and 7.6% of variance (LOD = 3.08), respectively (Table 5, Supplementary Table 3).
Discussion
TSC and reducing sugar content in potato tubers are quantitative traits. Genes contributing to the phenotypic effects are located on all twelve potato chromosomes (Menendez et al. 2002). In the mapping population 12-3, the QTLs for TSC mapped to similar positions as QTLs for reducing sugar content (this study and Śliwka et al. 2016). The effects on both traits can be controlled either by the same genes with pleiotropic effects or by genes located within physical proximity of each other (genetically linked) (Gebhardt 2005). To determine the QTLs directly associated with chip color and diminish the influence of starch, starch correction was applied to chip color data, calculated from the regression of the chip color traits on TSC.
We recognized that the QTLs on chromosomes I and IV were significant and reproducible for all chip color traits (AH, CS, and RC). Chromosome I was previously described as important for AH and RC in another chipping population 11–40; the main QTLs spanned 44.7–50.7 cM and 66.0–72.9 cM, respectively, and corresponded with the regions of the QTLs for AH and RC in population 12-3 (Sołtys-Kalina et al. 2015). The significance of the QTLs on chromosome IV for reducing sugar content and tuber fry color after CS was previously detected in diploid and tetraploid potatoes (Menendez et al. 2002; Massa et al. 2018). Chromosomes I and IV harbor genes encoding enzymes influencing starch content and involved in direct carbohydrate metabolism, e.g., β-galactosidase and β-amylase (chromosome I) and maltose content as phosphoglucoisomerase (chromosome IV); carbohydrate transport, e.g., triose-phosphate/phosphate translocator (chromosome I) or genes regulating cell wall biosynthesis, such as glycosyltransferase (chromosome I); transcription regulation, e.g., transcriptional factor B3 (Schönhals et al. 2017; Van Harsselaar et al. 2017). Despite most QTLs on genetic map 12-3 sharing similar locations for at least two chip color traits, we identified chromosomes with QTL for only one trait. We detected QTLs on chromosome III significant for AH and SCAH and on chromosome VI significant only for CS and SCCS (Fig. 1). This finding is in agreement with the results of other studies, where measuring the expression of the genes involved in starch and sugar metabolism in tubers provided markers for AH but not for CS (Neilson et al. 2017). Other studies have suggested that tendencies for the accumulation of reducing sugars after CS and RC are genetically independent, and reliable mapping of the QTLs allows dissection of the biological function of each QTL (Xiao et al. 2018).
We demonstrated that all analyzed chip color (AH, CS, and RC) traits and TSC were genotype dependent and correlated with each other with significant Pearson’s correlation coefficients (Tables 2, 3). Such interconnections within the examined traits indirectly inform us about the genetic linkage or multidirectional functions of genes involved in carbohydrate metabolism, which determine chip color. However, genotype dependency indicates high heritability of a trait. Among clones derived from the diploid potato hybrid S. phureja–S. stenotomum, broad-sense heritability for chip color after storage was estimated at 68% (Haynes 2008). According to Haynes (2008), such heritability makes clones a valuable source of an acceptable chip color in crosses 4 × –2 × . In our study, broad-sense heritability coefficients for CS and SCCS were 81.9% and 72.9%, respectively, and were even higher than the heritability of TSC (66.7%).
Higher starch content can cause an increased accumulation of reducing sugars during CS. This phenomenon is due mainly to the activation of starch-degrading enzymes (Zhang et al. 2017). This outcome is expected because the starch biosynthesis pathway and the starch degradation pathway remain in reciprocal relationships. Additionally, starch metabolism genes are organized into gene families, and their similar genomic location does not necessarily correspond to identical functions in starch metabolism (Van Harsselaar et al. 2017). It was proven that isoenzymes encoded by the genes at the same genomic location play distinct roles in the formation of transitory and storage starch in potatoes (Van Harsselaar et al. 2017). To minimize starch effect on chip color, we applied starch correction to chip color data. After correcting chip color values, the QTLs common for all traits were still detectable. Most chromosome segments harboring QTLs for chip color were also significant for starch-corrected traits. The QTLs for all starch-corrected traits were detected on chromosomes I and IV (as well as for all chip color traits). Nevertheless, the QTLs on nine chromosomes differed in length of QTL region, particularly at chromosome I (all starch-corrected traits), chromosome III (SCAH), chromosome IV (all starch-corrected traits), chromosome VI (SCCS), chromosome VIII (SCAH and SCCS), chromosome IX (SCAH and SCRC) chromosome X (SCCS) and chromosome XI (SCCS). Starch correction revealed new QTLs for SCAH on chromosome III at 10.3–11.5 cM, on chromosome IX at 43.6–60.4 cM and on chromosome XII at 74.8–77.4 cM (Fig. 1, Table 4).
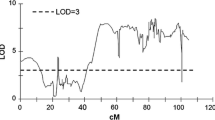
In previous studies, QTLs for starch and reducing sugar contents were mapped to the same region of potato chromosomes (Gebhardt 2005). This finding is in agreement with our results (see the comparison of the QTLs for all chip color and TSC traits on chromosome I) (Fig. 2). The effect of starch content on chip color is also evident after comparing the significance of the candidate gene markers developed in population 12-3. Three markers, β-amyl, AOX1a and AOXb, that localized on chromosome VIII are significant both for TSC (Śliwka et al. 2016) and chip color after CS and RC (this study). After starch correction, the linkage between trait loci and these markers became irrelevant. We observed only one locus, PPCK1a (chromosome IV), that is associated with chip color after starch correction (SCAH and SCRC) (Table 5, Supplementary Table 3). Interestingly, the AGPaseS-a1334 marker of the main rotary enzyme involved in starch turnover that mapped in population 12-3 to chromosome I was associated with TSC (Śliwka et al. 2016) but none of the chip color traits (Supplementary Table 3). In contrast, in other study, the pleiotropic effects of the alleles of the same gene had an antagonistic or a synergistic influence on the potato tuber traits (Schönhals et al. 2017). The AGPaseS alleles AGPsS-9a and AGPsS-10a were positively and negatively correlated with both total starch content and chip color AH and after CS, respectively (Li et al. 2013).

Overview of QTL for chip color and TSC (Śliwka et al. 2016) detected on chromosome I in the diploid potato mapping population 12-3
QTL mapping allows for the identification of genomic regions which are highly statistically correlated with the quantitative trait, but does not identify their molecular basis (Chen et al. 2001). Functional complementation analysis in conjunction with sequence and expression studies of candidate genes can provide information on their function as quantitative trait loci.
Conclusions
Results from this study confirm polygenic character of chip color traits, genotype dependency and high broad-sense heritability value of these traits. Starch correction of chip color data allows separation of genetic factors responsible for chip color and starch content. Our analysis confirms influence of starch content on chip color that can modify the landscape of obtained QTLs for chip color.
References
Barichello V, Yada RY, Coffin RH, Stanley DW (1990) Low temperature sweetening in susceptible and resistant potatoes: starch structure and composition. J Food Sci 55:1054–1059. https://doi.org/10.1111/j.1365-2621.1990.tb01596.x
Barichello V, Yada RY, Coffin RH (1991) Starch properties of various potatoes (Solanum tuberosum L.) cultivars susceptible and resistant to low temperature sweetening. J Sci Food Agric 56:385–397. https://doi.org/10.1002/jsfa.2740560314
Bormann CA, Rickert AM, Castillo Ruiz RA, Paal J, Lübeck J, Strahwald J, Buhr K, Gebhardt C (2004) Tagging quantitative trait loci for maturity-corrected late blight resistance in tetraploid potato with PCR-based candidate gene markers. Mol Plant-Microbe Interact 17:1126–1138. https://doi.org/10.1094/MPMI.2004.17.10.1126
Braun SR, Endelman JB, Haynes KG, Jansky SH (2017) Quantitative trait loci for resistance to common scab and cold-induced sweetening in diploid potato. Plant Genome 10:3. https://doi.org/10.3835/plantgenome2016.10.0110
Chen X, Salamini F, Gebhardt C (2001) A potato molecular-function map for carbohydrate metabolism and transport. Theor Appl Genet 102:284–295. https://doi.org/10.1007/s001220051645
Dong S, Beckles DM (2019) Dynamic changes in the starch-sugar interconversion within plant source and sink tissues promote a better abiotic stress response. J Plant Physiol 234–235:80–93. https://doi.org/10.1016/j.jplph.2019.01.007
Douches DS, Freyre R (1994) Identification of genetic factors influencing chip color in diploid potato (Solanum spp.). Am Pot J 71:581–590. https://doi.org/10.1007/BF02851523
Duplessis PM, Marangoni AG, Yada RY (1996) A mechanism for low temperature induced sugar accumulation in stored potato tubers: the potential role of the alternative pathway and invertase. Am Pot J 73:483–494. https://doi.org/10.1007/BF02849671
Ellis RP, Cochrane MP, Dale MFB, Duffus CM, Lynn A, Morrison IM, Prentice RDM, Swanston JS, Tiller SA (1998) Starch production and industrial use. J Sci Food Agric 77:289–311. https://doi.org/10.1002/(SICI)1097-0010(199807)77:3%3C289:AID-JSFA38%3E3.0.CO;2-D
Fuller TJ, Hughes JC (1984) Factors influencing the relationships between reducing sugars and fry colour of potato tubers of cv. Record. Int J Food Sci Tech 19:455–467. https://doi.org/10.1111/j.1365-2621.1984.tb00369.x
Gebhardt C (2005) Potato Genetics: molecular maps and more. In: Nagata T, Lörz H, Widholm JM (eds) Molecular marker systems in plant breeding and crop improvement, vol 55. Springer, Berlin, pp 221–224 (Biotechnology in agriculture and forestry. Lörz, G. Wenzel)
Guo Z, Chen D, Schnurbusch T (2015) Variance components, heritability and correlation analysis of anther and ovary size during the floral development of bread wheat. J Exp Bot 66:3099–3111. https://doi.org/10.1093/jxb/erv117
Hara-Skrzypiec A, Śliwka J, Jakuczun H, Zimnoch-Guzowska E (2018) Quantitative trait loci for tuber blackspot bruise and enzymatic discoloration susceptibility in diploid potato. Mol Genet Genomics 293:331–342. https://doi.org/10.1007/s00438-017-1387-0
Haynes KG (2008) Heritability of chip color and specific gravity in a long-day adapted Solanum phureja–S. stenotomum population. Am J Pot Res 85:361–366. https://doi.org/10.1007/s12230-008-9036-0
Isherwood FA (1973) Starch-sugar interconversion in Solanum tuberosum. Phytochemistry 12:2579–2591. https://doi.org/10.1016/0031-9422(73)85060-5
Jansky SH, Fajardo DS (2014) Tuber starch amylose content is associated with cold-induced sweetening in potato. Food Sci Nutr 2:628–633. https://doi.org/10.1002/fsn3.137
Krusiewicz D, Jakuczun H, Wasilewicz-Flis I, Strzelczyk-Żyta D, Marczewski W (2011) Molecular mapping of the AOX1a and β-AmyI genes in potato. Plant Breed 130:500–502. https://doi.org/10.1111/j.1439-0523.2011.01846.x
Kumar D, Singh BP, Kumar P (2004) An overview of the factors affecting sugar content of potatoes. Ann Appl Biol 145:247–256. https://doi.org/10.1111/j.1744-7348.2004.tb00380.x
Li L, Paulo MJ, Strahwald J, Lübeck J, Hofferbert HR, Tacke E, Junghans H, Wunder J, Draffehn A, van Eeuwijk F, Gebhardt C (2008) Natural DNA variation at candidate loci is associated with potato chip color, tuber starch content, yield and starch yield. Theor Appl Genet 116:1167–1181. https://doi.org/10.1007/s00122-008-0746-y
Li L, Paulo MJ, van Eeuwijk F, Gebhardt C (2010) Statistical epistasis between candidate gene alleles for complex tuber traits in an association mapping population of tetraploid potato. Theor Appl Genet 121:1303–1310. https://doi.org/10.1007/s00122-010-1389-3
Li L, Eckhard T, Hofferbert H-R, Lübeck J, Strahwald J, Draffehn AM, Walkemeier B, Gebhardt C (2013) Validation of candidate gene markers for marker-assisted selection of potato cultivars with improved tuber quality. Theor Appl Genet 126:1039–1052. https://doi.org/10.1007/2Fs00122-012-2035-z
Lunden PA (1956) Underldokerd over forholder mellom popetens spesifikka vekt og deres torvstoff og Stivelsesinhold Forhl. Forsok Landbruket 7:81–107
MacNeill GJ, Mehrpouyan S, Minow MAA, Patterson JA, Tetlow IJ, Emes MJ (2017) Starch as a source, starch as a sink: the bifunctional role of starch in carbon allocation. J Exp Bot 68:4433–4453. https://doi.org/10.1093/jxb/erx291
Massa MN, Manrique-Carpintero NC, Coombs J, Haynes KG, Bethke BC, Brandt TL, Gupta SK, Yencho GC, Novy RG, Douches DS (2018) Linkage analysis and QTL mapping in a tetraploid russet mapping population of potato. BMC Genet 19:87. https://doi.org/10.1186/s12863-018-0672-1
Menendez CM, Ritter E, Schäfer-Pregl R, Walkemeier B, Kalde A, Salamini F, Gebhardt C (2002) Cold-sweetening in diploid potato: mapping QTL and candidate genes. Genetics 162:1423–1434
Neilson J, Lagüe M, Thomson S, Aurousseau F, Murphy AM, Bizimungu B, Deveaux V, Bègue Y, Jacobs JME, Tai HH (2017) Gene expression profiles predictive of cold-induced sweetening in potato. Funct Integr Genomics 17:459–476. https://doi.org/10.1007/s10142-017-0549-9
O’Donoghue EP, Marangoni AG, Yada RY (1996) The relationship of crisps color with structural parameters of starch. Am Potato J 73:545–558
Schönhals EM, Ding J, Ritter E, Paulo MJ, Cara N, Tacke E, Hofferbert H-R, Lübeck J, Strahwald J, Gebhardt C (2017) Physical mapping of QTL for tuber yield, starch content and starch yield in tetraploid potato (Solanum tuberosum L.) by means of genome wide genotyping by sequencing and the 8.3 K SolCAP SNP array. BMC Genomics 18:642. https://doi.org/10.1186/s12864-017-3979-9
Schreiber L, Nader-Nieto AC, Schönhals EM, Walkemeier B, Gebhardt C (2014) SNPs in genes functional in starch-sugar interconversion associate with natural variation of tuber starch and sugar content of potato (Solanum tuberosum L). G3 Genes. Genomes Genet 4:1797–1811. https://doi.org/10.1534/g3.114.012377
Śliwka J, Sołtys-Kalina D, Szajko K, Wasilewicz-Flis I, Strzelczyk-Żyta D, Zimnoch-Guzowska E, Jakuczun H, Marczewski W (2016) Mapping of quantitative trait loci for tuber starch and leaf sucrose contents in diploid potato. Theor Appl Genet 129:131–140. https://doi.org/10.1007/s00122-015-2615-9
Sołtys-Kalina D, Szajko K, Sierocka I, Śliwka J, Strzelczyk-Żyta D, Wasilewicz-Flis I, Jakuczun H, Szweykowska-Kulinska Z, Marczewski W (2015) Novel candidate genes AuxRP and Hsp90 influence the chip color of potato tubers. Mol Breed 35:224. https://doi.org/10.1007/2Fs11032-015-0415-1
Thalmann M, Santelia D (2017) Starch as a determinant of plant fitness under abiotic stress. New Phytol 214:943–951. https://doi.org/10.1111/nph.14491
Van Harsselaar JK, Lorenz J, Senning M, Sonnewald U, Sonnewald S (2017) Genome-wide analysis of starch metabolism genes in potato (Solanum tuberosum L.). BMC Genomics 18:37. https://doi.org/10.1186/2Fs12864-016-3381-z
Van Ooijen JW (2009) JoinMap 6, software for the calculation of the genetic linkage maps in experimental populations. Kyazma company, Wageningen
Verma SC, Sharma TR, Joshi KC (1975) Relation between specific gravity, starch and nitrogen content of potato tubers. Pot Res 18:120–122. https://doi.org/10.1007/BF02361783
Werij JS, Furrer H, van Eck HJ, Visser RGF, Bachem GWB (2012) A limited set of starch related genes explain several interrelated traits in potato. Euphytica 186:501–516. https://doi.org/10.1007/s10681-012-0651-y
Xiao G, Huang W, Cao H, Tu W, Wang H, Zheng X, Liu J, Song B, Xie C (2018) Genetic loci conferring reducing sugar accumulation and conversion of cold-stored potato tubers revealed by QTL analysis in a diploid population. Front Plant Sci 9:315. https://doi.org/10.3389/fpls.2018.00315
Zhang H, Hou J, Liu J, Zhang J, Song B, Xie C (2017) The roles of starch metabolic pathways in the cold-induced sweetening process in potatoes. Starch/Stärke 69:1600194. https://doi.org/10.1002/star.201600194
Acknowledgements
The research was supported by The National Science Centre in Poland (No. UMO-2015/19/B/NZ9/00776).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sołtys-Kalina, D., Szajko, K., Wasilewicz-Flis, I. et al. Quantitative trait loci for starch-corrected chip color after harvest, cold storage and after reconditioning mapped in diploid potato. Mol Genet Genomics 295, 209–219 (2020). https://doi.org/10.1007/s00438-019-01616-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01616-1