
Abstract
Despite decades of research, accurate diagnosis of Parkinson’s disease remains a challenge, and disease-modifying treatments are still lacking. Research into the early (presymptomatic) stages of Parkinson’s disease and the discovery of novel biomarkers is of utmost importance to reduce this burden and to come to a more accurate diagnosis at the very onset of the disease. Many have speculated that non-motor symptoms could provide a breakthrough in the quest for early biomarkers of Parkinson’s disease, including the visual disturbances and retinal abnormalities that are seen in the majority of Parkinson’s disease patients. An expanding number of clinical studies have investigated the use of in vivo assessments of retinal structure, electrophysiological function, and vision-driven tasks as novel means for identifying patients at risk that need further neurological examination and for longitudinal follow-up of disease progression in Parkinson’s disease patients. Often, the results of these studies have been interpreted in relation to α-synuclein deposits and dopamine deficiency in the retina, mirroring the defining pathological features of Parkinson’s disease in the brain. To better understand the visual defects seen in Parkinson’s disease patients and to propel the use of retinal changes as biomarkers for Parkinson’s disease, however, more conclusive neuropathological evidence for the presence of retinal α-synuclein aggregates, and its relation to the cerebral α-synuclein burden, is urgently needed. This review provides a comprehensive and critical overview of the research conducted to unveil α-synuclein aggregates in the retina of Parkinson’s disease patients and animal models, and thereby aims to aid the ongoing discussion about the potential use of the retinal changes and/or visual symptoms as biomarkers for Parkinson’s disease.
Similar content being viewed by others

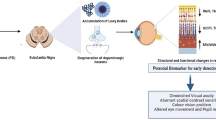
Introduction
In 2016, the number of Parkinson’s disease (PD) patients was estimated at over 10 million, making PD the second most common neurodegenerative disease worldwide [43]. Moreover, due to our increasingly ageing society, age-related neurodegenerative diseases, such as PD, will become even more prevalent [149, 150]. Up until now, PD cannot be prevented, slowed or cured, adding to an augmenting societal burden. The lack of disease-modifying treatments, poor success rate of clinical trials, and the shortage of techniques for patient screening and early diagnosis highlight the urgent need for intensive research to unravel the complex interplay of disease mechanisms underlying PD and to better understand the early manifestations of PD [5, 83].
The defining neuropathological hallmarks of PD are the progressive loss of dopaminergic neurons in the substantia nigra, on one hand, resulting in reduced striatal dopamine levels, and the accumulation of proteinaceous cytoplasmic inclusions mainly consisting of aggregated alpha-synuclein (αSYN), called Lewy bodies, on the other hand [15, 41]. Dopaminergic depletion leads to the development of progressive motor dysfunction [25], of which the cardinal symptoms can be summarised in the acronym TRAP, which stands for tremor, rigidity, akinesia (or bradykinesia), and postural instability [60, 112].
Importantly, recent clinical, neuropathological, and imaging evidence suggests an earlier initiation of PD pathology, before onset of these cardinal motor symptoms [60, 77, 93]. This prodromal phase in PD is particularly interesting for the development of early stage diagnosis and intervention [77, 93]. Therefore, over the past years, the importance of early, non-motor manifestations of PD is increasingly recognised [112]. These include reduced smell, sleep disturbances, mood changes, and gastrointestinal deficits that may emerge years or even decades before motor symptoms [25, 49, 77, 107], and could fit in the emerging interest for biological, biomarker-based diagnosis. In this recent view, diseases are detected by scans and biochemical assays that assess key pathological features (e.g., protein aggregates, neurodegeneration) even before symptoms set in. In the case of PD, this can further be complemented with assessments of early non-motor symptoms. By focusing on the presymptomatic phases of disease, a time window for early, more adequate treatment can be breached and patients at risk may be identified before irreversible damage has occurred. Importantly, biomarkers are essential to identify these preclinical or prodromal patients, as well as for monitoring disease progression and the effect of a disease-modifying treatment. This stresses the need for well-validated biomarkers and biomarker combinations. Because the retina might provide a window to the pathological processes that are ongoing in the brain, more research into the retinal manifestations of PD is highly needed to explore whether visual tests and/or retinal imaging techniques could be used to monitor retinal biomarkers.
In recent years, there has been an expansion of studies investigating the use of in vivo assessments of retinal structure, electrophysiological, and visual function as novel means for identifying patients at risk that need further neurological examination and for longitudinal follow-up of disease progression in PD patients. These have been extensively reviewed elsewhere (e.g., [7, 10, 22, 49, 70, 138, 148]) and fall beyond the scope of this review. Often, the results of these studies have been related to dopaminergic depletion and αSYN accumulation in the retina. However, the neuropathological evidence for the manifestation of these defining PD hallmarks in the retina is scarce and inconclusive, hampering our understanding of the retinal/visual abnormalities seen in PD patients and their validation as retinal biomarkers. In summary, there is evidence that retinal dopamine deficiency underlies some of the vision problems observed in PD patients, which can be temporarily alleviated with L-DOPA [7, 148, 151]. Furthermore, immunohistological studies on post-mortem retinas of PD patients suggest that pathological αSYN accumulations are present in the retina and/or visual system [15, 20, 53, 98], although it remains elusive whether the functional deficits and structural neurodegenerative changes in the retina are related to these αSYN deposits. Finally, information on the presence of retinal αSYN deposits in animal models is still scarce, as PD pathology in these models is mainly studied in the brain, while the retina is largely overlooked [34, 93, 113, 120]. Only in toxin-induced models, retinal PD manifestations were studied in the retina [56, 93, 141], while in more recent viral vector- and αSYN fibril-based studies, this is not the case yet [26, 96, 102, 142]. In transgenic rodent models, only two papers report on retinal αSYN accumulation in two different transgenic mice: one expressing human A53T αSYN under the control of the Prnp promoter (TgM83) [78], the other expressing fused αSYN::GFP under the PDGFβ promoter [113]. In this review, new preliminary data on the presence of αSYN and phospho-αSYN in the retina of (Thy-1)-h[A30P] αSYN mice are provided. However, the direct impact of αSYN accumulation on the structure and function of the retina remains to be studied.
The aim of this review is to provide a comprehensive and critical overview of the research conducted to unveil α-synuclein aggregates in the retina of PD patients and animal models and to outline outstanding questions and future challenges. It may thereby aid the ongoing discussion about the potential use of visual symptoms and retinal abnormalities as biomarkers for diagnosis and disease monitoring of PD and the implementation of the retina as a model organ to study PD processes.
The eye as a window to the brain
The strong link between the eye and the brain has led to an emerging concept in neurobiological research: “the eye as a window to the brain” [34, 75]. Indeed, the retina is an integral part of the CNS, displaying striking similarities to the other CNS structures in terms of anatomy, response to insult, and immunology [75]. This strong interplay between the brain and the retina is underscored by the presence of retinal manifestations in many neurodegenerative diseases or insults, e.g., Alzheimer’s disease, Huntington’s disease, multiple sclerosis, and stroke [3, 29, 86, 120, 124].
Furthermore, the retina is the most accessible part of the CNS, and the unique properties of the eye in terms of in vivo imaging, manipulation, and administration of compounds, can be exploited for both fundamental and clinical research of CNS diseases [34, 75]. In addition, some retinal manifestations are suggested to be present and/or detectable before typical cerebral manifestations [34, 93, 125, 138]. Indeed, given that the resolution of current retinal imaging techniques is at least an order of magnitude higher than available brain imaging techniques, they might be able to detect more subtle changes (i.e., earlier disease stages). The eye thus can offer an alternative approach, which can complement brain examinations/research, especially in terms of early diagnosis and disease monitoring [77, 93].
Thanks to the accessibility and the transparency of the anterior segment of the eye (in contrast to the bony skull that covers the brain), an array of technologies allows non-invasive, high-resolution imaging of the retina, e.g., confocal scanning laser ophthalmoscopy (cSLO), optical coherence tomography (OCT), adaptive optics, and oximetry [13]. In combination with electroretinography (ERG), visual-evoked potentials, and visual function tests, these techniques are routinely used to detect (early) changes in retinal morphology, blood flow, electrophysiology, and visual performance. Especially relevant, or most commonly used, to the study of PD in the retina, are OCT, cSLO, and ERG. Optical coherence tomography provides structural information on ocular tissues with 1-to-10 μm resolution, and enables direct observation of remodelling of the retinal tissue and morphometric measurements of retinal layer thickness [6]. A second non-invasive, diagnostic imaging technique for the retina is confocal scanning laser ophthalmoscopy, which produces an en face view of the retina on which changes in the optic nerve head, vasculature, and retinal tissue morphology can be assessed [147]. In addition, cSLO can be used to detect fluorescent signals, an application upon which the ‘Detection of Apoptosing Retinal Cells’ (DARC) technique for single-cell imaging of apoptotic cells is based [14, 44]. Electroretinography, finally, together with visual-evoked potentials (VEP), has proven very sensitive in revealing alterations in the electrical responses of various retinal cell types, thereby pinpointing the exact circuitries affected. Pattern electroretinography (PERG), more specifically, uses pattern-reversal stimuli to capture retinal ganglion cell activity [46].
Retinal manifestations of Parkinson’s disease
Visual symptoms in Parkinson’s disease patients
Almost 80% of PD patients report at least one visual symptom, resulting from either defects in primary vision, such as visual acuity [8, 54, 61, 85], spatial contrast sensitivity [19, 21], and colour vision [52, 114, 122, 130], or deficits in more complex visual tasks [8, 9, 11, 49, 110, 135, 148]. One of the most prevalent visual dysfunctions among PD patients is impaired colour vision, which is reported to manifest several years before diagnosis and to correlate with disease progression (i.e., motor function) [38, 52, 114, 122]. Next, one in three PD patients has issues with seeing fine detail due to reduced visual acuity and visuospatial processing, a feature that is also correlated with motor performance [61, 85]. Third, some of the earliest visual abnormalities due to PD, together with colour recognition, are poor vision at dim light conditions and the disability to perceive small increments of light versus dark, which are manifestations of reduced contrast sensitivity. Given its progression and correlation with disease severity, reduced contrast sensitivity has been suggested to be a valuable biomarker for early PD [19, 38, 49, 52, 109, 114, 134, 138, 146]. Notably, changes in colour discrimination and contrast sensitivity have shown a discriminatory power for early diagnosis that outweighs non-motor symptoms such as hyposmia and sleep disturbance [148]. Besides these manifestations in the neuroretina of PD patients, other parts of the eye are reported to be affected as well, e.g., the pupil and lens, leading to blurred vision due to disordered pupil reactivity and cataract, respectively [94].
While visual dysfunctions experienced by PD patients are at least in part to be attributed to defects in subcortical and cortical areas for visual processing [16, 86], electrophysiological tests, and structural imaging prove that they are (also) the result of local retinal pathogenesis. Indeed, several OCT studies have demonstrated retinal nerve fibre layer thinning in PD patients as compared to age-matched controls [12, 24, 70, 75, 84]. In addition, studies comparing retinal nerve fibre layer thickness in the four retinal quadrants demonstrated that the temporal quadrant is most commonly affected. This is of particular interest, given that this quadrant is typically affected in mitochondrial optic neuropathies [34, 68, 79] and that PD has been linked to mitochondrial dysfunction [4, 58, 68, 91]. PD patients also display thinning of the inner retinal layers: the ganglion cell layer, inner plexiform layer, and inner nuclear layer [1, 22, 71, 128, 133]. Together, these observations are suggestive of a loss of retinal ganglion cells (cfr. retinal nerve fibre layer, ganglion cell layer, and inner plexiform layer changes) and a thinning of the dopaminergic plexus (cfr. inner plexiform layer and inner nuclear layer changes) [19, 32, 88, 139]. These findings are corroborated by ERG studies, which have shown significant changes in ERG responses in PD patients. On one hand, decreases in the amplitude of the photopic b-wave and scotopic oscillatory potentials were observed [27, 48, 57, 95], which are diagnostic indicators of the functioning of retinal interneurons, which comprise, amongst many other cells, dopaminergic amacrine cells [10]. Of note, decreased dopamine levels have been shown to affect scotopic oscillatory responses [95]. On the other hand, a reduced amplitude and delayed latency of the P50 component of the PERG were recorded [45, 48, 69, 92, 103,104,105, 121], for which there is also evidence that these can be correlated with disturbed dopamine signalling affecting the retinal ganglion cells [10, 120].
Histopathological evidence
In the brain, the defining neuropathological features of PD are loss of dopaminergic neurons and accumulation of αSYN inclusions, both of which are confirmed post-mortem via histopathological examination. While in vivo evidence from OCT and ERG studies points to dopaminergic degeneration in the retina as well, only one paper reveals post-mortem evidence for a reduction in dopamine levels to support and better understand these findings [30, 39, 51]. Nonetheless, dopaminergic involvement is underscored by the transient improvement in visual function tests and PERG responses upon administration of L-DOPA and by post-mortem studies revealing reduced retinal tyrosine hydroxylase expression [7, 51, 61, 120]. In addition, a recent study reported reduced melanopsin-immunoreactive retinal ganglion cell (mRGC) density and complexity of the melanopsin plexus in the retina of PD patients. While being involved in circadian rhythm, commonly affected in PD, a subset of these mRGCs also receives dopaminergic input, and their loss may thus be linked to a diminution of retinal dopamine levels (i.e., a loss of their main synaptic inputs) [99]. Furthermore, little histopathological proof for retinal accumulation of αSYN inclusions has been gathered thus far.
Studies on αSYN and phosphorylated αSYN in the retina of Parkinson’s disease patients
Between 2014 and 2018, four studies have investigated the presence of αSYN or phospho-αSYN in the retina of PD patients, in a total of 27 PD patients and 24 age-matched controls [15, 20, 53, 98]. In the years before, endogenous αSYN had already been described for the healthy retina [72, 82]. The methodology and primary findings of these studies are summarised in Table 1 and Fig. 1. First of all, in healthy subjects, two independent studies described the presence of endogenous αSYN in the inner nuclear layer and in the ganglion cell layer [72, 82]. In addition, the outer segments of the photoreceptors in the photoreceptor layer and their terminals in the outer plexiform layer were strongly immunoreactive for αSYN, and αSYN-positive cell bodies and neurites were detected in the inner nuclear layer and the inner plexiform layer, respectively [82]. These observations might be reconciled with αSYN in its monomeric form, which is predominantly located in the cytoplasm of presynaptic nerve terminals of both dopaminergic and non-dopaminergic neurons, and of which the physiological function has been suggested to be related to synaptic plasticity, vesicle trafficking, and neurotransmission [41].

Schematic overview of a αSYN expression in healthy retinas and b (phospho-)αSYN expression and related pathological processes in the retina of PD patients. a Post-mortem examinations on cross sections of the retina reveal the presence of αSYN in four retinal layers in non-PD subjects. b In PD patients, in addition, pathological αSYN accumulation is found, which presents as phospho-αSYN and Lewy-like inclusions and neurites. Furthermore, decreased dopamine levels in the total retina and decreased TH immunoreactivity in the IPL are observed. OCT studies report thinning of the inner retina. PERG studies show (#1) reduced amplitude and delayed latency of the P50 component in PD patients, which are indicators of affected retinal ganglion cell (yellow) function. ERG studies demonstrate (#2) a decrease in the amplitude of the photopic b-wave, a read-out of bipolar cell (pink) malfunction; and (#3) reduced scotopic oscillatory potentials, indicating disturbed neuronal activity of the amacrine cells (orange). αSYN alpha-synuclein, p-αSYN phosphorylated serine-129 αSYN, OCT optical coherence tomography, (P)ERG (pattern) electroretinography, TH tyrosine hydroxylase, DA dopamine, RNFL retinal nerve fibre layer, GCL ganglion cell layer, INL inner nuclear layer, IPL inner plexiform layer, ONL outer nuclear layer, OPL outer plexiform layer, PL photoreceptor layer
In PD retinas, however, additional αSYN was found that had accumulated into insoluble aggregates, undergone conformational changes, and/or abnormal phosphorylation. First, Bodis-Wollner et al. [20] demonstrated a distinct pattern of intense αSYN immunoreactivity in different retinal layers: upon immunostaining for native αSYN, 8–10-μm globular inclusions were reported in inner nuclear layer neurons, αSYN-positive neurites were seen in the inner plexiform layer, and both intra- and extracellular αSYN-positive inclusions were found in the ganglion cell layer. Importantly, this immunoreactivity pattern was seen in the central and peripheral retina and was consistent for all eight PD retinas investigated. Furthermore, morphometric analysis of the retina pointed to a thinner inner retina, especially the inner nuclear layer. Although the authors suggest that this latter change might be due to a loss of dopaminergic cells, one should consider that the fraction of dopaminergic cell bodies in the inner nuclear layer is limited and, therefore, unlikely to account for substantial inner nuclear layer thinning. In fact, there is more evidence pointing to the loss of dopaminergic processes in the inner plexiform layer, which constitute a large plexus and, therefore, may account for substantial inner retinal thinning when lost [19, 32, 88, 139].
More evidence for the accumulation of αSYN deposits in the retina was gathered by Ortuno-Lizaran et al. [98] and Beach et al. [15], who both investigated the presence of αSYN phosphorylated at serine-129. These studies investigated nine PD patients each, and found phospho-αSYN immunoreactivity in all nine patients and in seven out of nine PD patients, respectively. Immunostaining for phospho-αSYN was also found in a subset of incidental Lewy body disease patients (three of four and one of three, respectively), yet was absent in all control retinas (four and six subjects, respectively). Ortuno-Lizaran et al. [98] described that phospho-αSYN deposits were present in axonal fibres, dendrites, and neuronal perikarya. Although the cell morphology and soma size of the affected neurons varied, double stainings confirmed that these cells were all retinal ganglion cells, and no inclusions were seen in (dopaminergic) amacrine cells [98]. In contrast, Beach et al. did not observe any intraneuronal inclusions, except for one case, and concluded that the phospho-αSYN immunopositive fibres seen in their study must be centrifugal/retinopetal fibres originating from the brain or retinal ganglion cell (RGC) axons [15].
Strikingly, both Bodis-Wollner et al. [20] and Ortuno-Lizaran et al. [98] claimed that the retinal αSYN inclusions that they found displayed characteristics reminiscent of classic Lewy bodies and neurites. According to Bodis-Wollner et al., intracytoplasmic αSYN staining in the inner nuclear layer and ganglion cell layer was condensed into globular inclusions replacing other cell components, while Lewy-like neurites were observed in the IPL. Ortuno-Lizaran et al. performed a phospho-αSYN staining and revealed deposits in neuronal perikarya, as well as phospho-αSYN-positive neurites resembling Lewy neurites with typical dystrophic morphology. Finally, abnormal beading, swollen axonal segments, and increased tortuosity and swelling of the dendrites suggested that the phospho-αSYN-positive neurons found in this study were dysfunctional and/or undergoing neurodegeneration [20, 98]. Importantly, in the study by Bodis-Wollner et al., an antibody for total non-modified αSYN was used, while Ortuno-Lizaran et al. used a phospho-αSYN specific antibody. Given that phospho-αSYN is an indicator of increased αSYN aggregation/toxicity and more relevant than total or native (unphosphorylated) αSYN to evaluate Lewy body pathology (as highlighted in Table 2), caution is thus warranted when interpreting the findings of Bodis-Wollner et al., and overall, one can assume phospho-αSYN to have a higher value as PD biomarker.
Although Ortuno-Lizaran et al. [98] reported that phospho-αSYN accumulation was sparse, with relatively few retinal ganglion cells affected, quantification of the number of immunopositive retinal ganglion cells in the nasal-inferior quadrant revealed a strong positive correlation between Lewy‐type synucleinopathy density in the retina and brain. Furthermore, retina Lewy‐type synucleinopathy density score correlated with the Unified Parkinson’s disease pathology stage. Importantly, retinas from patients with incidental Lewy body disease were included in this study, besides cases of moderate-to-severe PD [81]. Hereby, this study was the first to present evidence that phospho-αSYN accumulates in the retina in parallel with the brain, including in the early stages preceding development of clinical signs of Parkinsonism or dementia.
Notably, Ortuno-Lizaran et al. [98] also looked at native αSYN, albeit using a different antibody than Bodis-Wollner et al. [20], and found ubiquitous expression in the photoreceptor outer segments and the amacrine cells in the inner nuclear layer, that was identical in PD patients and healthy subjects [98]. Likewise, Ho et al. [53] reported diffuse cytoplasmic staining of native αSYN in the inner nuclear layer and ganglion cell layer that was comparable in six PD patients and six age-matched controls. Both studies failed to find Lewy bodies in the retina of PD patients. On the one hand, this reconciles the notion that phospho-αSYN is the preferred neuropathological marker of disease (cfr. above and Table 2). On the other hand, these contradicting findings point out the shortcomings of the studies that are currently available. First of all, due to the difficulty in obtaining high-quality post-mortem human retinas, the number of patients in each of the studies is relatively low, and at this point still does not allow to make extrapolations of the prevalence and specificity of retinal αSYN deposits to the general population. Second, different αSYN species (native versus phosphorylated) have been investigated, being indicative of a distinct pathological status; total αSYN antibodies stain both soluble and aggregated forms of αSYN, while phosphorylation-specific antibodies identify αSYN species with increased aggregation propensity and toxicity (Table 2). Third, different methodologies might underlie the different results. These include differences in and/or lack of immunohistochemical methods, comprehensive retinal tissue processing and wholemount techniques, and quality of available αSYN antibodies [31]. In particular, for αSYN immunostainings, the time between death of the subject and fixation of the tissue, the fixation time, and antigen retrieval method have been shown to lead to variability in staining results [2, 66, 106]. Furthermore, whereas most antibodies against αSYN are directed against an epitope at the C terminus of αSYN (Table 1) [37], the exact amino acid sequences differ, and this may (co-)account for variability in the staining results. Finally, it has been suggested that the sparse retinal αSYN inclusions can easily be missed when evaluating a limited number of retinal sections; thus, retinal wholemount preparations are preferred over sections [97, 98].
Figure 2 shows a series of unpublished immunostainings for total, unmodified αSYN and phospho-αSYN on retinal sections and flatmount preparations of PD subjects and healthy controls, according to the methods published by Ortuno-Lizaran et al. [98]. As described in their recent manuscript, phospho-αSYN staining was found in axonal fibres, dendrites, and neuronal perikarya of PD retinas only, and phospho-αSYN-positive neurites displayed features of a dystrophic morphology (Fig. 2c–g). Importantly, immunostainings for native αSYN revealed identical staining patterns in PD and control retinas (Fig. 2a, b), again corroborating phospho-αSYN but not total/native αSYN as a potential biomarker of PD.

Immunostainings for total and phosphorylated αSYN on retinal sections and flatmounts of a PD patient and healthy control subject. a, b Immunostaining for total αSYN on retinal sections of a control and b PD subjects reveals αSYN expression in the photoreceptor outer segments, in cell bodies of the INL and GCL, and in their stratifications in the IPL, not displaying differences in the staining pattern between both groups. c Phospho-αSYN staining in a PD retinal section shows expression in axons (arrowheads) and cell body in the GCL (arrow). d–g Phospho-αSYN in retinal PD flatmounts is found in d ganglion cells (cell body and dendrites), e in axons in the NFL, and in f Lewy neurites (arrowheads) and f, g Lewy bodies located in the GCL. Scale bar 50 µm. NFL nerve fibre layer, GCL ganglion cell layer, INL inner nuclear layer, IPL inner plexiform layer, ONL outer nuclear layer, OPL outer plexiform layer, ONL outer nuclear layer, αSYN alpha-synuclein, p-αSYN αSYN phosphorylated at serine-129
In conclusion, half a dozen immunohistological studies have investigated the presence of native and phosphorylated αSYN in the retina. Their major findings are summarised in Fig. 1. They showed that native αSYN in healthy control retinas is most abundant in the photoreceptor outer segments, in the outer plexiform layer and in neurons and their processes in the inner nuclear layer and inner plexiform layer. Results from studies looking at PD retinas are somewhat contradictory, yet overall, they do provide evidence for the presence of phospho-αSYN-positive cell bodies and neurites in the retinal nerve fibre layer, ganglion cell layer, and inner plexiform layer. Several of these studies suggest that ganglion cells and (dopaminergic) amacrine cells are preferentially affected and that this also corresponds to the structural and functional changes that have been reported in OCT and ERG studies in PD patients [12, 24, 75, 84]. It currently remains unclear, however, how the significant structural effects can be reconciled with the relatively low numbers of dopaminergic amacrine cells in the retina, yet very large dopaminergic plexus, and detailed histological studies are warranted to univocally demonstrate the involvement of dopaminergic retinal neurons. Finally, earlier this year, Ortuno-Lizaran et al. were the first to provide evidence that PD progression in the retina and brain is related and that retinal phospho-αSYN inclusions may provide information about the disease severity in the brain, already in an early presymptomatic stage of PD.
The retina in animal models of Parkinson’s disease
The observation that retinal αSYN inclusions can be found in PD patients raises many new research questions, including whether the mechanistic pathway that leads to retinal neurodegeneration and/or -dysfunction is related to αSYN accumulation, whether retinal αSYN burden can be used as a biomarker for improved diagnosis and disease monitoring, how these αSYN inclusions can be visualised non-invasively in vivo, etc. Further research into αSYN pathology in the retina is thus highly needed, not only in PD patients, but also in animal models. As in the patient studies, PD research in animal models has been mainly focused on the brain, while the retina, and more specifically αSYN in the retina, is understudied [34, 120]. Nevertheless, several animal models are available to study PD, ranging from toxin-induced models to transgenic models to viral vector-based models, each having their specific strengths and limitations [18, 33, 59, 141], and thereby unique characteristics to address particular research questions [40, 47]. In the following section, we give an overview of the retinal manifestations that have been studied in these different animal models.
Toxin-induced models
Classical neurotoxin-induced rodent and primate models of PD make use of systemic administration or intracranial injection of drugs, such as 6-hydroxydopamine, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone or paraquat. Treated animals typically display dopaminergic neurodegeneration in the substantia nigra and striatal dopamine depletion, with subsequent motor syndrome [59, 141]. Although increased αSYN immunoreactivity was detected in the brain of 6-hydroxydopamine lesioned rats [55, 154, 158] and in MPTP mouse [156] and macaque monkey [50] models, actual Lewy bodies have not been detected [59, 141]. Only in the rat rotenone model, Lewy body-like inclusions are formed in the cytoplasm of nigral neurons [17, 59, 127].
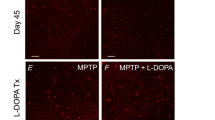
The failure of most toxin-induced models to display Lewy body pathology in the brain logically results in a limited number of studies looking at αSYN inclusions in the retina. For the rat rotenone model, one study by Normando et al. [93] revealed an increase in diffuse αSYN immunostaining in all retinal layers of treated rats, 60 days post rotenone administration [93]. Furthermore, in vivo OCT and DARC measurements showed that retinal neurodegeneration peaks at day 20 post rotenone administration in this model, while histological evidence for dopaminergic degeneration in the substantia nigra and striatum becomes only visible at day 60. As such, this was the first paper to report that significant alterations in the retina occur before the classical PD symptoms in the brain and to claim that the retina can be used as a potential biomarker tissue for early diagnosis [93]. Inner retinal layer thinning was also confirmed in other studies using the rodent rotenone model, in addition to decreased expression of dopaminergic cell markers and photoreceptor loss [42, 119, 155, 157]. While little attention has been devoted to retinal αSYN deposits, retinal function and structure have been studied more intensively in toxin-induced models, and has unveiled the importance of dopaminergic signalling in the retina. In 6-OHDA models, diminished dopamine levels were reported in rats [87], as well as abnormal PERG responses of the retinal ganglion cells in monkeys [23]. In MPTP-injected mice, rabbits, and monkeys, reductions in the number of tyrosine hydroxylase-immunopositive amacrine cells and in retinal dopamine levels were found [32, 56, 136, 152], leading to abnormal VEP and ERG responses and a decline in visual acuity and contrast sensitivity [23, 56]. In the MPTP monkey model, also retinal nerve fibre layer thinning, a decreased macula volume and foveal thickness were shown using OCT [125], similar to what is seen in PD patients. Finally, the potential of the retina as a site of novel biomarkers was underscored by a study applying cSLO imaging of retinal astrocytes in transgenic GFAP–GFP reporter mice that had received MPTP. Besides an increase in reactive gliosis that coincided with dopaminergic degeneration, this study also showed that this technology can be used to evaluate anti-inflammatory PD drugs [67].
Transgenic models
Besides toxin-induced models, genetic rodent models of PD carrying a wide array of mutations that have been linked to PD are used, including transgenic models with SNCA or LRRK2 mutations, and knockout mice of Smad-3, Parkin, PINK-1, and DJ-1 [33, 47]. Transgenic αSYN animals often develop proteinaceous inclusions in the brain, but lack robust neurodegeneration [33, 47, 141]. Even though a multitude of transgenic mice and rats carrying SNCA (αSYN) gene constructs has been created [18, 41, 59, 141], information on αSYN expression in the retina of these rodents is limited. In a recent publication of Mammadova et al. (2018), αSYN was shown to accumulate in the inner and outer retina of 8-month-old TgM83 transgenic mice, expressing A53T human αSYN under the control of the Prnp promoter, while phospho-αSYN was only present in the outer nuclear layer. In addition, transgenic mice of 8 months of age exhibited increased microglial activation, followed by increased GFAP immunoreactivity at later ages. No differences in retinal tyrosine hydroxylase immunoreactivity were observed [78]. Visual function abnormalities have not yet been characterised in this model.
Given the lack of well-characterised mouse models to study the retinal manifestations of PD, we performed αSYN and phospho-αSYN immunohistochemistry in the (Thy-1)-h[A30P]αSYN transgenic mouse eye. While the cerebral phenotype of these mice is thoroughly studied, the retinal phenotype is not yet touched upon. First, endogenous mouse αSYN expression (clone 42, BD Transduction Laboratories) was observed in sparse cell bodies in the ganglion cell layer and inner nuclear layer and in the inner plexiform layer of wild-type mice (Fig. 3a). Analysis of immunostainings for human native αSYN (clone Syn211, Millipore) and phospho-Ser-129 αSYN (clone 11A5, Elan Pharmaceuticals) revealed expression of the αSYN transgene in the inner retinal layers of (Thy-1)-h[A30P]αSYN mice, of which a proportion was phosphorylated (Fig. 3c–f). More in detail, prominent neuronal αSYN expression was observed in the ganglion cell layer, where the ganglion cells and displaced amacrine cells reside, as well as in their neurites in the retinal nerve fibre layer and inner plexiform layer. Moreover, dispersed cell bodies in the inner nuclear layer were stained, which, based on their morphology and location at the border of the inner plexiform and nuclear layers, are presumed to be amacrine cells (Fig. 3c). Whether this constitutes a subpopulation of dopaminergic amacrine cells remains to be investigated. Phospho-αSYN immunoreactivity was found in cell bodies in the ganglion cell layer and in the inner nuclear layer, bordering the inner plexiform layer; and in neurites in the inner plexiform layer and nerve fibre layer (Fig. 3d–f). Notably, these distribution patterns are strikingly similar to the ones observed after αSYN and phospho-αSYN immunostainings in human PD retinas (Fig. 2) (Table 1).

Immunostainings for total and phosphorylated αSYN on retinal sections and flatmounts of the Thy1-h[A30P]αSYN transgenic PD mouse model, at 15 months of age. a, b Immunostainings of retinal cross sections with an antibody recognising both human and mouse αSYN (clone 42, BD Transduction Laboratories) reveal a endogenous mouse αSYN expression in sparse cell bodies in the GCL and INL, and in the IPL of a wild-type mouse and b increased αSYN levels in Thy1-h[A30P]αSYN mice (versus wild-type) due to a combination of endogenous mouse αSYN and transgenic human αSYN expression. c Representative microscopic image of a retinal section immunostained for total (unmodified) human αSYN (clone Syn211, Millipore) shows expression in somata in the GCL and INL, and neurites/synapses in the IPL. d Phospho-αSYN immunostaining (clone 11A5, Elan Pharmaceuticals) on a retinal cross section reveals phospho-αSYN in cell bodies in the GCL and INL (arrow heads), in neurites/synapses in the IPL, and in ganglion cell axons (on top of the GCL). e, f Confocal images of a retinal flatmount at different focus planes along the z-axis, showing phospho-αSYN expression in e interneurons in the INL, f ganglion cells and displaced amacrine cells, ganglion cell axons (*), and dendrites (arrow) in the GCL. Of note, no human (transgenic) αSYN or phospho-αSYN is found in the wild types (data not shown). Scale bar 50 µm. GCL ganglion cell layer, INL inner nuclear layer, IPL inner plexiform layer, ONL outer nuclear layer, OPL outer plexiform layer, PL photoreceptor layer, αSYN alpha-synuclein, hαSYN human alpha-synuclein, p-αSYN αSYN phosphorylated at serine-129
In addition, a retinal imaging study in mice overexpressing the αSYN-eGFP fusion protein under the control of the PDGFβ promoter revealed an age-dependent increasing accumulation of αSYN-GFP in the retinal ganglion cells [113]. As such, this study presents a proof-of-concept for longitudinal in vivo imaging of the retina to monitor αSYN accumulation over time, and thus to evaluate potential therapies [113]. This further supports the possibility of using the eye for early diagnosis and follow-up of PD progression by monitoring αSYN accumulation in the retina, given that non-invasive imaging techniques for αSYN deposits become available (cfr. below) [98, 113].
Viral vector- and αSYN pre-formed fibril-based models
Animal models of diseases are inherently linked to their advantages and disadvantages, e.g., many of the transgenic models do not display dopaminergic degeneration and/or an L-DOPA responsive motor phenotype. In a counter reaction to limitations of the latter models, the use of αSYN-expressing viral vectors and/or inoculation of αSYN-pre-formed fibrils to induce local pathology is rising [123].
Local viral vector-mediated overexpression of αSYN in the mouse and rat substantia nigra induces progressive dopaminergic neurodegeneration, αSYN aggregate formation and motor impairment within a time span of 12 weeks [96, 102, 140]. Alternatively, in the inducible pre-formed αSYN fibril model, pre-formed fibrils, mimicking aggregated forms of αSYN found in Lewy bodies [26, 101, 108, 142], are injected into the striatum, substantia nigra, or other brain areas, inducing robust Lewy body and Lewy neurite formation, leading to loss of tyrosine hydroxylase-positive neurons and motor symptoms [108, 142, 143]. Importantly, this novel approach has been pivotal in the research into the prion-like behaviour of αSYN, and αSYN oligomers/fibrils have been shown to seed αSYN aggregation and spreading to more distant brain regions [36, 116]. Furthermore, a combinatorial approach using injection of fibrillary αSYN species in addition to viral vector injections can be used to speed up the pathological process and induce robust Lewy-like synucleinopathy, consistent neuroinflammation, progressive dopaminergic cell loss, and motor dysfunction [102, 137]. The viral vector-based models and in particular the pre-formed fibril models are quite novel, and therefore, it is not surprising that none of the papers that reported on these models included any investigation of the retina [96, 102, 108, 137, 142, 143]. However, the retina and the visual system should not be overlooked, especially since they constitute an ideal model system to study αSYN seeding/spreading. This was very recently shown in a study by Mammadova et al., in which retinal pathology in TgM83 mice was shown to be accelerated upon cerebral inoculation with brain homogenate from clinically ill transgenic mice. More in detail, compared to non-inoculated mice, the retinas of inoculated mice had accelerated and increased phospho-αSYN accumulation and microglia activation [78]. Another illustration of this concept comes from the field of prion disease research, where it was shown that mice inoculated with brain homogenate of a Creutzfeldt–Jakob disease patient displayed spreading of prion disease-related lesions along the visual pathway [74, 115].
Future perspectives
Reconciling the recent advances in our understanding of PD, including our increased knowledge on the underlying pathological mechanisms and the non-motor aspect of PD, new diagnostic criteria for PD have been defined by the International Parkinson Disease and Movement Disorders Society in 2017. Noteworthy is that the probability that any individual will develop PD (i.e., the criteria for prodromal PD) is calculated based on a weighted calculation of PD predictors. “These criteria represent only a first step in the correct description of early stages of PD, and will require constant updating as more information becomes available” [80, 111]. In this respect, the eye can be a window to the brain and the use of measures of visual function, retinal electrophysiology, neurodegeneration, and αSYN deposition, as PD biomarkers or criteria for prodromal PD are currently under intense investigation [138]. While the specificity and predictive value of OCT and ERG changes in PD patients are still under debate, due to overlap with normal ageing and other neurological and ophthalmological diseases, the presence of αSYN deposits in the retina may have high potential for early diagnosis of PD.
Two major challenges, however, still need to be tackled before in vivo, non-invasive imaging of retinal αSYN inclusions can be brought into practice. First, there remains a need for further studies confirming the presence of phospho-αSYN in the retina of PD patients and the timing of their occurrence in the retina relative to the brain. The limited number of neuropathological studies available does not allow to draw any final conclusions about the prevalence and specificity of retinal αSYN inclusions in PD, due to the low numbers of patients and differences in study protocols. In addition, future research should include studies with patients in different stages of PD, with different subtypes of PD, as well as age-matched controls, to assess the potential of retinal αSYN burden for monitoring and prediction of disease progression, and to evaluate to what extent it can differentiate between different α-synucleinopathies and normal ageing. Importantly, these trials should also concentrate on correlating retinal αSYN burden with ante mortem gold standard measures for PD diagnosis.
Second, an outstanding question is how to visualise retinal αSYN inclusions in living subjects. In this respect, the Alzheimer’s research field is already one step ahead. Albeit the neuropathological evidence for the presence of amyloid β plaques in the Alzheimer’s disease retina is as limited and controversial as for PD [97], several approaches for non-invasive, high-resolution visualisation of retinal amyloid β have already been developed [64, 132]. First, in vivo confocal scanning laser ophthalmoscopy imaging following systemic administration of curcumin, a fluorochrome that is safe to use by humans and labels amyloidogenic proteins, has shown promising results in Alzheimer’s disease patients, with a retinal amyloid β index that correlated well with cerebral amyloid β plaques [62, 63, 65, 126]. A second development is the use of BluePeak autofluorescence imaging that reveals metabolic stress in the retina using lipofuscin as an indicator, and thereby points to inclusion bodies. Inclusions were found in individuals with significant neocortical amyloid burden, but not in individuals without substantial amyloid aggregation in their brain [132]. Finally, hyperspectral imaging has been put forward as a promising approach for label-free amyloid imaging, potentially with a higher specificity than the two aforementioned techniques. This technique has been shown to detect a decrease in spectral reflectance at wavelengths between 460 and 570 nm that is indicative of increased Rayleigh scattering due to the presence of amyloid β. Comparing these spectra to a database with amyloid aggregates spectra, it is shown that hyperspectral imaging can accurately visualise retinal amyloid aggregates ex vivo in retinas of patients and in vivo in rodents [89, 90]. These three approaches are still in various stages of (pre-)clinical development and validation, yet may lead to a practical approach for large-scale Alzheimer’s diagnosis and monitoring [64, 89, 132].
Similar technologies are needed in the PD research field, to visualise retinal αSYN and/or phospho-αSYN and/or Lewy bodies in vivo. A recent advance in the PD imaging field is the development of bimolecular fluorescence complementation of αSYN, in which overexpression and oligomerisation of αSYN are detected in the brain via conjugated fluorescence. However, this experimental technology has not yet been translated to the retina, and is still very far from clinical development [28, 118]. Alternatively, with a specificity for amyloidogenic proteins rather than exclusively amyloid β, curcumin has been shown to bind to oligomeric αSYN species as well. In addition, the amount of curcumin binding is positively correlated with the extent of αSYN oligomerisation [131]. Together, this suggests that it may be an interesting candidate for in vivo labelling of pathogenic αSYN in the retina, in analogy with its use for amyloid labelling in the Alzheimer’s retina [64, 131]. Alternatively, the Anle138b compound may be used to detect αSYN aggregation. Animal studies have shown that, upon systemic administration, Anle138b binds aggregate-specific αSYN sequences, thereby reducing protein aggregation and conferring neuroprotection [73, 144, 145]. Remarkably, upon binding of oligomers, Anle138b shows a strong increase in fluorescence intensity around 335 nm. Although there might be some concerns about the specificity of Anle138b for αSYN, this suggests that it could be used in combination with confocal scanning laser ophthalmoscopy to visualise retinal αSYN deposits [35, 117, 145]. Finally, although not yet explored in any study, the theory of hyperspectral imaging could also be applied to αSYN, given that it also reveals a specific spectral signature as is the case for amyloid β.
In summary, besides the urgency for additional studies gathering more conclusive evidence for the manifestation of retinal deposits in the PD retina, there is also need for novel imaging techniques to visualise and quantify retinal αSYN burden. In the latter quest, animal models of PD will be pivotal, and therefore, there is also a demand for well-characterised models to study retinal alternations in PD. Alternatively, given the opportunities of the eye as a research model organ, studies focusing on the retina in this mouse line might help to unravel the complex interplay of disease mechanisms underlying PD.
Conclusion
In the search for improved methods for diagnosis and disease monitoring, PD experts have high expectations of novel approaches integrating multiple biomarkers and employing novel technologies to increase diagnostic yield. Compelling evidence for neuropathological, structural, and electrophysiological alterations in the PD retina has led to the suggestion that retinal biomarkers could fit in this approach. One of the most promising research tracks in this field is the manifestation of phospho-αSYN inclusions in the PD retina. To further propel research into the use of retinal αSYN burden as a diagnostic biomarker, however, a more solid base of evidence for the presence of retinal αSYN deposits in PD is urgently needed. Together with technological investments in the development of non-invasive, high-resolution, preferably label-free, αSYN-imaging techniques, this would allow to finally assess the potential of the retina as a site to monitor extracerebral αSYN. Notably, to tackle these challenges, preclinical studies in animal models of PD seem indispensable, and therefore, animal research looking at retinal manifestations of PD should not lag behind.
References
Adam CR, Shrier E, Ding Y, Glazman S, Bodis-Wollner I (2013) Correlation of inner retinal thickness evaluated by spectral-domain optical coherence tomography and contrast sensitivity in Parkinson disease. J Neuroophthalmol 33:137–142. https://doi.org/10.1097/WNO.0b013e31828c4e1a
Alafuzoff I, Parkkinen L, Al-Sarraj S, Arzberger T, Bell J, Bodi I et al (2008) Assessment of alpha-synuclein pathology: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol 67:125–143. https://doi.org/10.1097/nen.0b013e3181633526
Alonso R, Gonzalez-Moron D, Garcea O (2018) Optical coherence tomography as a biomarker of neurodegeneration in multiple sclerosis: a review. Mult Scler Relat Disord 22:77–82. https://doi.org/10.1016/j.msard.2018.03.007
Altintas O, Iseri P, Ozkan B, Caglar Y (2008) Correlation between retinal morphological and functional findings and clinical severity in Parkinson’s disease. Doc Ophthalmol 116:137–146
Alzheimer’sAssociation (2017) Alzheimer’s disease facts and figures. Alzheimer’s Dement 13:325–373
Ang M, Tan ACS, Cheung CMG, Keane PA, Dolz-Marco R, Sng CCA et al (2018) Optical coherence tomography angiography: a review of current and future clinical applications. Graefes Arch Clin Exp Ophthalmol 256:237–245. https://doi.org/10.1007/s00417-017-3896-2
Archibald NK, Clarke MP, Mosimann UP, Burn DJ (2009) The retina in Parkinson’s disease. Brain 132:1128–1145. https://doi.org/10.1093/brain/awp068
Archibald NK, Clarke MP, Mosimann UP, Burn DJ (2011) Visual symptoms in Parkinson’s disease and Parkinson’s disease dementia. Mov Disord 26:2387–2395. https://doi.org/10.1002/mds.23891
Armstrong RA (2011) Visual signs and symptoms of progressive supranuclear palsy. Clin Exp Optom 94:150–160. https://doi.org/10.1111/j.1444-0938.2010.00504.x
Armstrong RA (2011) Visual symptoms in Parkinson’s disease. Parkinsons Dis 2011:908306. https://doi.org/10.4061/2011/908306
Armstrong RA (2015) Oculo-visual dysfunction in parkinson’s disease. J Parkinsons Dis 5:715–726. https://doi.org/10.3233/JPD-150686
Aydin TS, Umit D, Nur OM, Fatih U, Asena K, Nefise OY et al (2018) Optical coherence tomography findings in Parkinson’s disease. Kaohsiung J Med Sci 34:166–171. https://doi.org/10.1016/j.kjms.2017.11.006
Bajwa A, Aman R, Reddy AK (2015) A comprehensive review of diagnostic imaging technologies to evaluate the retina and the optic disk. Int Ophthalmol 35:733–755. https://doi.org/10.1007/s10792-015-0087-1
Balendra SI, Normando EM, Bloom PA, Cordeiro MF (2015) Advances in retinal ganglion cell imaging. Eye (London, England) 29:1260–1269. https://doi.org/10.1038/eye.2015.154
Beach TG, Carew J, Serrano G, Adler CH, Shill HA, Sue LI et al (2014) Phosphorylated alpha-synuclein-immunoreactive retinal neuronal elements in Parkinson’s disease subjects. Neurosci Lett 571:34–38. https://doi.org/10.1016/j.neulet.2014.04.027
Bertrand JA, Bedetti C, Postuma RB, Monchi O, Genier Marchand D, Jubault T et al (2012) Color discrimination deficits in Parkinson’s disease are related to cognitive impairment and white-matter alterations. Mov Disord 27:1781–1788. https://doi.org/10.1002/mds.25272
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3:1301–1306. https://doi.org/10.1038/81834
Bezard E, Yue Z, Kirik D, Spillantini MG (2013) Animal models of Parkinson’s disease: limits and relevance to neuroprotection studies. Mov Disord 28:61–70. https://doi.org/10.1002/mds.25108
Bodis-Wollner I (2013) Foveal vision is impaired in Parkinson’s disease. Parkinsonism Relat Disord 19:1–14. https://doi.org/10.1016/j.parkreldis.2012.07.012
Bodis-Wollner I, Kozlowski PB, Glazman S, Miri S (2014) alpha-synuclein in the inner retina in parkinson disease. Ann Neurol 75:964–966. https://doi.org/10.1002/ana.24182
Bodis-Wollner I, Marx MS, Mitra S, Bobak P, Mylin L, Yahr M (1987) Visual dysfunction in Parkinson’s disease. Loss in spatiotemporal contrast sensitivity. Brain 110(Pt 6):1675–1698
Bodis-Wollner I, Miri S, Glazman S (2014) Venturing into the no-man’s land of the retina in Parkinson’s disease. Mov Disord 29:15–22. https://doi.org/10.1002/mds.25741
Bodis-Wollner I, Tzelepi A (1998) The push-pull action of dopamine on spatial tuning of the monkey retina: the effects of dopaminergic deficiency and selective D1 and D2 receptor ligands on the pattern electroretinogram. Vision Res 38:1479–1487
Boeke A, Rosen D, Mastrianni J, Xie T, Bernard J (2016) Optical coherence tomography as potential biomarker in parkinson’s disease and Alzheimer’s disease (P5.177). Neurology 86:P5–P177
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134. https://doi.org/10.1007/s00441-004-0956-9
Brundin P, Melki R (2017) Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 37:9808–9818. https://doi.org/10.1523/JNEUROSCI.1788-16.2017
Burguera JA, Vilela C, Traba A, Ameave Y, Vallet M (1990) The electroretinogram and visual evoked potentials in patients with Parkinson’s disease. Arch Neurobiol (Madr) 53:1–7
Cai W, Feng D, Schwarzschild MA, McLean PJ, Chen X (2018) Bimolecular fluorescence complementation of alpha-synuclein demonstrates its oligomerization with dopaminergic phenotype in mice. EBioMedicine 29:13–22. https://doi.org/10.1016/j.ebiom.2018.01.035
Cheung CY, Ikram MK, Chen C, Wong TY (2017) Imaging retina to study dementia and stroke. Prog Retin Eye Res 57:89–107. https://doi.org/10.1016/j.preteyeres.2017.01.001
Chorostecki J, Seraji-Bozorgzad N, Shah A, Bao F, Bao G, George E et al (2015) Characterization of retinal architecture in Parkinson’s disease. J Neurol Sci 355:44–48. https://doi.org/10.1016/j.jns.2015.05.007
Croisier E, Elfant D, Deprez M, Goldring K, Dexter DT, Pearce RK et al (2006) Comparative study of commercially available anti-alpha-synuclein antibodies. Neuropathol Appl Neurobiol 32:351–356. https://doi.org/10.1111/j.1365-2990.2006.00722.x
Cuenca N, Herrero MT, Angulo A, de Juan E, Martinez-Navarrete GC, Lopez S et al (2005) Morphological impairments in retinal neurons of the scotopic visual pathway in a monkey model of Parkinson’s disease. J Comp Neurol 493:261–273. https://doi.org/10.1002/cne.20761
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909
De Groef L, Cordeiro MF (2018) Is the eye an extension of the brain in central nervous system disease? J Ocul Pharmacol Ther 34:129–133. https://doi.org/10.1089/jop.2016.0180
Deeg AA, Reiner AM, Schmidt F, Schueder F, Ryazanov S, Ruf VC et al (2015) Anle138b and related compounds are aggregation specific fluorescence markers and reveal high affinity binding to alpha-synuclein aggregates. Biochim Biophys Acta 1850:1884–1890. https://doi.org/10.1016/j.bbagen.2015.05.021
Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S et al (2015) Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol 14:855–866. https://doi.org/10.1016/s1474-4422(15)00006-x
Dhillon JS, Riffe C, Moore BD, Ran Y, Chakrabarty P, Golde TE et al (2017) A novel panel of α-synuclein antibodies reveal distinctive staining profiles in synucleinopathies. PLoS One 12:e0184731. https://doi.org/10.1371/journal.pone.0184731
Diederich NJ, Raman R, Leurgans S, Goetz CG (2002) Progressive worsening of spatial and chromatic processing deficits in Parkinson disease. Arch Neurol 59:1249–1252
Djamgoz MB, Hankins MW, Hirano J, Archer SN (1997) Neurobiology of retinal dopamine in relation to degenerative states of the tissue. Vision Res 37:3509–3529. https://doi.org/10.1016/s0042-6989(97)00129-6
Duty S, Jenner P (2011) Animal models of Parkinson’s disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol 164:1357–1391. https://doi.org/10.1111/j.1476-5381.2011.01426.x
Eschbach J, Danzer KM (2014) alpha-Synuclein in Parkinson’s disease: pathogenic function and translation into animal models. Neurodegener Dis 14:1–17. https://doi.org/10.1159/000354615
Esteve-Rudd J, Fernandez-Sanchez L, Lax P, De Juan E, Martin-Nieto J, Cuenca N (2011) Rotenone induces degeneration of photoreceptors and impairs the dopaminergic system in the rat retina. Neurobiol Dis 44:102–115. https://doi.org/10.1016/j.nbd.2011.06.009
Frost, Sullivan (2017) Therapeutic breakthroughs in Alzheimer’s and Parkinson’s disease. http://www.frost.com/sublib/display-report.do?id=D7B4-01-00-00-00
Galvao J, Davis BM, Cordeiro MF (2013) In vivo imaging of retinal ganglion cell apoptosis. Curr Opin Pharmacol 13:123–127. https://doi.org/10.1016/j.coph.2012.08.007
Garcia-Martin E, Rodriguez-Mena D, Satue M, Almarcegui C, Dolz I, Alarcia R et al (2014) Electrophysiology and optical coherence tomography to evaluate Parkinson disease severity. Invest Ophthalmol Vis Sci 55:696–705. https://doi.org/10.1167/iovs.13-13062
Ghilardi MF, Bodis-Wollner I, Onofrj MC, Marx MS, Glover AA (1988) Spatial frequency-dependent abnormalities of the pattern electroretinogram and visual evoked potentials in a parkinsonian monkey model. Brain 111(Pt 1):131–149
Giraldez-Perez R, Antolin-Vallespin M, Munoz M, Sanchez-Capelo A (2014) Models of alpha-synuclein aggregation in Parkinson’s disease. Acta Neuropathol Commun 2:176. https://doi.org/10.1186/s40478-014-0176-9
Gottlob I, Schneider E, Heider W, Skrandies W (1987) Alteration of visual evoked potentials and electroretinograms in Parkinson’s disease. Electroencephalogr Clin Neurophysiol 66:349–357
Guo L, Normando EM, Shah PA, De Groef L, Cordeiro MF (2018) Oculo-visual abnormalities in Parkinson’s disease: possible value as biomarkers. Mov Disord 33:1390–1406. https://doi.org/10.1002/mds.27454
Halliday G, Herrero MT, Murphy K, McCann H, Ros-Bernal F, Barcia C et al (2009) No Lewy pathology in monkeys with over 10 years of severe MPTP Parkinsonism. Mov Disord 24:1519–1523. https://doi.org/10.1002/mds.22481
Harnois C, Di Paolo T (1990) Decreased dopamine in the retinas of patients with Parkinson’s disease. Invest Ophthalmol Vis Sci 31:2473–2475
Haug BA, Trenkwalder C, Arden GB, Oertel WH, Paulus W (1994) Visual thresholds to low-contrast pattern displacement, color contrast, and luminance contrast stimuli in Parkinson’s disease. Mov Disord 9:563–570. https://doi.org/10.1002/mds.870090510
Ho CY, Troncoso JC, Knox D, Stark W, Eberhart CG (2014) Beta-amyloid, phospho-tau and alpha-synuclein deposits similar to those in the brain are not identified in the eyes of Alzheimer’s and Parkinson’s disease patients. Brain Pathol 24:25–32. https://doi.org/10.1111/bpa.12070
Holroyd S, Currie L, Wooten GF (2001) Prospective study of hallucinations and delusions in Parkinson’s disease. J Neurol Neurosurg Psychiatry 70:734–738
Huang L, Deng M, He Y, Lu S, Liu S, Fang Y (2016) beta-asarone increases MEF2D and TH levels and reduces alpha-synuclein level in 6-OHDA-induced rats via regulating the HSP70/MAPK/MEF2D/Beclin-1 pathway: chaperone-mediated autophagy activation, macroautophagy inhibition and HSP70 up-expression. Behav Brain Res 313:370–379. https://doi.org/10.1016/j.bbr.2016.07.028
Huang YM, Yin ZQ (2011) Minor retinal degeneration in Parkinson’s disease. Med Hypotheses 76:194–196. https://doi.org/10.1016/j.mehy.2010.09.016
Ikeda H, Head GM, Ellis CJ (1994) Electrophysiological signs of retinal dopamine deficiency in recently diagnosed Parkinson’s disease and a follow up study. Vision Res 34:2629–2638
Inzelberg R, Ramirez JA, Nisipeanu P, Ophir A (2004) Retinal nerve fiber layer thinning in Parkinson disease. Vision Res 44:2793–2797. https://doi.org/10.1016/j.visres.2004.06.009
Jagmag SA, Tripathi N, Shukla SD, Maiti S, Khurana S (2015) Evaluation of models of Parkinson’s disease. Front Neurosci 9:503. https://doi.org/10.3389/fnins.2015.00503
Jankovic J (2008) Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79:368–376. https://doi.org/10.1136/jnnp.2007.131045
Jones RD, Donaldson IM, Timmings PL (1992) Impairment of high-contrast visual acuity in Parkinson’s disease. Mov Disord 7:232–238. https://doi.org/10.1002/mds.870070308
Kayabasi U, Sergott RC, Rispoli M (2014) Retinal examination for the diagnosis of Alzheimers disease. Int J Ophthalmol Clin Res. https://doi.org/10.23937/2378-346X/1410002
Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL et al (2011) Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54(Suppl 1):S204–S217. https://doi.org/10.1016/j.neuroimage.2010.06.020
Koronyo Y, Biggs D, Barron E, Boyer DS, Pearlman JA, Au WJ et al (2017) Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight. https://doi.org/10.1172/jci.insight.93621
Koronyo Y, Salumbides BC, Black KL, Koronyo-Hamaoui M (2012) Alzheimer’s disease in the retina: imaging retinal abeta plaques for early diagnosis and therapy assessment. Neurodegener Dis 10:285–293. https://doi.org/10.1159/000335154
Kovacs GG, Wagner U, Dumont B, Pikkarainen M, Osman AA, Streichenberger N et al (2012) An antibody with high reactivity for disease-associated α-synuclein reveals extensive brain pathology. Acta Neuropathol (Berl) 124:37–50. https://doi.org/10.1007/s00401-012-0964-x
Kumar S, Ho G, Zhang Y, Zhuo L (2010) In vivo imaging of retinal gliosis: a platform for diagnosis of PD and Screening of anti-PD compounds. Conf Proc 2010:3049–3052. https://doi.org/10.1109/iembs.2010.5626122
La Morgia C, Barboni P, Rizzo G, Carbonelli M, Savini G, Scaglione C et al (2013) Loss of temporal retinal nerve fibers in Parkinson disease: a mitochondrial pattern? Eur J Neurol 20:198–201. https://doi.org/10.1111/j.1468-1331.2012.03701.x
Langheinrich T, Tebartz van Elst L, Lagreze WA, Bach M, Lucking CH, Greenlee MW (2000) Visual contrast response functions in Parkinson’s disease: evidence from electroretinograms, visually evoked potentials and psychophysics. Clin Neurophysiol 111:66–74
Lee JY, Ahn J, Kim TW, Jeon BS (2014) Optical coherence tomography in Parkinson’s disease: is the retina a biomarker? J Parkinsons Dis 4:197–204
Lee JY, Kim JM, Ahn J, Kim HJ, Jeon BS, Kim TW (2014) Retinal nerve fiber layer thickness and visual hallucinations in Parkinson’s Disease. Mov Disord 29:61–67. https://doi.org/10.1002/mds.25543
Leger F, Fernagut PO, Canron MH, Leoni S, Vital C, Tison F et al (2011) Protein aggregation in the aging retina. J Neuropathol Exp Neurol 70:63–68. https://doi.org/10.1097/NEN.0b013e31820376cc
Levin J, Schmidt F, Boehm C, Prix C, Bötzel K, Ryazanov S et al (2014) The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol 127:779–780. https://doi.org/10.1007/s00401-014-1265-3
Liberski PP, Yanagihara R, Gibbs CJ Jr, Gajdusek DC (1990) Spread of Creutzfeldt-Jakob disease virus along visual pathways after intraocular inoculation. Arch Virol 111:141–147
London A, Benhar I, Schwartz M (2013) The retina as a window to the brain-from eye research to CNS disorders. Nat Rev Neurol 9:44–53
Ma MR, Hu ZW, Zhao YF, Chen YX, Li YM (2016) Phosphorylation induces distinct alpha-synuclein strain formation. Sci Rep 6:37130. https://doi.org/10.1038/srep37130
Mahlknecht P, Seppi K, Poewe W (2015) The concept of prodromal Parkinson’s disease. J Parkinsons Dis 5:681–697. https://doi.org/10.3233/JPD-150685
Mammadova N, Summers CM, Kokemuller RD, He Q, Ding S, Baron T et al (2018) Accelerated accumulation of retinal alpha-synuclein (pSer129) and tau, neuroinflammation, and autophagic dysregulation in a seeded mouse model of Parkinson’s disease. Neurobiol Dis 121:1–16. https://doi.org/10.1016/j.nbd.2018.09.013
Maresca A, la Morgia C, Caporali L, Valentino ML, Carelli V (2013) The optic nerve: a “mito-window” on mitochondrial neurodegeneration. Mol Cell Neurosci 55:62–76. https://doi.org/10.1016/j.mcn.2012.08.004
Marsili L, Rizzo G, Colosimo C (2018) Diagnostic criteria for parkinson’s disease: from James Parkinson to the concept of prodromal disease. Front Neurol 9:156. https://doi.org/10.3389/fneur.2018.00156
Martinez-Martin P, Rodriguez-Blazquez C, Mario A, Arakaki T, Arillo VC, Chana P et al (2015) Parkinson’s disease severity levels and MDS-Unified Parkinson’s Disease Rating Scale. Parkinsonism Relat Disord 21:50–54. https://doi.org/10.1016/j.parkreldis.2014.10.026
Martinez-Navarrete GC, Martin-Nieto J, Esteve-Rudd J, Angulo A, Cuenca N (2007) Alpha synuclein gene expression profile in the retina of vertebrates. Mol Vis 13:949–961
Mathur S, DeWitte S, Robledo I, Isaacs T, Stamford J (2015) Rising to the challenges of clinical trial improvement in Parkinson’s disease. J Parkinsons Dis 5:263–268. https://doi.org/10.3233/jpd-150541
Matlach J, Wagner M, Malzahn U, Schmidtmann I, Steigerwald F, Musacchio T et al (2018) Retinal changes in Parkinson’s disease and glaucoma. Parkinsonism Relat Disord. https://doi.org/10.1016/j.parkreldis.2018.06.016
Matsui H, Udaka F, Tamura A, Oda M, Kubori T, Nishinaka K et al (2006) Impaired visual acuity as a risk factor for visual hallucinations in Parkinson’s disease. J Geriatr Psychiatry Neurol 19:36–40. https://doi.org/10.1177/0891988705284739
Mazzarella J, Cole J (2016) All eyes on neurodegenerative diseases. Rev Optometry. https://www.reviewofoptometry.com/article/all-eyes-on-neurodegenerative-disease
Meng T, Zheng ZH, Liu TT, Lin L (2012) Contralateral retinal dopamine decrease and melatonin increase in progression of hemiparkinsonium rat. Neurochem Res 37:1050–1056. https://doi.org/10.1007/s11064-012-0706-4
Miri S, Shrier EM, Glazman S, Ding Y, Selesnick I, Kozlowski PB et al (2015) The avascular zone and neuronal remodeling of the fovea in Parkinson disease. Ann Clin Transl Neurol 2:196–201. https://doi.org/10.1002/acn3.146
More SS, Beach JM, Vince R (2016) Early detection of amyloidopathy in Alzheimer’s mice by hyperspectral endoscopy. Invest Ophthalmol Vis Sci 57:3231–3238. https://doi.org/10.1167/iovs.15-17406
More SS, Vince R (2015) Hyperspectral imaging signatures detect amyloidopathy in Alzheimer’s mouse retina well before onset of cognitive decline. ACS Chem Neurosci 6:306–315. https://doi.org/10.1021/cn500242z
Moschos MM, Tagaris G, Markopoulos I, Margetis I, Tsapakis S, Kanakis M et al (2011) Morphologic changes and functional retinal impairment in patients with Parkinson disease without visual loss. Eur J Ophthalmol 21:24–29
Nightingale S, Mitchell KW, Howe JW (1986) Visual evoked cortical potentials and pattern electroretinograms in Parkinson’s disease and control subjects. J Neurol Neurosurg Psychiatry 49:1280–1287
Normando EM, Davis BM, De Groef L, Nizari S, Turner LA, Ravindran N et al (2016) The retina as an early biomarker of neurodegeneration in a rotenone-induced model of Parkinson’s disease: evidence for a neuroprotective effect of rosiglitazone in the eye and brain. Acta Neuropathol Commun 4:86. https://doi.org/10.1186/s40478-016-0346-z
Nowacka B, Lubinski W, Honczarenko K, Potemkowski A, Safranow K (2014) Ophthalmological features of Parkinson disease. Med Sci Monit 20:2243–2249. https://doi.org/10.12659/MSM.890861
Nowacka B, Lubiński W, Honczarenko K, Potemkowski A, Safranow K (2015) Bioelectrical function and structural assessment of the retina in patients with early stages of Parkinson’s disease (PD). Doc Ophthalmol 131:95–104. https://doi.org/10.1007/s10633-015-9503-0
Oliveras-Salva M, Van der Perren A, Casadei N, Stroobants S, Nuber S, D’Hooge R et al (2013) rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol Neurodegener 8:44. https://doi.org/10.1186/1750-1326-8-44
Ong SS, Doraiswamy PM, Lad EM (2018) Controversies and Future directions of ocular biomarkers in Alzheimer disease. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2018.0602
Ortuno-Lizaran I, Beach TG, Serrano GE, Walker DG, Adler CH, Cuenca N (2018) Phosphorylated alpha-synuclein in the retina is a biomarker of Parkinson’s disease pathology severity. Mov Disord. https://doi.org/10.1002/mds.27392
Ortuno-Lizaran I, Esquiva G, Beach TG, Serrano GE, Adler CH, Lax P et al (2018) Degeneration of human photosensitive retinal ganglion cells may explain sleep and circadian rhythms disorders in Parkinson’s disease. Acta Neuropathol Commun 6:90. https://doi.org/10.1186/s40478-018-0596-z
Oueslati A (2016) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade? J Parkinsons Dis 6:39–51. https://doi.org/10.3233/jpd-160779
Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ et al (2015) Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82:185–199. https://doi.org/10.1016/j.nbd.2015.06.003
Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M et al (2015) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. https://doi.org/10.1038/nature14547
Peppe A, Stanzione P, Pierantozzi M, Semprini R, Bassi A, Santilli AM et al (1998) Does pattern electroretinogram spatial tuning alteration in Parkinson’s disease depend on motor disturbances or retinal dopaminergic loss? Electroencephalogr Clin Neurophysiol 106:374–382
Peppe A, Stanzione P, Pierelli F, De Angelis D, Pierantozzi M, Bernardi G (1995) Visual alterations in de novo Parkinson’s disease: pattern electroretinogram latencies are more delayed and more reversible by levodopa than are visual evoked potentials. Neurology 45:1144–1148
Peppe A, Stanzione P, Pierelli F, Stefano E, Rizzo PA, Tagliati M et al (1992) Low contrast stimuli enhance PERG sensitivity to the visual dysfunction in Parkinson’s disease. Electroencephalogr Clin Neurophysiol 82:453–457
Pikkarainen M, Martikainen P, Alafuzoff I (2010) The effect of prolonged fixation time on immunohistochemical staining of common neurodegenerative disease markers. J Neuropathol Exp Neurol 69:40–52. https://doi.org/10.1097/NEN.0b013e3181c6c13d
Poewe W (2008) Non-motor symptoms in Parkinson’s disease. Eur J Neurol 15(Suppl 1):14–20. https://doi.org/10.1111/j.1468-1331.2008.02056.x
Polinski NK, Volpicelli-Daley LA, Sortwell CE, Luk KC, Cremades N, Gottler LM et al (2018) Best practices for generating and using alpha-synuclein pre-formed fibrils to model Parkinson’s disease in rodents. J Parkinsons Dis. https://doi.org/10.3233/jpd-171248
Polo V, Satue M, Rodrigo MJ, Otin S, Alarcia R, Bambo MP et al (2016) Visual dysfunction and its correlation with retinal changes in patients with Parkinson’s disease: an observational cross-sectional study. BMJ Open 6:e009658. https://doi.org/10.1136/bmjopen-2015-009658
Possin KL (2010) Visual spatial cognition in neurodegenerative disease. Neurocase 16:466–487. https://doi.org/10.1080/13554791003730600
Postuma RB, Berg D (2017) The new diagnostic criteria for parkinson’s disease. Int Rev Neurobiol 132:55–78. https://doi.org/10.1016/bs.irn.2017.01.008
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W et al (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30:1591–1601. https://doi.org/10.1002/mds.26424
Price DL, Rockenstein E, Mante M, Adame A, Overk C, Spencer B et al (2016) Longitudinal live imaging of retinal alpha-synuclein:GFP deposits in a transgenic mouse model of Parkinson’s Disease/Dementia with Lewy Bodies. Sci Rep 6:29523. https://doi.org/10.1038/srep29523
Price MJ, Feldman RG, Adelberg D, Kayne H (1992) Abnormalities in color vision and contrast sensitivity in Parkinson’s disease. Neurology 42:887–890
Rahimi J, Milenkovic I, Kovacs GG (2015) Patterns of Tau and alpha-Synuclein pathology in the visual system. J Parkinsons Dis 5:333–340. https://doi.org/10.3233/JPD-140485
Recasens A, Dehay B (2014) Alpha-synuclein spreading in Parkinson’s disease. Front Neuroanat 8:159. https://doi.org/10.3389/fnana.2014.00159
Reiner AM, Schmidt F, Ryazanov S, Leonov A, Weckbecker D, Deeg AA et al (2017) Photophysics of diphenyl-pyrazole compounds in solutions and α-synuclein aggregates. BBA Gen Subj 22:22. https://doi.org/10.1016/j.bbagen.2017.12.007
Richter F (2018) Lighting up alpha-synuclein oligomers. EBioMedicine 29:3–4. https://doi.org/10.1016/j.ebiom.2018.02.016
Rojas JC, Saavedra JA, Gonzalez-Lima F (2008) Neuroprotective effects of memantine in a mouse model of retinal degeneration induced by rotenone. Brain Res 1215:208–217. https://doi.org/10.1016/j.brainres.2008.04.001
Santano C, Perez de Lara M, Pintor J (2011) Retinal disturbances in patients and animal models with Huntington’s, Parkinson’s and Alzheimer’s disease. In: Basu S, Wiklund L (eds) Oxidative stress in applied basic research and clinical practice—studies on experimental models. Humana, New York, pp 221–250
Sartucci F, Orlandi G, Bonuccelli U, Borghetti D, Murri L, Orsini C et al (2006) Chromatic pattern-reversal electroretinograms (ChPERGs) are spared in multiple system atrophy compared with Parkinson’s disease. Neurol Sci 26:395–401. https://doi.org/10.1007/s10072-006-0522-1
Sartucci F, Porciatti V (2006) Visual-evoked potentials to onset of chromatic red-green and blue-yellow gratings in Parkinson’s disease never treated with L-dopa. J Clin Neurophysiol 23:431–435. https://doi.org/10.1097/01.wnp.0000216127.53517.4d
Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B et al (2017) APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 36:2473–2487. https://doi.org/10.15252/embj.201797397
Satue M, Rodrigo MJ, Otin S, Bambo MP, Fuertes MI, Ara JR et al (2016) Relationship between visual dysfunction and retinal changes in patients with multiple sclerosis. PLoS One 11:e0157293. https://doi.org/10.1371/journal.pone.0157293
Schneider JS, Ault ME, Anderson DW (2014) Retinal pathology detected by optical coherence tomography in an animal model of Parkinson’s disease. Mov Disord 29:1547–1551. https://doi.org/10.1002/mds.25974
Shah TM, Gupta SM, Chatterjee P, Campbell M, Martins RN (2017) Beta-amyloid sequelae in the eye: a critical review on its diagnostic significance and clinical relevance in Alzheimer’s disease. Mol Psychiatry 22:353–363. https://doi.org/10.1038/mp.2016.251
Sherer TB, Kim JH, Betarbet R, Greenamyre JT (2003) Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol 179:9–16
Shrier EM, Adam CR, Spund B, Glazman S, Bodis-Wollner I (2012) Interocular asymmetry of foveal thickness in Parkinson disease. J Ophthalmol 2012:728457. https://doi.org/10.1155/2012/728457
Shults CW (2006) Lewy bodies. Proc Natl Acad Sci USA 103:1661–1668. https://doi.org/10.1073/pnas.0509567103
Silva MF, Faria P, Regateiro FS, Forjaz V, Januario C, Freire A et al (2005) Independent patterns of damage within magno-, parvo- and koniocellular pathways in Parkinson’s disease. Brain 128:2260–2271. https://doi.org/10.1093/brain/awh581
Singh PK, Kotia V, Ghosh D, Mohite GM, Kumar A, Maji SK (2013) Curcumin modulates alpha-synuclein aggregation and toxicity. ACS Chem Neurosci 4:393–407. https://doi.org/10.1021/cn3001203
Snyder PJ, Johnson LN, Lim YY, Santos CY, Alber J, Maruff P et al (2016) Nonvascular retinal imaging markers of preclinical Alzheimer’s disease. Alzheimers Dement (Amst) 4:169–178. https://doi.org/10.1016/j.dadm.2016.09.001
Spund B, Ding Y, Liu T, Selesnick I, Glazman S, Shrier EM et al (2013) Remodeling of the fovea in Parkinson disease. J Neural Transm (Vienna) 120:745–753. https://doi.org/10.1007/s00702-012-0909-5
Stenc Bradvica I, Mihaljevic I, Butkovic-Soldo S, Kadojic D, Titlic M, Bradvica M et al (2015) Transcranial sonography and the pocket smell test in the differential diagnosis between parkinson’s disease and essential tremor. Neurol Sci 36:1403–1410. https://doi.org/10.1007/s10072-015-2152-y
Tagliati M, Bodis-Wollner I, Kovanecz I, Stanzione P (1994) Spatial frequency tuning of the monkey pattern ERG depends on D2 receptor-linked action of dopamine. Vis Res 34:2051–2057
Tatton WG, Kwan MM, Verrier MC, Seniuk NA, Theriault E (1990) MPTP produces reversible disappearance of tyrosine hydroxylase-containing retinal amacrine cells. Brain Res 527:21–31
Thakur P, Breger LS, Lundblad M, Wan OW, Mattsson B, Luk KC et al (2017) Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci US A 114:E8284–E8293. https://doi.org/10.1073/pnas.1710442114
Turcano P, Chen JJ, Bureau BL, Savica R (2018) Early ophthalmologic features of Parkinson’s disease: a review of preceding clinical and diagnostic markers. J Neurol. https://doi.org/10.1007/s00415-018-9051-0
Ucak T, Alagoz A, Cakir B, Celik E, Bozkurt E, Alagoz G (2016) Analysis of the retinal nerve fiber and ganglion cell—inner plexiform layer by optical coherence tomography in Parkinson’s patients. Parkinsonism Relat Disord 31:59–64. https://doi.org/10.1016/j.parkreldis.2016.07.004
Van der Perren A, Toelen J, Casteels C, Macchi F, Van Rompuy AS, Sarre S et al (2015) Longitudinal follow-up and characterization of a robust rat model for Parkinson’s disease based on overexpression of alpha-synuclein with adeno-associated viral vectors. Neurobiol Aging 36:1543–1558. https://doi.org/10.1016/j.neurobiolaging.2014.11.015
Vingill S, Connor-Robson N, Wade-Martins R (2017) Are rodent models of Parkinson’s disease behaving as they should? Behav Brain Res. https://doi.org/10.1016/j.bbr.2017.10.021
Volpicelli-Daley LA, Kirik D, Stoyka LE, Standaert DG, Harms AS (2016) How can rAAV-alpha-synuclein and the fibril alpha-synuclein models advance our understanding of Parkinson’s disease? J Neurochem 139(Suppl 1):131–155. https://doi.org/10.1111/jnc.13627
Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A et al (2011) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. https://doi.org/10.1016/j.neuron.2011.08.033
Wagner J, Krauss S, Shi S, Ryazanov S, Steffen J, Miklitz C et al (2015) Reducing tau aggregates with anle138b delays disease progression in a mouse model of tauopathies. Acta Neuropathol (Berl) 130:619–631. https://doi.org/10.1007/s00401-015-1483-3
Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F et al (2013) Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol 125:795–813. https://doi.org/10.1007/s00401-013-1114-9
Watson R, Blamire AM, Colloby SJ, Wood JS, Barber R, He J et al (2012) Characterizing dementia with Lewy bodies by means of diffusion tensor imaging. Neurology 79:906–914. https://doi.org/10.1212/WNL.0b013e318266fc51
Webb RH, Hughes GW, Delori FC (1987) Confocal scanning laser ophthalmoscope. Appl Opt 26:1492–1499. https://doi.org/10.1364/ao.26.001492
Weil RS, Schrag AE, Warren JD, Crutch SJ, Lees AJ, Morris HR (2016) Visual dysfunction in Parkinson’s disease. Brain 139:2827–2843. https://doi.org/10.1093/brain/aww175
Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC et al (2013) The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement 9:e111–e194. https://doi.org/10.1016/j.jalz.2013.05.1769
Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J (2011) Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol 26(Suppl 1):S1–58. https://doi.org/10.1007/s10654-011-9581-6
Witkovsky P (2004) Dopamine and retinal function. Doc Ophthalmol 108:17–40
Wong C, Ishibashi T, Tucker G, Hamasaki D (1985) Responses of the pigmented rabbit retina to NMPTP, a chemical inducer of parkinsonism. Exp Eye Res 40:509–519
Xu Y, Deng Y, Qing H (2015) The phosphorylation of alpha-synuclein: development and implication for the mechanism and therapy of the Parkinson’s disease. J Neurochem 135:4–18. https://doi.org/10.1111/jnc.13234
Yin WL, Yin WG, Huang BS, Wu LX (2017) Neuroprotective effects of lentivirus-mediated cystathionine-beta-synthase overexpression against 6-OHDA-induced parkinson’s disease rats. Neurosci Lett 657:45–52. https://doi.org/10.1016/j.neulet.2017.07.019
Zhang L, Liu L, Philip AL, Martinez JC, Guttierez JC, Marella M et al (2015) Long-term evaluation of Leber’s hereditary optic neuropathy-like symptoms in rotenone administered rats. Neurosci Lett 585:171–176. https://doi.org/10.1016/j.neulet.2014.12.004
Zhang QS, Wang ZH, Zhang JL, Duan YL, Li GF, Zheng DL (2016) Beta-asarone protects against MPTP-induced Parkinson’s disease via regulating long non-coding RNA MALAT1 and inhibiting alpha-synuclein protein expression. Biomed Pharmacother 83:153–159. https://doi.org/10.1016/j.biopha.2016.06.017
Zhang X, Jones D, Gonzalez-Lima F (2006) Neurodegeneration produced by rotenone in the mouse retina: a potential model to investigate environmental pesticide contributions to neurodegenerative diseases. J Toxicol Environ Health A 69:1681–1697. https://doi.org/10.1080/15287390600630203
Zhang Y, Long H, Zhou F, Zhu W, Ruan J, Zhao Y et al (2017) Echinacoside’s nigrostriatal dopaminergic protection against 6-OHDA-Induced endoplasmic reticulum stress through reducing the accumulation of Seipin. J Cell Mol Med 21:3761–3775. https://doi.org/10.1111/jcmm.13285
Acknowledgements
We acknowledge support from Research Foundation Flanders (fellowships to Lies De Groef and Lien Veys).
Funding
LV and LDG are supported by the Research Foundation Flanders (fellowships 1S51718N and 12I3817N). IOL acknowledges financial support from the Ministerio de Educación, Spain (FPU 14/03166). NC acknowledges financial support from the Ministerio de Economía y Competitividad, Spain (MINECO-FEDER-BFU2015-67139-R), Generalitat Valenciana (Prometeo 2016/158), and Instituto Carlos III (ISCIII RETICS-FEDER RD16/0008/0016). The Brain and Body Donation Program has been supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026), the National Institute on Aging (P30 AG19610), the Arizona Department of Health Services, the Arizona Biomedical Research Commission, and the Michael J. Fox Foundation for Parkinson’s Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Veys, L., Vandenabeele, M., Ortuño-Lizarán, I. et al. Retinal α-synuclein deposits in Parkinson’s disease patients and animal models. Acta Neuropathol 137, 379–395 (2019). https://doi.org/10.1007/s00401-018-01956-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-01956-z