
Abstract
Chronic traumatic encephalopathy (CTE) is a neurodegeneration characterized by the abnormal accumulation of hyperphosphorylated tau protein within the brain. Like many other neurodegenerative conditions, at present, CTE can only be definitively diagnosed by post-mortem examination of brain tissue. As the first part of a series of consensus panels funded by the NINDS/NIBIB to define the neuropathological criteria for CTE, preliminary neuropathological criteria were used by 7 neuropathologists to blindly evaluate 25 cases of various tauopathies, including CTE, Alzheimer’s disease, progressive supranuclear palsy, argyrophilic grain disease, corticobasal degeneration, primary age-related tauopathy, and parkinsonism dementia complex of Guam. The results demonstrated that there was good agreement among the neuropathologists who reviewed the cases (Cohen’s kappa, 0.67) and even better agreement between reviewers and the diagnosis of CTE (Cohen’s kappa, 0.78). Based on these results, the panel defined the pathognomonic lesion of CTE as an accumulation of abnormal hyperphosphorylated tau (p-tau) in neurons and astroglia distributed around small blood vessels at the depths of cortical sulci and in an irregular pattern. The group also defined supportive but non-specific p-tau-immunoreactive features of CTE as: pretangles and NFTs affecting superficial layers (layers II–III) of cerebral cortex; pretangles, NFTs or extracellular tangles in CA2 and pretangles and proximal dendritic swellings in CA4 of the hippocampus; neuronal and astrocytic aggregates in subcortical nuclei; thorn-shaped astrocytes at the glial limitans of the subpial and periventricular regions; and large grain-like and dot-like structures. Supportive non-p-tau pathologies include TDP-43 immunoreactive neuronal cytoplasmic inclusions and dot-like structures in the hippocampus, anteromedial temporal cortex and amygdala. The panel also recommended a minimum blocking and staining scheme for pathological evaluation and made recommendations for future study. This study provides the first step towards the development of validated neuropathological criteria for CTE and will pave the way towards future clinical and mechanistic studies.
Similar content being viewed by others

Introduction
In 1928, the pathologist and medical examiner, Harrison Stanford Martland, introduced the term ‘punch-drunk’ to describe the clinical features of a distinct neuropsychiatric syndrome that affected boxers [26]; a condition that later came to be known as ‘dementia pugilistica’ [33]. Case reports and small series describing the neuropathologic features of the condition appeared in the 1950s and 1960s [3, 6, 16, 27, 35, 41]. Although the histological techniques varied, the most common pathological findings were cerebral atrophy, neuronal loss, gliosis and argyrophilic neurofibrillary tangles. In the seminal 1973 monograph on the clinicopathological features of dementia pugilistica in 15 former male boxers, Corsellis, Bruton, and Freeman-Browne described cerebral atrophy, enlargement of the lateral and third ventricles, thinning of the corpus callosum, cavum septum pellucidum with fenestrations, cerebellar scarring, and argyrophilic neurofibrillary degeneration using cresyl violet and Von Braunmühl’s silver stains [5]. Subsequent re-examination of Corsellis’ original series of boxers and additional cases using beta-amyloid (Aβ) immunohistochemistry determined that 95 % of CTE cases showed widespread diffuse Aβ deposits [43, 46].
Over the following decades, it was recognized that the condition affected men and women with a broad range of exposure to brain trauma, including physical abuse [42], head-banging [13, 18], poorly controlled epilepsy, “dwarf-throwing” [48], and rugbyFootnote 1 [13]. Eventually, the term “chronic traumatic encephalopathy” or “CTE”, introduced by Critchley in 1949 [8], became the preferred designation for the condition.
Coincident with the use of more refined methodology, the early pathology of CTE was reported in several young subjects [13, 14, 18]. Hof reported a single case of repetitive head-banging in a young autistic patient with numerous perivascular clusters of thioflavin and Gallyas-positive neurofibrillary tangles (NFTs) and neurites at the depths of the cerebral sulci and in the superficial layers of the inferior temporal, entorhinal and perirhinal cortices in the absence of Aβ plaques [18]. Hof and colleagues also quantitatively demonstrated the preferential distribution of the NFTs in superficial layers II and III in CTE, a laminar predilection characteristic of two other environmentally acquired tauopathies, post-encephalitic parkinsonism and Guamanian parkinsonism dementia complex (GPDC), but not found in Alzheimer’s disease (AD) [19]. Geddes and colleagues further described argyrophilic, hyperphosphorylated tau (p-tau) immunopositive neocortical NFTs and neuropil threads strikingly arranged in groups around small cortical blood vessels, in addition to diffuse granular cytoplasmic immunopositivity in some neurons [13]. Geddes also noted that the topography of the p-tau pathology principally involved the depths of sulci and that there was no Aß deposition in the 5 young cases that formed the basis of their manuscript [13].
Omalu and colleagues were the first to report CTE in a professional American football player [38, 39] and a professional wrestler [37]. Recent neuropathological studies have identified CTE in athletes who played soccer, baseball, ice hockey and rugby, as well as in military personnel exposed to explosive blast [15, 29, 31, 32, 36, 45]. P-tau pathology, with some features of CTE, has also been described following exposure to single moderate or severe traumatic brain injury, together with Aβ plaques [20, 44].
In 2013, McKee and colleagues described a spectrum of p-tau pathology in 68 male subjects with a history of exposure to repetitive brain trauma with neuropathological evidence of CTE, ranging in age from 17 to 98 years (mean 59.5 years). In young subjects with the mildest forms of CTE, focal perivascular epicenters of NFTs and astrocytic tangles (ATs) were found clustered at the depths of the cortical sulci; in subjects with severe disease, a profound tauopathy involved widespread brain regions [32]. Other abnormalities encountered in advanced disease included abnormal deposits of phosphorylated TAR DNA-binding protein of 43 kDa (TDP-43) protein that occasionally co-localized with p-tau, varying degrees of Aβ pathology, axonal dystrophy and neuroinflammation [30, 32]. Based on these findings, preliminary criteria for the neuropathological diagnosis of CTE were proposed, as follows:
-
1.
Perivascular foci of p-tau immunoreactive NFTs and ATs in the neocortex
-
2.
Irregular distribution of p-tau immunoreactive NFTs and ATs at the depths of cerebral sulci
-
3.
NFTs in the cerebral cortex located preferentially in the superficial layers (often most pronounced in temporal cortex)
-
4.
Supportive, non-diagnostic features: Clusters of subpial ATs in the cerebral cortex, most pronounced at the sulcal depths.
In March 2013, the National Institutes of Health (NIH), supported by the Foundation for NIH’s Sports Health Research Program with funding from the National Football League (NFL), launched a major effort to define the neuropathological characteristics of CTE. Two projects were initiated on the neuropathology of CTE and the delayed effects of traumatic brain injury. One of the initial objectives was to convene a consensus meeting to define the neuropathological criteria for the diagnosis of CTE. The primary objective for the first meeting was to determine whether CTE was a distinctive tauopathy that could be reliably distinguished from other tauopathies using the preliminary criteria. The study design was modeled after previous successful NIH-sponsored consensus conferences for other tauopathies, specifically progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) [10, 17, 25].
Materials and methods
Individuals not directly involved in the pathological evaluation (ACM, VEA, KFB, JFC) selected 25 cases of the various tauopathies. The selected cases were considered to be representative of the disease and of at least moderate disease severity. The cases included 10 recently acquired cases of suspected CTE that were donated as part of the NINDs-funded traumatic brain injury (TBI) brain bank at Boston University School of Medicine (BUSM), including 7 cases with Aβ plaques and 3 cases without Aß plaques. Five cases of AD, Braak stage V-VI, 2 cases of PSP and 2 cases of CBD were selected from the Alzheimer’s Disease Center (ADC) brain bank at BUSM. Two cases of GPDC and 2 cases of argyrophilic grain disease (AGD) were selected from the Alzheimer’s Disease Research Center (ADRC) brain bank at Mayo Clinic-Jacksonville, and 2 cases of primary age-related tauopathy (PART) were selected from the ADRC brain bank at Columbia University. Paraffin-embedded tissue blocks from 12 brain regions from each case were sent to the TBI Brain Bank at BUSM for uniform processing, staining and immunohistochemistry (IHC) (Table 1); 2 of the selected cases were missing the superior temporal block. Sections were stained with Luxol fast blue counterstained with hematoxylin and eosin (LHE) and Bielschowsky silver impregnation; IHC was performed using anti-Aβ42 (Aß 1-42, EMD Millipore, 1:2000, pretreated with 88 % formic acid for 2 min; or Aβ-4G8, Bio Legend, 1:100,000, pretreated with formic acid); anti-p-tau (AT8, Thermo Fisher Scientific/Pierce, 1:2000, pretreated with formic acid) and anti-p-TDP-43 (Anti-TDP-43, phospho, 1:2000, pretreated with formic acid) for a total of 27 slides per case (25 slides in 2 cases). An individual blinded to the origin and identity of the cases (KFB) scanned the 671 glass pathology slides into digital images at the Mayo Clinic Jacksonville using an Aperio scanner (Leica Biosystems, Buffalo Grove, IL). The digitized images were organized into folders labeled with only the case number (#1-25), brain region, stain and IHC and provided to the evaluating neuropathologists on portable hard drives as well as on an online slide-hosting website (Leica Biosystems-Aperio). No clinical or demographic information was provided to the evaluating neuropathologists—including no information regarding the subjects’ age, gender, clinical symptoms, diagnosis or athletic exposure. No information was supplied regarding the gross neuropathological features of the brains. The neuropathologists were given a tauopathy criteria guide that provided the provisional criteria for CTE [32] as well as published criteria for the other tauopathies (See supplementary material for full tauopathy criteria guide) [4, 7, 11, 21, 25, 34, 40, 47]. Although the neuropathologists knew that the selected cases represented presumptive CTE, AD, PSP, CBD, AGD, PART, and GPDC, they did not know how many cases representing each diagnosis were to be evaluated.
Seven neuropathologists with experience in neurodegenerative diseases, including the tauopathies, participated in the evaluation of the digitized images (NJC, DWD, RDF, CDK, DPP, TDS, JPV). The neuropathologists evaluated the cases independently, at their own pace, and completed an evaluation form that included the pathological diagnosis and a 4-scale level of certainty (1, unsure; 2, possible; 3, probable; 4, definite). After the initial evaluations were sent to BUSM for analysis, the evaluator was provided the gross neuropathological findings and clinical summaries for each case, and asked to reevaluate the diagnosis and provide a second level of conviction. The results of all evaluations were analyzed prior to the face-to-face meeting held on February 25–26, 2015.
Statistical analysis
To evaluate the agreement among the neuropathologists who reviewed the cases, two sets of Cohen’s kappa statistics were calculated. The first kappa coefficient measured the agreement among the overall neuropathological diagnoses; the second kappa coefficient measured the agreement among neuropathologists regarding the specific diagnosis of CTE. The overall kappa coefficient combines the neuropathologist-level estimates of kappa into an overall estimate of the common agreement. Kappa values of 0.81–1.0 indicate very good agreement, kappa values of 0.61–0.80 show good agreement, while kappa values of 0.41–0.60 indicate moderate agreement [12]. All statistical analyses were done using SAS 9.4 (SAS Institute Inc., Cary, North Carolina, USA) software.
At the face-to-face consensus meeting, a larger panel that included ACM2, WS,Footnote 2 IL, WAG and members of the NINDs TBI/CTE group reviewed the results of the neuropathological evaluations, digitized images and glass pathology slides and discussed the cases as a group. Discussions led to refinements in the neuropathological criteria for CTE, as well as “best practice” recommendations for neuropathologists examining brains for evidence of CTE.
Results
There was good agreement regarding the overall neuropathological diagnosis of all 25 cases (Cohen’s kappa, 0.67), and even better agreement regarding the specific diagnosis of CTE (Cohen’s kappa, 0.78), using the proposed criteria. In evaluating the 10 cases submitted with the presumptive diagnosis of CTE (Supplementary Table 1), 64 of the 70 reviewers responses (91.4 %) indicated CTE as the diagnosis. There was a significant decrease of errors that paralleled the sequence of cases evaluated. The log of the expected errors significantly decreased by 0.43 for each case of CTE reviewed (p value = 0.024). There were common additions to the CTE diagnosis, including “Changes of Alzheimer’s disease” (ADC) and AD in the cases with Aß plaques (cases #4–10). Other co-morbid diagnoses included hippocampal sclerosis (HS), AGD and PART. In the 15 other tauopathy cases (cases submitted for review with diagnoses other than CTE) (Supplementary Table 2), the reviewers generally agreed with the submission diagnoses of AD (97.1 % of responses), CBD (92.8 %), and PART (78.5 %); however, there were frequent discrepancies in cases with the presumptive diagnoses of PSP, AGD and GPDC (Supplementary Table 2). The evaluators reported a significantly increased degree of certainty (t test = 4.36, p value <0.001) in the diagnosis of CTE from an overall mean of 3.1 in a 4-point scale of conviction (1, unsure; 2, possible; 3, probable; 4, definite) to a mean of 3.7 after the gross neuropathological features and clinical features of the cases were provided to the evaluator. Three initial diagnoses of non-CTE were changed to CTE and 9 diagnoses of co-morbid CTE in non-CTE cases were changed to no CTE after revealing the clinical and gross neuropathological features.
Diagnostic neuropathological features of CTE
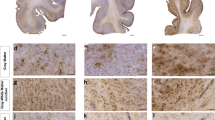
The group defined a neuropathological lesion specific to CTE that distinguished the disorder from other tauopathies. The pathognomonic lesion of CTE consists of p-tau aggregates in neurons, astrocytes, and cell processes around small vessels in an irregular pattern at the depths of the cortical sulci (Figs. 1, 2; Table 2). The group also noted that the distinctively irregular spatial pattern of p-tau in CTE was often visible with low-power inspection (Fig. 1). Although other abnormalities in p-tau were also found, especially in the more severely affected brains, the pathognomonic lesion was distinct and not found in the other degenerative tauopathies (Fig. 2). In addition, the group observed frequent evidence of other pathologies in CTE, including TDP-43-immunoreactive neuronal cytoplasmic inclusions, Aβ plaques and amyloid angiopathy, and hippocampal neurofibrillary degeneration, including extracellular tangles best seen with silver stains.

Low magnification inspection of p-tau-stained slides often revealed the irregular spatial pattern of CTE pathology. AT8-stained slides of cerebral cortex in 3 cases of CTE showing irregular patches of p-tau pathology most dense at the depths of the sulci

The microscopic features of the pathognomonic lesion of CTE. The pathognomonic feature of CTE is a perivascular accumulation of p-tau aggregates in neurons, astrocytes and cell processes in an irregular spatial pattern in the cerebral cortex and found preferentially at the depths of the sulci. a A large perivascular p-tau lesion is found at the sulcal depths in a subject with CTE. b–f Multiple perivascular foci are often found in the cortex in CTE. g The p-tau aggregates in CTE include strikingly rounded structures in the neuropil that often are most dense in the areas surrounding the vessel. h The rounded p-tau immunoreactive cell processes are more densely distributed than those found in argyrophilic grain disease. All sections immunostained for AT8, bars indicate 100 µm, except in g and h where the small bars indicate 10 µm

Hippocampal pathology in CTE. Examples of hippocampal pathology in 2 cases of CTE of moderate severity. In example 1 (a–f), there is a mild hippocampal atrophy, b mild neuronal loss in CA1, c sparse NFTs in CA1, Bielschowsky silver stain, d sparse NFTs in CA1, AT8 immunostain, e moderate numbers of diffusely immunopositive AT8 stained neurons in CA4, and f occasional AT8 immunopositive NFTs in the dentate gyrus. In example 2 (g–l), there is g more severe hippocampal atrophy, h clear neuronal loss in CA1, i moderate density of NFTs in CA1, Bielschowsky silver stain, j moderate density of NFTs in CA1, AT8 immunostain, k high numbers of AT8-stained neurons and NFTs in CA4, and l moderate numbers of AT8 immunopositive NFTs in the dentate gyrus. Bars indicate 100 µm

pTDP-43 pathology in CTE. a pTDP-43 neuronal inclusions in the amygdala. b p-TDP-43 inclusions and dot-like neurites in CA1. c p-TDP-43 dot-like neurites in entorhinal cortex. d pTDP-43 inclusions and dot-like neurites in the dentate granule cell layer. All sections immunostained for p-TDP-43, bars indicate 100 µm
Supportive neuropathological features of CTE
The group defined supportive pathological features for CTE. These features were commonly found in the CTE cases in addition to the required criteria, but were not considered diagnostic in isolation (Table 2).
Exclusions to the sole diagnosis of CTE
The presence of changes compatible with the diagnosis of another neurodegenerative disease excludes CTE as a single diagnosis, and indicates the presence of co-morbid pathology. These features include CA1-predominant neurofibrillary degeneration in the hippocampus in association with Aβ plaques consistent with AD [34]; prominent cerebellar dentate nucleus cell loss, coiled bodies in oligodendroglia, and tufted astrocytes as seen in PSP [25]; severe involvement of the striatum and pallidum with extensive astrocytic plaques in cortical and subcortical structures as seen in CBD [21] or globular astrocytic inclusions of globular glial tauopathy [1].
Discussion
The consensus panel of neuropathologists found that the p-tau pathology of CTE is clearly distinct from other tauopathies. The panel concluded that there is a pathognomonic lesion of CTE that consists of an accumulation of abnormal tau in neurons and astroglia distributed around small blood vessels at the depths of sulci in the cortex in an irregular spatial pattern. Other supportive features of CTE include abnormal p-tau immunoreactive pretangles and NFTs preferentially affecting superficial layers (layers II–III), pretangles, NFTs or extracellular tangles primarily in CA2 and CA4 of the hippocampus, NFTs in subcortical nuclei, including the mammillary bodies and other hypothalamic nuclei, amygdala, nucleus accumbens, thalamus, midbrain tegmentum, isodendritic core (nucleus basalis of Meynert, raphe nuclei, substantia nigra and locus coeruleus), p-tau immunoreactive thorned astrocytes at the glial limitans in the subpial and periventricular regions, p-tau immunoreactive large grain-like and dot-like structures, and TDP-43 immunoreactive neuronal cytoplasmic inclusions and dot-like structures in the hippocampus, anteromedial temporal cortex and amygdala. While this was only the first meeting to address the neuropathological diagnosis of CTE, and more research is needed to determine the nature and degree of brain injury necessary to cause this neurodegeneration, the panel members also noted that the pathognomonic lesion of CTE has, thus far, only been found in individuals who were exposed to brain trauma, typically multiple episodes.
The panel also determined that the pathognomonic lesion of CTE is distinct from age-related tau astrogliopathy (ARTAG), a morphological spectrum of astroglial pathology detected by p-tau immunohistochemistry that may coexist in the same brain with other disorders and is of unclear etiology (Fig. 5). P-tau-immunoreactive astrocytes in ARTAG include thorn-shaped astrocytes in the subpial, subependymal, and perivascular regions of the white and gray matter (Kovacs, in press). Changes of ARTAG may be present in CTE, but in isolation, are non-specific and non-diagnostic.

Age-related p-tau astrogliopathy that may be present. a and b. Subpial p-tau immunopositive astrocytes may be found at the glial limitans in the sulcal depths but are non-specific and non-diagnostic for CTE (asterisks). c However, p-tau immunopositive subpial astrocytes accompanied by perivascular foci of p-tau positive neurons and astrocytes (arrowhead) at the depths of the sulci are diagnostic for CTE. d and e p-Tau immunopositive astrocytes surrounding small venules in the deep white matter of the temporal lobe are not diagnostic for CTE and are often found in association with aging [22]. f p-Tau positive astrocytes may also be found in the crests of the white matter of the frontal and temporal lobes with aging and other conditions that are not diagnostic for CTE [22]. All sections immunostained for AT8, bars indicate 100 µm
Although 9 of the 10 subjects diagnosed with CTE in this study were former American football players and only one was a former professional boxer, previous data has shown that the pathological features of CTE associated with boxing (often referred to as “dementia pugilistica”) are similar to the pathological features of CTE associated with football [28, 32]. Furthermore, the cortical areas most likely to show early focal CTE pathology in boxers are similar to American football players. While initial reports of boxers with CTE described cerebellar scarring, atrophy and loss of Purkinje cells [5], recent studies of pugilists find that cerebellar pathology is rare aside from p-tau NFTs and neurites in the dentate nucleus, Purkinje cells and roof of the 4th ventricle [28, 32].
Future directions
Using criteria from this consensus meeting, Bieniek and colleagues reviewed the clinical records and brains of 1721 cases donated to the Mayo Clinic Brain Bank over the past 18 years, and found CTE pathology in 32 % of contact sport athletes [2]. No cases of CTE were found in 162 control brains without a history of brain trauma or in 33 cases with a history of a single traumatic brain injury. Of the 21 with CTE pathology, 19 had participated in football or boxing, and many were multiple sport athletes including rugby, wrestling, basketball, and baseball. One athlete played only baseball, and another athlete only played basketball. Similarly, Ling and colleagues screened 268 cases of neurodegenerative disease and controls in the Queen Square Brain Bank for Neurological Disorders using the preliminary McKee criteria [32] and found changes of CTE in 11.9 % of neurodegenerative disorders and 12.8 % of elderly controls. Of the cases with changes of CTE, 93.8 % had a history of TBIs, 34 % had participated in high-risk sports including rugby, soccer, cricket, lacrosse, judo and squash, and 18.8 % were military veterans [24]. However, it is unclear if all the cases with CTE changes described by Ling and colleagues would have met strict criteria for CTE using these newly defined NINDS guidelines. Furthermore, the relationship between non-diagnostic, non-specific astrocytic p-tau pathology and a history of traumatic exposure remains to be determined (Kovacs, in press).
At the present time, CTE remains a diagnosis that can only be made definitively upon neuropathological examination of the brain. Because the pathological diagnosis requires p-tau immunohistochemistry and the lesions are irregularly distributed, the detection of CTE in autopsy cohorts may require additional sampling compared to routine practices. The consensus panel’s minimum recommended sampling for CTE is found in Table 3. Sampling follows the protocol recommended by Alzheimer Disease Centers (National Institute on Aging-Alzheimer’s Association (NIA-AA) [34]) with the further recommendation that all cortical sections be taken to include the region at the depths of the cortical sulci. This has been shown in pilot studies to detect 80 % of CTE cases; however, 20 % of CTE cases, all early stage, would be missed by this sampling scheme [9]. Of the NIA-AA sampling guidelines, the following blocks are most valuable for detecting CTE: sulcal depths of the superior and middle frontal gyrus, superior and middle temporal gyrus and inferior parietal gyrus (Fig. 6). Of note, the Bielschowsky silver stain does not always detect the diagnostically significant focal perivascular cortical tau lesions, and the panel recommended p-tau immunohistochemistry for the diagnosis of CTE using AT8 immunostaining or equivalent p-tau antibody (CP-13 or PHF-1). The question of how extensive the sampling must be to “rule out” CTE was discussed, but no data were available to make this determination.

Minimum recommended brain regions for evaluation for CTE. The following sections from the NIA-AA blocking scheme are recommended for p-tau immunostaining in evaluation for CTE (blue rectangles). In the cortical sections (blocks 1–5, 12, 13), the depths of the cortical sulci should be included in the section. 1 Middle frontal gyrus, 2 superior and middle temporal gyri, 3 inferior parietal lobule, 4 hippocampus, 5 amygdala and entorhinal cortex, 6 basal ganglia at level of anterior commissure with basal nucleus of Meynert, 7 thalamus, 8 midbrain with substantia nigra, 9 pons with locus coeruleus, 10 medulla oblongata, 11 cerebellar cortex and dentate nucleus; additional sections if high suspicion of CTE (red rectangles): 12 superior frontal gyrus, 13 temporal pole, 14 hypothalamus and mammillary body
These criteria are the beginning of the process to fully characterize the pathology of CTE, and this is only the first of a series of consensus conferences on the subject funded by the U01 NINDS research initiative. Many important questions were not addressed in this first consensus panel, including the degree of neuronal cell loss, gliosis, inflammation, and hemosiderin deposition, and the diagnosis of CTE in the presence of comorbid pathologies, including AD. Future directions will include further validation of the neuropathological criteria for CTE, including staging of the severity of p-tau pathology and characterization of early disease. More pathological characterization will also be necessary to delineate the involvement of the other subcortical regions, including amygdala, globus pallidus, subthalamic nucleus, accumbens, neostriatum, thalamus, midbrain, cerebellum, spinal cord and white matter. It will also be important to determine the differential hippocampal p-tau pathology in CTE compared to AD, whether the TDP-43 pathology is distinctive for CTE and the contribution of hippocampal sclerosis and TDP-43 deposition to the clinical and pathological features. Population isolates that develop unusual p-tau pathologies will need to be distinguished from CTE pathology in addition to Guam, such as the Kii peninsula of Japan [23]. In addition, the contributions of other proteinopathies, including β-amyloidosis (diffuse and dense core Aβ plaques and amyloid angiopathy) and alpha-synuclein will be important to determine. Similarly, the role of microvascular pathology, iron deposition, axonal injury, neuroinflammation and astrocytosis to the pathogenesis of CTE pathology needs resolution.
Future investigation will be needed to understand the relationship of the pathology to the clinical symptoms, genetics, neuroimaging and other biomarkers (including p-tau positron emission tomography (PET) imaging and cerebrospinal fluid (CSF) and blood biomarkers), metabolomics, proteomics, and epigenetics. It will also be important to determine whether specific “tau strains” are involved in the development of CTE. Furthermore, more information is needed regarding the frequency, severity, and nature of the traumatic exposures, length of survival after trauma, as well as factors such as the age at first and last exposure to trauma, and the effects of military compared to civilian brain trauma.
The limitations of the present study include the relatively small sample set, the use of digitized images, the selection of suspected CTE cases by a single source, the use of representative cases of moderate-to-late stage severity of CTE, and presence of some age-related co-morbidities. However, these limitations are offset by the fact that all evaluating neuropathologists were evaluating the exact same digital images, the cases were all uniformly prepared by a central laboratory, and the evaluation was performed blinded to all clinical or demographical data and gross neuropathological findings. Other limitations to the present study include the lack of data regarding TBI history in the non-CTE cases under evaluation. Future studies are being designed to specifically address the contribution of TBI at all levels of severity to neurodegenerative pathologies.
Conclusion
A consensus panel of 7 neuropathologists blinded to all clinical conditions and demographics evaluated the identical digitized images of 25 cases representing various tauopathies and concluded that the pathology of CTE is distinct from other tauopathies. In addition, the panel described the pathognomonic lesion of CTE as an accumulation of abnormal tau in neurons and astroglia distributed perivascularly at the depths of sulci in the isocortex in an irregular pattern. Future consensus meetings will address validation of the criteria among a wider group of neuropathologists using cases submitted from multiple sources. In addition, future meetings will address the identification of comorbid CTE when other neurodegenerative diseases and other diseases are present. Furthermore, additional research will be necessary to determine the contribution of p-tau and other pathologies to the development of clinical symptoms of CTE.
The incidence and prevalence of CTE remain unknown and will likely require methods of in vivo detection and diagnosis to make a clear determination. This first consensus conference on the pathological criteria for CTE represents the first step along the path to standardizing the neuropathology of CTE and paving the way for future determinations of specific clinical symptomatology and refinements in clinical diagnosis.
Notes
Although often referred to as a soccer player, the young subject with CTE described by Geddes et al. 1999 as the “keen amateur footballer who frequently “headed” the ball while playing” [13], was an amateur rugby player (personal communication, T. Revesz).
Neuropathologist present at the face-to-face panel discussion, but did not participate in the slide evaluations.
References
Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B et al (2013) Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 126:537–544
Bieniek KF, Ross OA, Cormier KA, Walton RL, Soto-Ortolaza A, Johnston AE et al (2015) Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 130:877–889. doi:10.1007/s00401-015-1502-4
Brandenburg W, Hallervorden J (1954) Dementia pugilistica with anatomical findings. Virchows Arch 325:680–709
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM-Y, Hatanpaa KJ et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol 114:5–22
Corsellis J, Bruton C, Freeman-Browne D (1973) The aftermath of boxing. Psychol Med 3:270–303
Courville CB (1962) Punch drunk. Its pathogenesis and pathology on the basis of a verified case. Bull Los Angel Neuro Soc 27:160–168
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I et al (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128(6):755–766. doi:10.1007/s00401-014-1349-0
Critchley M (1949) Punch-drunk syndromes: the chronic traumatic encephalopathy of boxers. Hommage a Clovis Vincent (ed) Maloine, Paris
Darby A, Adams J, Babcock K, Avarez V, Stein T, McKee A (2015) Detection of CTE in autopsy cohorts using restricted cortical sampling. In: Journal of Neuropathology and Experimental Neurology, vol 6. Lippincott Williams and Wilkins Two Commerce SQ, 2001 Market ST, Philadelphia, pp 615–615
Dickson D, Bergeron C, Chin S, Duyckaerts C, Horoupian D, Ikeda K et al (2002) Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946
Dickson DW (2009) Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol 3:1–23
Fleiss JL, Levin B, Paik MC (2003) Statistical methods for rates and proportions. Wiley series in probability and statistics, 3rd edn. J. Wiley, Hoboken
Geddes J, Vowles G, Nicoll J, Revesz T (1999) Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 98:171–178
Geddes JF, Vowles GH, Robinson SF, Sutcliffe JC (1996) Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol 22:12–16
Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA et al (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4:134ra160. doi:10.1126/scitranslmed.3003716
Grahmann H, Ule G (1956) Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica and traumatic encephalopathy of boxers). Psychiatr Neurol (Basel) 134:261–283
Hauw J-J, Daniel S, Dickson D, Horoupian D, Jellinger K, Lantos P et al (1994) Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44:2015–2015
Hof P, Knabe R, Bovier P, Bouras C (1991) Neuropathological observations in a case of autism presenting with self-injury behavior. Acta Neuropathol 82:321–326
Hof PR, Bouras C, Buee L, Delacourte A, Perl DP, Morrison JH (1992) Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol 85:23–30
Johnson VE, Stewart W, Smith DH (2012) Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 22:142–149
Kouri N, Whitwell JL, Josephs KA, Rademakers R, Dickson DW (2011) Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol 7:263–272
Kovacs GG, Milenkovic I, Wöhrer A, Höftberger R, Gelpi E, Haberler C et al (2013) Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 126:365–384
Kuzuhara S, Kokubo Y, Sasaki R, Narita Y, Yabana T, Hasegawa M et al (2001) Familial amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Kii peninsula of Japan: clinical and neuropathological study and tau analysis. Ann Neurol 49:501–511
Ling H, Holton JL, Shaw K, Davey K, Lashley T, Revesz T (2015) Histological evidence of chronic traumatic encephalopathy in a large series of neurodegenerative diseases. Acta Neuropathol. doi:10.1007/s00401-015-1496-y
Litvan I, Hauw J, Bartko J, Lantos P, Daniel S, Horoupian D et al (1996) Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 55:97–105
Martland HS (1928) Punch drunk. J Am Med Assoc 91:1103–1107. doi:10.1001/jama.1928.02700150029009
Mawdsley C, Ferguson F (1963) Neurological disease in boxers. Lancet 282:795–801
McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE et al (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735. doi:10.1097/NEN.0b013e3181a9d503
McKee AC, Daneshvar DH, Alvarez VE, Stein TD (2014) The neuropathology of sport. Acta Neuropathol 127:29–51
McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW et al (2010) TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69:918–929. doi:10.1097/NEN.0b013e3181ee7d85
McKee AC, Robinson ME (2014) Military-related traumatic brain injury and neurodegeneration. Alzheimer’s Dementia 10:S242–S253
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. doi:10.1093/brain/aws307
Millspaugh J (1937) Dementia pugilistica. US Naval Med Bull 35:297Y303
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123:1–11
Neubuerger KT, Sinton DW, Denst J (1959) Cerebral atrophy associated with boxing. AMA Arch Neurol Psychiatry 81:403–408
Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G et al (2011) Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus 31:E3
Omalu BI, Bailes J, Hammers JL, Fitzsimmons RP (2010) Chronic traumatic encephalopathy, suicides and parasuicides in professional American athletes: the role of the forensic pathologist. Am J Forensic Med Pathol 31:130–132
Omalu BI, DeKosky ST, Hamilton RL, Minster RL, Kamboh MI, Shakir AM et al (2006) Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery 59:1086–1093
Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH (2005) Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57:128–134
Oyanagi K, Hashimoto T, Yamazaki M (2011) Parkinsonism–dementia complex of guam. neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Wiley-Blackwell, Oxford, pp 171–177
Payne E (1968) Brains of boxers. Neurochirurgia (Stuttg) 11:173–188
Roberts G, Whitwell H, Acland PR, Bruton C (1990) Dementia in a punch-drunk wife. Lancet 335:918–919
Roberts GW, Allsop D, Bruton C (1990) The occult aftermath of boxing. J Neurol Neurosurg Psychiatry 53:373–378
Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211–221
Stewart W, McNamara P, Lawlor B, Hutchinson S, Farrell M (2015) Chronic traumatic encephalopathy: a potential late and under recognized consequence of rugby union? QJM: hcv070
Tokuda T, Ikeda S, Yanagisawa N, Ihara Y, Glenner G (1991) Re-examination of ex-boxers’ brains using immunohistochemistry with antibodies to amyloid β-protein and tau protein. Acta Neuropathol 82:280–285
Tolnay M, Clavaguera F (2004) Argyrophilic grain disease: a late-onset dementia with distinctive features among tauopathies. Neuropathology 24:269–283
Williams DJ, Tannenberg AE (1996) Dementia pugilistica in an alcoholic achondroplastic dwarf. Pathology 28:102–104
Acknowledgments
The authors gratefully acknowledge the use of the resources and facilities at the Edith Nourse Rogers Memorial Veterans Hospital (Bedford, MA, USA), the Boston University School of Medicine, and the Mayo Clinic Jacksonville. We also gratefully acknowledge the help of all members of the Chronic Traumatic Encephalopathy Program at Boston University School of Medicine, the Boston VA, as well as the individuals and families whose participation and contributions made this work possible. This work was supported by the National Institute of Neurological Disorders and Stroke (1U01NS086659-01, R01NS078337, R56NS078337, R01NS095252), Department of Defense (W81XWH-13-2-0064, W81XWH-14-1-0399), Department of Veterans Affairs, the Veterans Affairs Biorepository (CSP 501), the National Institute of Aging Boston University Alzheimer’s Disease Center (P30AG13846; supplement 0572063345–5), Department of Defense Peer Reviewed Alzheimer’s Research Program (DoD-PRARP #13267017), the National Institute of Aging Boston University Framingham Heart Study (R01AG1649), the National Operating Committee on Standards for Athletic Equipment and the Concussion Legacy Foundation. This work was also supported by unrestricted gifts from the Andlinger Foundation, the World Wrestling Entertainment and the National Football League. This work was supported by grants P50 AG05681 and P01 AG03991 from the National Institute on Aging (NJC).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
ACM, NJC, DWD, RDF, CDK, IL, DPP, TDS, JPVS, WS, YT, JFC, KFB, KDO, VEA and WAG have no conflicts of interest to disclose.
Additional information
The members representing TBI/CTE group are listed in the Appendix.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix: TBI/CTE group
Appendix: TBI/CTE group
Elissa Flannery, Daniel H. Daneshvar, Patrick T. Kiernan, Jesse Mez, Lauren Murphy, Todd M. Solomon, Debra Babcock, Patrick S. F. Bellgowan, and Walter J. Koroshetz.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
McKee, A.C., Cairns, N.J., Dickson, D.W. et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131, 75–86 (2016). https://doi.org/10.1007/s00401-015-1515-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1515-z