
Abstract
The exact mechanism underlying amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) associated with the GGGGCC repeat expansion in C9orf72 is still unclear. Two gain-of-function mechanisms are possible: repeat RNA toxicity and dipeptide repeat protein (DPR) toxicity. We here dissected both possibilities using a zebrafish model for ALS. Expression of two DPRs, glycine–arginine and proline–arginine, induced a motor axonopathy. Similarly, expanded sense and antisense repeat RNA also induced a motor axonopathy and formed mainly cytoplasmic RNA foci. However, DPRs were not detected in these conditions. Moreover, stop codon-interrupted repeat RNA still induced a motor axonopathy and a synergistic role of low levels of DPRs was excluded. Altogether, these results show that repeat RNA toxicity is independent of DPR formation. This RNA toxicity, but not the DPR toxicity, was attenuated by the RNA-binding protein Pur-alpha and the autophagy-related protein p62. Our findings demonstrate that RNA toxicity, independent of DPR toxicity, can contribute to the pathogenesis of C9orf72-associated ALS/FTD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A GGGGCC hexanucleotide repeat in the 5′ end of the C9orf72 gene is the most frequent genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [16, 22, 38]. ALS is characterized by motor neuron degeneration in the spinal cord, brainstem and motor cortex, and presents with progressive muscle weakness and wasting. It is rapidly progressive and usually fatal within 3–5 years after diagnosis. FTD presents with behavioral and/or language abnormalities, caused by degeneration of neurons in the frontal and temporal cortex. ALS and FTD form a continuum and are thought to be different clinical expressions of one disease [39].
The GGGGCC expansion in C9orf72 underlies ~ 40% of familial ALS and ~ 25% of familial FTD cases. It is also found in nearly 10 and 5% of patients with apparent non-familial ALS or FTD, respectively [37]. Control individuals most often carry 2–8 repeats, whereas patients have hundreds and even up to several thousands of repeats [46, 24]. At the cellular level, ALS/FTD associated with the C9orf72 expansion mutation (here called C9 ALS/FTD) is characterised by nuclear, and to a lesser extent cytoplasmic, RNA foci that contain sense GGGGCC or antisense CCCCGG repeats [26]. Furthermore, repeat-associated non-ATG (RAN) translation products (called dipeptide repeat proteins or DPRs) have been identified in the p62-positive, TDP43-negative inclusions that are characteristically found in C9 ALS/FTD patients. RAN translation is an unusual type of translation that generates aggregation-prone repeats of dipeptides from all three reading frames from both sense and antisense repeat RNA [19, 26].
The exact pathogenic mechanism underlying C9 ALS/FTD remains unknown. Because of reduced expression of C9orf72 transcripts in brain tissue and cells [3, 50], a loss-of-function mechanism has been proposed. Knockdown of C9orf72 in zebrafish and C. elegans indeed resulted in a neuronal phenotype [8, 44]. However, several mouse models with a general or neuron-specific deletion of C9orf72 did not develop clinical or pathological signs of motor neuron disease [2, 27, 28, 35, 42]. Instead, some models developed splenomegaly, lymphadenopathy, auto-immune disorders and neoplastic events, indicative of a primary role of the C9orf72 protein in immunity [2, 35, 42]. Interestingly, the C9orf72 protein has been shown to be a key player in regulation of autophagy [49]. Moreover, homozygosity for the repeat expansion does not generate a phenotype in humans that is worse than heterozygosity [18]. Altogether, the current data make loss-of-function rather unlikely to be a causal mechanism, although it remains to be investigated how loss-of-function mechanisms might contribute to the disease.
In contrast, mice overexpressing the hexanucleotide expansion developed a motor neuron syndrome accompanied by the same pathological hallmarks as observed in C9 ALS patients [7, 32]. Furthermore, treatment of iPSC-derived neurons (iPSNs) from C9 patients with C9 antisense oligonucleotides at least partially corrected their phenotype, all suggesting a gain-of-function pathogenic mechanism [17, 41].
Two such mechanisms have been proposed. First, a pathogenic gain-of-function mechanism relates to the toxicity of DPRs generated through RAN translation, and will be called ‘DPR toxicity’. The arginine-containing DPRs, PR (proline–arginine) and GR (glycine–arginine), have been shown to be toxic in yeast and Drosophila, whereas PR, GR, as well as GA (glycine–alanine) and GP (glycine–proline) are able to induce toxicity in a variety of cells [19]. Moreover, overexpression of GA might induce neurodegeneration in mice [53]. However, on postmortem examination of C9orf72 ALS patients, the pattern of DPR deposition poorly correlates with the pathological hallmarks of ALS (especially TDP43 pathology and motor neuron predominance) or with several clinical parameters [19, 26].
Alternatively, the GGGGCC repeat RNA has been hypothesized to be toxic itself (‘RNA toxicity’). Several mechanisms underlying such toxicity are possible, one of them being sequestration of proteins essential for normal RNA processing, which leads to widespread errors in RNA metabolism and eventually to neuronal dysfunction. This is similar to what has been suggested to occur in other RNA repeat diseases such as myotonic dystrophy. In vivo and in vitro studies identified a list of RNA-binding proteins that bind to C9 repeats [26]. Indeed, C9 repeats have been shown to induce splicing dysregulation [10], especially by sequestration of hnRNPH [9]. Additionally, C9 repeats might interfere with mRNA transport granules in vitro [6]. However, data supporting RNA toxicity in a vertebrate model are currently lacking.
While a gain-of-function mechanism seems to be evident, so far it is still unclear whether this relies on DPR toxicity or RNA toxicity, or a combination of both. Here, we dissect the two proposed gain-of-function mechanisms in an in vivo zebrafish model for C9 ALS. This model is similar to previously established ALS zebrafish models [29, 31] which have generated robust, relevant and translational insights into the pathogenesis of ALS [47].
Materials and methods
Plasmids and in vitro RNA transcription
Constructs containing 3, 4 or 10 GGGGCC repeats were synthesized (IDT, Leuven, Belgium), digested with EcoRI and subcloned into the pCMV6-Entry vector (Origene, Rockville, USA), containing a T7 promoter, to generate the p3-S, p4-S and p10-S plasmids. The p35-S and p35-AS plasmids, containing ~ 35 GGGGCC or ~ 35 CCCCGG repeats, were generated by EcoRI digestion of the pATG75 construct [25] and ligation into the pCMV6-Entry vector. Plasmids pUAST91, containing 91 GGGGCC hexanucleotide repeats, and pUAST108RO [33], containing 108 GGGGCC stop codon-interrupted repeats, were digested with EcoRI/NotI and KpnI, respectively, and the repeat was subcloned into the pCMV6-Entry vector to generate the p90-S and the p108RO-S plasmids. In both plasmids, a reverse complementary sequence of the T3 promoter was inserted 30 bp after the repeats to generate antisense transcripts. During propagation of these plasmids, shorter constructs containing ~ 70 non-stop (70S) or 70 stop codon-interrupted (70RO) repeats were obtained by spontaneous repeat deletions. Direction of the inserted repeats was controlled by Sanger sequencing (LGC Genomics, Berlin, Germany) and repeat length was estimated by restriction analysis. All final constructs were transformed and propagated in an E. coli Stbl3 background (Life Technologies, Gent, Belgium) to minimize spontaneous contractions/expansions of the repeat.
The plasmids encoding the DPRs GA, GR, PR and PA, all containing 50 repeats and a C-terminal FLAG tag, as well as the Pur-alpha deletion constructs, all containing a C-terminal FLAG tag, were synthesized (Genscript, Piscataway, USA) and subcloned into the pCMV6-Entry vector. The construct containing 50 GP repeats was obtained through spontaneous contraction of a construct containing 100 GP repeats [kind gift from M. Yamakawa (Keio University School of Medicine, Tokyo, Japan)] [52]. Sequences from all constructs can be provided upon request.
The human Pur-alpha FLAG-tagged encoding plasmid (RC206280) and the human p62 FLAG-tagged encoding plasmid (RC203214) were purchased from Origene.
To synthesize RNA, all plasmids were linearized by restriction digestion, transcribed with the mMESSAGE mMACHINE®T7 or T3 kit (Ambion, Huntingdon, UK) and the resulting RNA purified with the MEGAclear™ Kit (Ambion). RNA concentration was determined by spectrophotometry (Nanodrop, Thermo Fisher Scientific). RNA quality and length was verified by RNA gel electrophoresis.
Repeat RNA was fluorescently labeled with the Ulysis™ AlexaFluor™ 594 Nucleic Acid Labeling Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions and subsequently purified using Micro Bio-Spin® Columns with Bio-Gel® P-30 (Bio-Rad) according to the manufacturer’s instructions.
ATG blocking morpholinos against Danio rerio sqstm1 (transcript NM_001312913.1; morpholino sequence 5′-AGCTTTCACTGTCATCGACATCGTA-3′) and Danio rerio pur-alpha (transcript NM_001001846.1; morpholino sequence 5′-CCACTGTCTCTGTCCGCCATGATGC-3′) were designed and generated by Gene Tools (Philomath, USA). The standard control oligo provided by Gene Tools was used as negative control.
Zebrafish injections, SV2 immunohistochemistry (IHC) and phenotyping
Zebrafish work was performed as previously described [31]. One- to two-cell stage zebrafish embryos from the AB strain were injected in the yolk sac with the indicated amounts of the different RNAs. Injected embryos were kept in a 28 °C incubator. After 24-h post-fertilization (hpf) the embryos were manually dechlorinated using a forceps. Subsequently, the embryos underwent a rigorous visual inspection for dysmorphic features. Only morphologically normal embryos were retained. At 30 hpf, the morphologically normal fish were manually deyolked and subsequently fixed overnight at 4 °C in 4% formaldehyde in phosphate-buffered saline (PBS). Fish were permeabilized with acetone for 1 h at − 20 °C, blocked with 1% BSA/1% DMSO/PBS for 1 h at room temperature and immunostained with mouse anti-SV2 (1/200; AB2315387 (http://antibodyregistry.org), Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, USA) and secondary Alexa Fluor 555 anti-mouse antibody (1/500; Molecular Probes, Eugene, USA).
For phenotyping, 15 consecutive embryos per condition were analyzed with imaging (Leica DM 3000 LED microscope; DMK 33UX250 USB3.0 monochrome industrial camera, The Imaging Source, Bremen, Germany) using Lucia software (version 4.60, Laboratory Imaging, Prague, Czech Republic) by a blinded observer. For the axonal length, a standardized method was used; five predefined and consecutive motor axons (i.e., the 8th up to the 12th axon on one side) were measured in all 15 embryos. Data for axonal length were normalized to the control condition. For the abnormal branching, a predefined set of 20 consecutive motor axons (i.e., the 8th up to the 17th axon on both sides) in these same 15 embryos was analyzed visually. Motor axons were considered abnormal when axons branched at or before the ventral edge of the notochord. An embryo was considered as having ‘abnormal branching’ when at least two of these 20 axons were abnormal.
Number of biological replicates (‘n’) is indicated in each figure legend. Number of biological replicates equals at least three, which has been shown previously to be adequate to measure an effect [31]. For each experiment, GFP-encoding RNA was used as control at the same amount as the highest dose of the tested RNA.
Dot blot
Fish were homogenized in RIPA buffer supplemented with protease inhibitors (Complete, Roche) using sonication (Branson, model 102C) at 30 hpf. After centrifugation (10 min, 12,000 rpm) supernatant was collected and heated at 95 °C for 5 min. One to ten microliters of this supernatant was applied on a nitrocellulose membrane with pore size 0.2 µm (Amersham, GE Healthcare Life Sciences) and air-dried for 25 min. After overnight blocking in 5% non-fat dry milk in Tris-buffered saline (TBST; 10 mM Tris, 150 mM NaCl, pH 7.6) at 4 °C, membranes were incubated with the appropriate primary antibodies (rabbit anti-PR and rabbit anti-GP were obtained from Thermo Scientific (custom-made), rabbit anti-GR (23978-1-AP, validation data reported on the manufacturer’s website) antibody was obtained from Proteintech (Manchester, UK) at a dilution of 1/1000 in TBST for 2 h at room temperature. After three-time 15-min washing with TBST, blots were incubated with secondary antibody (polyclonal goat anti-rabbit-immunoglobulins/HRP, DAKO, 1/5000 in TBST) at room temperature. After two-time 15-min washing with TBST and one wash with TBS, blots were visualized by chemiluminescence (ECL Western Blotting Substrate or SuperSignal West Femto Maximum Sensitivity Substrate, Thermo Fisher Scientific; ImageQuant Las 4000, GE Healthcare Life Sciences).
Western blot
Fish samples were homogenized in RIPA buffer supplemented with protease inhibitors using sonication. After protein quantification (Micro BCA™ Protein Assay Kit #23235, Thermo Fisher Scientific, Waltham, USA) appropriate amounts of sample were resolved on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinyldifluoride membrane (Immobilon®-P, Merck Millipore, Billerica, USA) using a semi-dry blotting apparatus (TE70XP, Hoefer, San Francisco, USA). After blocking with 5% non-fat dry milk in TBST for 1 h at room temperature, the membranes were incubated overnight at 4 °C with the primary antibody (mouse anti-FLAG antibody, Sigma #F1804 (http://antibodyregistry.com AB_262044), 1/500; rabbit anti-p62, MBL International #PM045, 1/1000; mouse anti-beta-actin, Sigma #A5441 (http://antibodyregistry.com AB_476744), 1/1000; rabbit anti-Pur-alpha, Abcam #ab79936, 1/500) in TBST, washed three times with TBST and incubated with secondary antibody (polyclonal goat anti-rabbit/mouse-immunoglobulins/HRP, DAKO, 1/5000 in TBST) during 1 h. After two-time 15-min washing with TBST and one wash with TBS, blots were visualized by chemiluminescence (cf. supra).
GP immunoassay
Fish were homogenized in RIPA buffer supplemented with protease inhibitors using sonication. Levels of soluble poly(GP) were measured using a previously described sandwich immunoassay using a poly(GP) antibody [21].
qPCR
Total RNA was extracted with Trizol (Thermo Fisher Scientific, Waltham, USA) and precipitated with isopropyl alcohol according to the manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA with SuperScript III Reverse transcriptase (Thermo) and random hexamer primers, according to the manufacturer’s instructions. Quantitative PCR was performed using StepOnePlus™ (Life Technologies) with TaqMan® Fast Universal PCR Master Mix (Life Technologies). Gene expression assays were purchased from Life Technologies: Danio rerio p62, Dr03114502_m1; Danio rerio Acta 1b, Dr03440340_m1. Relative gene expression was determined by the 2−ΔΔct method and normalized to the average of the control group. Graphs represent gene expression relative to Acta1b expression.
RNA pull-down assay
Biotinylated RNAs used were generated by in vitro biotinylation (Pierce RNA 3′ END Biotinylation Kit, Thermo Scientific), according to the manufacturer’s instructions, from different constructs, indicated where appropriate. Adult mouse brain was homogenized in T-PER buffer (Thermo Fisher Scientific) supplemented with protease inhibitors by mechanical disruption for 30 s (MagNA Lyser, Roche). HEK cells were transiently transfected with the different Pur-alpha constructs using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions, lysed 48 h post-transfection with T-PER buffer and analyzed for protein concentration. Lysates were precleared by incubation with magnetic streptavidin-coated beads (Life Technologies) for 1 h at 4 °C. Ten picomoles of biotinylated repeat RNA were denatured at 95 °C for 10 min and incubated overnight at 4 °C with 1 mg of lysate. RNA and bound proteins were pulled down with 40 µl of magnetic streptavidin-coated beads for 1 h at 4 °C. Samples were washed five times in ice-cold PBS and denatured in SDS containing loading buffer and separated by SDS-PAGE followed by Western blotting.
Whole-mount zebrafish immunohistochemistry
Whole-mount zebrafish staining of embryos injected with fluorescently labeled repeat RNA or codon-optimized DPR constructs was similar to SV2 staining (cf. supra). Antibodies used were anti-FLAG (Sigma #F1804, 1/250), anti-eif3e (Abcam #ab36766, 1/250) and anti-Pur-alpha (Abcam #ab79936, 1/250). Subsequently, nuclei were visualized using NucBlue counterstaining (Thermo Fisher Scientific). Embryos were mounted using ProLong Gold antifade reagent (Life Technologies). Images were taken using Leica TCS SP8 confocal microscope equipped with Leica HyD™ detector and Leica Las X (version 3.1.1.15751) software. Quantification of RNA foci was performed using ImageJ software.
Statistical analysis
Statistics were performed using GraphPad Prism software (GraphPad Software, San Diego, USA) or RStudio [RStudio Team (2015). RStudio: Integrated Development for R. RStudio, Inc., Boston, MA URL (http://www.rstudio.com/)]. For zebrafish experiments axonal length was analyzed with one-way ANOVA (GraphPad Prism), Shapiro–Wilk normality test (GraphPad Prism) was used to check for normality. For multiple comparisons, Tukey’s post hoc correction was applied. Axonal length is represented as bar graphs with indication of mean and 95% confidence interval. Abnormal branching was pooled and analyzed using logistic regression (RStudio) and represented as a pooled proportion, with indication of 95% confidence interval. Protein expression assessed by Western blot was analyzed by two-tailed ratio paired t test (GraphPad Prism), Shapiro–Wilk normality test (GraphPad Prism) was used to check for normality, and represented as dot plots with indication of mean ± SEM. RNA levels assessed by qPCR were analyzed with one-way ANOVA (GraphPad Prism), Shapiro–Wilk normality test (GraphPad Prism) was used to check for normality, and represented as dot plots with indication of mean ± SEM. Significance level was defined at 0.05. Significance levels are indicated in the figures as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Results
Sense and antisense RNA repeats induce motor axon abnormalities in zebrafish
We first investigated whether GGGGCC repeat RNA was toxic to zebrafish motor axons. We injected RNA consisting of ~ 70 GGGGCC repeats in zebrafish embryos and compared motor axonal outgrowth to the control condition by injecting 3 GGGGCC repeat RNA. RNA of ~ 70 GGGGCC repeats dose dependently induced a motor axonopathy consisting of reduced axonal outgrowth and aberrant branching at 30 hpf (Fig. 1a–d). These abnormalities were very similar to those described for mutant SOD1 and TDP43 [29, 31], two other genetic causes of ALS. Next, we injected RNA consisting of 3, 4, 10, ~ 35, ~ 70 and ~ 90 GGGGCC repeats, in equimolar concentration to the highest dose (0.844 µM) used in Fig. 1c, d to determine a repeat length-dependent effect. Ten or less repeats did not affect axon morphology (Fig. 1e, f). From ~ 35 repeats on, a clear axonopathy was induced, which did not increase in severity with repeat length (Fig. 1e, f).

Zebrafish axonopathy induced by C9orf72 sense and antisense repeats. a, b Visualization of motor axons by immunohistochemistry (SV2 antibody) of 30 hpf zebrafish embryos injected with 3S (a) or ~ 70S (b) repeat RNA. c, d Axonal length (c) and aberrant axonal branching (d) of zebrafish embryos injected with increasing doses of 3 and ~ 70 sense repeat RNA, compared to GFP control RNA, n = 5 experiments. c Data represent mean ± 95% confidence interval (CI) one-way ANOVA, F(6, 754) = 18.87, ****p < 0.0001. d Data represent mean ± 95% CI logistic regression (z values compared to GFP − 0.796, − 0.877, − 0.200, 3.025, 0.104, 5.703; z values for 3S vs 70S: 1.373, 2.480, 4.538), *p < 0.05, **p < 0.01, ***p < 0.001. e, f Axonal length (e) and aberrant axonal branching (f) of zebrafish embryos injected with sense repeat RNA with a length of 3 (n = 4 experiments), 4 (n = 4 experiments), 10 (n = 4 experiments), ~ 35 (n = 4 experiments), ~ 70 (n = 24 experiments) and ~ 90 (n = 4 experiments) repeats at equimolar dose (0.844 µM). e Data represent mean ± 95% CI one-way ANOVA, F(6, 1140) = 57.16, ****p < 0.0001. f Data represent mean ± 95% CI logistic regression (z values compared to GFP 1.130, − 0.573, 1.130, 3.035, 3.063, 2.565), *p < 0.05, **p < 0.01. g, h Axonal length (g) and axonal branching (h) of zebrafish embryos injected with antisense repeat RNA with a length of ~ 35 (n = 4 experiments) and ~ 70 (n = 12 experiments) repeats at equimolar dose (0.844 µM). g Data represent mean ± 95% CI one-way ANOVA, F(2, 474) = 35.66, ****p < 0.0001. h Data represent mean ± 95% CI logistic regression (z values compared to GFP 2.450, 3.576), *p < 0.05, **p < 0.01. i–k Whole-mount staining (SV2 antibody) of 30 hpf zebrafish embryos injected with fluorescently labeled 3S, ~ 90S and ~ 70AS repeat RNA
Because the expanded repeat in patients is also transcribed in the antisense direction, we next studied the effect of expanded antisense repeats, in particular ~ 35 and ~ 70 CCCCGG repeats. Interestingly, injection of RNA consisting of ~ 70 antisense repeat RNA (0.844 µM) also induced a motor axonopathy in zebrafish (Fig. 1g, h). Of note, the ~ 35 antisense repeat RNA was less toxic to the fish (Fig. 1g, h) than the ~ 70 antisense repeat RNA (no effect on axonal length and a more limited effect on branching) indicating a higher threshold for toxicity compared to sense repeats.
These data implicate that both sense and antisense C9orf72 hexanucleotide repeat RNA induced motor neuron toxicity when injected into zebrafish embryos. Moreover, this toxicity displayed a length-dependent threshold effect and the antisense repeats were less toxic than the sense repeats.
Next, we investigated the cellular distribution of repeat RNA by injecting fluorescently labeled 3 sense, ~ 90 sense and ~ 70 antisense repeat RNA. All three RNAs were found to be diffusely present in the cytoplasm, and mainly in neural tissue (Online Resource 1a–h). In addition, they clearly formed cytoplasmic RNA foci (Fig. 1i–k), both in neural and non-neural tissue. Of note, these RNA foci did not colocalize with stress granules (Online Resource 1i–l).
Arginine-containing RAN translation products induce motor axon abnormalities in zebrafish
To determine whether RAN translation products mediate the toxicity of sense and antisense repeat RNA, we first investigated whether DPR proteins themselves are toxic to motor axons in the fish. To this end, DPR expression constructs were designed that generate the five possible DPRs observed in C9 ALS/FTD, i.e., GA, GR, GP, PR and PA (proline–alanine), all with a length of 50 repeats. These constructs contained an ATG start codon and were codon optimized [4]. This was obtained by choosing codons so that they encode the correct amino acid, but without repeats in the RNA, thus avoiding the potential toxicity of the repeat RNA. The expression of four DPRs (GR, PR, GA and GP), but not PA, was detectable in zebrafish using Western blot or dot blot (Fig. 2a).

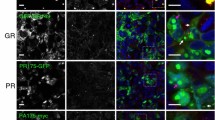
DPR-induced axonopathy in zebrafish. a Western blot using anti-FLAG antibody to detect formation of GA in fish injected with GA-coding mRNA (upper panel) and dot blots using DPR-specific antibodies to detect PR, GR and GP, respectively (three lower panels; pictures for GR and GP are derived from the dot blots in Fig. 3c), n = 3 biological replicates. b, c Quantification of axonal length (b) and aberrant axonal branching (c) of fish injected with equimolar amounts (0.844 µM) of codon-optimized RNA encoding a single DPR, compared to GFP control RNA (n = 18 experiments). b Data represent mean ± 95% CI one-way ANOVA, F(5, 1390) = 55.07, ****p < 0.0001. c Data represent mean ± 95% CI logistic regression (z values compared to GFP 1.693, 7.090, − 0.177, 7.410, − 0.533), ***p < 0.001. d, e Quantification of axonal length (d) and aberrant axonal branching (e) of fish injected with increasing doses of PR-encoding RNA with a mutation of the ATG start codon (‘PRmut’), n = 3 experiments. d Data represent mean ± 95% CI one-way ANOVA, F(3, 179) = 0.3464. e Data represent mean ± 95% CI logistic regression (z values compared to GFP 0.828, 0.558, 0.854). f–h Whole-mount staining (FLAG antibody) of 30 hpf zebrafish embryos injected with GA-coding mRNA
Expression of GR and PR in zebrafish induced dose-dependent toxicity (Fig. 2b, c and Online Resource 2). In contrast, expression of GP, GA or PA did not result in any motor axon abnormalities (Fig. 2b, c and Online Resource 2).
As an additional control to exclude an RNA-mediated contribution to the phenotype observed, we deleted the ATG start codon in the PR construct to abolish PR production (Fig. 2a). Injection of this RNA in zebrafish embryos did not induce any abnormalities of axonal length or branching (Fig. 2d, e). Altogether, these data suggest that arginine-containing DPRs (i.e., GR or PR) can induce motor neuron toxicity in the zebrafish model.
To investigate the cellular distribution of the DPRs in zebrafish embryos, we performed whole-mount staining using FLAG antibody. GA formed abundant cytoplasmic aggregates in varying locations in the embryo, both in neural and non-neural tissue (Fig. 2f–h). GR, GP, PR and PA did not form detectable aggregates on whole-mount staining (data not shown).
Repeat RNA toxicity is independent of DPR toxicity
To investigate whether the effects caused by the expanded sense and antisense repeats (Fig. 1) were a direct effect of RNA toxicity or whether they were (at least partially) mediated by DPR production through RAN translation, we next investigated the expression of DPRs in the injected fish.
First, we assessed whether PR, which is the only toxic DPR in our model that was generated from the antisense strand, was present in fish injected with antisense repeat RNA. Using an anti-PR dot blot assay, PR was not detectable in fish injected with the clearly toxic dose of ~ 70 antisense RNA (0.844 µM), while it was readily detectable in fish injected with decreasing doses of PR RNA (Fig. 3a–c). Interestingly, although in low amounts, PR was even detectable in non-toxic PR conditions (0.211 and 0.106 µM) (Fig. 3a–c). Combined, these data indicate that toxicity in our ~ 70 antisense RNA model is unlikely to be caused by PR generated through RAN translation.

DPRs are not detected in C9orf72 repeat RNA injected zebrafish. a, b Axonal toxicity of decreasing concentrations of PR-encoding mRNA and ~ 70AS (0.844 µM), statistics are compared to GFP condition (n = 6 experiments). a Data represent mean ± 95% CI one-way ANOVA, F(6,1009) = 21.09, ****p < 0.0001. b Data represent mean ± 95% CI logistic regression (z values compared to GFP; 4.554, 2.750, − 0.013, − 0.013, 4.900), **p < 0.01, ****p < 0.0001. c Dot blots of zebrafish samples (n = 3 biological replicates) at 30 hpf, all injected with equimolar amounts (0.844 µM) of indicated RNA. The upper panel represents a dot blot stained with anti-PR antibody of zebrafish samples of the same conditions as (a) and (b). The lower two panels represent dot blots, stained with GR- and GP-antibody, respectively. d Immunoassay using GP-antibody of zebrafish at 30 hpf (n = 3), all injected with equimolar amounts (0.844 µM) of indicated RNA (NI non-injected embryos)
Second, we investigated whether GR, the only toxic DPR generated from the sense strand in our model, is formed in fish injected with sense repeat RNA. Using an anti-GR dot blot assay, GR was not detected in any of the sense RNA conditions, while it was readily detectable in fish injected with RNA encoding GR protein (Fig. 3c). This suggests that sense toxicity in this model is unlikely to be caused by GR generated through RAN translation.
Third, we assessed the presence of GP, a DPR generated from the sense and/or antisense strand, which is not toxic in our model. GP was not detected, by dot blot assay, in any of the sense or antisense conditions, while it was detected in fish injected with GP RNA (Fig. 3c). GP was detected by immunoassay in fish injected with the toxic sense repeat RNA, but only at very low levels (marginally above the limit of detection) compared to fish injected with GP RNA (Fig. 3d).
We next excluded the contribution of low levels of DPRs acting synergistically with the toxicity induced by repeat RNA. This is of interest since in C9 patients more than one DPR may co-exist in the same cell. To investigate this, we first co-expressed low levels (0.211 µM, Online Resource 2) of all four DPRs of which the expression could be demonstrated using dot blot or Western blot (Fig. 2a). Coexpression of these DPRs did not result in synergistic toxicity of these DPRs (Fig. 4a, b). Second, since DPRs also co-exist with repeat RNA in cells of C9 patients, we also co-injected the combination of DPRs with low non-toxic levels (0.211 µM, Fig. 1c, d) of ~ 70 sense repeat RNA. Again, no increase in toxicity was noted (Fig. 4a, b). Third, low non-toxic levels (0.211 µM) of ~ 90 sense or ~ 70 antisense repeat RNA were co-injected with high non-toxic levels (0.844 µM) of GA- or GP-coding RNA. Again, neither GA nor GP were found to act synergistically with sense (Fig. 4c, d) or antisense (Fig. 4e, f) repeat RNA. Altogether, these data suggest that the toxicity of repeat RNA is not mediated through a synergistic effect with several DPRs, further supporting the existence of RNA toxicity.

Supporting evidence for RNA toxicity. a, b Axonal length (a) and aberrant axonal branching (b) upon injection with a low dose (0.211 µM) of a single DPR-encoding mRNA, a combination of four DPRs at the same dose or these four DPRs together with a low dose (0.211 µM) of ~ 70S repeat RNA (n = 6 experiments). a Data represent mean ± 95% CI one-way ANOVA, F(6826) = 6.5, *p < 0.05. b Data represent mean ± 95% CI logistic regression (z values compared to GFP 1.688, 1.543, 0.454, − 0.014, 2.166, 2.005), *p < 0.05. c, d Axonal length (c) and aberrant axonal branching (d) upon injection with a high dose (0.844 µM) of GA- or GP-encoding mRNA with a low dose (0.211 µM) of ~ 90S repeat RNA (n = 4 experiments). c Data represent mean ± 95% CI one-way ANOVA, F(5, 459) = 2.55. d Data represent mean ± 95% CI logistic regression (z value of GA + GFP vs GA + 91S: 0, z value of GP + GFP vs GP + 91S − 0.467). e, f Axonal length (e) and aberrant axonal branching (f) upon injection with high dose (0.844 µM) of GA- or GP-encoding mRNA with low dose (0.211 µM) of ~ 70AS repeat RNA (n = 4 experiments). e Data represent mean ± 95% CI one-way ANOVA, F(5, 464) = 1.099. f Data represent mean ± 95% CI logistic regression (z value of GA + GFP vs GA + 70AS: 0.388, z value of GP + GFP vs GP + 70AS − 0.939). g, h Axonal length (g) and aberrant axonal branching (h) upon injection with stop codon-interrupted sense (white bars) and antisense (red bars) repeat RNA at equimolar dose (0.844 µM), n = 7 experiments. g Data represent mean ± 95% CI one-way ANOVA, F(6,1659) = 40.94, *p < 0.05, ***p < 0.001, ****p < 0.0001. h Data represent mean ± 95% CI logistic regression (z values compared to GFP 8.507, 1.287, 4.823, 4.573, − 1.259, 0.113; z value of 70S vs 70RO − 5.487; z value of 70AS vs 70RO AS − 2.843), **p < 0.01, ***p < 0.001, ****p < 0.0001
Finally, we studied the effect of stop codon-interrupted repeat RNA, designated ‘RNA only’ (RO) repeat RNA. It has previously been shown that these constructs are not RAN translated and do not generate DPRs, while they still adopt the same stable conformation as the uninterrupted GGGGCC repeat RNA [33]. Interestingly, in spite of not being able to generate DPRs, sense RO repeat RNA (70RO and 108RO) still significantly affected axonal length and axonal branching, although to a somewhat lesser degree than the uninterrupted repeat RNA (Fig. 4g, h). Similarly, antisense RO repeat RNA (70RO AS and 108RO AS) was still toxic to the fish but to a lesser extent than uninterrupted repeat RNA (Fig. 4g, h). Thus, the presence of stretches of either sense or antisense C9orf72 hexanucleotide repeat RNA in the complete absence of any DPRs was sufficient to cause motor neuron toxicity in our zebrafish model, supporting the existence of RNA toxicity.
Altogether, our results indicate that expanded sense and antisense repeats are directly toxic to motor axons, independent of the formation of DPRs.
RNA-binding protein Pur-alpha prevents the axonopathy induced by sense and antisense repeat RNAs
In patients, RNA toxicity of C9orf72 hexanucleotide repeat expansions has been hypothesized to be due to the sequestration of several RNA-binding proteins [23]. Therefore, we investigated whether different RNA-binding proteins known to bind GGGGCC repeats, such as Pur-alpha [12, 41, 51], hnRNPH1 [30] and hnRNPA1 [41], are also able to bind the toxic RNA repeats used in our model. Pur-alpha indeed bound to the toxic sense (~ 70S, 108RO) repeat RNA and, albeit with less affinity, to the toxic antisense (~ 70AS, 108RO AS) repeat RNA (Online Resource 3a). Moreover, hnRNPA1 and hnRNPH1 also bound the ~ 70S repeat RNA (Online Resource 3a).
Next, we studied the effect of these RNA-binding proteins on the axonal phenotype induced by toxic repeat RNA in the zebrafish. Expression of Pur-alpha dose dependently prevented the axonal abnormalities induced by the ~ 70 sense repeat RNA (Fig. 5a, b). In contrast, expression of hnRNPA1 and hnRNPH1 at its highest non-lethal concentration had no effect on the toxic effect of sense repeat RNA (Fig. 5c, d), indicating that the protective effect of Pur-alpha was specific, rather than a general effect of any RNA-binding protein. Interestingly, overexpression of Pur-alpha induced a 50% decrease in ~ 90 sense RNA foci (Fig. 5e–g), whereas Pur-alpha was only occasionally found to colocalize with 3 sense, ~ 90 sense and ~ 70 antisense RNA foci (Online Resource 3g–j). Of note, Pur-alpha also prevented the toxicity induced by the ~ 70 antisense repeat RNA (Online Resource 3b, c) and the 108RO sense repeat RNA (Online Resource 3d, e), which is DPR independent.

Pur-alpha prevents repeat induced axonopathy in the zebrafish. a, b Dose-dependent effect of injection of Pur-alpha mRNA (0.072–0.143–0.287–0.573 µM) on ~ 70S (0.844 µM) axonal toxicity, statistics are compared to the second condition, i.e., 70S (n = 3 experiments). a Data represent mean ± 95% CI one-way ANOVA, F(6, 310) = 21.56, ****p < 0.0001. b Data represent mean ± 95% CI logistic regression (z values − 3.516, − 0.393, − 1.272, − 1.594, − 2.007, − 2.909), *p < 0.05, **p < 0.01, ***p < 0.001. c, d Effect of hnRNPA1 (‘A1’; 0.108 µM) and hnRNPH1 (‘H1’; 0.030 µM) on ~ 90S (0.844 µM) axonal toxicity, statistics are compared to the second condition, i.e., 90S + GFP (n = 8 experiments). c Data represent mean ± 95% CI one-way ANOVA, F(3, 460) = 29.63, ****p < 0.0001. d Data represent mean ± 95% CI logistic regression (z values − 3.157, 0.393, − 0.624), **p < 0.01. e, f Whole-mount staining (Hoechst) of 30 hpf zebrafish embryos injected with fluorescently labeled ~ 90S repeat RNA together with GFP (e) or Pur-alpha (f) Mrna, respectively. g Quantification of number of RNA foci per cell in conditions of panel e and f, n = 8 embryos, data represent mean ± SEM, unpaired t test (t14 = 5.677), ****p < 0.0001. h, i Effect of Pur-alpha (0.573 µM) on PR and GR (0.844 µM) axonal toxicity (n = 6 experiments). h Data represent mean ± 95% CI one-way ANOVA, F(4, 444) = 18.49, (PR + GFP vs PR + Pur-alpha: p = 0.7575; GR + GFP vs GR + Pur-alpha: p = 0.9235). i Data represent mean ± 95% CI logistic regression (PR + GFP vs PR + Pur-alpha: z value = − 1.340, p = 0.18; GR + GFP vs GR + Pur-alpha: z value = − 1.123, p = 0.261)
Finally, we tested the effect of Pur-alpha on DPR toxicity. Despite being able to bind the RNA encoding GR (Online Resource 3f), Pur-alpha overexpression did not prevent the axonopathy induced by it, nor by the RNA encoding PR (Fig. 5h, i). Thus, in our model Pur-alpha prevents repeat RNA toxicity but does not modify DPR toxicity. This differential effect of Pur-alpha again underscores the conclusion that a different pathophysiological mechanism underlies these two forms of toxicity.
The protective effect of Pur-alpha is mediated by its G-rich and PUR2 domain
To further explore the protective role of Pur-alpha, we generated and expressed five different Pur-alpha constructs in the zebrafish, each lacking one of the five functional domains [14] (Fig. 6a, b) and tested their ability to prevent the toxicity induced by toxic sense repeat RNA.

The protective effect of Pur-alpha is mediated by its G-rich and PUR2 domain. a Pur-alpha protein and the five deletion mutants; numbers indicate amino acid position in N- to C-terminal order. b Confirmation of in vivo translation of mRNA encoding the different Pur-alpha mutants in 30 hpf zebrafish embryos by Western blot (FLAG antibody), n = 2 biological replicates. c, d Binding affinity of the different Pur-alpha deletion mutants to ~ 70S RNA as assessed by RNA pull-down (‘RNA PD’) followed by Western blotting using FLAG antibody, n = 4 biological replicates (c—upper panel). Input; immunoprecipitation (FLAG antibody) of HEK cells transfected with the different Pur-alpha deletion mutants followed by Western blot using FLAG antibody (c–lower panel). d Quantification of RNA pull-down, data are normalized to input. Data represent mean ± SEM, one-way ANOVA, F(5, 17) = 3.471. e, f Effect of the different Pur-alpha deletion mutants at equimolar dose (0.573 µM) on ~ 90S (0.844 µM) axonal toxicity (n = 5 experiments), statistics are compared to the second condition (90S + GFP). e Data represent mean ± 95% CI one-way ANOVA, F(7, 827) = 23.37, ****p < 0.0001. f Data represent mean ± 95% CI logistic regression (z values − 3.350, − 2.634, − 0.559, − 2.506, − 0.816, − 1.439, − 2.584), *p < 0.05, **p < 0.01, ***p < 0.001
First, the binding affinity of these recombinant Pur-alpha proteins to sense repeat RNA was assessed by RNA pull-down using biotinylated ~ 90 sense repeat RNA. Interestingly, all Pur-alpha deletion proteins were still able to bind sense repeat RNA to a similar extent as the wild-type Pur-alpha protein (Fig. 6c, d).
Next, we co-injected toxic sense RNA either with RNA encoding GFP (control), wild-type Pur-alpha or one of the five Pur-alpha deletion proteins. We found that the G-rich domain and the PUR2 domain, but not the PUR1, the PUR3 or the EQ domains, are necessary for Pur-alpha to have its effect on repeat RNA-induced toxicity, as their deletion abolished the protective effect (Fig. 6e, f). As the binding of the ΔG and ΔPUR2 proteins to the ~ 90 sense RNA is not altered, these data suggest that the G-rich domain and the PUR2 domain mediate a downstream effect of the Pur-alpha protein which is responsible for the protection against repeat RNA toxicity.
p62 is upregulated by Pur-alpha and prevents repeat RNA toxicity
As p62 protein levels have previously been shown to be increased in C9orf72 iPSC-derived neurons [1, 13], we investigated the possible involvement of p62 in our zebrafish model. Injection of sense repeat RNA did not modify the protein (Fig. 7a, Online Resource 4a) and mRNA (Online Resource 4b) levels of p62. However, overexpression of Pur-alpha increased protein levels of p62 without affecting the p62 mRNA levels, while p62 protein levels were not increased upon overexpression of ΔG and ΔPUR2 (Fig. 7b, Online Resource 4c). Since ΔG and ΔPUR2 also failed to prevent RNA toxicity (Fig. 6e, f), this suggests that p62 induction is necessary for Pur-alpha to have its protective effect. Indeed, upon morpholino-mediated p62 knockdown (Online Resource 4d, e), the protective effect of Pur-alpha was abolished (Fig. 7c, d). Interestingly, morpholino-mediated Pur-alpha knockdown resulted in a decrease in p62 levels (Fig. 7e, Online Resource 4f), again indicating that Pur-alpha modulates the p62 protein level.

p62 is necessary and sufficient for the protective effect of Pur-alpha on RNA toxicity. a Quantification of Danio rerio p62 protein levels in 30 hpf zebrafish embryos upon injection of ~ 70S (0.844 µM) repeat RNA as assessed by Western blot. Data represent mean ± SEM, statistics are compared to GFP condition, n = 8 experiments, ratio paired t test (t7 = 0.2229). b Quantification of Danio rerio p62 protein levels in 30 hpf zebrafish embryos upon injection of PURA, ΔG or ΔPUR2 mRNA at equimolar dose (0.573 µM) as assessed by Western blot. Data represent mean ± SEM, statistics are compared to GFP condition, n = 7–11 experiments, ratio paired t test (PURA t10 = 3.316, ΔG t6 = 2.64, ΔPUR2 t6 = 3.142), *p < 0.05, **p < 0.01. c, d Effect of morpholino (0.30 mM)-mediated p62 knockdown on the protective effect of Pur-alpha on RNA toxicity (n = 11 experiments; p62 MO p62 morpholino, co MO control morpholino). c Data represent mean ± 95% CI one-way ANOVA, F(4, 789) = 28.24, ***p < 0.001, ****p < 0.0001. d Data represent mean ± 95% CI logistic regression (z value of condition 2 vs 3: − 2.363; z value of condition 4 vs 5: 3.027), *p < 0.05, **p < 0.01. e Quantification of Danio rerio pura and p62 protein levels in 30 hpf zebrafish embryos upon morpholino (0.05 mM)-mediated pura knockdown (purMO pura morpholino). Data represent mean ± SEM, statistics are compared to control morpholino condition, n = 10 experiments, ratio paired t test (pura t9 = 3.717, p62 t9 = 3.532), **p < 0.01. f, g Effect of injection of p62 mRNA (0.153 µM) on ~ 70S (0.844 µM) axonal toxicity (n = 6 experiments). f Data represent mean ± 95% CI one-way ANOVA, F(2, 258) = 18.46, ****p < 0.0001. g Data represent mean ± 95% CI logistic regression (z values compared to GFP 2.613, 0.596; z value of 70S + GFP vs 70S + p62 − 2.165), *p < 0.05, **p < 0.01. h, i Effect of p62 (0.153 µM) on GR (0.844 µM) axonal toxicity (n = 7 experiments). h Data represent mean ± 95% CI one-way ANOVA, F(2, 309) = 6.546, *p < 0.05, **p < 0.01. i Data represent mean ± 95% CI logistic regression (z values compared to GFP 3.205, 2.120; z value of GR + GFP vs GR + p62 − 1.246), *p < 0.05, **p < 0.01
To assess whether overexpression of p62 had a protective effect on RNA toxicity, p62 RNA was co-injected with sense repeat RNA in zebrafish oocytes. Indeed, overexpression of p62 almost completely prevented repeat RNA toxicity (Fig. 7f, g). Interestingly and similar to Pur-alpha, overexpression of p62 did not affect the toxicity induced by GR (Fig. 7h, i), again highlighting the different underlying mechanisms of RNA versus DPR toxicity. Altogether these data suggest that the protective effect of Pur-alpha is mediated by its modulating effect on p62.
Discussion
We generated a novel C9 ALS/FTD repeat-associated model in zebrafish to investigate the mechanism through which this repeat causes toxicity. We found that injection of sense ~ 70 GGGGCC repeat RNA was dose dependently toxic to motor axons in this model. It induced a phenotype similar to that of overexpression of mutant SOD1 and TDP-43 as we have shown before [29, 31]. When testing a range of repeat lengths, we found the threshold of sense toxicity to be between 10 and ~ 35 repeats. Interestingly, above this threshold the phenotype was not repeat length dependent, as the ~ 35, ~ 70 and the ~ 90 sense repeat RNA induced a similar phenotype. Both findings are in line with a presumed threshold of pathogenicity around 30 repeats in C9 ALS/FTD patients on the one hand and the lack of a clear correlation between repeat length and phenotype on the other hand [26, 48]. However, no firm conclusions can be drawn since repeat lengths in the range of hundreds to thousands, as is mostly the case in C9orf72 ALS patients, could not be investigated in this model due to the inherent instability of these constructs.
We also report, for the first time, direct in vivo toxicity of the antisense repeat RNA. In our model, the threshold for the antisense repeat to induce toxicity was higher as compared to that of the sense toxicity; it was between ~ 35 and ~ 70 repeats, while the latter is between 10 and ~ 35 repeats.
Using this model, we investigated whether expanded GGGGCC repeat RNA can be directly toxic, independent of the generation of DPRs. We first tested DPR toxicity by generating DPRs from codon-optimized constructs. These constructs generate DPRs without the need for repeat RNA using the freedom afforded by the wobble base principle. When testing all DPRs in our zebrafish model, GR and PR induced motor axon toxicity, whereas PA, GA and GP induced no obvious toxicity. It is unlikely that RNA toxicity contributes to the PR- and GR-induced axonopathy because of the absence of repeats in the RNA sequence. Furthermore, elimination of the ATG initiation codon abolished the toxic effect of the PR construct. The finding that mostly arginine-containing DPRs display neurotoxicity in this in vivo model is in line with the findings in Drosophila [4, 33] and provides evidence for a potential role of these DPRs in C9 ALS/FTD pathology. Despite the observation of GA forming abundant aggregates, it failed to induce motor axon toxicity in our model. Hence, this in vivo model does not support the relationship between the aggregating aspect of GA and its toxicity as was suggested by others [53].
Based on our findings from DPR-expressing fish, one could hypothesize that the neurotoxicity observed in repeat RNA injected fish is due to the formation of toxic arginine-containing DPRs through RAN translation; GR in the sense direction and PR in the antisense direction. Our data provide several lines of evidence that suggest otherwise.
First, using DPR-specific antibodies we failed to show the formation of PR, GR and GP in zebrafish injected with either sense or antisense repeat RNA. It may be argued that the level of expression in the zebrafish is below the detection limit. However, we were able to detect PR expression even below the threshold for toxicity. Therefore, it is unlikely that their abundance is below the detection limit when injecting the toxic ~ 70 antisense RNA. It could be argued that a combination of DPRs mediates toxicity, while they each separately remain below the detection limit. We could, however, exclude such a synergistic toxicity, both at the DPR–DPR level and at the DPR–RNA level. Moreover, based on these findings, RAN translation might not occur in this early developmental stage of zebrafish, as was suggested by others [36], or only at a very low level as suggested by our immunoassay findings.
Second, both sense and antisense ‘RNA only’ constructs of a length of 70 and 108 repeats induced clear toxicity in our model. By inserting stop codons in all reading frames in both directions, RAN translation is completely ruled out with these constructs. Hence, toxicity arising from these RNAs is likely to be due to a direct effect of the repeat RNA itself. However, we did observe that the presence of interruptions slightly diminished the toxicity of the repeat constructs. This may be due to an alteration of the RNA–protein interaction affinities caused by the interruptions, which may compromise the toxic potential of these RNAs as was suggested by others [6]. Alternatively, this may indicate a residual effect of DPR toxicity arising from non-interrupted repeat RNA. However, as explained above, this is not supported by our data. Of note, while these ‘RNA only’ constructs display toxicity in zebrafish, they do not in Drosophila [33], suggesting a different sensitivity to RNA toxicity in zebrafish versus Drosophila models.
Third, we observed a differential effect of overexpressing Pur-alpha on the toxicity of the repeat RNA versus that of the DPRs. Pur-alpha reduced the toxicity of sense and antisense repeat RNA, but had no effect on GR or PR toxicity. The finding that Pur-alpha also robustly abrogated the toxicity of the interrupted repeat RNAs excludes the possibility that the protective effect of Pur-alpha relies on inhibition of RAN translation. This means that the toxicity of sense and antisense repeat RNA must, at least partially, rely on pathophysiological mechanisms other than the toxicity observed in GR and PR, and is hence indirect evidence for the existence of RNA toxicity.
Next, we aimed to elucidate some aspects of the mechanism of this RNA toxicity. Sequestration of several RNA binding proteins by abnormally long GGGGCC repeats has been suggested to be an important pathogenic factor in C9 ALS/FTD, similar to what is thought to happen in myotonic dystrophy [45]. In this disease, the sequestration of the protein muscleblind by the CUG repeat may explain the phenotype. We studied three proteins (hnRNPA1, hnRNPH1 and Pur-alpha) which were shown to bind the ~ 70S repeat RNA. Pur-alpha has already been shown to be able to rescue the eye abnormalities in Drosophila expressing a GGGGCC repeat [51]. Concordantly, we observed in our zebrafish model that Pur-alpha overexpression protected against the RNA toxicity arising from both the sense and antisense strand, supporting the hypothesis of RNA-binding protein sequestration as a possible pathogenic mechanism. hnRNPA1 and hnRNPH1, despite being able to bind the sense RNA repeats, showed no clear rescuing effect, strengthening the specificity of the Pur-alpha findings in our model.
Since sense and antisense repeat RNA foci are a pathological hallmark of C9orf72 ALS, we investigated its presence in our zebrafish model. First, we found both toxic (~ 90 sense and ~ 70 antisense) and non-toxic (3 sense) repeat RNA to form RNA foci, suggesting that, at least in our model, there is no clear correlation between presence of RNA foci and toxicity. Second, both sense and antisense repeat RNA were found to be mainly localized to the cytoplasm, both diffusely as well as in RNA foci. While in C9orf72 patients most RNA foci have been found to be located in the nucleus, cytoplasmic RNA foci have also been found in postmortem tissue [11, 12, 34], as well as in patient-derived cell cultures [17]. These data suggest that RNA toxicity might be mediated by cytoplasmic repeat RNA, which might explain why no correlation between extent of nuclear RNA foci and neurodegeneration has been found in postmortem tissue [15]. Third, we observed a decrease in RNA foci upon Pur-alpha overexpression while Pur-alpha did not colocalize with RNA foci. This suggests that Pur-alpha mainly interacts with soluble repeat RNA, which, therefore, may be the main mediator of RNA toxicity rather than RNA foci are.
To investigate the underlying mechanisms of RNA toxicity, we further examined the protective effect of Pur-alpha. The 322-amino acid (AA)-containing protein Pur-alpha can roughly be divided into five functional domains [14]: an N-terminal glycine-rich domain (G-rich; AA7-53), two regions with high affinity for purine rich RNA/DNA (PUR1; AA59-123–PUR2; AA142-209), a region also with purine affinity but implicated in Pur-alpha dimerization (PUR3; AA218-278) and a C-terminal glutamine–glutamate-rich domain (EQ; AA282-322). We found that deleting the G-rich or PUR2 domain abolishes the effect. Interestingly, deleting these domains does not interfere with the binding of the mutant Pur-alpha protein to repeat RNA. This suggests that the protective effect does not only result from interfering with a sequestration process, e.g., through shielding of repeat RNA by exogenous Pur-alpha protein. Instead, Pur-alpha must exert its protective effect also through specific functions of its G-rich and PUR2 domain; hence, investigating these functions might provide important insights into the mechanism of RNA toxicity.
We investigated this in our RNA toxicity zebrafish model. First, as opposed to findings in C9orf72 iPSC-derived neurons [1, 13], we observed no increased levels of p62. However, p62 was upregulated by overexpression of Pur-alpha but not by the Pur-alpha deletion constructs lacking the G-rich or PUR2 domain. Thus, these two domains appear to be necessary for Pur-alpha to increase p62 expression and to be protective. Moreover, when p62 was knocked down, the protective effect of Pur-alpha was abolished. This suggests that p62 upregulation is necessary for the protective effect of Pur-alpha. Second, overexpression of p62 almost completely prevented RNA toxicity in our zebrafish model. This suggests that the inductive effect on p62 is sufficient for Pur-alpha to have its protective effect. This beneficial effect of p62 is in line with the observation of loss-of-function mutations in SQSTM1 (the gene coding the p62 protein) as a cause of ALS [43]. Moreover, p62 was also able to reduce TDP43 aggregation [5]. Similarly, overexpression of p62 is beneficial in other neurodegenerative diseases [20, 40]. Hence, these data suggest that the observed increase in p62 in some C9orf72 models probably represents an attempted beneficial compensatory upregulation by the cell. Altogether, these data, unexpectedly, relate RNA toxicity to autophagy. Interestingly, since the C9orf72 protein itself is involved in autophagy as well, these findings support a multiple-hit model of C9 ALS in which reduced autophagy by loss of C9orf72 function might exacerbate RNA toxicity [49].
Altogether, our findings show that expanded C9orf72 hexanucleotide repeat-associated neurodegeneration arises, at least in part, from RNA toxicity independent of DPRs. This has important translational implications, especially for the design of therapeutic approaches. Assuming that both RNA and DPR toxicity play a role in the disease mechanism, approaches targeting both modalities, such as antisense oligonucleotides or small molecules, have the highest therapeutic potential as opposed to approaches targeting DPR toxicity only.
References
Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, DeGroot S et al (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 126:385–399. https://doi.org/10.1007/s00401-013-1149-y
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee H-C et al (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep 6:23204. https://doi.org/10.1038/srep23204
Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK et al (2013) Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol 126:895–905. https://doi.org/10.1007/s00401-013-1199-1
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A et al (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 6:20877. https://doi.org/10.1038/srep20877
Brady OA, Meng P, Zheng Y, Mao Y, Hu F (2011) Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem 116:248–259. https://doi.org/10.1111/j.1471-4159.2010.07098.x
Burguete AS, Almeida S, Gao F-B, Kalb R, Akins MR, Bonini NM (2015) GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife 4:e08881. https://doi.org/10.1017/CBO9781107415324.004
Chew J, Gendron TF, Prudencio M, Zhang Y, Castanedes-casey M, Chris W et al (2015) C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss and behavioral deficits. Science 5:1151–1154
Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A et al (2013) Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol 74:180–187. https://doi.org/10.1002/ana.23946
Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA et al (2016) The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS patient brains. Elife 5:e17820. https://doi.org/10.7554/eLife.17820
Cooper-Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C et al (2015) C9ORF72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS One 10:e0127376. https://doi.org/10.1371/journal.pone.0127376
Cooper-Knock J, Higginbottom A, Stopford MJ, Highley JR, Ince PG, Wharton SB et al (2015) Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol 130(1):63–75. https://doi.org/10.1007/s00401-015-1429-9
Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D et al (2014) Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 137:2040–2051. https://doi.org/10.1093/brain/awu120
Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H et al (2016) C9orf72 hexanucleotide expansions are associated with altered ER calcium homeostasis and stress granule formation in iPSC-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34:2063–2078. https://doi.org/10.1002/stem.2388
Darbinian N, White MK, Gallia GL, Amini S, Rappaport J, Khalili K (2004) Interaction between the pura and E2F-1 transcription factors. Anticancer Res 24:2585–2594
DeJesus-Hernandez M, Finch NA, Wang X, Gendron TF, Bieniek KF, Heckman MG et al (2017) In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol 134(2):255–269. https://doi.org/10.1007/s00401-017-1725-7
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Donnelly CJ, Zhang P-W, Pham JT, Heusler AR, Mistry NA, Vidensky S et al (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80:415–428. https://doi.org/10.1016/j.neuron.2013.10.015
Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J et al (2013) Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol 126:401–409. https://doi.org/10.1007/s00401-013-1147-0
Freibaum BD, Taylor JP (2017) The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front Mol Neurosci 10:1–9. https://doi.org/10.3389/fnmol.2017.00035
Geethaa T, Zhenga C, McGregor W, White B, Diaz-Mecoc M, Moscatc J et al (2012) TRAF6 and p62 inhibit amyloid β-induced neuronal death through p75 neurotrophin receptor. Neurochem Int 61:1289–1293
Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK et al (2015) Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol 130:559–573. https://doi.org/10.1007/s00401-015-1474-4
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G et al (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11:54–65. https://doi.org/10.1016/S1474-4422(11)70261-7
Gitler AD, Tsuiji H (2016) There has been an awakening: emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res 1647:19–29. https://doi.org/10.1016/j.brainres.2016.04.004
Gómez-Tortosa E, Gallego J, Guerrero-López R, Marcos A, Gil-Neciga E, José Sainz M et al (2013) C9ORF72 hexanucleotide expansions of 20–22 repeats are associated with frontotemporal deterioration. Neurology 80:366–370. https://doi.org/10.1212/WNL.0b013e31827f08ea
Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim M-S et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507:195–200. https://doi.org/10.1038/nature13124
Haeusler AR, Donnelly CJ, Rothstein JD (2016) The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci 17:383–395. https://doi.org/10.1038/nrn.2016.38
Koppers M, Blokhuis AM, Westeneng H, Margo L, Zundel CAC, Vieira de Sá R et al (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78:426–438
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li H-R et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. PNAS 110:E4530–E4539. https://doi.org/10.1073/pnas.1318835110
Laird AS, Van Hoecke A, De Muynck L, Timmers M, Van Den Bosch L, Van Damme P et al (2010) Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One 5:e13368. https://doi.org/10.1371/journal.pone.0013368
Lee Y-B, Chen H-J, Peres JN, Gomez-Deza J, Attig J, Stalekar M et al (2013) Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5:1178–1186. https://doi.org/10.1016/j.celrep.2013.10.049
Lemmens R, Van Hoecke A, Hersmus N, Geelen V, D’Hollander I, Thijs V et al (2007) Overexpression of mutant superoxide dismutase 1 causes a motor axonopathy in the zebrafish. Hum Mol Genet 16:2359–2365. https://doi.org/10.1093/hmg/ddm193
Liu Y, Pattamatta A, Zu T, Borchelt DR, Yachnis AT, Ranum LPW (2016) C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 4:521–534. https://doi.org/10.1016/j.neuron.2016.04.005
Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345:1192–1195
Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P et al (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 126:845–857. https://doi.org/10.1007/s00401-013-1200-z
O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AKMG et al (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science 351:1324–1329
Ohki Y, Wenninger-weinzierl A, Hruscha A, Asakawa K, Kawakami K, Haass C et al (2017) Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Mol Neurodegener 12:6. https://doi.org/10.1186/s13024-016-0146-8
Renton AE, Chiò A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17:17–23. https://doi.org/10.1038/nn.3584
Renton AE, Majounie E, Waite A, Simón-sánchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010.A
Robberecht W, Philips T (2013) The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14:248–264. https://doi.org/10.1038/nrn3430
Saitoh Y, Fujikake N, Okamoto Y, Popiel HA, Hatanaka Y, Ueyama M et al (2014) p62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies. J Biol Chem 290:1442–1453. https://doi.org/10.1074/jbc.M114.590281
Sareen D, O’Rourke J, Meera P, Muhammad A, Grant S, Simpkinson M et al (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with C9ORF72 repeat expansion. Sci Transl Med 5:208ra. https://doi.org/10.1126/scitranslmed.3007529.Targeting
Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng H-J, Zundel CA et al (2016) Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol 132:145–147. https://doi.org/10.1007/s00401-016-1581-x
Teyssou E, Takeda T, Lebon V, Boillée S, Doukouré B, Bataillon G et al (2013) Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 125:511–522. https://doi.org/10.1007/s00401-013-1090-0
Therrien M, Rouleau GA, Dion PA, Parker JA (2013) Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One 8:e83450. https://doi.org/10.1371/journal.pone.0083450
Udd B, Krahe R (2012) The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 11:891–905. https://doi.org/10.1016/S1474-4422(12)70204-1
van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN et al (2013) Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 12:978–988. https://doi.org/10.1016/S1474-4422(13)70210-2
Van Hoecke A, Schoonaert L, Lemmens R, Timmers M, Staats KA, Laird AS et al (2012) EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med 18:1418–1422. https://doi.org/10.1038/nm.2901
Van Mossevelde S, van der Zee J, Cruts M, Van Broeckhoven C (2017) Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev 44:117–124. https://doi.org/10.1016/j.gde.2017.02.008
Webster CP, Smith EF, Grierson AJ, De Vos KJ (2016) C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 0:1–10. https://doi.org/10.1080/21541248.2016.1240495
Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C et al (2013) Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet 92:981–989. https://doi.org/10.1016/j.ajhg.2013.04.017
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL et al (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. PNAS 110:7778–7783. https://doi.org/10.1073/pnas.1219643110
Yamakawa M, Ito D, Honda T, Kubo K, Noda M, Nakajima K et al (2014) Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet 24:1630–1645
Zhang Y-J, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu Y-F et al (2016) C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 19:668–677. https://doi.org/10.1038/nn.4272
Acknowledgements
This work was supported by the European Research Council under the European’s Seventh Framework Programme (FP7/2007–2013) under the Euro-MOTOR project, Grant agreement no. 259867, and the ERC Grant agreement no. 340429, the Research Foundation Flanders (FWO Flanders, G.0983.14N), the University of Leuven [GOA/11/014, C14/17/107 and the ‘Opening the Future’ fund], the Interuniversity Attraction Poles program [P7/16] of the Belgian Federal Science Policy Office, the ALS Liga (Belgium) and the Association Belge contre les Maladies Neuro-Musculaires (ABMM). WR is supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders and the ‘Hart voor ALS’ Fund, KU Leuven. SB was supported by the Agency for Innovation by Science and Technology (IWT) Flanders. BS is a PhD Fellow, EB was a Postdoctoral Fellow and both PVD and RL are Senior Clinical Investigators of FWO Flanders. We are grateful to Dr. M. Yamakawa (Keio University School of Medicine, Tokyo, Japan) for sharing the GP construct.
Author information
Authors and Affiliations
Contributions
BS and AB-A performed most of the experiments, analyzed the data and wrote the manuscript. SB and EB performed additional experiments and analyzed the data. TFG and LP performed the immunoassay experiment. RN, MT, WS and NH provided technical support. JW, SM and AMI provided essential material to perform some of the experiments. RL, PVD, LVDB and WR supervised the experiments, discussed the results and wrote the manuscript. All authors contributed to and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Swinnen, B., Bento-Abreu, A., Gendron, T.F. et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol 135, 427–443 (2018). https://doi.org/10.1007/s00401-017-1796-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1796-5