
Abstract
Complex characters of plants such as starch and sugar content of seeds, fruits, tubers and roots are controlled by multiple genetic and environmental factors. Understanding their molecular basis will facilitate diagnosis and combination of superior alleles in crop improvement programs (“precision breeding”). Association genetics based on candidate genes is one approach toward this goal. Tetraploid potato varieties and breeding clones related by descent were evaluated for 2 years for chip quality before and after cold storage, tuber starch content, yield and starch yield. Chip quality is inversely correlated with tuber sugar content. A total of 36 loci on 11 potato chromosomes were evaluated for natural DNA variation in 243 individuals. These loci included microsatellites and genes coding for enzymes that function in carbohydrate metabolism or transport (candidate loci). The markers were used to analyze population structure and were tested for association with the tuber quality traits. Highly significant and robust associations of markers with 1–4 traits were identified. Most frequent were associations with chip quality and tuber starch content. Alleles increasing tuber starch content improved chip quality and vice versa. With two exceptions, the most significant and robust associations (q < 0.01) were observed with DNA variants in genes encoding enzymes that function in starch and sugar metabolism or transport. Comparing linkage and linkage disequilibrium between loci provided evidence for the existence of large haplotype blocks in the breeding materials analyzed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most characters important for crop quality show quantitative phenotypic variation, due to the fact that they are controlled by natural DNA variation at multiple loci and by environmental factors. Knowing the molecular basis of the genetic components of this variation will facilitate the selection of improved cultivars with DNA-based markers, which are diagnostic for superior alleles of the underlying genes.
Genetic dissection of plant complex traits in quantitative trait loci (QTL) first became possible with the advent of DNA-based markers (Osborn et al. 1987). Since then, the first genes and their allelic variants underlying plant QTL have been identified by positional cloning (reviewed in Salvi and Tuberosa 2005). Positional QTL cloning requires the generation and analysis of large experimental mapping populations. This is a labor and time-intensive process, which is feasible in inbreeding species with short generation time such as Arabidopsis, rice or tomato, but is rather prohibitive in polyploid, non-inbred species such as potato or in species with long generation time such as trees. An alternative to positional QTL cloning is the candidate gene approach, which is based on the knowledge of a gene’s function in controlling a character of interest on the one hand, and genetic co-localization of a functional candidate gene with QTL for the character of interest on the other (Pflieger et al. 2001). DNA variation of genes fulfilling these criteria is examined in natural populations of individuals related by descent for associations with positive or negative character values (Li et al. 2005; González-Martínez et al. 2007). Finding such associations indicates that DNA variation either at the candidate locus itself or at a physically linked locus is causal for the phenotypic variation. Indirect associations due to physical linkage depend on the extent of the linkage disequilibrium (LD) present in the population under study. Functional complementation analysis using candidate gene alleles provides direct evidence that a candidate gene underlies a QTL (Salvi and Tuberosa 2005). Association mapping can produce diagnostic markers that are useful for plant breeding, irrespective of whether the association is direct or indirect.
Quantitative traits linked to carbohydrate contents can serve as model for the candidate gene approach to QTL identification in crops. Carbohydrates are the primary products of photosynthetic CO2 fixation and major constituents of wood and storage compounds. Starch and sugars deposited in seeds, roots, tubers, fruits or berries are the basis of human and animal nutrition. Carbohydrate metabolism has been intensively studied in plants at the physiological, biochemical and molecular level. The enzymes catalyzing the principal anabolic and katabolic reactions are ubiquitous in plants, and the coding genes have been cloned and characterized in several species (reviewed in Beck and Ziegler 1989; Frommer and Sonnewald 1995; Winter and Huber 2000; Salerno and Curatti 2003). Functional candidate genes are therefore available in abundance.
In the potato (Solanum tuberosum), complex traits linked to carbohydrate composition are the starch and sugar content of the tubers, which are together with tuber yield important determinants of tuber quality. With 10–25% of the fresh weight, starch is the major storage compound of the tuber. The sugars sucrose, glucose and fructose are minor tuber constituents that do not have a role as storage compounds. Glucose and fructose accumulate, however, in tubers in response to low temperature exposure, a phenomenon called “cold sweetening” (Burton 1969; Isherwood 1973). The tuber content of the reducing sugars fructose and glucose determines the color of deep fried products such as potato chips and French fries, which is the result of a Maillard reaction between amino acids and reducing sugars at high temperatures. High reducing sugar content results in dark chip color due to polyphenol formation (Habib and Brown 1957; Townsend and Hope 1960). As this is an unwanted character, potato variety development aims at selecting genotypes with low reducing sugar content during tuber storage, preferably at low temperature to prevent sprouting.
On the one hand, QTL for tuber starch and sugar content have been mapped to potato chromosomes in experimental, diploid populations (Schäfer-Pregl et al. 1998; Menendez et al. 2002). On the other hand, cloned and characterized genes of potato have also been mapped, which function in starch biosynthesis (e.g., ADP-glucose pyrophosphorylase, starch synthases, branching enzyme), starch degradation (e.g., starch phosphorylases, debranching enzyme, α-amylase), sucrose metabolism (e.g., sucrose phosphate synthase, sucrose synthases, invertases) or transport (e.g., sucrose transporters) (Chen et al. 2001; Menendez et al. 2002). Genes functional in starch and sugar metabolism, which co-localized with QTL for tuber starch and sugar content were selected as marker loci for association studies in populations of tetraploid potato individuals related by descent. A first association has been found between chip color and natural DNA variation at the invertase locus invGE/GF on potato chromosome IX (Li et al. 2005) that co-localized with a QTL for tuber sugar content (Menendez et al. 2002). Invertase converts sucrose in the reducing sugars glucose and fructose. Invertase encoding genes are therefore obvious functional candidates. The invGE/GF locus is a candidate for one of several QTL for tuber starch and sugar content, which have been localized on each of the 12 potato chromosomes (Schäfer-Pregl et al. 1998; Menendez et al. 2002; Gebhardt et al. 2005). To further dissect the molecular basis of these complex traits and to obtain diagnostic markers for breeding, we conducted an association study based on DNA variation at candidate loci in a population of tetraploid genotypes used for potato variety development.
Materials and methods
Plant material
Three populations of 100 tetraploid breeding clones each (SAR, BNA, NOR) and 36 varieties as standards were sampled from the breeding programs for chips, starch and table potatoes of the companies SAKA-RAGIS Pflanzenzucht (SAR clones), Böhm-Nordkartoffel Agrarproduktion (BNA clones) and NORIKA (NOR clones). The genotypes resulted from a number of crosses among different varieties and breeding clones and were selected to represent the variation for chip quality, tuber yield, starch content and starch yield present in advanced commercial breeding materials in Germany. The standard varieties were: Albatros, Apart, Artis, Aula, Christa, Diana, Eldena, Fasan, Goldika, Ilona, Karlena, Kolibri, Leyla, Likaria, Marabel, Marlen, Melina, Milva, Molli, Novara, Orlando, Panda, Pirol, Ponto, Satina, Saturna, Sempra, Sirius, Solara, Solist, Terra, Theresa, Tomensa, Valisa, Velox and Vitara. Historical pedigree information for most of the varieties is available at http://potatodbase.dpw.wau.nl/potatopedigree (van Berloo et al. 2007). According to this, the standard varieties included three pairs of half sibs (Karlena and Likaria, Marabel and Milva, Satina and Velox), one pair of full sibs (Artis and Sempra) and three varieties, which were parent of another variety in the set (Saturna is parent of Marlen, Solara of Vitara and Panda of Artis, Sempra and Sirius). The pedigree structure of varieties and breeding clones used for developing new varieties is similar. A total of 20 plants per plot were propagated in the field in two consecutive years under standard phytosanitary regimes. The sample populations SAR, BNA and NOR were grown and evaluated at the breeding stations of Saka-Ragis Pflanzenzucht GbR, BNA Zuchtgesellschaft mbH and NORIKA Ltd in Windeby, Ebstorf and Groß-Lüsewitz (Germany), respectively. The standards were grown and evaluated at all three locations (Table 1). Balanced phenotypic data from 2 years were obtained for 259 genotypes (36 standards, 100 SAR and BNA breeding clones each, 23 NOR breeding clones). Based on genetic similarity analysis among the 259 genotypes (Li et al. 2005), 16 highly similar pairs of individuals were identified. One individual of each similar pair was therefore removed from the sample populations. The remaining 243 genotypes (34 standards, 90 BNA, 96 SAR and 23 NOR) constituted the population ALL.
Phenotyping
Chip quality was assessed by visually scoring the chip color after deep frying of 1.2–2.0 mm tuber slices in oil at 160–180°C for 2–3 min (Putz 1989), using a 1–9 color scale (1 = very dark chip color, very bad chip quality; 9 = very light yellow chip color, very good chip quality), first time after harvest in autumn (CQA) and second time after 3–4 months storage at 4°C (CQS). After cold storage, the average chip quality decreased. To further differentiate chip quality at the lower end of the scale, chip scores for the SAR population were extended to include negative values. Tuber starch content (TSC, percent fresh weight) was determined by specific gravity (Von Scheele et al. 1937). Tuber yield (TY, dt/ha = deciton per hectare, 1 dt = 100 kg) was determined by the tuber weight. Tuber starch yield (TSY, dt/ha) is the product of TSC × TY.
Genotyping
Genomic DNA was isolated from freeze-dried leaf tissue as described (Li et al. 2005). DNA fragments were amplified by PCR (polymerase chain reaction) according to Li et al. (2005) using primers and annealing temperatures specified in Table 2, and adjusting the extension time to amplicon length (from 30 s to 2 min). DNA polymorphisms in the amplicons were detected by SSCP (single strand conformation polymorphism) analysis as described (Li et al. 2005), or by agarose gel electrophoresis of amplicons with (CAPS = cleaved amplified polymorphic sequence) or without (SCAR = sequence characterized amplified region, ASA = allele specific amplification) restriction enzyme digestion. SSR (simple sequence repeat) alleles were separated on Spreadex gels (Elchrom Scientific, CH-6330 Cham, Switzerland) according to the supplier’s instructions. DNA fragments of all marker types were recorded in each individual as 1 for presence, 0 for absence or as missing value in unclear cases.
Data analysis
Analysis of genetic similarity was done as described (Li et al. 2005), based on 182 DNA fragments. Population substructure was evaluated using the software STRUCTURE (Pritchard et al. 2000). The dominant markers were coded into sets of four haplotypes, to correctly represent the tetraploid individuals. Within STRUCTURE, we chose to represent DNA fragments when present by (1, *, *, *) for each individual, because of the dominant scoring, whereas absence was coded by (0, 0, 0, 0).We chose a model with admixture and independent allele frequencies. The number of subpopulations (K) was set to vary between 2 and 30. For each fixed K, two independent MCMCs (Markov Chain Monte Carlo) were run using 600,000 iterations for each. The first 100,000 iterations were discarded as burn-in. The likelihood of the data given K was saved for each K value. The underlying number of subpopulations in the sample was estimated by the K value that had the highest likelihood.
LD between all pairs of DNA fragments was calculated using Fisher’s exact test on two-by-two contingency tables (DNA fragment present or absent), using GenStat (GENSTAT 2005). The P values were corrected for multiple testing to correspond to a false discovery rate (FDR) equal or smaller than 0.05 (see below). DNA fragments with a frequency (fraction of individuals having the fragment) less than 10% or higher than 90% were excluded from the association analysis.
Association analysis was carried out using various regression and mixed models in GenStat (GENSTAT 2005). First, the adjusted means of each trait were extracted in a model that corrects the observations for the conditions of a specific trial (company performing the trial and year of the trial):
The factor trial had six levels describing differences in environmental conditions related to location, soil type and scoring rules. Each level of trial corresponded to the measurements performed by the same company and in the same year (Table 1). Genotype is a factor that identifies breeding clones or varieties as measured in the different trials. Adjusted genotypic means from the model above were used as response variable in the next model that tests for marker-trait associations:
where y * stands for adjusted means saved from the previous model. Origin is a factor with four classes to identify the origin of each genotype in the sample: three corresponding to the genotype groups “SAR”, “BNA” and “NOR”, and the fourth to the group “Standards”. Marker is a factor with two levels, for DNA fragment present or absent. Its association with the trait was tested by a (partial) t test, testing the additional variation explained by a marker after correction for origin. The resulting P values were converted into q values (see below). The proportion of genetic variation explained by each marker was calculated as the relative increase in R 2 when the marker is added to the model. Finally, we constructed multi-QTL models by applying stepwise selection to the set of significant markers found in the previous single-marker regression model (1):
To identify multi-QTL models, we used an F value for entering the regression model, F in = 4, and an F value for dropping out of the model, F out = 4 (Montgomery and Peck 1982).
In addition, we investigated the influence of possible population substructure, by fitting two mixed models, (2) and (3), that were more complex than model (1) and attempted to correct for population substructure in more sophisticated ways, as described by Yu et al. (2006) and Malosetti et al. (2007). The mixed model (2) included a marker-based genetic similarity matrix to account for kinship between genotypes:
with fixed marker and random genotype. The covariance between two genotypes i and j is given by \( \theta _{ij} .\sigma _g^2 \) with \( \sigma _g^2 \) being the genetic variance, and θ ij the pairwise genetic similarity coefficients calculated as Jaccard indices based on DNA information. Mixed model (3) corrected for both kinship and origin. For comparison, association analysis was also performed without correcting for kinship and origin (model 4).
The statistical tests for LD between DNA fragments and the association analysis in the population ALL were corrected to control the false discovery rate (FDR). The FDR is the expected proportion of false associations in the total set of significant associations. The P values were adjusted according to the Two-Stage Linear Step-Up Procedure (Benjamini et al. 2005), from which q values were obtained. These q values provide each test with an individual measure of significance in terms of an FDR of 0.05 (5% false associations in the total set of significant associations).
Genetic mapping
The diploid population H94A was used for mapping, where linkage maps have been constructed for all chromosomes based on AFLP (amplified fragment length polymorphism) and RFLP (restriction fragment length polymorphism) markers (Menendez et al. 2002). The same primers as used for association mapping were used to amplify markers from DNA of the parents and 150 F1 individuals of the H94A population. The amplicons were analyzed for SSCPs. Segregating SSCP alleles were scored as present or absent and tested for linkage with AFLP or RFLP markers of known map position. Genetic distance in centimorgan between linked marker loci was estimated as described (Menendez et al. 2002).
Results
Populations and phenotypic data
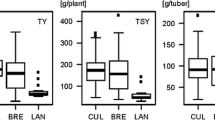
Chip quality after harvest in autumn (CQA) and in spring after cold storage (CQS), tuber yield (TY), tuber starch content (TSC) and tuber starch yield (TSY) were evaluated in the tetraploid breeding clones of Saka-Ragis (SAR), Böhm-Nordkartoffel Agrarproduktion (BNA) and NORIKA (NOR) and in the standard varieties (Table 1). The adjusted genotypic means of each trait in the sub-populations BNA, SAR, NOR and Standards, and in the ALL population (the combination of SAR, BNA, NOR clones and standard varieties after removal of potential duplicates) are shown in Fig. 1. Chip quality was generally lower after storage at 4°C, due to the accumulation of reducing sugars at low temperature (cold sweetening). The analysis of residuals, resulting from the association models (see below), showed normal distribution and independence for all traits, and the variance was homogeneous (not shown).

Box plots of the adjusted genotypic means of 2 years of traits CQA (chip quality autumn), CQS (chip quality spring), TSC (tuber starch content), TY (tuber yield) and TSY (tuber starch yield) in sample populations BNA (90 individuals), NOR (23 individuals), SAR (96 individuals) and Standards (Std., 34 individuals), and in the ALL population (243 individuals). Based on genetic similarity 16 individuals were excluded from the populations . The boxes span the interquartile range of the trait values, so that the middle 50% of the data lay within the box, with a line indicating the median. Whiskers extend beyond the ends of the box as far as the minimum and maximum values. Chip quality was scored from 1 to 9, score 1 corresponding to bad chip quality. To evaluate chip quality after cold storage with a similar range, the scale was extended to scores <1, resulting in whiskers extending into negative values in the CQS box plot. Tuber starch content is given in % (w/w), and yield and starch yield in dt/ha (1 dt = 100 kg)
Genotypic data and population structure
The SAR, BNA and NOR individuals and the standards were scored for 188 polymorphic DNA fragments generated at 36 loci on all potato chromosomes except chromosome I (Fig. 2, Table 2). From 1–17 polymorphic DNA fragments were scored at each locus, derived from 1–8 amplicons generated with different primer pairs, which annealed to various regions of a gene’s sequence (Table 2). A total of 23 loci encoded genes with known function, mostly in carbohydrate metabolism (Table 3). The remaining 13 loci were genomic fragments of unknown coding capacity or gene fragments of unknown function. The DNA fragments were used to analyze genetic similarity between the individuals and to evaluate population substructure (Pritchard et al. 2000). We did not find evidence for population substructure in the ALL population. The likelihood of our sample kept increasing with K for all tested values of K (Fig. 3), suggesting no obvious structure in the analyzed individuals.

Map positions of genotyped loci and marker-trait associations. Potato chromosomes are represented by 12 linkage groups based on genetic distances between RFLP markers shown on the left, which were mapped in reference molecular maps (Gebhardt et al. 2001, 2003; https://gabi.rzpd.de/projects/Pomamo/). The loci genotyped in the populations are shown in blue. Genetic distances are indicated in centimorgan (cM). Marker-trait associations are indicated by orange circles (online) for chip quality, blue circles (online) for tuber starch content, red circles (online) for tuber yield and purple circles (online) for starch yield. Circles with larger diameter indicate the associations with the largest effects (Table 4)

Analysis of substructure in the ALL population using STRUCTURE (Pritchard et al. 2000). The number of subpopulations (K) was set to vary between 2 and 30. For each fixed K, two independent MCMCs (Markov Chain Monte Carlo) were run. The likelihood of the data given K (ln P(X|K)) is plotted for each value of K
Linkage and linkage disequilibrium (LD) between markers
LD was estimated for all pairs of DNA fragments (Fig. 4). Highest LD values were observed between DNA fragments originating from the same locus, e.g., from different regions of the same gene. One locus corresponded to approximately 500–8,000 base pairs genomic DNA sequence. In a number of cases, DNA fragments generated from different regions of the same gene showed very high LD to each other, due to the fact that the same allele was detected by more than one DNA fragment (e.g., Stp23 and Pain1; Table 4). Some DNA fragments were in high LD because they appeared mutually exclusive. Despite similar fragment frequencies, individuals heterozygous for both DNA fragments were very rare or absent in the population (not shown). Examples for mutually exclusive alleles are Sut2-7c and Sut2-7b on chromosome V, both associated with tuber starch content but with opposite effect (Supplementary Table 2). This observation violates the assumption that the alleles combine at random in the population and genotype frequencies are determined only by the allele frequencies (Hardy-Weinberg equilibrium). We cannot rule out the possibility, however, that the mutually exclusive alleles resulted from preferential PCR amplification of one allele versus the other in heterozygous individuals.

LD matrix between DNA fragments in the ALL population. DNA fragments originating from the same locus are framed with thin black lines. The loci are arranged according to their physical order on the chromosome. Locus names are shown on the left and right of the matrix. Loci on the same chromosome (chromosome number indicated at the top) are framed with bold black lines. Black cells LD between DNA fragments is significant at P < 0.000013. Dark gray cells LD between DNA fragments is significant at 0.000013 < P < 0.00013. Light gray cells LD between DNA fragments is significant at 0.00013 < P < 0.0013. The threshold value P < 0.0013 was derived from the multiple testing correction (FDR ≤ 0.05)
LD was also observed among DNA fragments at physically linked loci on the same chromosome and with low frequency between unlinked loci on different chromosomes (Fig. 4). The extent of LD was assessed by estimating the recombination frequency between four loci on chromosome III (Stp23, Pain1, SssI, StKI), three loci each on chromosomes VII (Sps, Pha2, Sus3), IX (STM1052, Inv-ap-b, STM3012), X (Rca, Inv-ap-a, STM1106) and XI (Sut1, STM0037, Dbe) by genetic mapping in a diploid experimental population. Loci CT120, GP250, St1.1 and cLET5E4 on chromosome XI are tightly linked within 2 cM (Huang et al. 2004). The genetic distances between these loci are shown in Fig. 2. LD was observed between DNA fragments at loci that were up to 14 cM apart (Figs. 2, 4). For example, alleles at the Pain1 locus on chromosome III showed LD with alleles at Stp23 located 6 cM in the distal and SssI located 14 cM in the proximal direction. Some alleles at loci Sps, Pha2 and Sus located within 13 cM on chromosome VII were in LD with each other. This indicated the existence of large haplotype blocks in the genetic material analyzed, at least in some regions of the potato genome.
Marker-trait associations
A total of 150 DNA fragments with frequencies between 10 and 90% in the ALL population were tested for association with traits CQA, CQS, TSC, TY and TSY using four different models. All DNA fragments associated with any trait at q < 0.05 with any of the four models are reported in Supplementary Table 1. All models tested gave similar results concerning major marker-trait associations. The most relevant marker-trait associations obtained with the multiple regression model (1) are summerized in Table 4. Further details such as allele frequency, direction of effect and variance explained for all marker-trait associations found with model (1) (q < 0.05) are reported in Supplementary Table 2. A total of 66 DNA fragments (44%) were associated with one or more traits at q < 0.05. When DNA fragments of the same locus with highly similar distribution in the population and consequently similar associations were considered as single alleles or haplotypes, 46 alleles at 22 loci on eight chromosomes were associated with one or more traits (Supplementary Table 2; Fig. 2). Twenty-five marker alleles each were associated with CQA and CQS, respectively; twenty-seven alleles were associated with TSC, one with TY and nine with TSY. Except markers GP171 and STM0037, the most significant associations (q < 0.01) were detected with DNA polymorphisms at eight loci, which encode the enzymes glucose-6-phosphate dehydrogenase (G6pdh), the starch phosphorylases Stp23 and StpL, the soluble starch synthase SssI, the invertases Pain1 and Inv-ap-a, plasma membrane H+ -ATPase 2 (Pha2) and ribulose bisphosphate carboxylase activase (Rca) (Table 4; Supplementary Table 1).
Half of the 46 alleles were associated with more than one trait. Most frequent were associations both with chip quality and tuber starch content (Fig. 2, Table 4; Supplementary Table 2). Without exception, the direction of the effect, whether increasing or decreasing the trait mean, was the same for all alleles associated with chip quality and tuber starch content (Table 4). In contrast, the allele StpL-3e associated with tuber starch content and yield showed an opposite effect. Higher starch content reduced yield and vice versa (Table 4). Six loci were associated with chip quality, tuber starch content and starch yield, Stp23, PainI, SssI, Pha2, St1.1 and STM0037 (Fig. 2, Table 4). Alleles at the StpL locus were associated with all traits evaluated (Fig. 2, Table 4). Different alleles at the same locus could be associated with different effects. For example, allele StpL-3b decreased chip quality, starch content and starch yield, whereas allele StpL-3e increased chip quality and starch content but lowered yield. StpL-3d affected negatively only chip quality (CQS), whereas StpL-3c had a negative effect only on tuber starch content. At the Pain1 locus, allele PainI-5b had negative, and PainI-9a positive effects on the associated traits. The positive association with chip quality of invertase alleles InvGE-6f and InvGF-4b at the Inv-ap-b locus on chromosome IX found previously in independent populations (Li et al. 2005), was again observed in the ALL population (Table 4).
Stepwise selection identified 10, 8, 8 and 6 marker alleles for CQA, CQS, TSC and TSY, respectively, which are highlighted in bold letters in Table 4. These markers collectively explained 39.7, 49.0, 54.9 and 26.1%, respectively, of the total variation in the ALL population.
Discussion
Our experiment is among the most comprehensive association studies for important agronomic traits that are currently available for crop plants, comprising association tests for multiple traits at multiple loci in advanced varieties and breeding materials (Wilson et al. 2004; Breseghello and Sorrells 2006; Wei et al. 2006; González-Martínez et al. 2007). Chip quality, tuber starch content and yield were evaluated at three breeding stations with the standard methods used for variety selection. Proprietary breeding clones were evaluated at single locations. To facilitate joint data evaluation, the same set of varieties was included in the trials at all locations. This experimental design may serve as model in association studies based on proprietary breeding materials that cannot be shared. Marker-trait associations were detected using multiple regression and mixed models, which took into account trials, locations, years, clone origin and kinship. Pedigree information as suggested (Malosetti et al. 2007) was not available for most of the breeding clones. Instead, a marker-based genetic similarity matrix was included in models 2 and 3 to account for kinship, similarly as in Yu et al. (2006). Clone origin was included in models 1 and 2, because the SAR, BNA, NOR and “Standards” populations represented four independent samples from the tetraploid germplasm adapted to potato cultivation in temperate European climate. The marker-trait associations with the largest effects were consistent across different models. The results were more variable, depending on the model, for associations with minor effects. The minor association of invertase alleles InvGE-6f and InvGF-4b with chip quality, found in the ALL population, confirmed the previous results obtained in independent samples of breeding materials (Li et al. 2005). This provides the first evidence for the reproducibility of a marker-trait association in different samples. As observed in other studies with similar genetic materials (Li et al. 2005; Simko et al. 2006; Malosetti et al. 2007), middle European potato varieties and advanced breeding clones so far represent unstructured populations, probably due to a rather homogeneous genetic background.
At least one marker allele at 22 of the 36 examined loci showed association with one or more traits. This was an unexpected high number of associations, when considering the small proportion of the potato genome that was sampled. There are two explanations for this finding. First, allelic variation at some loci may indeed be causal for the trait variation. About two-thirds of the loci were selected based on co-localization of functional candidate genes with QTL for tuber sugar and/or starch content (Beck and Ziegler 1989; Frommer and Sonnewald 1995; Winter and Huber 2000; Salerno and Curatti 2003; Schäfer-Pregl et al. 1998; Chen et al. 2001; Menendez et al. 2002; Gebhardt et al. 2005). Except GP171 and STM0037 on chromosome VIII and XI, respectively, the most significant and robust associations were observed with candidate gene alleles. Second, LD between alleles at loci several centimorgans apart could result in indirect associations, thereby increasing the proportion of the potato genome that was actually tagged. This is indicated by associations found with genomic and SSR markers that did not encode any candidate gene. The large haplotype blocks observed in populations of advanced potato breeding clones and varieties likely result from a limited number of meiotic recombination events separating the individuals and/or selection (Gebhardt et al. 2004; Simko et al. 2006; Malosetti et al. 2007). Based on these observations, we propose that genome-wide association studies should be feasible in potato breeding materials with, on average, one marker locus per centimorgan, amounting to 500–1,000 loci in total. Large haplotype blocks are favorable for identification of diagnostic markers for breeding purposes, but are less favored when aiming at the verification of candidate genes by association mapping. However, candidate gene alleles associated with positive or negative trait values can be isolated from carrier individuals and validated by comparative complementation analysis in heterologous model systems such as yeast, as demonstrated for tomato invertase alleles (Fridman et al. 2004).
LD was also observed between unlinked markers on different chromosomes. With a threshold of 5% false positives among all significant LD pairs, a background of false positive LD pairs is expected, particularly among pairs in the lowest significance category. Interestingly, a few unlinked markers showed very strong LD. This might result from adaptive co-selection of epistatic alleles linked to these markers in the germplasm studied. Epistatic marker-trait associations have been identified in the ALL population (manuscript in preparation).
Most loci associated with chip quality were also associated with tuber starch content, suggesting that these complex traits are controlled, in part, by the same genes. The pleiotropic effects of individual alleles always had the same direction, either positive (more starch, better chip color, less reducing sugars) or negative (less starch, worse chip color, more reducing sugars). Starch and sugars in dormant tubers are interconvertible, and the energy for this conversion is provided by respiration. The temperature-dependent balance between starch and sugars shifts toward sugars at low temperature (Isherwood 1973). Less efficient and/or cold labile alleles of katabolic enzymes such as invertases and starch phosphorylases might shift the balance toward higher starch and lower sugar content, whereas more active and/or cold stable alleles would have the opposite effect. The StpL locus on chromosome V is an example where both types of alleles were observed. The molecular basis of such differences remains to be elucidated. Possibilities are the variation of enzyme activity, stability or post-translational modifications due to amino acid changes, which alter protein conformation or modification sites, or the variation in expression level due to DNA polymorphisms in cis-regulatory sequences.
Cold sweetening has been correlated with increased enzymatic activity of invertase and starch hydrolyzing enzymes, or with the cold lability of glycolytic enzymes (Pollock and Ap Rees 1975; Pressey and Shaw 1966; Zrenner et al. 1996; Cottrell et al. 1993). In our experiment, the majority of the alleles associated with chip quality after cold storage was also associated with chip quality after harvest, before the onset of cold storage. The genes controlling sugar content in the final stages of tuber development and in the dormant, cold-stored tuber may be largely identical, and low temperature was not required for differentiating their allelic variants. Alternatively, alleles specifically effective during cold storage might have escaped detection in the association test due to low frequency in the breeding material analyzed.
In conclusion, our results demonstrate that association genetics in advanced breeding populations of the potato, a polyploid, non-inbred crop, is a valuable approach toward elucidating the molecular basis of complex agronomic traits and for developing diagnostic DNA-based markers for “precision breeding” of improved varieties.
References
Barker L, Kühn C, Weise A, Schulz A, Gebhardt C, Hirner B, Hellmann H, Schulze W, Ward JM, Frommer WB (2000) A sucrose transporter homolog in sieve elements is a candidate gene for QTLs controlling starch and yield. Plant Cell 12:1153–1164
Beck E, Ziegler P (1989) Biosynthesis and degradation of starch in higher plants. Annu Rev Plant Physiol Plant Mol Biol 40:95–117
Benjamini Y, Krieger A, Yekutieli D (2005) Two staged linear step up FDR controlling procedure. Unpublished report. http://www.math.tau.ac.il/~ybenja
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Burton WG (1969) The sugar balance in some British potato varieties during storage II. The effects of tuber age, previous storage temperature, and intermittent refrigeration upon low-temperature sweetening. Eur Potato J 12:81–95
Chen X, Salamini F, Gebhardt C (2001) A potato molecular function map for carbohydrate metabolism and transport. Theor Appl Genet 102:284–295
Copeland L, Turner JF (1987) The regulation of glycolysis and the pentose-phosphate pathway. In: Stumpf PK, Conn EE (eds) The biochemistry of plants. Academic Press, New York, pp 107–125
Cottrell JE, Duffus CM, Paterson L, Mackay GR, Allison MJ, Main H (1993) The effect of storage temperature on reducing sugar concentration and the activities of three amylolytic enzymes in tubers of the cultivated potato, Solanum tuberosum L. Potato Res 36:107–117
Fridman E, Carrari F, Liu YS, Fernie AR, Zamir D (2004) Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science 305:1786–1789
Frommer WB, Sonnewald U (1995) Molecular analysis of carbon partitioning in solanaceous species. J Exp Bot 46:587–607
Gebhardt C, Ritter E, Salamini F (2001) RFLP map of the potato. In: Phillips RL, Vasil IK (eds) DNA-based markers in plants. Advances in cellular and molecular biology of plants, 2nd edn. Kluwer Academic Publishers, Dordrecht, pp 319–336
Gebhardt C, Walkemeier B, Henselewski H, Barakat A, Delseny M, Stüber K (2003) Comparative mapping between potato (Solanum tuberosum) and Arabidopsis thaliana reveals structurally conserved domains and ancient duplications in the potato genome. Plant J 34:529–541
Gebhardt C, Ballvora A, Walkemeier B, Oberhagemann P, Schüler K (2004) Assessing genetic potential in germ plasm collections of crop plants by marker-trait association: a case study for potatoes with quantitative variation of resistance to late blight and maturity type. Mol Breed 13:93–102
Gebhardt C, Menendez C, Chen X, Li L, Schäfer-Pregl R, Salamini F (2005) Genomic approaches for the improvement of tuber quality traits in potato. Acta Hort 684:85–91
GenStat (2005) GenStat, 8th edn. Release 8.1. VSN International Ltd., Oxford, UK
Glaczinski H, Heibges A, Salamini F, Gebhardt C (2002) Members of the Kunitz-type protease inhibitor gene family of potato inhibit soluble tuber invertase in vitro. Potato Res 45:163–176
González-Martínez SC, Wheeler NC, Ersoz E, Nelson CD, Neale DB (2007) Association genetics in Pinus taeda L. I. Wood property traits. Genetics 175:399–409
Habib A, Brown HD (1957) The role of reducing sugars and amino acids in browning of potato chips. Food Technol 11:85–89
Huang S, Vleeshouwers GAA, Werij JS, Hutten RCB, van Eck HJ, Visser RGF, Jacobsen E (2004) The R3 resistance to Phytophthora infestans in potato is conferred by two closely linked R genes with distinct specificities. Mol Plant Microbe Interact 17:428–435
Isherwood FA (1973) Starch-sugar interconversion in Solanum tuberosum. Phytochemistry 12:2579–2591
Li L, Strahwald J, Hofferbert H-R, Lübeck J, Tacke E, Junghans H, Wunder J, Gebhardt C (2005) DNA variation at the invertase locus invGE/invGF is associated with tuber quality traits in populations of potato breeding clones. Genetics 170:813–821
Malosetti M, van der Linden CG, Vosman B, van Eeuwijk FA (2007) A mixed-model approach to association mapping using pedigree information with an illustration of resistance to Phytophthora infestans in potato. Genetics 175:879–889
Menendez CM, Ritter E, Schäfer-Pregl R, Walkemeier B, Kalde A, Salamini F, Gebhardt C (2002) Cold-sweetening in diploid potato. Mapping QTL and candidate genes. Genetics 162:1423–1434
Milbourne D, Meyer RC, Collins AJ, Ramsay LD, Gebhardt C, Waugh R (1998) Isolation, characterisation and mapping of simple sequence repeat loci in potato. Mol Gen Genet 259:233–245
Montgomery DC, Peck EA (1982) Introduction to linear regression analysis, 2nd edn. Wiley & Sons, New York
Mori H, Tanizawa K, Fukui T (1991) Potato tuber type-H phosphorylase isozyme. Molecular cloning, nucleotide sequence and expression of a full-length cDNA in Escherichia coli. J Biol Chem 266:18446–18453
Osborn TC, Alexander DC, Fobes JF (1987) Identification of restriction fragment length polymorphisms linked to genes controlling soluble solids content in tomato fruit. Theor Appl Genet 73:350–356
Pflieger S, Lefebvre V, Causse M (2001) The candidate gene approach in plant genetics: a review. Mol Breed 7:275–291
Pollock CJ, ap Rees T (1975) Activities of enzymes of sugar metabolism in cold-stored tubers of Solanum tuberosum. Phytochemistry 14:613–617
Pressey R, Shaw R (1966) Effect of temperature on invertase, invertase inhibitor, and sugars in potato tubers. Plant Physiol 41:1657–1661
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Putz B (1989) Technologie der Chipsherstellung. In: Kartoffeln, Züchtung, Anbau, Verwertung. Behr’s, Hamburg, pp 219
Salerno GL, Curatti L (2003) Origin of sucrose metabolism in higher plants: when, how and why? Trends Plant Sci 8:63–69
Salvi S, Tuberosa R (2005) To clone or not to clone plant QTLs: present and future challenges. Trends Plant Sci 10:297–304
Schäfer-Pregl R, Ritter E, Concilio L, Hesselbach J, Lovatti L, Walkemeier B, Thelen H, Salamini F, Gebhardt C (1998) Analysis of quantitative trait loci (QTL) and quantitative trait alleles (QTA) for potato tuber yield and starch content. Theor Appl Genet 97:834–846
Simko I, Haynes KG, Jones RW (2006) Assessment of linkage disequilibrium in potato genome with single nucleotide polymorphism markers. Genetics 173:2237–2245
Townsend LR, Hope GW (1960) Factors influencing the color of potato chips. Can J Plant Sci 40:58–64
Van Berloo R, Hutten RCB, van Eck HJ, Visser RGF (2007) An online potato pedigree database resource. Potato Res 50:45–57
Von Scheele C, Svensson G, Rasmusson J (1937) Die Bestimmung des Stärkegehalts und der Trockensubstanz der Kartoffel mit Hilfe des spezifischen Gewichts. Landw Vers Station 127:67–96
Wei X, Jackson PA, McIntyre CL, Aitken KS, Croft B (2006) Associations between DNA markers and resistance to diseases in sugarcane and effects of population substructure. Theor Appl Genet 114:155–164
Wilson LM, Whitt SR, Ibáñez AM, Rocheford TR, Goodman MM, Buckler ES (2004) Dissection of maize kernel composition and starch production by candidate gene association. Plant Cell 16:2719–2733
Winter H, Huber SC (2000) Regulation of sucrose metabolism in higher plants: Localization and regulation of activity of key enzymes. Crit Rev Biochem Mol Biol 35:253–289
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovitch S, Buckler ES (2006) A unified mixed.model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics 38:204–208
Zrenner R, Schüler K, Sonnewald U (1996) Soluble acid invertase determines the hexose-to-sucrose ratio in cold-stored potato tubers. Planta 198:246–252
Acknowledgments
This work was funded by the German Ministry for Research and Education under the GABI (Genome analysis in the biological system plant) program (grant no. 0313038, GABI-CHIPS) and by the Max-Planck society. Part of the work was carried out in The Department for Plant Breeding and Genetics, headed by Maarten Koornneef. The authors thank David Turra for providing Kunitz-type inhibitor primers and M. Koornneef for critically reading the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Yu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Li, L., Paulo, MJ., Strahwald, J. et al. Natural DNA variation at candidate loci is associated with potato chip color, tuber starch content, yield and starch yield. Theor Appl Genet 116, 1167–1181 (2008). https://doi.org/10.1007/s00122-008-0746-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-008-0746-y