
Abstract
Astrocytes are specialized glial cells that outnumber neurons by over fivefold. They contiguously tile the entire central nervous system (CNS) and exert many essential complex functions in the healthy CNS. Astrocytes respond to all forms of CNS insults through a process referred to as reactive astrogliosis, which has become a pathological hallmark of CNS structural lesions. Substantial progress has been made recently in determining functions and mechanisms of reactive astrogliosis and in identifying roles of astrocytes in CNS disorders and pathologies. A vast molecular arsenal at the disposal of reactive astrocytes is being defined. Transgenic mouse models are dissecting specific aspects of reactive astrocytosis and glial scar formation in vivo. Astrocyte involvement in specific clinicopathological entities is being defined. It is now clear that reactive astrogliosis is not a simple all-or-none phenomenon but is a finely gradated continuum of changes that occur in context-dependent manners regulated by specific signaling events. These changes range from reversible alterations in gene expression and cell hypertrophy with preservation of cellular domains and tissue structure, to long-lasting scar formation with rearrangement of tissue structure. Increasing evidence points towards the potential of reactive astrogliosis to play either primary or contributing roles in CNS disorders via loss of normal astrocyte functions or gain of abnormal effects. This article reviews (1) astrocyte functions in healthy CNS, (2) mechanisms and functions of reactive astrogliosis and glial scar formation, and (3) ways in which reactive astrocytes may cause or contribute to specific CNS disorders and lesions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Until quite recently, the prevailing view regarding the biology and pathology of astrocytes among those studying clinical disorders and structural lesions of the central nervous system (CNS) was most likely to be that (1) astrocytes are supportive glial cell components in neural tissue and (2) reactive astrogliosis is a reliable and sensitive marker of diseased tissue. The notion that dysfunctions of astrocytes or reactive astrocytes might contribute to the presentation of clinical signs or to mechanisms leading to pathological changes in CNS tissue was generally not considered. However, this viewpoint is gradually changing as a result of steadily increasing interest in and study of the biology and pathology of astrocytes. Over the past 25 years it has become clear that astrocytes are responsible for a wide variety of complex and essential functions in the healthy CNS, including primary roles in synaptic transmission and information processing by neural circuit functions. The functions and mechanisms of reactive astrogliosis and glial scar formation are gradually being elucidated. There is a growing body of evidence that points towards the potential for loss of normal astrocyte functions or gain of abnormal effects to contribute to, or play primary roles in, disease processes, and there are now numerous examples of astrocyte contributions to clinical and pathological mechanisms [15, 52, 212, 224–226, 237]. This article summarizes recent advances in (1) astrocyte functions in healthy tissue, (2) the gradations, mechanisms, functions, and effects of reactive astrogliosis, and (3) the appearance and potential roles of reactive astrocytes in a variety of CNS disorders and pathologies.
Astrocyte biology in healthy CNS
Basic astrocyte morphology
Since the late nineteenth century, astrocytes have been divided into two main subtypes, protoplasmic or fibrous, on the basis of differences in their cellular morphologies and anatomical locations [191]. These two main categories retain their validity and usefulness today. Protoplasmic astrocytes are found throughout all gray matter and, as first demonstrated using classical silver impregnation techniques, exhibit a morphology of several stem branches that give rise to many finely branching processes in a uniform globoid distribution (Fig. 1a). Fibrous astrocytes are found throughout all white matter and exhibit a morphology of many long fiber-like processes [191]. Classical and modern neuroanatomical studies also indicate that both astrocyte subtypes make extensive contacts with blood vessels (Fig. 1a, b). Electron microscopic analyses of the mid-twentieth century revealed that the processes of protoplasmic astrocytes envelop synapses and that the processes of fibrous astrocytes contact nodes of Ranvier, and that both types of astrocytes form gap junctions between distal processes of neighboring astrocytes [183]. These basic attributes of astrocyte morphology and some of their functional interactions are summarized schematically in (Fig. 2a).

Astrocyte morphology and interactions with blood vessels in healthy and diseased tissue. a Protoplasmic astrocyte giving rise to a dense network of finely branching processes throughout its local gray matter neuropil, as well as to a large stem branch that extends foot processes along a blood vessel (bv). b Two color fluorescence showing astrocytes in healthy mouse gray matter stained immunohistochemically for GFAP (red) as well as the transgene-derived reporter molecule GFP (green). Note that in these transgenic mice [95], GFP reporter is present in all of the fine processes of the protoplasmic astrocytes throughout the neuropil, whereas the GFAP is present only in the large stem astrocyte processes and endfeet (which appear yellow where green and red staining overlap). Note that endfeet from many astrocytes contact and envelop bv. c Two color fluorescence showing dense accumulations of GFAP-positive (green) endfeet and processes of reactive astrocytes lining up along perivascular cuffs or clusters (pvc) of inflammatory cells stained with DAPI (blue) in a mouse with experimental autoimmune encephalomyelitis (EAE). Transgenic disruption of this reactive astrocyte barrier leads to widespread invasion of inflammatory cells away from perivascular clusters into CNS parenchyma during EAE [251]. d Two color brightfield staining showing human autopsy specimen with reactive astrocytes lining their processes along perivascular cuffs of inflammatory cells as if forming perivascular scar-like barriers similar to those observed in experimental animal models [251]. Scale bars a 3 μm, b 7.5 μm, c 15 μm, d 5 μm

Schematic representations that summarize, a astrocyte functions in healthy CNS, and b triggers and molecular regulators of reactive astrogliosis
Molecular markers and proteomic characterization
Immunohistochemical techniques that enable the detection of specific molecular markers at the single-cell level are essential tools for identifying and characterizing cells in healthy and pathological tissue. Expression of glial fibrillary acid protein (GFAP) has become a prototypical marker for immunohistochemical identification of astrocytes. Nevertheless, it is important to recognize the appropriate uses and limitations of GFAP as an astrocyte marker. GFAP was first isolated as a protein highly concentrated in old demyelinated plaques from multiple sclerosis patients and was then found to be associated immunohistochemically with reactive astrocytes in such plaques and in other pathological contexts [62, 63]. In line with this original mode of identification, GFAP expression can be regarded as a sensitive and reliable marker that labels most, if not all, reactive astrocytes that are responding to CNS injuries (Fig. 3). However, GFAP is not an absolute marker of all non-reactive astrocytes and is often not immunohistochemically detectable in astrocytes in healthy CNS tissue or remote from CNS lesions (Figs. 3a, 4a). Although GFAP is immunohistochemically detectable in many astrocytes throughout the healthy CNS, it is clear from double staining with multiple markers (including transgenic reporter proteins) that many mature astrocytes in healthy CNS tissue do not express detectable levels of GFAP and that GFAP expression by astrocytes exhibits both regional and local variability that is dynamically regulated by a large number of inter- and intra-cellular signaling molecules [226].

Astrocyte morphology remote from CNS lesions and with different gradations of reactive astrogliosis. Brightfield immunohistochemistry for GFAP counterstained with haematoxylin (H) in human autopsy specimens, surveys with details (boxed areas). a Appearance of astrocytes in tissue remote from a lesion and presumed healthy. Note that the territories of astrocyte processes do not overlap and that many astrocytes do not express detectable levels of GFAP. b Moderately reactive astrogliosis in which most (if not all) astrocytes have up regulated expression of GFAP and exhibit cellular hypertrophy, but with preservation of individual astrocyte domains and without pronounced overlap of astrocyte processes. c Severe diffuse reactive astrogliosis with pronounced up regulation of GFAP expression, astrocyte hypertrophy, astrocyte proliferation and pronounced overlap of astrocyte processes resulting in disruption of individual astrocyte domains. Scale bars surveys = 25 μm, details = 10 μm

Schematic representations that summarize different gradations of reactive astrogliosis. a Astrocytes in healthy CNS tissue. b Mild to moderate reactive astrogliosis comprises variable changes in molecular expression and functional activity together with variable degrees of cellular hypertrophy. Such changes occur after mild trauma or at sites distant from a more severe injury, or after moderate metabolic or molecular insults or milder infections or inflammatory activation. These changes vary with insult severity, involve little anatomical overlap of the processes of neighboring astrocytes and exhibit the potential for structural resolution if the triggering insult is removed or resolves. c Severe diffuse reactive astrogliosis includes changes in molecular expression, functional activity and cellular hypertrophy, as well newly proliferated astrocytes (with red nuclei in figure), disrupting astrocyte domains and causing long-lasting reorganization of tissue architecture. Such changes are found in areas surrounding severe focal lesions, infections or areas responding to chronic neurodegenerative triggers. d Severe reactive astrogliosis with compact glial scar formation occurs along borders to areas of overt tissue damage and inflammation, and includes newly proliferated astrocytes (with red nuclei in figure) and other cell types (gray in figure) such as fibromeningeal cells and other glia, as well as deposition of dense collagenous extracellular matrix. In the compact glial scar, astrocytes have densely overlapping processes. Mature glial scars tend to persist for long periods and act as barriers not only to axon regeneration but also to inflammatory cells, infectious agents, and non-CNS cells in a manner that protects healthy tissue from nearby areas of intense inflammation
GFAP has been studied extensively. GFAP is one of a family of intermediate filament proteins, including vimentin, nestin, and others, that serve largely cyto-architectural functions [179]. Studies in transgenic mice indicated that the expression of GFAP is not essential for the normal appearance and function of most astrocytes in healthy CNS of transgenic mice, but is essential for the process of reactive astrogliosis and glial scar formation [95, 179, 180]. There are different isoforms and splice variants of GFAP including GFAP α, β, γ, δ, and κ, and these may be expressed in a heterogeneous manner in both healthy CNS and in pathological specimens including glioma, but the differential distribution and roles of GFAP isoforms are only beginning to be studied [4, 20, 195].
At the single-cell level, in line with its structural role, GFAP is not present throughout astrocyte cytoplasm, and GFAP immunohistochemistry does not label all portions of the astrocyte but only in the main stem branches (Fig. 1b). GFAP is entirely absent from the finely branching astrocyte processes and is often not detectably present in the cell body (Figs. 1b, 3a). Consequently, GFAP immunohistochemistry can markedly underestimate the extent of astrocyte branching and territory in comparison with other means of detection such as Golgi staining (Fig. 1a) or expression of reporter proteins such as GFP or β-galactosidase (Fig. 1b), or filling with fluorescent dyes [34].
In addition, as regards the use of GFAP as an astrocyte marker, it is important to note that GFAP expression is not exclusive to protoplasmic and fibrous astrocytes. Within the CNS, GFAP is also expressed by a number of cells that can be considered part of an extended astroglial family that is described in more detail below. Outside of the CNS, GFAP is expressed widely in many tissues by a variety of cell types, also discussed below.
Other molecular markers that have been used for immunohistochemical identification of astrocytes and reactive astrocytes include glutamine synthetase and S100β [85, 165], but these molecules are not entirely exclusive to astrocytes. Several recent studies have conducted large-scale genetic analysis of the astrocyte transcriptome in rodents and humans, and have identified large numbers of molecules enriched in astrocytes as compared with other neural cells such as neurons and oligodendrocytes [35, 129]. Such studies will provide a rich resource for identifying molecules for mechanistic analyses of functions and roles of astrocytes and reactive astrocytes. In addition, such studies are identifying potential candidates for additional, and better, molecular markers with which to identify astrocytes. One such candidate appears to be the protein Aldh1L1, whose promoter reliably targets the expression of reporter molecules such as GFP (green fluorescent protein) to astrocytes and whose identification by immunohistochemistry may provide a sensitive chemical marker for most if not all astrocytes in healthy tissue [15].
Anatomical organization
Astrocytes tile the entire CNS in a contiguous and essentially non-overlapping manner that is orderly and well organized (Fig. 3a). There are no CNS regions devoid of astrocytes or closely related cells. Dye filling of individual cells and other anatomical techniques have demonstrated that in healthy CNS, individual protoplasmic astrocytes have essentially non-overlapping domains in gray matter, such that only the most distal tips of processes from individual astrocytes interdigitate with one another and thereby provide the substrate for formation of the gap junctions detected at the ultrastructural level [34, 91, 162, 172]. Similar individual astrocyte domains appear likely to exist in white matter, but this has not yet been as extensively reported on. Individual protoplasmic astrocytes typically extend from five to ten main stem branches, each of which gives rise to many finely branching processes that are evenly distributed throughout the astrocyte domain in the gray matter. In hippocampus or cortex many finely branching processes from a single astrocyte are estimated to contact several hundred dendrites from multiple neurons and to envelope 100,000 or more synapses [34, 91, 172]. It is also noteworthy that human astrocytes are larger, structurally more complex, and more diverse than astrocytes in rodents [170].
Astrocyte physiology
Astrocytes express potassium and sodium channels and can exhibit evoked inward currents, but unlike neurons, astrocytes do not ‘fire’ or propagate action potentials along their processes [162, 212]. However, this does not mean that astrocytes are physiologically ‘silent’. Astrocytes exhibit regulated increases in intracellular calcium concentration [Ca++]i that represent a form of astrocyte excitability [39, 46]. A large body of evidence is now available that these regulated increases in astrocyte [Ca++]i are of functional significance in astrocyte–astrocyte as well as in astrocyte–neuron intercellular communication. Astrocyte [Ca++]i elevations can (1) occur as intrinsic oscillations resulting from Ca++ released from intracellular stores, (2) be triggered by transmitters (including glutamate and purines) released during neuronal activity, (3) elicit the release from astrocytes of transmitters such as glutamate into the extracellular space and thereby trigger receptor mediated currents in neurons, and (4) be propagated to neighboring astrocytes [89, 162, 182, 215, 249]. Evidence that calcium signaling enables astrocytes to play a direct role in synaptic transmission is discussed below. It is also noteworthy that astrocytes can couple to neighboring astrocytes through gap junctions (Fig. 2a) formed by connexins, and that the gap junctional coupling of astrocytes into multicellular networks may play a role in both normal function and CNS disorders [162, 212].
Roles in development
The developmental generation of astrocytes tends to occur after the initial production of neurons in many CNS regions. Nevertheless, astrocytes exert a number of important functions during development of both gray and white matter. Molecular boundaries formed by astrocytes take part in guiding the migration of developing axons and certain neuroblasts [186]. In addition, substantive evidence is accumulating that astrocytes are essential for the formation and function of developing synapses by releasing molecular signals such as thrombospondin [15, 43, 243]. Astrocytes appear also to influence developmental synaptic pruning by releasing signals that induce expression of complement C1q in synapses and thereby tag them for elimination by microglia [15, 230]. As regards the development of white matter, the loss or dysfunction of astrocyte connexins and gap junctions leads to dysmelination [131].
Regulation of blood flow
Astrocytes make extensive contacts with (Fig. 1a, b) and have multiple bidirectional interactions with blood vessels, including regulation of local CNS blood flow. Recent findings show that astrocytes produce and release various molecular mediators, such prostaglandins (PGE), nitric oxide (NO), and arachidonic acid (AA), that can increase or decrease CNS blood vessel diameter and blood flow in a coordinated manner [86, 102]. Moreover, astrocytes may be primary mediators of changes in local CNS blood flow in response to changes in neuronal activity [112]. Astrocytes have processes in contact with both blood vessels and synapses (Fig. 2a). Via these contacts, astrocytes titrate blood flow in relation to levels of synaptic activity, as demonstrated recently in the visual cortex where fMRI detected changes in blood flow in response to visual stimuli were shown to be dependent on astrocyte function [211, 260].
Fluid, ion, pH, and transmitter homeostasis
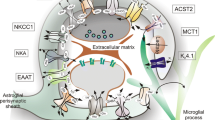
Astrocyte processes envelop essentially all synapses [27, 183] and exert essential functions in maintaining the fluid, ion, pH, and transmitter homeostasis of the synaptic interstitial fluid in a manner that is critical for healthy synaptic transmission (Fig. 2a). Astrocyte processes are rich in the aquaporin 4 (AQP4) water channel and in transporters for the uptake of K+ [212, 220, 264]. Astrocyte membranes have different means of proton shuttling, including the Na+/H+ exchanger, bicarbonate transporters, monocarboxylic acid transporters, and the vacuolar-type proton ATPase [168]. AQP4 water channels are densely clustered along astrocyte processes that contact blood vessels and play a critical role in regulating fluid homeostasis in healthy CNS and play roles in both vasogenic and cytotoxic edema as discussed below. Astrocyte processes at synapses also play essential roles in transmitter homeostasis by expressing high levels of transporters for neurotransmitters such as glutamate, GABA, and glycine that serve to clear the neurotransmitters from the synaptic space [205, 212]. After uptake into astrocytes, the transmitters are converted by enzymes such as glutamine synthetase into precursors such as glutamine and recycled back to synapses for reconversion into active transmitters (Fig. 2a). Networks of astrocytes linked together by gap junctions are thought to be able to rapidly dissipate small molecules such as potassium and glutamate and prevent their potentially detrimental accumulation (Fig. 2a) [212].
Roles in synapse function
There is now steadily accumulating evidence that astrocytes play direct roles in synaptic transmission through the regulated release of synaptically active molecules including glutamate, purines (ATP and adenosine), GABA, and D-serine [89, 162, 182, 215]. The release of such gliotransmitters occurs in response to changes in neuronal synaptic activity, involves astrocyte excitability as reflected by increases in astrocyte [Ca++]i, and can alter neuronal excitability [89, 162, 182, 215]. Such evidence has given rise to the ‘tripartite synapse’ hypothesis, which posits that astrocytes play direct and interactive roles with neurons during synaptic activity in a manner that is essential for information processing by neural circuits [5, 89, 182].
In addition to having direct effects on synaptic activity via the release of gliotransmitters, astrocytes have the potential to exert powerful and long-term influences on synaptic function through the release of growth factors and related molecules. As discussed above, molecular mechanisms have been identified through which astrocytes play a role in the formation, maintenance, and pruning of synapses during development [15, 43, 230]. Such mechanisms may also provide astrocytes with the means of exerting powerful influences on synaptic remodeling and pruning in the healthy adult CNS or in response to CNS disorders [15]. Cytokines such as tumor necrosis alpha (TNFα) have been shown to influence homeostatic synaptic scaling by inducing the insertion of AMPA receptors at post-synaptic membranes [229]. Although it is not certain whether astrocytes or microglia are primary sources of TNFα in the CNS in vivo, the effects on synaptic function of astrocyte-derived growth factors and cytokines warrant further study. Astrocytes are also sources of neuroactive steroids (neurosteroids), including estradiol, progesterone and various intermediaries and metabolites that can have synaptic effects, particularly at GABAA receptors [80].
Energy and metabolism
A growing body of evidence also now indicates that astrocytes make important contributions to CNS metabolism. Astrocytes, which have processes that on the one hand contact blood vessels and on the other hand contact neuronal perikarya, axons (at nodes of Ranvier), and synapses, are well positioned to take up glucose from blood vessels and furnish energy metabolites to different neural elements in gray and white matter (Fig. 2a). Although it was known for many years that astrocytes are the principal storage sites of glycogen granules in the CNS and that the greatest accumulation of astrocytic glycogen occurs in areas of high synaptic density [183, 184], the functional contribution of these stores was originally discounted [27]. Compelling evidence now demonstrates that astrocytic glycogen utilization can sustain neuronal activity during hypoglycemia and during periods of high neuronal activity [27, 231]. In this regard, it is noteworthy that astrocyte glycogen content can be modulated by transmitters such as glutamate [27] and that glucose metabolites can be passed across gap junctions in a manner that is regulated by glutamate and neuronal activity [200]. Other lines of evidence indicate that during hypoglycemia, astrocyte glycogen breaks down to lactate that is transferred to adjacent neural elements (both synapses in gray matter and axons in white matter) where it is used aerobically as fuel [27, 28, 181, 252]. In addition, computer-based modeling studies suggest that during periods of high neuronal activity, inhibition of phosphofructokinase leads to impairment of neuronal glycolysis, with the consequence that lactate effluxed from astrocytes becomes the preferred energy substrate for neurons [171].
Blood brain barrier
The blood brain barrier (BBB) is a diffusion barrier that impedes the influx into brain parenchyma of certain molecules on the basis of polarity and size [1, 11]. The principal cellular constituents of the BBB are cerebral capillary endothelial cells that form tight junctions and are surrounded by a basal lamina, perivascular pericytes, and astrocyte endfeet. The role of pericytes in BBB function is not well studied and the role of astrocytes is controversial. The main functional components of the BBB are the endothelial tight junctions [1, 11]. Numerous lines of in vitro evidence indicate that astrocytes can induce barrier properties in cerebral and other endothelial cells as well as in related epithelial, arguing in favor of a role for astrocytes in BBB induction [1, 11, 17, 208]. In contrast, certain aspects of the BBB become functional in vivo before the appearance of astrocytes during development [93, 206]. In this regard, it is interesting to note that embryonic neural progenitor cells are able to induce BBB properties in cerebral endothelial cells [255]. Other studies suggest a role for astrocytes in regulating BBB properties of cerebral endothelial in vivo in adult mice through specific BMP signaling mechanisms on astrocyte endfeet that when disrupted cause BBB leaks [6]. In addition, both astrocytes and related enteric glial cells are able to induce barrier properties in gut epithelial cells and the molecule GSNO has been identified as a molecular mediator of this effect [208]. Additional studies will be needed to clarify the roles of astrocytes and pericytes in BBB function and to identify molecular mediators that may induce BBB properties in cerebral endothelia. It is possible that different cell types provide such factors at different times during development and in healthy or injured CNS.
Astrocyte heterogeneity
The increasing awareness of the complexity, importance, and diversity of astrocyte functions is giving rise to a growing interest in the potential for specialization and heterogeneity among astrocytes [96]. The notion of astrocyte heterogeneity is not new. Numerous anatomical distinctions have long been recognized among gray (protoplasmic) and white (fibrous) matter astrocytes in different CNS regions [191], but these have long been over looked or discounted. In addition, there is a long-standing recognition of an extended family of astroglial cells that share similarities with, but also exhibit differences to, protoplasmic and fibrous astrocytes, including Müller glia in the retina, Bergmann glia of the cerebellum, tanycytes at the base of the third ventricle, pituicytes in the neurohypophysis, cribrosocytes at the optic nerve head, and others. These different cell types express various astrocyte-related molecules such as GFAP, S100β, glutamine synthetase and others, and exert functions similar to astrocytes in manners specialized to their locations. In addition, these different types of astroglial cells share with astrocytes the ability to become reactive in response to CNS insults and these cells have the potential to play important roles in pathological changes in their specific locations as discussed below. As regards protoplasmic and fibrous astrocytes, various lines of evidence suggest that there is considerable molecular, structural, and potentially functional diversity of astrocytes at both the regional and local levels, but such investigations are at an early stage [10, 96]. In this context it is interesting to note that the number, complexity, and diversity of astroglial cells associated with neurons has increased considerably with evolution, such that the ratio of astrocytes to neurons is 1:6 in worms, 1:3 in rodent cortex, and 1.4:1 in human cortex, implying that astrocyte roles increase in importance with sophistication of neural tissue [162]. The human cerebral neocortex contains multiple subtypes of astrocytes, including types that do not appear to exist in rodent cortex [170]. As molecular markers become more sophisticated, it seems likely that astrocytes will be revealed to be considerably more heterogeneous than thus far imagined. Such information is likely to impact on concepts about astrocyte roles in both health and disease.
GFAP-expressing multipotent radial neural stem cells in juvenile and adult brain
Recent studies have identified specific morphological and chemical phenotypes of neural stem cells (NSC) in embryonic, juvenile, and adult brain. In the embryonic brain, it is now clear that radial cells are NSC that give rise to cortical pyramidal neurons [116, 134, 164]. As these radial cells mature, they adopt GFAP expression and some give rise to GFAP-expressing radial NSC that persist in juvenile and adult forebrain, while others become astrocytes [116, 122, 142]. Some GFAP-expressing radial NSC remain constitutively active throughout life in the subependymal (or subventricular) zone of the lateral ventricles and in the subgranular zone of the hippocampal dentate gyrus, where they are the predominant source of adult neurogenesis [56, 78, 103, 214]. Based almost entirely on the observation that these adult NSC express GFAP, they have been referred to by some authors as a ‘sub-type’ of astrocyte [56, 116], but it is not clear that such a nomenclature is either warranted or useful [14, 84, 153]. There is ample evidence that GFAP is not an exclusive marker of astrocytes. Moreover, as described above, protoplasmic and fibrous astrocytes are highly differentiated and specialized cells that exert a wide variety of complex activities essential for neural function in the healthy CNS, and at present there is no evidence that GFAP-expressing adult radial NSC exert any of the complex functions of mature differentiated protoplasmic and fibrous astrocytes. In addition, there is clear evidence that mature adult astrocytes are not neurogenic. Differentiated astrocytes in uninjured mature cerebral cortex do not exhibit neurogenic potential either in vivo or in vitro [104]. Even proliferating reactive astrocytes after CNS trauma give rise only to other astrocytes and do not spontaneously exhibit multipotent neurogenic potential unless they are reprogrammed genetically or by exposure to high doses of specific growth factors in vitro [29, 30]. In addition, GFAP-expressing NSC differ in morphology (radial phenotype), chemical phenotype (LeX and nestin expression), and physiological characteristics from protoplasmic and fibrous astrocytes [78, 104, 126]. Thus, the majority of currently available evidence argues that juvenile and adult NSC share more similarities with embryonic radial NSC than with mature astrocytes. There seems to be no reason to label the multipotent neural progenitors that persist throughout life as a subtype of astrocyte, which risks the blurring of clear functional distinctions among cell types. Instead, it seems that clarity would best be served by referring to these progenitors simply as either juvenile or adult radial NSC, in keeping with their derivation and phenotype. It has been argued that it may be useful to return to the nomenclature of early neuroanatomists who referred to such radial progenitor cells as ‘neuroepithelial cells’ rather than glia [14, 153]. Regardless of nomenclature, the presence in certain forebrain areas of GFAP-expressing cells that have constitutive multipotent NSC potential throughout life has important implications for concepts regarding various CNS disorders, in particular CNS neoplasms, as discussed below.
Astrocyte related cells outside of the CNS
There are many cells that have morphological and functional similarities to astrocytes outside of the CNS in both neural and non-neural tissues. Peripheral ganglia contain satellite cells that express GFAP and envelope neurons. Peripheral nerves contain non-myelinating Schwann cells that express GFAP and surround non-myelinated axons. The enteric nervous system contains a vast population of GFAP-expressing enteric glia that extends throughout the different neural plexuses. Enteric glia surround neuronal cell bodies and axons, as well as contact blood vessels and epithelial cells, and are likely to take part in many gut functions in manners that are similar to astrocytes [32, 202]. GFAP is expressed widely in many tissues by a variety of cell types including mesenchymal stellate cells that share structural and functional similarities with astrocytes in many organs, including liver, kidney, pancreas, lungs, and testes [32, 124, 266]. The functional roles of these cells and their potential similarities with astrocytes are only beginning to be explored, but some appear to share with astrocytes functions in tissue repair and scar formation, as well as newly discovered roles as regulators of local immune and inflammatory responses.
Reactive astrogliosis and glial scar formation
Descriptive features of reactive astrogliosis and glial scars
Although reactive astrogliosis is used widely as a pathological hallmark of diseased CNS tissue, definitions of reactive astrogliosis can vary considerably among authors and there are no widely accepted categories of intensity or severity. Based on a large body of observations in experimental animals, a definition of reactive astrogliosis has recently been proposed [226] that encompasses four key features: (1) reactive astrogliosis is a spectrum of potential molecular, cellular and functional changes in astrocytes that occur in response to all forms and severities of CNS injury and disease including subtle perturbations, (2) the changes undergone by reactive astrocytes vary with severity of the insult along a gradated continuum of progressive alterations in molecular expression, progressive cellular hypertrophy, and in severe cases, proliferation and scar formation, (3) the changes of reactive astrogliosis are regulated in a context-specific manner by inter- and intra-cellular signaling molecules, (4) the changes undergone during reactive astrogliosis have the potential to alter astrocyte activities both through gain and loss of functions that can impact both beneficially and detrimentally on surrounding neural and non-neural cells [226]. Here, we propose to apply and extend this definition to identifying different gradations of reactive astrogliosis and glial scar formation that may be encountered in histopathological examinations of human disorders (Figs. 3, 4). According to this definition, reactive astrogliosis is not an all-or-none response, nor is it a single uniform process, nor is it ubiquitously synonymous with scar formation. Instead, reactive astrogliosis is a finely gradated continuum of progressive changes in gene expression and cellular changes. Although the increasing severities of reactive astrogliosis transition seamlessly along a continuum, it is convenient for purposes of description and classification to recognize three broad categories.
Mild to moderate reactive astrogliosis
In mild or moderate reactive astrogliosis there is variable up regulation of expression of GFAP and other genes [226], as well as hypertrophy of cell body and processes that can vary in degree but that occurs within the domains of individual astrocytes [257] without substantive intermingling or overlap of processes of neighboring astrocytes or loss of individual domains (Figs. 3b, 4b) [226]. There is little or no astrocyte proliferation in mild or moderate reactive astrogliosis; however, the up regulation of GFAP expression in astrocytes that do not express detectable levels of GFAP in healthy tissue (Figs. 3a, 4a) can lead to the staining of more cells (Figs. 3b, 4b), sometimes giving the false impression of proliferation. Mild or moderate reactive astrogliosis is generally associated with mild non-penetrating and non-contusive trauma, diffuse innate immune activation (viral infections, system bacterial infections), and in areas that are some distance to focal CNS lesions. Because there is little or no reorganization of tissue architecture, if the triggering mechanism is able to resolve, then mild or moderate reactive astrogliosis exhibits the potential for resolution in which the astrocytes return to an appearance similar to that in healthy tissue [226]. The physiological consequences of mild or moderate reactive astrogliosis are not well understood.
Severe diffuse reactive astrogliosis
In severe diffuse reactive astrogliosis there is pronounced up regulation of expression of GFAP and other genes, together with pronounced hypertrophy of cell body and processes, as well as astrocyte proliferation, resulting in considerable extension of processes beyond the previous domains of individual astrocytes. As a result, there is substantive intermingling and overlapping of neighboring astrocyte processes with blurring and disruption of individual astrocyte domains (Figs. 3c, 4c). These changes can result in long-lasting reorganization of tissue architecture that can extend diffusely over substantive areas without the formation of dense, compact barriers as found in glial scars along borders to necrotic tissue (see below). Severe diffuse reactive astrogliosis is generally found in areas surrounding severe focal lesions, infections or areas responding to chronic neurodegenerative triggers.
Severe reactive astrogliosis with compact glial scar formation
Severe reactive astrogliosis with compact glial scar formation includes changes associated with milder forms, such as pronounced up regulation of GFAP and other genes, and pronounced hypertrophy of cell bodies and processes. In addition, glial scar formation progresses beyond these changes with groups of reactive astrocytes that exhibit pronounced overlapping of reactive astrocyte processes, obliteration of individual astrocyte domains, evidence of substantive astrocyte proliferation, and the clear formation of dense, narrow, and compact glial scars (Fig. 4d). Recent experimental evidence indicates that these astrocyte scars act as neuroprotective barriers to inflammatory cells and infectious agents, and that they form in particular along borders to severe tissue damage, necrosis, infection or autoimmune-triggered inflammatory infiltration (Fig. 4d) [33, 59, 68, 95, 226, 251]. An important feature of these glial scars is the interaction of reactive astrocytes with other cell types, in particular fibromeningeal and other glial cells (Fig. 4d) [31, 95], and the deposition of a dense collagenous extracellular matrix that contains many molecular cues that inhibit axonal and cellular migration [219]. Triggering insults include penetrating trauma, severe contusive trauma, invasive infections or abscess formation, neoplasm, chronic neurodegeneration, systemically triggered inflammatory challenges. It is noteworthy that the glial scar formation is associated with substantive tissue reorganization and structural changes that are long lasting and persist long after the triggering insult may have resolved.
The findings summarized here show that there are pronounced differences along the continuum of potential responses of reactive astrocyte to insults of different kinds and severities. These differences are likely to be of consequence when considering the functions and impact of reactive astrogliosis on CNS disorders and pathologies, as discussed below.
The role of astrocyte proliferation in reactive astrogliosis and glial scar formation
The contribution of astrocyte proliferation to reactive astrogliosis warrants specific consideration. The widespread and often exclusive use of GFAP as a marker for astrocytes has led to a tendency to overestimate the contribution of astrocyte proliferation to reactive astrogliosis. As discussed above, many astrocytes in healthy CNS do not express GFAP at immunohistochemically detectable levels, or express only very low levels (Figs. 3a, 4a). During mild or moderate astrogliosis, there is a marked up regulation of GFAP expression as well as cellular hypertrophy in essentially all astrocytes, which can lead to the false impression of an increase in astrocyte number because more astrocytes are immunohistochemically stained and the larger astrocytes seem more densely packed (Figs. 3b, 4b). In healthy CNS tissue, astrocyte turnover is low and there are few proliferating or newly generated astrocytes and it appears that the majority of astrocytes are post-mitotic and long-lived [33, 45, 97, 167]. Experimental analysis of cell proliferation indicates that GFAP up regulation and hypertrophy can occur in mild or moderate astrogliosis in the absence of proliferation and increase in cell number [226]. The appearance of newly proliferated astrocytes occurs in severe diffuse reactive astrogliosis and severe reactive astrogliosis with compact glial scar formation as discussed above (Figs. 3c, 4c, d). In human specimens, actively dividing reactive astrocytes have been particularly reported in association with infection and acute demyelinating lesions [45]. The source of newly divided scar-forming astrocytes is not well established, and may include mature astrocytes that re-enter the cell cycle as well as progenitor cells in the local parenchyma or in the periventricular regions [30, 33, 37, 77, 133, 141, 226].
Molecular triggers and regulators of reactive astrogliosis and glial scar formation
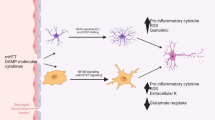
Many different types of intercellular signaling molecules are able to trigger reactive astrogliosis or to regulate specific aspects of reactive astrogliosis, including (1) large polypeptide growth factors and cytokines such as IL6, LIF, CNTF, TNFα, INFγ, Il1, Il10, TGFβ, FGF2, etc., (2) mediators of innate immunity such as lipopolysaccharide (LPS) and other Toll-like receptor ligands, (3) neurotransmitters such as glutamate and noradrenalin, (4) purines such as ATP, (5) reactive oxygen species (ROS) including nitric oxide (NO), (6) hypoxia and glucose deprivation, (7) products associated with neurodegeneration such as β-amyloid, (8) molecules associated with systemic metabolic toxicity such as NH4 +, and (9) regulators of cell proliferation such endothelin-1, as reviewed in detail elsewhere [226]. Such molecular mediators of reactive astrogliosis can be released by all cell types in CNS tissue, including neurons, microglia, oligodendrocyte lineage cells, pericytes, endothelia, and other astrocytes, in response to all forms of CNS insults, ranging from subtle cellular perturbations that release some of the specific factors just listed, to cell stretching as might be encountered during acceleration/deceleration CNS injury and which releases ATP, to intense tissue injury and cell death that release various intracellular molecules that signal intense tissue damage (Fig. 2b) [226]. It is also becoming clear that different molecular, morphological, and functional changes in reactive astrocytes are specifically controlled by inter- and intra-cellular signaling mechanisms that reflect the specific contexts of the stimuli and produce specific and gradated responses of reactive astrogliosis [226]. For example, the different intracellular signaling pathways associated with STAT3, NFκB, SOCS3, Nrf2, cAMP, Olig2 are implicated in mediating different aspects or different degrees of reactive astrogliosis such as GFAP up regulation, cell hypertrophy, proliferation, and pro- or anti-inflammatory effects [226]. Molecular triggers that lead to proliferation of reactive astrocytes in vivo are incompletely characterized but include EGF, FGF, endothelin 1, and ATP [77, 121, 161].
Functions of reactive astrogliosis and glial scar formation
Concepts about reactive astrogliosis have long been dominated by the recognition over 100 years ago that scars formed by reactive astrocytes inhibit axon regeneration and by the interpretation that this scar formation was the main impediment to functional recovery after CNS injury or disease [192]. The ensuing over 100-year-long emphasis on glial scar formation as an inhibitor of axon regeneration has often led to a generalized negative view of reactive astrogliosis per se, and there has been a tendency among certain authors to typecast the entire process of reactive astrogliosis as a uniformly negative and maladaptive phenomenon that unavoidably causes neurotoxicity, inflammation, or chronic pain. This stereotyped viewpoint has sometimes led to the simplistic notion that total inhibition of reactive astrogliosis can be regarded as a therapeutic strategy. This absolutely negative viewpoint of reactive astrogliosis is no longer tenable and it is now clear from many different lines of experimental evidence that there is a normal process of reactive astrogliosis that exerts essential beneficial functions and does not do harm. As reviewed in detail elsewhere [226], many studies using transgenic and experimental animal models provide compelling evidence that reactive astrocytes protect CNS cells and tissue by (1) uptake of potentially excitotoxic glutamate [33, 198, 234], (2) protection from oxidative stress via glutathione production [42, 216, 234, 245], (3) neuroprotection via adenosine release [125], (4) protection from NH4 + toxicity [193], (5) neuroprotection by degradation of amyloid-beta peptides [113], (6) facilitating blood brain barrier repair [33], (7) reducing vasogenic edema after trauma, stroke or obstructive hydrocephalus [33, 264], (8) stabilizing extracellular fluid and ion balance and reducing seizure threshold [264], and (9) limiting the spread of inflammatory cells or infectious agents from areas of damage or disease into healthy CNS parenchyma [33, 59, 68, 95, 123, 156, 175, 251].
Reactive astrogliosis, glial scar formation and CNS inflammation
Of particular interest as regards functions of reactive astrocytes, is recent evidence that reactive astrogliosis and glial scar formation play essential roles in regulating CNS inflammation [226]. In response to different kinds of stimulation, reactive astrocytes can make many different kinds of molecules with either pro- or anti-inflammatory potential [60, 108] and reactive astrocytes can exert both pro- and anti-inflammatory effects on microglia [67, 148]. A large body of experimental studies indicates that reactive astrocytes can exert both pro- and anti-inflammatory regulatory functions in vivo in a context dependent manner that is regulated by specific molecular signaling pathways [226]. A functional model, which may reconcile the apparent paradox that reactive astrocytes have the potential to exert both pro- and anti-inflammatory activities, is that reactive astrocytes exert different activities at different times after insults, or in different geographical locations in relation to lesions, as determined by context-specific signaling mechanisms. For example, reactive astrocytes may exert pro-inflammatory roles at early times after insults and in the center or immediate vicinity of lesions, but exert anti-inflammatory roles at later times and at the borders between lesions and healthy tissue. In cases of severe lesions, reactive astrocytes may form scars that act as cell migration barriers around the borders of areas where intense inflammation is needed and thereby restrict the spread of inflammatory cells and infectious agents into adjacent healthy tissue (Fig. 4d) [225, 226]. Together, these findings indicate that there is a normal process of reactive astrogliosis and glial scar formation that exerts various beneficial functions including protecting neural cells and function, restricting the spread of inflammation and infection, and promoting tissue repair. It is important to distinguish this normal process from the potential for dysfunction of reactive astrocytes to contribute to CNS disorders.
Mechanisms for contributions of reactive astrogliosis to CNS disorders and pathologies
Astrocytes and reactive astrocytes have the potential to impact on essentially all aspects of neural function through regulation of blood flow, provision of energy substrates, or by influencing synaptic function and plasticity. Increasing evidence indicates that dysfunctions of the process of reactive astrogliosis and scar formation have the potential to contribute to, or to be primary causes of, CNS disease mechanisms either through loss of normal functions or through gain of detrimental effects [226].
Potential for loss of essential functions by reactive astrocytes
As discussed earlier, in healthy neural tissue, astrocytes play critical roles in many functions, including regulation of blood flow, homeostasis of extracellular fluid, ions and transmitters, energy provision, and regulation of synapse function, and synaptic remodeling (Fig. 2a). In addition, different types of transgenic deletion studies show that reactive astrocytes exert a number of essential beneficial functions in response to CNS insults, including BBB repair, neural protection and restricting the spread of inflammatory cells, and infection. The disturbance or loss of these functions during the process of reactive astrogliosis has the potential to underlie neural dysfunction and pathology in various conditions including trauma, stroke, multiple sclerosis, and others. Genetic animal models are providing evidence for ways in which loss or attenuation of reactive astrocyte functions might worsen outcome after various kinds of CNS insults, for example, through excitotoxic neurodegeneration due to failure or attenuation of glutamate uptake [33, 198, 234] or increased inflammation or infection due to loss or failure of astrocyte barrier functions [33, 59, 68, 95, 251]. Clinical studies, discussed below, are also identifying a rapidly growing number of conditions that are precipitated by or exacerbated loss of functions by astrocytes or reactive astrocytes.
Potential for gain of detrimental effects by reactive astrocytes
In a manner analogous to inflammation, reactive astrogliosis also has the potential to exert detrimental effects. The prototypical gain of a detrimental effect by reactive astrogliosis and glial scar formation is the inhibition of axon regeneration, first proposed over 100 years ago and now supported by extensive molecular and cell-biological evidence [219]. In addition, there is also evidence from both clinical (discussed below) and experimental studies that, under specific circumstances, astrocytes or reactive astrocytes have the potential to exert a variety of other detrimental effects through gain of abnormal effects. For example, reactive astrocytes can be stimulated by specific signaling cascades (Fig. 2b) to gain of detrimental effects such as (1) exacerbating inflammation via cytokine production [22, 23], (2) producing neurotoxic levels of reactive oxygen species [92, 234], (3) releasing potentially excitotoxic glutamate [238], (4) potential contribution to seizure genesis [106, 240], (5) compromising blood brain barrier function due to VEGF-production [7], (6) causing cytotoxic edema during trauma and stroke through AQP4 over activity [264], and (7) contributing to chronic pain [147]. There also appears to be the potential for combinatorial signaling cascades resulting from exposure to microbial antigens such as lipopolysaccharide (LPS), to markedly increase the production of potentially toxic molecules by astrocytes [67, 92]. Context-dependent acute or chronic combinatorial signaling events (Fig. 2b) involving different cell types might thus stimulate reactive astrocytes to produce potentially cytotoxic levels of molecules or might lead to chronic inflammation or neuropathic pain. A greater understanding of such combinatorial signaling could facilitate targeted therapeutic strategies that preserve the beneficial and attenuate the potentially detrimental aspects of reactive astrogliosis. In this regard, it will also be interesting in future studies to look for genetic polymorphisms that may alter reactive astrocyte functions in certain individuals and thereby predispose them to certain disease processes.
Molecular dissection of reactive astrogliosis using transgenic technologies
Many experimental investigations of mechanisms and functions of reactive astrogliosis and glial scar formation make use of transgenic technologies combined with experimental animal models, which has revolutionized the study of neural injury and disease by allowing in vivo dissection of molecular and cellular mechanisms [226]. Nevertheless, in interpreting results derived from studies using transgenic mice it is important to understand associated caveats [226]. For example, different promoters (human or mouse GFAP, GLAST, etc.) may have different targeting properties/efficiencies as regards astrocytes and related cells. Although not all astrocytes express GFAP or GFAP-regulated transgenes in healthy tissue, most if not all appear to do so after CNS injury or other insults [33, 95, 251]. It is essential that authors define transgene expression levels individually and specifically in every tissue region and experimental model being studied, as well as control for any possible confounding effects that may result from transgene expression in other cells both within and outside of the CNS [33, 95, 226, 251].
Astrocyte appearance and roles in CNS disorders and pathology
CNS trauma
Traumatic brain and spinal cord injury are major causes of premature death, permanent physical disability, or long-lasting neuropsychiatric impairment [138]. Reactive astrogliosis and glial scar formation are prominent features of CNS trauma and are increasingly implicated as playing important roles in determining long-term clinical outcome [41, 226]. The appearance of reactive astrogliosis after CNS trauma (whether in the brain or spinal cord) varies with the (1) type of the trauma, i.e., whether it is penetrating, contusional, or diffuse (as in the case of diffuse axonal injury), (2) severity of the trauma, (3) length of time that has elapsed between injury and biopsy or autopsy, and (4) distance of the tissue examined from the site of injury/trauma. The same specimen may exhibit different severities of reactive astrogliosis in different locations, such as a distinct glial scar along areas of damaged tissue after penetrating or severe contusive trauma (Fig. 4d), which may be surrounded by areas of decreasing severity of reactive astrogliosis (Fig. 4c) as distance away from the lesion center increases (Fig. 4b). Tissue at a considerable distance from the site of direct injury may show sequelae of transient edema and/or downstream effects such as Wallerian or trans-synaptic degeneration, or a combination of the two, significantly contributing to neurologic disability.
Severe reactive astrogliosis and glial scar formation at sites of CNS trauma are well documented to inhibit axonal regeneration [219] and are widely regarded as detrimental to clinical outcome. Nevertheless, recent studies using transgenic mouse models indicate that scar-forming reactive astrocytes exert essential neuroprotective functions after both brain and spinal cord trauma [33, 68, 95, 144, 175, 226] as discussed earlier.
Diffuse traumatic brain injury can lead to mild or moderate diffuse astrogliosis without obvious scar formation, in which reactive astrocytes hypertrophy and up regulate gene expression, including GFAP, but in which there is little or no loss of individual astrocyte domains (Figs. 3b, 4b). In animal experiments, mild to moderate reactive astrogliosis of this type exhibits the potential for resolution [226], but the functional consequences of these changes are not well understood. A cerebral concussion almost certainly impacts on astrocyte structure and function, but such injury is not life threatening, results in minimal if any abnormalities on neuroimaging, and is thus (by definition) not usually the subject of neuropathological review or investigation.
Stroke and cerebrovascular disease
Brain parenchyma and the blood vessels that supply it are not, as they once were, viewed as separate ‘compartments’ within the CNS. Rather, the concept of the ‘neurovascular unit’ takes an integrated view of a given brain parenchymal blood vessel and the glial elements that surround it [38, 139, 206]. Cellular elements in the vessel wall influence surrounding CNS parenchyma, just as the surrounding parenchymal elements affect vascular morphology and function. The ‘neurovascular unit’ idea becomes especially important when considering functional recovery after an ischemic infarct or ‘stroke’.
From a practical diagnostic perspective, the appearance of astrocytes around a region of cystic encephalomalacia within brain that has resulted from an ischemic infarct or hemorrhage can be used to ‘date’ such a lesion (i.e., estimate how long it occurred prior to death), though not very accurately. The sequence of cellular events in an ischemic infarct progresses from intense neuronal eosinophilia (hours to 1–2 days after the insult), to influx of polymorphonuclear leukocytes (variable intensity, but usually within the same time frame), then ingress of macrophages/histiocytes (from blood or activated resident microglia) as liquefactive necrosis occurs (beginning 3–5 days after the infarct), and finally proliferation of an abundance of astrocytes that marginate and form a scar (Fig. 4d) along the borders of an infarct from about 7–10 days after ischemia and then persist for life of the patient (Fig. 5). None of these time frames are precise, particularly in aged individuals, where the process of aging, as well as the exposure to other conditions, may influence the level of reactive astrogliosis that is observed. ‘Chronic cystic infarcts’ are usually associated with astrocytic gliosis [247], and a characteristic finding in a cortical cystic infarct is the preservation of the relatively paucicellular molecular layer overlying it and located immediately beneath the meninges, which then develops a dense collection of astrocytes, usually with gemistocytic phenotype. Astrocytic processes usually traverse the cystic cavity left by the death of brain tissue, in a delicate meshwork, with persistence of foamy histiocytes among the glial fibers.

Reactive astrogliosis demarcates cerebral microinfarcts. Survey image of cerebral cortex of an elderly individual showing microinfarcts (arrows) highlighted by dense clusters of prominently reactive astrocytes that stain intensely for GFAP. Fibrous astrocytes within subcortical white matter (wm) exhibit GFAP staining, whereas GFAP is not detectable in most protoplasmic gray matter astrocytes remote from the lesions in this specimen. H haematoxylin counterstain. Scale bar 180 μm
The role of reactive astrogliosis in the evolution of ischemic brain lesions is at present uncertain, but recent studies suggest that reactive astrocytes provide essential metabolic support to neurons during transient ischemia and that failure of astrocyte functions may contribute to neuronal degeneration [196, 237]. In addition, experimental disruption of astroglial scar formation in transgenic mice after stroke is associated with loss of barrier functions along the margins of infarcts, resulting in increased spread of inflammation and increased lesion volume [123].
Stroke may also induce neurogenesis from periventricular neural progenitor cells that express GFAP [173]. Newly ‘born’ neuroblasts and immature neurons migrate to the infract region where they associate intimately with the cerebral microvasculature in and around the infarct, which itself is undergoing intense post-stroke remodeling. It remains to be seen whether this interesting interplay of neurogenic and vascular elements will become relevant in the treatment of human strokes.
Infection
Reactive astrogliosis is prominent in most CNS infections. Astrocytes can be both targets of, as well as responders to, infectious agents, especially viruses. Subpial astrocytes are especially prominent in individuals who have survived an episode of purulent or granulomatous meningitis, and their appearance of reactivity depends in large part on how quickly antibiotic therapy was initiated. The roles of reactive astrocytes during the response to infection are only beginning to be elucidated. For example, reactive and scar-forming astrocytes accumulate prominently around microbial abscesses, and recent evidence indicates that reactive astrocytes play essential roles in restricting the spread of invading microbial agents such as Toxoplasma gondii into CNS parenchyma [59]. These observations are consistent with the putative protective barrier functions of reactive astrocytes to infectious agents and inflammatory cells (Fig. 4d) as discussed above and elsewhere [226]. Reactive astrogliosis can also occur in response to peripheral infections that result in circulating bacterial antigens or endotoxins, such as LPS, due to peritonitis or sepsis without direct CNS infection [105, 213].
Reactive astrocytes are prominent in different types of viral encephalitis. Astrocytes are a main target and reservoir of human herpes virus 6 in the CNS, which can cause encephalitis in both healthy and immunsuppressed patients [57, 222]. Astrocytes are also a main target of human T-lymphotropic virus type-1, leading to disturbances in glutamate homeostasis and excitotoxicity [3, 235]. In addition to being targets of certain viruses, reactive astrocytes may take part more generally in regulating CNS inflammatory responses [226]. Perivascular cuffing, which is a common and essentially defining feature of different types of viral encephalitis, may occur as a result of barrier functions of reactive scar forming astrocytes that restrict the spread of inflammatory cells into CNS parenchyma (Fig. 1c, d) [226, 251]. Astrocyte endfeet prominently line blood vessels in healthy tissue (Fig. 1b) and become hypertrophic during encephalitis (Fig. 6a) and surround perivascular clusters of inflammatory cells (Fig. 1c, d), and experimental disruption of perivascular astrocytes leads to widespread infiltration of lymphocytes and other inflammatory cells into CNS parenchyma [251].

Reactive astrogliosis in two disorders with seizures. a High magnification image of cerebral cortex from an individual with Rasmussen encephalitis (RE). Immunohistochemical staining for GFAP shows moderate reactive astrogliosis that is especially prominent around small blood vessels. Note the intensely stained and hypertrophic astrocytic foot processes extending to and lining the adventitia of microvessels. b, c High magnification images of a resection specimen from an individual with severe seizures associated with severe focal cortical dysplasia stained for H&E (b) or GFAP (c). b In this unique case, abundant Rosenthal fibers were found throughout the resection specimen, especially in the subpial regions (arrows) and around blood vessels (arrowheads). The density of the Rosenthal fibers suggested the diagnosis of ‘focal’ Alexander’s disease. However, testing of the GFAP gene revealed no mutations (for details, see [111]). c Comparable section to b showing abundant GFAP immunoreactive astrocytes and associated Rosenthal fibers (arrows). Scale bars a 10 μm, b 20 μm, c 15 μm
Reactive astrocytes also play a role in the potential neurological complications resulting from direct HIV-1 infection of the brain, which commonly results from ‘neurotropic’ strains of HIV-1 gaining entry to the CNS. Such strains usually target microglia or macrophages (not astrocytes), and may gain entry to the brain by entering it within infected T-lymphocytes (the ‘Trojan horse’ hypothesis of CNS infection). This can eventually result in cognitive impairment, initially mild, which can progress to HIV-associated dementia (abbreviated as HAD, formerly described as AIDS dementia complex/ADC). Despite the relative efficacy of highly active antiretroviral therapy in controlling AIDS, no treatment has to date been found that consistently moderates the pathogenic events important in the evolution of HAD or that achieves neuronal protection from the effects of the virus. The majority of viral replication in the brain occurs in microglia. Astrocytes can be infected but do not appear to serve as sites of viral replication. Infected microglia and astrocytes are the main cause of neurotoxicity, which appears to result indirectly from signals exchanged between the two cell types, resulting in the secretion of potentially toxic molecules within the CNS parenchyma, both cerebral neocortex and white matter. Astrocyte dysfunction with loss of neurosupportive and neuroprotective functions, in particular disruption of glutamate homeostasis leading to synapse dysfunction and excitotoxicity are regarded as the most likely mechanisms in mediating HIV related cognitive impairment and neurodegeneration [100, 253].
HIV-1 infection of the CNS is arguably at present the most common form of brain viral infection. However, numerous other viruses can cause neurological morbidity and even mortality in individuals with a normal immune system, or in those who are immunosuppressed, regardless of whether said immunosuppression is the result of AIDS, aggressive therapy for solid tumors or hematologic malignancy, iatrogenic immunosuppression in a patient with organ or bone stem cell transplantation, or other (e.g., congenital immunodeficiency) disease. Almost all viruses, as with HIV-1 (see above), manifest tropism for various cell types within the brain or spinal cord; that tropism may include the astrocyte. Some of the herpes viruses, e.g., Herpes simplex and cytomegalovirus (the latter being a major opportunistic pathogen in the CNS of AIDS patients [248], infect a wide range of cell types within the CNS including astrocytes, ependyma, and microvascular endothelium. A mosquito-borne flavivirus that has recently appeared in North America, West Nile virus (WNV) infects a variety of tissues throughout the body, but within the CNS appears to favor neurons over astrocytes [8]. ‘Neuronotropism’ of viruses is shared by other agents, e.g., the rabies virus, which also appears to spare astrocytes, though the proliferation of uninfected reactive astrocytes is an inevitable result of rabies virus-associated encephalomyelitis. JC virus, a human polyomavirus of the Polyomaviridae family, shows selective infection and eventual destruction of oligodendroglia in immunosuppressed individuals, leading to neurologic deterioration caused by progressive multifocal leukoencephalopathy (PML). JC virus may also infect astrocytes, and in situ PCR techniques suggest an even broader repertoire of cells that are vulnerable to JC virus infection [132, 250]. In this context it is also interesting to note that effector CD8+ T cells form immunological synapses with virally infected astrocytes, resulting in pronounced changes in astrocyte morphology (and presumably in function as well) as well as potential clearance of infected cells [12, 13].
When bacterial (and some fungal) microorganisms cause a leptomeningitis, a clinical imperative is to prevent this meningitis from evolving into a meningo-encephalitis or cerebritis that leads to significant injury, necrosis, and inflammation of the brain parenchyma with far more serious neurologic morbidity than an infection isolated to the subarachnoid space. The anatomic structure largely responsible for preventing parenchymal injury in the presence of meningitis is the glia limitans, formed at the pial-subarachnoid interface by plump foot processes of reactive astrocytes, fibromeningeal cells, and an overlying basement membrane, in a manner that shares similarities with glia scar formation (Fig. 4d). A breach of the glia limitans may occur because of toxins released by the inflammatory infiltrate associated with a meningitis (especially the toxic molecules released by polymorphonuclear leukocytes/PMN) or frank necrosis of brain parenchyma underlying an inflammatory infiltrate resulting from occlusion/thrombosis of meningeal vessels.
Seizure disorders/epilepsy
Reactive astrogliosis is variable, but often prominent, in almost all forms of seizures, and is perhaps most notable in the setting of hippocampal sclerosis, the most common neuropathological substrate of intractable temporal lobe epilepsy [64]. Astrocytes proliferate in regions of pyramidal neuron loss, usually the endfolium and CA1 segments. In mouse models of severe epilepsy, reactive astrocytes lose their non-overlapping domains and exhibit a tenfold increase in overlapping processes, concurrent with an increase in synaptic spine density [169]. The nature of reactive astrogliotic changes varies with the intensity and duration of the seizure disorder.
The morphologic substrates of epileptic disorders are becoming understood as high-resolution neuroimaging techniques guide neurosurgeons to likely ‘epileptogenic foci’, thereby enabling surgical removal and providing novel lesions for pathologic examination from patients who are relatively ‘early’ in the course of their epileptic disorder [64, 150, 152]. The neuropathological findings within the foci of cortical dysplasia found in severe intractable epilepsy include pronounced dyslamination of the neocortex, neuronal dysmorphism and massive enlargement (‘giant’ neurons), gemistocyte-like ‘balloon cells’ [47]. Of interest is that some of these cellular elements show immunohistochemical evidence suggesting differentiation along both neuronal and astrocytic lines [47]. The most severe examples of focal cortical dysplasia (FCD) closely resemble the tubers found within the neocortex of individuals with tuberous sclerosis complex (TSC), and cells within severe FCD or TSC tubers exhibit distinct mechanisms for activating components of insulin-signaling pathways important in cell growth and metabolism, probably reflecting a modulating effect of TSC1 and TSC2 gene products on these pathways [152, 246]. It is also interesting to note that pronounced astrocytic abnormalities have been described in severe FCD with intractable pediatric epilepsy, including astrocytes with high densities of Rosenthal fibers (Fig. 6b, c), comparable to those seen in Alexander’s disease [111], a genetic disorder with mutation of the GFAP gene that is also associated with seizures and other symptoms (see below).
In addition to simply being passive responders to neuronal changes caused by seizure activity, various lines of recent evidence suggest that reactive astrocytes may play functional roles in mechanisms causing seizures and epilepsy, particularly in chronic relapsing forms. For example, various antiepileptic therapeutic agents block calcium-signaling in astrocytes [240]. In mesial temporal lobe epilepsy with hippocampal sclerosis, neuropathological ultrastructural evaluations suggest that reduced levels of AQP4 and dystrophin in perivascular foot-processes of astrocytes and reactive astrocytes may lead to perturbed water flux through astrocytes with impaired buffering of K+ and an increased propensity for seizures [61, 212]. In Rasmussen’s encephalitis, seizures are associated with perivascular reactive astrogliosis and pronounced hypertrophy of astrocyte endfeet (Fig. 6a), as well as with areas of apparent astrocyte apoptosis and loss, and certain evidence suggests that autoimmune induced (cytotoxic T lymphocyte-mediated) perturbation of astrocyte function and astrocyte degeneration may contribute to the seizure disorder [16, 256]. Reactive astrocytes may also play a role in focal epilepsy following traumatic, ischemic, or infectious brain injury. Experimental evidence and modeling simulations suggest that during the BBB breakdown incurred by such insults, exposure of reactive astrocytes to albumin and other serum proteins leads to reduced clearance by reactive astrocytes of synaptic glutamate and K+ that in turn facilitate seizure-like activity and epileptogenesis [51, 74].
Multiple sclerosis and autoimmune inflammatory disorders
The CNS is affected by a number of different autoimmune inflammatory conditions. Reactive astrogliosis is a not only a prominent feature of these conditions, but various lines of evidence point towards central roles for reactive astrocytes in key pathogenic disease mechanisms discussed below. In the most common (Charcot) type of multiple sclerosis, plaques of demyelination are interspersed with and surrounded by reactive astrocytes and there are widespread regions of focal reactive astrogliosis of varying intensity throughout white matter and in some regions of gray matter [117, 130]. Astrocytes in demyelinating disease may exhibit unusual nuclear and cytological features. They may contain multiple distinct nuclei, especially in chronic disease. Such enlarged multinucleated astrocytes are often referred to as ‘Creutzfeldt astrocytes’ or ‘Creutzfeldt–Peters cells’ [65, 117, 163, 185]. The multiple nuclei within these astrocytes may be fragmented, such that the appearance of a given cell resembles an atypical or ‘granular mitosis’ of the type often encountered in high grade gliomas. Demyelinating lesions may also exhibit astrocytes that label with markers of cell division [45]. The obvious diagnostic pitfall here is that a small biopsy from the edge of a demyelinating plaque may suggest a malignancy rather than multiple sclerosis (especially at the time of intraoperative consultation) [45, 65, 163]. Astrocytes in multiple sclerosis may also demonstrate the phenomenon of ‘emperipolesis’, i.e., an astrocyte apparently engulfing one or more other cells such as oligodendroglia [81] or lymphocytes [75]. The role of this process in the pathogenesis or progression/chronicity of multiple sclerosis is not certain. It should be emphasized that both multinucleation of astrocytes and astrocyte emperipolesis also occur in other disorders, including certain tumors and longstanding spongiform encephalopathy [45, 81, 155, 217].
The roles of astrocytes in multiple sclerosis and other autoimmune inflammatory disorders are under intense investigation and are likely to be complex. Astrocytes can produce a wide variety of pro- or anti-inflammatory molecules [58, 226], and can exert potent suppressive effects on inflammatory cells [114]. In the multiple sclerosis like model condition, experimental autoimmune encephalomyelitis (EAE), reactive astrocytes form scar like barriers around perivascular clusters of inflammatory cells (Fig. 1c), and transgenically targeted disruption of these perivascular astrocyte scars in EAE exacerbates the spread of inflammation, increases axonal degeneration, and worsens clinical signs [251]. Nevertheless, experimental evidence suggests that reactive astrocytes can exert both pro- and anti-inflammatory roles that are essential in EAE [23, 251] and other conditions [226]. While such findings may at first seem at odds with each other, they may be reconciled by evidence showing that reactive astrocytes take part both in attracting inflammatory cells to specific sites and in restricting inflammatory cells to those sites by limiting their spread into adjacent healthy CNS parenchyma [226]. Such findings further suggest that the loss of normal functions or the gain of abnormal effects by reactive astrocytes may contribute to disease mechanisms in multiple sclerosis and other autoimmune inflammatory conditions. Consistent with this idea is the recent observation that the majority (>90%) of patients with neuromyelitis optica, a CNS inflammatory/demyelinating disease that was once considered a rare variant of multiple sclerosis, have autoantibodies to aquaporin-4, which is expressed in the CNS only on astrocyte foot processes and that the severity of clinical signs and spinal cord lesions in such patients correlate with aquaporin-4 specific antibody titers [119, 236]. The potential involvement of astrocytes in other autoimmune inflammatory conditions that sometimes exhibit CNS involvement, such as systemic lupus erythematosus, has been suggested but has not been extensively investigated [242].
Edema
Astrocytes have several mechanisms through which they can influence water balance and the potential for edema, including ion channels and pumps, the water channel AQP4 and the production of molecules such as vascular endothelial growth factor (VEGF) or NO that influence endothelial cell permeability. The role of astrocytes in edema is complex and likely to be context dependent. Although excess uptake of water through AQP4 on astrocyte endfeet can lead to cytotoxic edema [135, 157, 264], the loss of astrocytes or the failure of astrocyte AQP4 channels can also lead to vasogenic edema or accumulation of extracellular fluid [33, 157, 177, 264].
Blood brain barrier integrity
Integrity of the BBB is lost after many types of CNS insults including trauma and stroke. The role of astrocytes in loss or repair of BBB functions is not well understood. A role for astrocytes is suggested by studies in transgenic mice in which ablation of reactive astrocytes after CNS trauma prevented the normally occurring repair of the BBB and grafts of normal astrocyte were able to restore BBB repair [33]. It is not yet known whether a primary dysfunction of astrocytes or reactive astrocytes could lead to loss of BBB integrity. It is possible that loss of astrocyte production of a potential BBB inductive molecule, such as GSNO [208], or astrocyte over production of BBB disrupting molecules might lead to loss of BBB integrity [69, 188].
Hepatic encephalopathy
Hepatic encephalopathy can lead to mild to severe neuropsychiatric symptoms, brain edema with increased intracranial pressure associated with cerebellar tonsillar, uncal and/or central diencephalic herniation, and coma. Astrocytes are now recognized as playing a central role in the pathophysiology of hepatic encephalopathy [166]. The brain edema is largely a result of astrocyte swelling, which in turn is caused by astrocytic uptake of NH4 +. Although astrocyte uptake of NH4 + is regarded initially as a neuroprotective mechanism, it also drives the synthesis of glutamine from glutamate by glutamine synthetase, resulting in changes in transmitter homeostasis that may contribute to behavioral symptoms. The accumulation of glutamine in astrocytes also leads to osmotic stress that results in astrocyte swelling and progressive cytotoxic edema [107, 166, 193].
Metabolic disorders
Niemann-Pick type C, is an inherited neurovisceral lipid storage disorder caused by loss of function mutations of the membrane-bound protein, NPC1, which is thought to play a role in trafficking of cholesterol and other lysosomal cargo. In the CNS, NPC1 is expressed predominantly in perisynaptic astrocyte processes that are closely associated with nerve terminals, the earliest site of degeneration in the disease, suggesting that disruption of NPC1-mediated vesicular trafficking in astrocytes may be linked to neuronal degeneration [178]. It is interesting that selective restoration of expression of Npc1 in GFAP-positive astrocytes in Npc1−/− mice enhanced survival and decreased neuronal storage of cholesterol in the CNS, while restoration of expression of Npc1 in GFAP-positive in enteric glial cells in Npc1−/− mice ameliorated enteric neuropathology [110, 265].
Aceruloplasminemia is an inherited disorder of iron metabolism caused by the lack of ceruloplasmin activity. The neuropathological hallmarks are excessive iron deposition, neuronal loss, bizarrely deformed astrocytes, and numerous ‘grumose’ or foamy spheroid bodies. In the CNS, ceruloplasmin is largely produced by perivascular astrocytes and exhibits a ferroxidase activity that inhibits iron-associated lipid peroxidation and hydroxyl radical formation [174]. Astrocytes play key roles in iron trafficking and the detoxification of iron-mediated free radicals. Lack of ceruloplasmin causes direct oxidative stress on astrocytes. In aceruloplasminemia, deformed astrocytes accumulate ferric iron and the antioxidative ability of astrocytes is compromised, leading to secondary neuronal cell death [174].
Astrocyte abnormalities and CNS malformations or lesions are also associated with the rare genetic disorders, congenital glutamine synthetase deficiency [88], and pyruvate carboxylase deficiency [79].
Alexander disease and other leukodystrophies
Alexander disease is a genetic disorder of astrocytes caused by a dominant, gain-of-function mutation of the gene encoding GFAP [25]. Astrocytes are hypertrophied and exhibit numerous, characteristic Rosenthal fibers that are aggregates of GFAP and are therefore prominently GFAP-immunoreactive [25, 247]. There is particular involvement of the white matter and patients exhibit macrocephaly, seizures, psychomotor disturbances, spasticity, and premature death. Over expression of GFAP in transgenic mice causes a fatal encephalopathy similar to the human condition [143]. It is interesting to note that high densities of Rosenthal fibers, comparable to those seen in Alexander’s disease, have been described in the context of pronounced astrocytic abnormalities in severe FCD with intractable pediatric epilepsy (Fig. 6b) [111] (see above).
Megaloencephalic leukoencephalopathy with subcortical cysts (MLC) is an autosomal recessive, progressive white matter disease in children, characterized by myelin splitting and intramyelinic vacuole formation and caused by mutations in the MLC1 gene. In the CNS, the MLC1 protein is specifically expressed in distal astroglial processes in perivascular, subependymal, and subpial regions. This localization plus the transmembrane domains of the protein, support the possible transport functions of MLC1, possibly across the blood–brain and brain-cerebrospinal fluid barriers [21, 210].
Vanishing white matter disease (VWM) is a rare autosomal recessive leukoencephalopathy linked to mutations in translation initiation factor 2B (eIF2B). VWM lesions in vivo lack GFAP expressing astrocytes. Mutations in the eIF2B genes are thought to impair the ability of cells to regulate protein synthesis. RNAi targeting of EIF2B5 severely compromised the generation of GFAP expressing astrocytes from normal human glial progenitors, raising the possibility that a deficiency in astrocyte function may contribute to the loss of white matter in VWM [55, 244].
Glaucoma
Glaucoma is characterized by unexplained loss of retinal ganglion neurons and by the reactive gliosis of astrocytes and related Müller cells in the retina, and the reactivity of astrocytes that surround ganglion cell axons in the optic nerve head. It is not clear whether the reactive gliosis in the retina is merely a response to a process afflicting the ganglion neurons or whether the gliosis contributes to that degeneration by loss of glial supportive functions, such as glutamate uptake, or contribution of degenerative effects such as production of reactive oxygen species [26, 109].
The optic nerve head, where the optic nerve emerges from the eye, is an area where several tissues meet and the area may be under particular stress and flux during increases in intraocular pressure as are often associated with glaucoma. Astrocytes in the optic nerve head differ from white and gray matter astrocytes and have a distinct appearance and morphology. In rodents, optic nerve astrocytes are spatially aligned to ensheathe axons in bundles in a unique arrangement that provides essential structural support for the axons [232]. In primates, optic head astrocytes exhibit a similarly specialized morphology and provide additional structural support by secreting an extracellular matrix referred to as the lamina cribrosa [94, 262]. Various lines of evidence suggest that an early precipitating event in glaucoma may be disturbances of axon–astrocyte interactions at the optic nerve head (perhaps as a consequence of increased stresses caused by raised intraocular pressure), which in turn lead to axonal damage and subsequent retrograde degeneration of retinal ganglion neurons [94, 98, 262].
Neuropathic pain
Compelling evidence now indicates that pain signaling in the spinal cord dorsal horn involves interactions among microglia, astrocytes and neurons [147]. As discussed earlier, astrocytes can influence synaptic activity in a variety of direct and indirect ways and astrocytes participate in regulation of CNS inflammation by responding to and producing a wide variety of cytokines. These mechanisms provide the basis for astrocyte involvement in both normal and pathological pain signaling. There is a growing body of evidence that astrocytes, which are chronically reactive in the dorsal horn due to local cellular interactions, have the potential to contribute to and exacerbate chronic and neuropathological pain in various ways, including release of neurotransmitters and neuromodulators such as glutamate, NO and PGE or through release of cytokines like TNFα that alter neuronal membrane receptor levels, all of which can amplify neuronal excitability [147]. The molecules involved in these emerging multi-cellular signaling interactions are providing novel targets for analgesic therapeutic strategies.
Migraine
Prolonged cortical spreading depression is thought to play a fundamental role in the pathophysiology of migraine and can be caused by excessive synaptic glutamate release, or by decreased removal of glutamate and potassium from the synaptic cleft [204]. Familial hemiplegic migraine type 2, an autosomal dominant form of migraine with aura, has been associated with four distinct mutations in the alpha2-subunit of the Na+,K+-ATPase that is expressed primarily in astrocytes. The mutations all result in decreased activity of the Na+,K+-pump in astrocytes [36], which in turn may lower the threshold for cortical spreading depression and migraine attacks.
Alzheimer’s disease
Reactive astrogliosis is a well-known feature of Alzheimer’s disease (AD), but its roles in AD are not well understood. Reactive astrogliosis tends to be focal in AD such that reactive astrocytes are intimately associated with amyloid plaques or diffuse deposits of amyloid and surround them with dense layers of processes as if forming miniature scars around them (Fig. 7a), perhaps to wall them off and act as neuroprotective barriers. Reactive astrocytes can contain substantial amounts of different forms of amyloid beta, including amyloid beta 1–42 (Aβ42) as well as truncated forms [159, 239]. Of considerable interest are reports that reactive astrocytes can take up and degrade extracellular deposits of Aβ42 [261] and that this function is attenuated in ApoE−/− astrocytes [113], suggesting that reactive astrocytes functions or dysfunctions could play a role in the progression and severity of AD. The intensity of reactive astrogliosis, as determined by GFAP levels, has been reported to increase in parallel with increasing progression of Braak stages in AD, while concomitantly the levels of astrocyte glutamate transporters have been reported to decline, which may raise the vulnerability of local neurons to excitotoxicity [221]. Reactive astrocytes also exhibit increased expression of presenilin in sporadic AD [101, 254], but the consequences of this expression are not known.

Reactive astrogliosis in two degenerative diseases. a High magnification image of autopsy specimen from a person with longstanding Alzheimer’s disease immunohistochemically stained for GFAP. Section of cerebral cortex shows an amyloid senile plaque with a pale unstained center (Aβ) ringed by dense layers of reactive astrocytic processes (arrows) that circumferentially surround the plaque as if forming a scar-like barrier around it. b High magnification image of autopsy specimen from a person with Creutzfeldt–Jakob disease (CJD) transmissible spongiform encephalopathy. Section of cerebral cortex shows pronounced neuron loss and severe diffuse reactive astrogliosis. Most of the cortex is packed with gemistocytic astrocytes, while spongiform change is relatively minimal in this area. Scale bars a, b 10 μm
Transmissible spongiform encephalopathies
Astrogliosis is prominent, diffuse, and intense throughout affected CNS regions in transmissible spongiform encephalopathies such as Creutzfeldt–Jakob disease (CJD) (Fig. 7b) and related prion diseases, especially if clinical symptoms have been of long duration. Although this astrogliosis exhibits some characteristics of severe diffuse reactive astrogliosis (Figs. 3c, 4c), it is not clear whether astrocytes play an active role in prion replication and disease pathogenesis or whether astrogliosis is largely reactive to the disease process in other cell types [115]. It is interesting that the hippocampal neuronal degeneration associated with experimental ablation of reactive astrocytes has a vacuolar appearance with similarities to spongiform changes [33].
Parkinson’s disease
The role of astrocytes in Parkinson’s disease and related syndromes is sparsely investigated and poorly understood [140]. Reactive astrogliosis is generally mild or moderate and rarely severe in autopsy specimens of substantia nigra from Parkinson’s disease patients [72, 149]. Astrocytes have been implicated as potentially exerting both neurotoxic and neuroprotective activities in Parkinson’s disease. Experimental studies show that astrocytes take up the Parkinson’s syndrome causing molecule, MPTP, from the blood stream and convert it to neurotoxic MPP+ and may similarly convert other environmental molecules that have been implicated in dopaminergic toxicity [54, 187]. On the other hand, activation of the transcription factor Nrf2 selectively in astrocytes protects mice from MPTP-induced Parkinsonism by activating anti-oxidative response pathways [40]. Rare mutations in Nurr1 lead to familial Parkinson’s disease, and expression of Nurr1 in astrocytes suppresses production of potentially toxic molecules and protects against loss of dopaminergic neurons [203]. Recent findings also show that subpopulations of astrocytes express disease-related proteins such as α-synuclein, parkin and phosho-tau to different levels and in different combinations in Parkinson’s disease, multiple-system atrophy, and progressive supranuclear palsy [227], but the roles of astrocytes in these conditions are not yet defined.
Amyotrophic lateral sclerosis (ALS)
Evidence is available suggesting two quite different potential roles for astrocytes in amyotrophic lateral sclerosis (ALS) or motor neuron disease, through either the loss of a neuroprotective function or the gain of a neurotoxic effect. Sporadic ALS is characterized by selective loss or dysfunction of astrocyte glutamate transporters in spinal cord and cerebral cortical areas that exhibit loss of lower and upper motor neurons [73, 136, 194, 197], suggesting that increased glutamate excitotoxicity may contribute to motor neuron death and raising the possibility of different types of potential interventions. A high-throughput screen of small molecules has identified that certain β-lactam antibiotics can stimulate the expression of glutamate transporters by astrocyte and thereby enhance glutamate uptake sufficiently to reduce excitotoxicity, and provide neuroprotection in animal models of stroke and ALS [199]. The β-lactam antibiotic, Ceftriaxone, began stage 3 clinical trials in May 2009 to determine efficacy in reducing excitotoxicity and neurodegeneration in ALS. Focal grafts of healthy astrocytes are reported to be neuroprotective in an animal model of ALS, suggesting that transplantation of astrocytes may be a potential therapeutic strategy [120]. From a different perspective, astrocytes may also exhibit neurotoxic dysfunction in ALS. Approximately 20% of familial ALS cases, and about 5% of apparently sporadic cases, exhibit missense mutations of the gene encoding superoxide dismutase (SOD) that are dominantly inherited [201]. Recent findings show that expression of ALS mutant SOD in astrocytes (but not other cell types) leads to production by astrocytes of soluble molecules that are selectively toxic to motor neurons but not to spinal cord interneurons [53, 158], but the nature of the toxic molecules and the mechanisms leading to their production are not yet clear.
Tauopathies
Tauopathies are primarily associated with intracellular filamentous deposits in neurons that lead to functional disturbances [83]. Nevertheless, there is evidence to suggest that astrocyte tau pathology may also contribute to disease mechanisms. Filamentous tau aggregates can be observed in astrocytes in human disease and in animal models, and in animal models can disturb glutamate uptake and other astrocyte functions leading to focal neuronal degeneration [48, 71].
Huntington’s disease
In Huntington’s disease, excitotoxicity has long been regarded as contributing to neuronal loss but the underlying mechanisms have been difficult to elucidate [82]. There is increasing investigation of the potential for astrocytes contributions to dysfunction and degeneration in Huntington’s disease through disturbances in glutamate uptake that can alter synaptic function and lead to excitotoxicity, or through abnormal production of neurotoxic molecules [127, 136].
Psychiatric disorders
As it becomes increasingly clear that astrocytes play essential roles in synaptic mechanisms (see above), there is growing evidence that astrocyte functions and dysfunctions may contribute to behavioral mechanisms and behavioral disorders [90]. For example, studies in experimental animals show that disruption of astrocyte-specific Na+,K+-ATPase results in increased anxiety-related behavior and impaired spatial learning in mice [154], and that astrocyte influences on synaptic functions are important in reward circuits, and may contribute to drug addiction [145]. Clinicopathological studies show that expression of astrocyte glutamate transporters has been found in prefrontal cortex of schizophrenic patients [137], and that the density of astrocytes is reduced in prefrontal cortex of patients with major depressive disorders [146, 189, 218]. Layer-specific reductions in GFAP-reactive have also been reported in astrocytes of the dorsolateral prefrontal cortex in schizophrenia [190] and astrocyte roles in synaptic function are now central to certain hypotheses regarding schizophrenic disease mechanisms [19, 151].
The study of cellular neuropathology in psychiatric disorders is fraught with roadblocks. Morphologic abnormalities associated with behavioral illness may be extremely subtle and difficult to detect. Individuals tend to die with a psychiatric disorder rather than of it (the exception to this being death by suicide), so that at the time of necropsy a past history of psychiatric illness may not be noted, or there may be changes in the brain secondary to longstanding other medical problems that ‘mask’ any intrinsic structural changes underlying a psychiatric illness. After many years of neuropsychiatric problems in a given patient, it may be difficult or impossible to distinguish structural sequelae of pharmacotherapy from structural changes that caused the illness. Astrocyte contributions to behavioral disorders are only beginning to be explored. In this regard, it should be considered that astrocytes may not show overt structural changes or changes in commonly used astrocyte markers in necropsy specimens. As histopathological molecular identification techniques expand in breadth and become more sensitive, it may become possible (keeping in mind the caveats just mentioned) to detect astrocyte specific changes in molecular expression that are associated with specific disorders and clinical conditions.
Brain tumors (primary or secondary)
Astrocytes play a major role in the evolution of many common primary brain tumors, gliomas, and a less prominent, though significant, role in the progression of metastases. From a practical diagnostic viewpoint, GFAP is a reliable immunohistochemical marker for staining surgically resected brain neoplasms to ascertain whether they have a significant component of cells differentiating along astrocytic pathways [247]. The likelihood that a neoplasm is an ‘astrocytoma’ increases if cells throughout the tumor manifest atypical nuclear features (which can vary considerably in their degree of atypia) and also show some degree of cytoplasmic immunoreactivity with GFAP (Fig. 8a, b). Alternatively, abundant reactive astrocytes may show GFAP immunoreactivity within or surrounding non-glial tumors such as primary CNS lymphoma (Fig. 9a) or medulloblastoma (Fig. 9b) [247]. When GFAP-positive reactive astrocytes align themselves in a scar-like fashion along a non-glial (usually metastatic) tumor, the impression of a secondary neoplasm is strongly supported.

GFAP immunoreactive cells in primary CNS neoplasms. a High magnification image of a subependymal giant cell astrocytoma (SEGA) immunohistochemically stained for GFAP. Note variable staining intensity within tumor cells, including some that show uniform cytoplasmic staining. b High magnification image of a high-grade glioma (glioblastoma, WHO grade IV) immunohistochemically stained for GFAP. Note the variable staining of tumor cells as well as the pronounced nuclear and cytological atypia. Scale bars a, b 10 μm

Reactive astrogliosis in response to CNS tumors. a High magnification image of autopsy specimen from a man with widely infiltrating primary CNS lymphoma. Two color immunohistochemistry for GFAP and the cell cycle marker Ki67, shows the cytoplasm of reactive astrocytes stained blue (arrows), with nuclei of infiltrating, malignant and highly proliferative lymphoid elements stained brown (arrowheads). Note that essentially none of the GFAP-immunoreactive astrocytes show Ki-67-immunoreactive nuclei and are thus not proliferative and exhibit only a moderate degree of reactive astrogliosis with cell hypertrophy and up regulation of GFAP, in spite of the presence of numerous infiltrating lymphoma cells. b High magnification image of a medulloblastoma immunohistochemically stained for GFAP and showing many processes of reactive astrocytes along one edge of the tumor (at right of figure). Scale bars a 15 μm, b 25 μm
Malignant astrocytic glioma, such as glioblastoma (astrocytoma WHO grade IV), are the most common and among the most lethal intracranial tumors [76, 128]. The cells of origin of astrocytic gliomas, whether potentially astrocytes, glial precursors, or stem cells, are the subject of intense investigation [76]. In a manner consistent with the cancer stem cell hypothesis, there is considerable evidence that only minor populations of cells in primary gliomas are capable of forming a tumor, [76, 223]. The presence throughout adult brain of periventricular adult NSC that express the astrocyte marker GFAP (see above), raises interesting questions regarding the possible contribution of these cells to astrocytoma initiation. Astrocytic gliomas contain small numbers of cells that exhibit stem cell-like properties in that they self-renew, are multipotent and form neurospheres in vitro and constitutively produce the different types of cells found within the parent tumors [160]. Growth properties of glioma-derived neurospheres in vitro were found to be significant predictors of tumor progression in vivo and of clinical outcome [118]. Nevertheless, the origins of glioma are not yet understood and could be heterogeneous. Astrocytic gliomas exhibit molecular heterogeneity, particularly as regards expression patterns of transcriptional regulators, tumor-suppressor proteins and kinase mutations, and molecular phenotyping of glioma is opening up the potential for molecularly targeted therapies [99]. Some properties of astroglioma appear related to astrocyte functions. For example, glutamate production by glioma may increase invasive growth properties [228]. Enhanced STAT3 signaling activity by astroglioma reduces inflammation and tumor-immune surveillance and increases cell proliferation [24] in a manner consistent with functions of STAT3 signaling in astrocytes [95].
Neurofibromatosis
Neurofibromatosis type 1 (NF1) is an inherited neurocutaneous disorder with neuropsychologic deficits and predisposition to Schwannomas and astrocytomas caused by mutations in the NF1 gene [2, 9]. Reduced expression of glutamate transporters have been reported in reactive astrocyte clusters in NF1 [263]. Changes in mTOR and PTEN-signaling pathways in Schwann cells and astrocytes may contribute to tumorigenesis in NF1 [50, 87].
Enteric glia and inflammatory bowel disorders
The gastrointestinal tract is highly innervated and neuropathology of the enteric nervous system is emerging as a central feature of many gut diseases. Although most considerations of the enteric nervous system have focused on neuronal dysfunction, a large population of astrocyte-like glia populates gut muscle layers and the intestinal mucosa, and mounting new evidence points toward enteric glia, which express GFAP and share functional similarities with astrocytes, as active participants in gut pathology [207]. Various lines of experimental and neuropathological evidence are consistent with the possibility that enteric glia constitute a previously unrecognized disease target in pathologies associated with intestinal barrier dysfunction, notably inflammatory bowel disease, necrotizing enterocolitis, irritable bowel syndrome, diabetes, autoimmune disease, and neurotropic virus infection of the gut pathology [44, 207]. Experimental ablation of enteric glia leads to inflammatory bowel disease [32]. Both astrocytes and related enteric glial cells are able to induce barrier properties in gut epithelial cells, and S-nitrosoglutathione (GSNO) has been identified as one molecular mediator of this effect [208]. Thus, enteric glia, like astrocytes, appear to play essential roles in barrier functions during disease processes, particularly as regards regulating the spread of inflammation [49, 207, 226, 251].
GFAP-expressing stellate cells within liver, pancreas, and kidney
There is now growing evidence that stellate shaped, GFAP-expressing cells in various tissues may have functions that are similar to those of astrocytes. For example, there is increasing evidence that GFAP-expressing pancreatic stellate cells play important roles in tissue repair, fibrosis, and scar formation [176]. Hepatic stellate cells not only express GFAP and have morphological appearance similar to astrocytes, but also contact hepatic sinusoids and blood vessels and may take part in hepatic immune processes [258, 259]. In this regard, it is particularly interesting that the liver, like the CNS, has a certain level of immune privilege [241], and that both CNS and liver have resident populations of macrophage-related cells (Kupfer cells in the liver and microglia in the CNS) as well as of stellate cells that participate in immune and inflammatory processes. In addition, there is evidence that GFAP-expressing mesangial cells in the kidney function as local modulators of innate and adaptive immune responses [209]. The increasing recognition that tissue-specific cells like astrocytes and stellate cells play essential roles in regulating local immune and inflammatory processes is likely to impact considerably on concepts about organ-specific vulnerability to autoimmunity and other inflammatory conditions.
Therapeutic strategies involving astrocytes
The actively ongoing dissection of molecular mechanisms of reactive astrogliosis is beginning to identify molecules whose functions might be enhanced or blocked in specific disease contexts as potential therapeutic strategies [66, 226]. For example, augmenting the function of the astrocyte glutamate transporter EAAT2 with parawexin 1, a molecule isolated from spider venom, protects retinal neurons from ischemic degeneration by enhancing glutamate uptake and reducing excitotoxicity [70]. Certain β-lactam antibiotics, identified by a high-throughput screen of small molecules, can stimulate the expression of astrocyte glutamate transporters sufficiently to reduce excitotoxicity and neuroprotection in models of stroke and ALS [199].
Transplantation strategies involving astrocytes are also under investigation. For example, grafts of stem or progenitor cells that mature into healthy astrocytes cells are reported to improve outcome in a mouse model of ALS in which host astrocytes are abnormal and express a mutant SOD [120]. A different strategy employs grafts of astrocytes that are genetically modified to produce specific molecules, such as growth factors, as therapeutic pumps to deliver those molecules in specific locations [18, 233]. In due course, grafted astrocytes may be tested in clinical contexts and become subject to neuropathological evaluations.
References
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7:41–53
Acosta MT, Gioia GA, Silva AJ (2006) Neurofibromatosis type 1: new insights into neurocognitive issues. Curr Neurol Neurosci Rep 6:136–143
Akaoka H, Szymocha R, Beurton-Marduel P, Bernard A, Belin MF, Giraudon P (2001) Functional changes in astrocytes by human T-lymphotropic virus type-1 T-lymphocytes. Virus Res 78:57–66
Andreiuolo F, Junier MP, Hol EM, Miquel C, Chimelli L, Leonard N, Chneiweiss H, Daumas-Duport C, Varlet P (2009) GFAPdelta immunostaining improves visualization of normal and pathologic astrocytic heterogeneity. Neuropathology 29:31–39
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215
Araya R, Kudo M, Kawano M, Ishii K, Hashikawa T, Iwasato T, Itohara S, Terasaki T, Oohira A, Mishina Y, Yamada M (2008) BMP signaling through BMPRIA in astrocytes is essential for proper cerebral angiogenesis and formation of the blood-brain-barrier. Mol Cell Neurosci 38:417–430
Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR (2009) VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA 106:1977–1982
Armah HB, Wang G, Omalu BI, Tesh RB, Gyure KA, Chute DJ, Smith RD, Dulai P, Vinters HV, Kleinschmidt-DeMasters BK, Wiley CA (2007) Systemic distribution of West Nile virus infection: postmortem immunohistochemical study of six cases. Brain Pathol 17:354–362
Arun D, Gutmann DH (2004) Recent advances in neurofibromatosis type 1. Curr Opin Neurol 17:101–105
Bachoo RM, Kim RS, Ligon KL, Maher EA, Brennan C, Billings N, Chan S, Li C, Rowitch DH, Wong WH, DePinho RA (2004) Molecular diversity of astrocytes with implications for neurological disorders. Proc Natl Acad Sci USA 101:8384–8389
Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis 16:1–13
Barcia C, Sanderson NS, Barrett RJ, Wawrowsky K, Kroeger KM, Puntel M, Liu C, Castro MG, Lowenstein PR (2008) T cells’ immunological synapses induce polarization of brain astrocytes in vivo and in vitro: a novel astrocyte response mechanism to cellular injury. PLoS One 3:e2977
Barcia C, Thomas CE, Curtin JF, King GD, Wawrowsky K, Candolfi M, Xiong WD, Liu C, Kroeger K, Boyer O, Kupiec-Weglinski J, Klatzmann D, Castro MG, Lowenstein PR (2006) In vivo mature immunological synapses forming SMACs mediate clearance of virally infected astrocytes from the brain. J Exp Med 203:2095–2107
Barres BA (2003) What is a glial cell? Glia 43:4–5
Barres BA (2008) The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60:430–440
Bauer J, Elger CE, Hans VH, Schramm J, Urbach H, Lassmann H, Bien CG (2007) Astrocytes are a specific immunological target in Rasmussen’s encephalitis. Ann Neurol 62:67–80
Beck DW, Vinters HV, Hart MN, Cancilla PA (1984) Glial cells influence polarity of the blood-brain barrier. J Neuropathol Exp Neurol 43:219–224
Behrstock S, Ebert AD, Klein S, Schmitt M, Moore JM, Svendsen CN (2008) Lesion-induced increase in survival and migration of human neural progenitor cells releasing GDNF. Cell Transplant 17:753–762
Bernstein HG, Steiner J, Bogerts B (2009) Glial cells in schizophrenia: pathophysiological significance and possible consequences for therapy. Expert Rev Neurother 9:1059–1071
Blechingberg J, Holm IE, Nielsen KB, Jensen TH, Jorgensen AL, Nielsen AL (2007) Identification and characterization of GFAPkappa, a novel glial fibrillary acidic protein isoform. Glia 55:497–507
Boor PK, de Groot K, Waisfisz Q, Kamphorst W, Oudejans CB, Powers JM, Pronk JC, Scheper GC, van der Knaap MS (2005) MLC1: a novel protein in distal astroglial processes. J Neuropathol Exp Neurol 64:412–419
Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR (2005) Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med 202:145–156
Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, Ivanov D, Nathanson L, Barnum SR, Bethea JR (2009) Transgenic inhibition of astroglial NF-kappaB improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol 182:2628–2640
Brantley EC, Nabors LB, Gillespie GY, Choi YH, Palmer CA, Harrison K, Roarty K, Benveniste EN (2008) Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res 14:4694–4704
Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A (2001) Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 27:117–120
Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A (2006) Muller cells in the healthy and diseased retina. Prog Retin Eye Res 25:397–424
Brown AM, Ransom BR (2007) Astrocyte glycogen and brain energy metabolism. Glia 55:1263–1271
Brown AM, Baltan Tekkok S, Ransom BR (2004) Energy transfer from astrocytes to axons: the role of CNS glycogen. Neurochem Int 45:529–536
Buffo A, Vosko MR, Erturk D, Hamann GF, Jucker M, Rowitch D, Gotz M (2005) Expression pattern of the transcription factor Olig2 in response to brain injuries: implications for neuronal repair. Proc Natl Acad Sci USA 102:18183–18188
Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Gotz M (2008) Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci USA 105:3581–3586
Bundesen LQ, Scheel TA, Bregman BS, Kromer LF (2003) Ephrin-B2 and EphB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J Neurosci 23:7789–7800
Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, Johnson MH, Sofroniew MV (1998) Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 93:189–201
Bush TG NP, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308
Bushong EA, Martone MA, Jones YZ, Ellisman MH (2002) Protoplasmic astrocytes in CA1 atratum radiatum occupy separate anatomical domains. J Neurosci 22:183–192
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278
Capendeguy O, Horisberger JD (2004) Functional effects of Na + , K + -ATPase gene mutations linked to familial hemiplegic migraine. Neuromolecular Med 6:105–116
Carlen M, Meletis K, Goritz C, Darsalia V, Evergren E, Tanigaki K, Amendola M, Barnabe-Heider F, Yeung MS, Naldini L, Honjo T, Kokaia Z, Shupliakov O, Cassidy RM, Lindvall O, Frisen J (2009) Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci 12:259–267
Carmichael ST (2006) Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann Neurol 59:735–742
Charles AC, Merrill JE, Dirksen ER, Sanderson MJ (1991) Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6:983–992
Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA (2009) Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc Natl Acad Sci USA 106:2933–2938
Chen Y, Swanson RA (2003) Astrocytes and brain injury. J Cereb Blood Flow Metab 23:137–149
Chen Y, Vartiainen NE, Ying W, Chan PH, Koistinaho J, Swanson RA (2001) Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J Neurochem 77:1601–1610
Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433
Cirillo C, Sarnelli G, Esposito G, Grosso M, Petruzzelli R, Izzo P, Cali G, D’Armiento FP, Rocco A, Nardone G, Iuvone T, Steardo L, Cuomo R (2009) Increased mucosal nitric oxide production in ulcerative colitis is mediated in part by the enteroglial-derived S100B protein. Neurogastroenterol Motil 21:1209–1212
Colodner KJ, Montana RA, Anthony DC, Folkerth RD, De Girolami U, Feany MB (2005) Proliferative potential of human astrocytes. J Neuropathol Exp Neurol 64:163–169
Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ (1990) Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247:470–473
Crino PB, Miyata H, Vinters HV (2002) Neurodevelopmental disorders as a cause of seizures: neuropathologic, genetic, and mechanistic considerations. Brain Pathol 12:212–233
Dabir DV, Robinson MB, Swanson E, Zhang B, Trojanowski JQ, Lee VM, Forman MS (2006) Impaired glutamate transport in a mouse model of tau pathology in astrocytes. J Neurosci 26:644–654
Daneman R, Rescigno M (2009) The gut immune barrier and the blood-brain barrier: are they so different? Immunity (Epub 20 October 2009)
Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH (2005) Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res 65:2755–2760
David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A (2009) Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci 29:10588–10599
De Keyser J, Mostert JP, Koch MW (2008) Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J Neurol Sci 267:3–16
Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K (2007) Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci 10:608–614
Di Monte DA, Royland JE, Irwin I, Langston JW (1996) Astrocytes as the site for bioactivation of neurotoxins. Neurotoxicology 17:697–703
Dietrich J, Lacagnina M, Gass D, Richfield E, Mayer-Proschel M, Noble M, Torres C, Proschel C (2005) EIF2B5 mutations compromise GFAP + astrocyte generation in vanishing white matter leukodystrophy. Nat Med 11:277–283
Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97:703–716
Donati D, Martinelli E, Cassiani-Ingoni R, Ahlqvist J, Hou J, Major EO, Jacobson S (2005) Variant-specific tropism of human herpesvirus 6 in human astrocytes. J Virol 79:9439–9448
Dong Y, Benveniste EN (2001) Immune function of astrocytes. Glia 36:180–190
Drogemuller K, Helmuth U, Brunn A, Sakowicz-Burkiewicz M, Gutmann DH, Mueller W, Deckert M, Schluter D (2008) Astrocyte gp130 expression is critical for the control of Toxoplasma encephalitis. J Immunol 181:2683–2693
Eddleston M, Mucke L (1993) Molecular profile of reactive astrocytes—implications for their role in neurological disease. Neuroscience 54:15–36
Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, Agre P, Ottersen OP, de Lanerolle NC (2005) Loss of perivascular aquaporin 4 may underlie deficient water and K + homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci USA 102:1193–1198
Eng LF, Gerstl B, Vanderhaeghen JJ (1970) A study of proteins in old multiple sclerosis plaques. Trans Am Soc Neurochem 1:42
Eng LF, Ghirnikar RS, Lee YL (2000) Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem Res 25:1439–1451
Engel J Jr (1996) Surgery for seizures. N Engl J Med 334:647–652
Erana-Rojas IE, Barboza-Quintana A, Ayala AG, Fuller GN (2002) Demyelinating pseudotumor. Ann Diagn Pathol 6:265–271
Escartin C, Bonvento G (2008) Targeted activation of astrocytes: a potential neuroprotective strategy. Mol Neurobiol 38:231–241
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145
Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24:2143–2155
Fisher M (2008) Injuries to the vascular endothelium: vascular wall and endothelial dysfunction. Rev Neurol Dis 5(Suppl 1):S4–S11
Fontana AC, de Oliveira Beleboni R, Wojewodzic MW, Ferreira Dos Santos W, Coutinho-Netto J, Grutle NJ, Watts SD, Danbolt NC, Amara SG (2007) Enhancing glutamate transport: mechanism of action of Parawixin1, a neuroprotective compound from Parawixia bistriata spider venom. Mol Pharmacol 72:1228–1237
Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, Trojanowski JQ (2005) Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci 25:3539–3550
Forno LS, DeLanney LE, Irwin I, Di Monte D, Langston JW (1992) Astrocytes and Parkinson’s disease. Prog Brain Res 94:429–436
Fray AE, Ince PG, Banner SJ, Milton ID, Usher PA, Cookson MR, Shaw PJ (1998) The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. Eur J Neurosci 10:2481–2489
Friedman A, Kaufer D, Heinemann U (2009) Blood-brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res 85:142–149
Furer M, Hartloper V, Wilkins J, Nath A (1993) Lymphocyte emperipolesis in human glial cells. Cell Adhes Commun 1:223–237
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21:2683–2710
Gadea A, Schinelli S, Gallo V (2008) Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J Neurosci 28:2394–2408
Garcia ADR, Doan NB, Imura T, Bush TG, Sofroniew MV (2004) GFAP-expressing progenitors are the principle source of constitutive neurogenesis in adult mouse forebrain. Nature Neurosci 7:1233–1241
Garcia-Cazorla A, Rabier D, Touati G, Chadefaux-Vekemans B, Marsac C, de Lonlay P, Saudubray JM (2006) Pyruvate carboxylase deficiency: metabolic characteristics and new neurological aspects. Ann Neurol 59:121–127
Garcia-Segura LM, Melcangi RC (2006) Steroids and glial cell function. Glia 54:485–498
Ghatak NR (1992) Occurrence of oligodendrocytes within astrocytes in demyelinating lesions. J Neuropathol Exp Neurol 51:40–46
Gil JM, Rego AC (2008) Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci 27:2803–2820
Goedert M, Jakes R (2005) Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta 1739:240–250
Goldman S (2003) Glia as neural progenitor cells. Trends Neurosci 26:590–596
Goncalves CA, Leite MC, Nardin P (2008) Biological and methodological features of the measurement of S100B, a putative marker of brain injury. Clin Biochem 41:755–763
Gordon GR, Mulligan SJ, MacVicar BA (2007) Astrocyte control of the cerebrovasculature. Glia 55:1214–1221
Gregorian C, Nakashima J, Dry SM, Nghiemphu PL, Smith KB, Ao Y, Dang J, Lawson G, Mellinghoff IK, Mischel PS, Phelps M, Parada LF, Liu X, Sofroniew MV, Eiber FC, Wu H (2009) PTEN dosage is essential for neurofibroma development and malignant transformation. Proc Natl Acad Sci USA 106:19479–19484
Haberle J, Gorg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, Gelot A, Suc AL, Hohne W, Schliess F, Haussinger D, Koch HG (2005) Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med 353:1926–1933
Halassa MM, Fellin T, Haydon PG (2007) The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med 13:54–63
Halassa MM, Fellin T, Haydon PG (2009) Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 57:343–346
Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG (2007) Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27:6473–6477
Hamby ME, Hewett JA, Hewett SJ (2006) TGF-beta1 potentiates astrocytic nitric oxide production by expanding the population of astrocytes that express NOS-2. Glia 54:566–577
Haseloff RF, Blasig IE, Bauer HC, Bauer H (2005) In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol 25:25–39
Hernandez MR, Miao H, Lukas T (2008) Astrocytes in glaucomatous optic neuropathy. Prog Brain Res 173:353–373
Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci 28:7231–7243
Hewett JA (2009) Determinants of regional and local diversity within the astroglial lineage of the normal central nervous system. J Neurochem 110:1717–1736
Horner PJ, Power AE, Kempermann G, Kuhn HG, Palmer TD, Winkler J, Thal LJ, Gage FH (2000) Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J Neurosci 20:2218–2228
Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW (2007) Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2 J glaucoma. J Cell Biol 179:1523–1537
Huang TT, Sarkaria SM, Cloughesy TF, Mischel PS (2009) Targeted therapy for malignant glioma patients: lessons learned and the road ahead. Neurotherapeutics 6:500–512
Hult B, Chana G, Masliah E, Everall I (2008) Neurobiology of HIV. Int Rev Psychiatry 20:3–13
Huynh DP, Vinters HV, Ho DH, Ho VV, Pulst SM (1997) Neuronal expression and intracellular localization of presenilins in normal and Alzheimer disease brains. J Neuropathol Exp Neurol 56:1009–1017
Iadecola C, Nedergaard M (2007) Glial regulation of the cerebral microvasculature. Nat Neurosci 10:1369–1376
Imura T, Kornblum HI, Sofroniew MV (2003) The predominant neural stem cell isolated from postnatal and adult forebrain but not from early embryonic forebrain expresses GFAP. J Neurosci 23:2824–2832
Imura T, Nakano I, Kornblum HI, Sofroniew MV (2006) Phenotypic and functional heterogeneity of GFAP-expressing cells in vitro: Differential expression of LeX/CD15 by GFAP-expressing multipotent neural stem cells and non-neurogenic astrocytes. Glia 53:277–293
Jacob A, Hensley LK, Safratowich BD, Quigg RJ, Alexander JJ (2007) The role of the complement cascade in endotoxin-induced septic encephalopathy. Lab Invest 87:1186–1194
Jansen LA, Uhlmann EJ, Crino PB, Gutmann DH, Wong M (2005) Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia 46:1871–1880
Jayakumar AR, Rama Rao KV, Tong XY, Norenberg MD (2009) Calcium in the mechanism of ammonia-induced astrocyte swelling. J Neurochem 109(Suppl 1):252–257
John GR, Lee SC, Brosnan CF (2003) Cytokines: Powerful regulators of glial cell activation. Neuroscientist 9:10–22
Johnson EC, Morrison JC (2009) Friend or foe? Resolving the impact of glial responses in glaucoma. J Glaucoma 18:341–353
Kapur R, Donohue C, Jelinek D, Erickson RP (2009) Amelioration of enteric neuropathology in a mouse model of Niemann-Pick C by Npc1 expression in enteric glia. J Neurosci Res 87:2994–3001
Khanlou N, Mathern GW, Mitchell WG, Salamon N, Pope WB, Yong WH, Vinters HV (2009) Cortical dysplasia with prominent Rosenthal fiber formation in a case of intractable pediatric epilepsy. Hum Pathol 40:1200–1204
Koehler RC, Roman RJ, Harder DR (2009) Astrocytes and the regulation of cerebral blood flow. Trends Neurosci 32:160–169
Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10:719–726
Kostianovsky AM, Maier LM, Anderson RC, Bruce JN, Anderson DE (2008) Astrocytic regulation of human monocytic/microglial activation. J Immunol 181:5425–5432
Kovacs GG, Budka H (2008) Prion diseases: from protein to cell pathology. Am J Pathol 172:555–565
Kriegstein A, Alvarez-Buylla A (2009) The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32:149–184
Kuhlmann T, Lassmann H, Bruck W (2008) Diagnosis of inflammatory demyelination in biopsy specimens: a practical approach. Acta Neuropathol 115:275–287
Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I, Yong WH, Vinters HV, Liau LM, Lazareff JA, Mischel PS, Cloughesy TF, Horvath S, Kornblum HI (2009) Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 27:980–987
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 202:473–477
Lepore AC, Rauck B, Dejea C, Pardo AC, Rao MS, Rothstein JD, Maragakis NJ (2008) Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci 11:1294–1301
Levison SW, Jiang FJ, Stoltzfus OK, Ducceschi MH (2000) IL-6-type cytokines enhance epidermal growth factor-stimulated astrocyte proliferation. Glia 32:328–337
Levitt P, Rakic P (1980) Immunoperoxidase localization of glial fibrillary acidic protein in radial glial cells and astrocytes of the developing rhesus monkey brain. J Comp Neurol 193:815–840
Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Stahlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, Nilsson M, Pekny M (2008) Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab 28:468–481
Lim MC, Maubach G, Zhuo L (2008) Glial fibrillary acidic protein splice variants in hepatic stellate cells–expression and regulation. Mol Cells 25:376–384
Lin JH, Lou N, Kang N, Takano T, Hu F, Han X, Xu Q, Lovatt D, Torres A, Willecke K, Yang J, Kang J, Nedergaard M (2008) A central role of connexin 43 in hypoxic preconditioning. J Neurosci 28:681–695
Liu X, Bolteus AJ, Balkin DM, Henschel O, Bordey A (2006) GFAP-expressing cells in the postnatal subventricular zone display a unique glial phenotype intermediate between radial glia and astrocytes. Glia 54:394–410
Lobsiger CS, Cleveland DW (2007) Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci 10:1355–1360
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M (2007) The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci 27:12255–12266
Love S (2006) Demyelinating diseases. J Clin Pathol 59:1151–1159
Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF (2009) Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci 29:7743–7752
Maginnis MS, Atwood WJ (2009) JC virus: an oncogenic virus in animals and humans? Semin Cancer Biol 19:261–269
Magnus T, Carmen J, Deleon J, Xue H, Pardo AC, Lepore AC, Mattson MP, Rao MS, Maragakis NJ (2008) Adult glial precursor proliferation in mutant SOD1G93A mice. Glia 56:200–208
Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Götz M (2003) Neuronal or glial progeny: Regional differences in radial glia fate. Neuron 37:751–764
Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS (2000) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6:159–163
Maragakis NJ, Rothstein JD (2006) Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2:679–689
Matute C, Melone M, Vallejo-Illarramendi A, Conti F (2005) Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia 49:451–455
McArthur DL, Chute DJ, Villablanca JP (2004) Moderate and severe traumatic brain injury: epidemiologic, imaging and neuropathologic perspectives. Brain Pathol 14:185–194
McCarty JH (2009) Cell adhesion and signaling networks in brain neurovascular units. Curr Opin Hematol 16:209–214
McGeer PL, McGeer EG (2008) Glial reactions in Parkinson’s disease. Mov Disord 23:474–483
Meletis K, Barnabe-Heider F, Carlen M, Evergren E, Tomilin N, Shupliakov O, Frisen J (2008) Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol 6:e182
Merkle FT, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A (2004) Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci USA 101:17528–17532
Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M (1998) Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol 152:391–398
Meyer EM, St.Onge E, Crews FT (1984) Effects of aging on rat cortical presynaptic cholinergic processes. Neurobiol Aging 5:315–317
Miguel-Hidalgo JJ (2009) The role of glial cells in drug abuse. Curr Drug Abuse Rev 2:72–82
Miguel-Hidalgo JJ, Baucom C, Dilley G, Overholser JC, Meltzer HY, Stockmeier CA, Rajkowska G (2000) Glial fibrillary acidic protein immunoreactivity in the prefrontal cortex distinguishes younger from older adults in major depressive disorder. Biol Psychiatry 48:861–873
Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10:23–36
Min KJ, Yang MS, Kim SU, Jou I, Joe EH (2006) Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci 26:1880–1887
Mirza B, Hadberg H, Thomsen P, Moos T (2000) The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 95:425–432
Mischel PS, Nguyen LP, Vinters HV (1995) Cerebral cortical dysplasia associated with pediatric epilepsy Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol 54:137–153
Mitterauer B (2005) Nonfunctional glial proteins in tripartite synapses: a pathophysiological model of schizophrenia. Neuroscientist 11:192–198
Miyata H, Chiang AC, Vinters HV (2004) Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol 56:510–519
Morest DK, Silver J (2003) Precursors of neurons, neuroglia, and ependymal cells in the CNS: what are they? Where are they from? How do they get where they are going? Glia 43:6–18
Moseley AE, Williams MT, Schaefer TL, Bohanan CS, Neumann JC, Behbehani MM, Vorhees CV, Lingrel JB (2007) Deficiency in Na, K-ATPase alpha isoform genes alters spatial learning, motor activity, and anxiety in mice. J Neurosci 27:616–626
Muller W, Dahmen HG (1978) Lymphocytes within glial cells (“emperipolesis”) in a case of a granular cell tumor. Acta Neuropathol 44:163–165
Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV (2006) Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129:2761–2772
Nag S, Manias JL, Stewart DJ (2009) Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol 118:197–217
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S (2007) Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10:615–622
Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC (2004) Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging 25:663–674
Nakano I, Kornblum HI (2009) Methods for analysis of brain tumor stem cell and neural stem cell self-renewal. Methods Mol Biol 568:37–56
Neary JT, Zimmermann H (2009) Trophic functions of nucleotides in the central nervous system. Trends Neurosci 32:189–198
Nedergaard M, Ransom B, Goldman SA (2003) New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci 26:523–530
Nishie M, Mori F, Ogawa M, Sannohe S, Tanno K, Kurahashi K, Kuroda N, Wakabayashi K (2004) Multinucleated astrocytes in old demyelinated plaques in a patient with multiple sclerosis. Neuropathology 24:248–253
Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR (2001) Neurons derived from radial glial cells establish radial units in neocortex. Nature 409:714–720
Norenberg MD (1979) Distribution of glutamine synthetase in the rat central nervous system. J Histochem Cytochem 27:756–762
Norenberg MD, Rao KV, Jayakumar AR (2005) Mechanisms of ammonia-induced astrocyte swelling. Metab Brain Dis 20:303–318
Norton WT, Aquino DA, Hozumi I, Chiu FC, Brosnan CF (1992) Quantitative aspects of reactive gliosis: a review. Neurochem Res 17:877–885
Obara M, Szeliga M, Albrecht J (2008) Regulation of pH in the mammalian central nervous system under normal and pathological conditions: facts and hypotheses. Neurochem Int 52:905–919
Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M (2008) Loss of astrocytic domain organization in the epileptic brain. J Neurosci 28:3264–3276
Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD, Pilcher W, Ojemann JG, Ransom BR, Goldman SA, Nedergaard M (2009) Uniquely hominid features of adult human astrocytes. J Neurosci 29:3276–3287
Occhipinti R, Somersalo E, Calvetti D (2009) Astrocytes as the glucose shunt for glutamatergic neurons at high activity: an in silico study. J Neurophysiol 101:2528–2538
Ogata K, Kosaka T (2002) Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience 113:221–233
Ohab JJ, Fleming S, Blesch A, Carmichael ST (2006) A neurovascular niche for neurogenesis after stroke. J Neurosci 26:13007–13016
Oide T, Yoshida K, Kaneko K, Ohta M, Arima K (2006) Iron overload and antioxidative role of perivascular astrocytes in aceruloplasminemia. Neuropathol Appl Neurobiol 32:170–176
Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H (2006) Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nature Med 12:829–834
Omary MB, Lugea A, Lowe AW, Pandol SJ (2007) The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest 117:50–59
Papadopoulos MC, Manley GT, Krishna S, Verkman AS (2004) Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 18:1291–1293
Patel SC, Suresh S, Kumar U, Hu CY, Cooney A, Blanchette-Mackie EJ, Neufeld EB, Patel RC, Brady RO, Patel YC, Pentchev PG, Ong WY (1999) Localization of Niemann-Pick C1 protein in astrocytes: implications for neuronal degeneration in Niemann- Pick type C disease. Proc Natl Acad Sci USA 96:1657–1662
Pekny M, Pekna M (2004) Astrocyte intermediate filaments in CNS pathologies and regeneration. J Pathol 204:428–437
Pekny M, Leveen P, Pekna M, Eliasson C, Berthold CH, Westermark B, Betsholtz C (1995) Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J 14:1590–1598
Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ (2007) Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55:1251–1262
Perea G, Navarrete M, Araque A (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32:t421–t431
Peters A, Palay SL, Webster HD (1991) The fine structure of the nervous system, Third edn. Oxford University Press, New York
Phelps CH (1972) Barbiturate-induced glycogen accumulation in brain. An electron microscopic study. Brain Res 39:225–234
Pittock SJ, Lucchinetti CF (2007) The pathology of MS: new insights and potential clinical applications. Neurologist 13:45–56
Powell EM, Geller HM (1999) Dissection of astrocyte-mediated cues in neuronal guidance and process extension. Glia 26:73–83
Przedborski S, Jackson-Lewis V, Djaldetti R, Liberatore G, Vila M, Vukosavic S, Almer G (2000) The parkinsonian toxin MPTP: action and mechanism. Restor Neurol Neurosci 16:135–142
Pun PB, Lu J, Moochhala S (2009) Involvement of ROS in BBB dysfunction. Free Radic Res 43:348–364
Rajkowska G, Miguel-Hidalgo JJ (2007) Gliogenesis and glial pathology in depression. CNS Neurol Disord Drug Targets 6:219–233
Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, Meltzer H, Overholser J, Stockmeier C (2002) Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophr Res 57:127–138
Ramon Y, Cajal S (1909) Histologie du systeme nerveux de l’homme et des vertebres. Maloine, Paris
Ramon y Cajal S (1928) Degeneration and regeneration of the nervous system. Oxford University Press, London
Rao KV, Panickar KS, Jayakumar AR, Norenberg MD (2005) Astrocytes protect neurons from ammonia toxicity. Neurochem Res 30:1311–1318
Rattray M, Bendotti C (2006) Does excitotoxic cell death of motor neurons in ALS arise from glutamate transporter and glutamate receptor abnormalities? Exp Neurol 201:15–23
Roelofs RF, Fischer DF, Houtman SH, Sluijs JA, Van Haren W, Van Leeuwen FW, Hol EM (2005) Adult human subventricular, subgranular, and subpial zones contain astrocytes with a specialized intermediate filament cytoskeleton. Glia 52:289–300
Rossi DJ, Brady JD, Mohr C (2007) Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci 10:1377–1386
Rothstein JD, Martin LJ, Kuncl RW (1992) Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med 326:1464–1468
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kunci RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16:675–686
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433:73–77
Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C (2008) Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322:1551–1555
Rowland LP, Shneider NA (2001) Amyotrophic lateral sclerosis. N Engl J Med 344:1688–1700
Ruhl A (2005) Glial cells in the gut. Neurogastroenterol Motil 17:777–790
Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK (2009) A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137:47–59
Sanchez-Del-Rio M, Reuter U, Moskowitz MA (2006) New insights into migraine pathophysiology. Curr Opin Neurol 19:294–298
Sattler R, Rothstein JD (2006) Regulation and dysregulation of glutamate transporters. Handb Exp Pharmacol 175:277–303
Saunders NR, Ek CJ, Habgood MD, Dziegielewska KM (2008) Barriers in the brain: a renaissance? Trends Neurosci 31:279–286
Savidge TC, Sofroniew MV, Neunlist M (2007) Starring roles for astroglia in barrier pathologies of gut and brain. Lab Invest 87:731–736
Savidge TC, Newman P, Pothoulakis C, Ruhl A, Neunlist M, Bourreille A, Hurst R, Sofroniew MV (2007) Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology 132:1344–1358
Schlondorff D, Banas B (2009) The mesangial cell revisited: no cell is an island. J Am Soc Nephrol 20:1179–1187
Schmitt A, Gofferje V, Weber M, Meyer J, Mossner R, Lesch KP (2003) The brain-specific protein MLC1 implicated in megalencephalic leukoencephalopathy with subcortical cysts is expressed in glial cells in the murine brain. Glia 44:283–295
Schummers J, Yu H, Sur M (2008) Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science 320:1638–1643
Seifert G, Schilling K, Steinhauser C (2006) Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci 7:194–206
Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT (2005) Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat 30:144–157
Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21:7153–7160
Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS (2008) Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci 28:6659–6663
Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH (2003) Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 23:3394–3406
Shintaku M, Yutani C (2004) Oligodendrocytes within astrocytes (“emperipolesis”) in the white matter in Creutzfeldt-Jakob disease. Acta Neuropathol 108:201–206
Si X, Miguel-Hidalgo JJ, O’Dwyer G, Stockmeier CA, Rajkowska G (2004) Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression. Neuropsychopharmacology 29:2088–2096
Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nature Rev Neurosci 5:146–156
Simard M, Nedergaard M (2004) The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 129:877–896
Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB (2008) Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging (Epub 1 July 2008)
Singh N, Paterson DL (2000) Encephalitis caused by human herpesvirus-6 in transplant recipients: relevance of a novel neurotropic virus. Transplantation 69:2474–2479
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Sofroniew MV (2000) Astrocyte failure as a cause of CNS dysfunction. Mol Psychiatry 5:230–232
Sofroniew MV (2005) Reactive astrocytes in neural repair and protection. Neuroscientist 5:400–407
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647
Song YJC, Halliday GM, Holton JL, Lashley T, O’Sullivan SS, McCann H, Lees AJ, Ozawa T, Williams DR, Lockhart PJ, Revesz TR (2009) Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J Neuropathol Exp Neurol 68:1073–1083
Sontheimer H (2008) A role for glutamate in growth and invasion of primary brain tumors. J Neurochem 105:287–295
Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178
Suh SW, Bergher JP, Anderson CM, Treadway JL, Fosgerau K, Swanson RA (2007) Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316, 819 ([R-R*, S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmet hyl)propyl]-1H-indole-2-carboxamide). J Pharmacol Exp Ther 321:45–50
Sun D, Lye-Barthel M, Masland RH, Jakobs TC (2009) The morphology and spatial arrangement of astrocytes in the optic nerve head of the mouse. J Comp Neurol 516:1–19
Suzuki M, McHugh J, Tork C, Shelley B, Klein SM, Aebischer P, Svendsen CN (2007) GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One 2:e689
Swanson RA, Ying W, Kauppinen TM (2004) Astrocyte influences on ischemic neuronal death. Curr Mol Med 4:193–205
Szymocha R, Akaoka H, Dutuit M, Malcus C, Didier-Bazes M, Belin MF, Giraudon P (2000) Human T-cell lymphotropic virus type 1-infected T lymphocytes impair catabolism and uptake of glutamate by astrocytes via Tax-1 and tumor necrosis factor alpha. J Virol 74:6433–6441
Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, Watanabe S, Shiga Y, Kanaoka C, Fujimori J, Sato S, Itoyama Y (2007) Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 130:1235–1243
Takano T, Oberheim N, Cotrina ML, Nedergaard M (2009) Astrocytes and ischemic injury. Stroke 40:S8–12
Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, Li Y, Yang J, Dienel G, Zielke HR, Nedergaard M (2005) Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci USA 102:16466–16471
Thal DR, Schultz C, Dehghani F, Yamaguchi H, Braak H, Braak E (2000) Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol 100:608–617
Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M (2005) An astrocytic basis of epilepsy. Nat Med 11:973–981
Tiegs G, Lohse AW (2009) Immune tolerance: what is unique about the liver. J Autoimmun (Epub 1 September 2009)
Tomita M, Khan RL, Blehm BH, Santoro TJ (2004) The potential pathogenetic link between peripheral immune activation and the central innate immune response in neuropsychiatric systemic lupus erythematosus. Med Hypotheses 62:325–335
Ullian EK, Sapperstein SK, Christopherson KS, Barres BA (2001) Control of synapse number by glia. Science 291:657–661
van Kollenburg B, van Dijk J, Garbern J, Thomas AA, Scheper GC, Powers JM, van der Knaap MS (2006) Glia-specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol 65:707–715
Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 28:13574–13581
Vinters HV, Miyata H (2006) Neuropathologic features of tuberous sclerosis. In: McLendon RE, Rosenblum MK, Bigner DD (eds) Russel and Rubenstein’s Pathology of Tumors of the Nervous System, 7th edn. Hodder Arnold, Oxford, pp 955–969
Vinters HV, Farrell MA, Mischel PS, Anders K (1998) Diagnostic Neuropathology. Marcel Dekker, Inc., New York
Vinters HV, Kwok MK, Ho HW, Anders KH, Tomiyasu U, Wolfson WL, Robert F (1989) Cytomegalovirus in the nervous system of patients with the acquired immune deficiency syndrome. Brain 112(Pt 1):245–268
Volterra A, Meldolesi J (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6:626–640
von Einsiedel RW, Samorei IW, Pawlita M, Zwissler B, Deubel M, Vinters HV (2004) New JC virus infection patterns by in situ polymerase chain reaction in brains of acquired immunodeficiency syndrome patients with progressive multifocal leukoencephalopathy. J Neurovirol 10:1–11
Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci 29:11511–11522
Voutsinos-Porche B, Bonvento G, Tanaka K, Steiner P, Welker E, Chatton JY, Magistretti PJ, Pellerin L (2003) Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron 37:275–286
Wang Z, Trillo-Pazos G, Kim SY, Canki M, Morgello S, Sharer LR, Gelbard HA, Su ZZ, Kang DC, Brooks AI, Fisher PB, Volsky DJ (2004) Effects of human immunodeficiency virus type 1 on astrocyte gene expression and function: potential role in neuropathogenesis. J Neurovirol 10(Suppl 1):25–32
Weggen S, Diehlmann A, Buslei R, Beyreuther K, Bayer TA (1998) Prominent expression of presenilin-1 in senile plaques and reactive astrocytes in Alzheimer’s disease brain. Neuroreport 9:3279–3283
Weidenfeller C, Svendsen CN, Shusta EV (2007) Differentiating embryonic neural progenitor cells induce blood-brain barrier properties. J Neurochem 101:555–565
Whitney KD, McNamara JO (2000) GluR3 autoantibodies destroy neural cells in a complement-dependent manner modulated by complement regulatory proteins. J Neurosci 20:7307–7316
Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, Ellisman MH, Pekny M (2006) Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci USA 103:17513–17518
Winau F, Quack C, Darmoise A, Kaufmann SH (2008) Starring stellate cells in liver immunology. Curr Opin Immunol 20:68–74
Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, Modlin RL, Liblau RS, Gressner AM, Kaufmann SH (2007) Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 26:117–129
Wolf F, Kirchhoff F (2008) Neuroscience. Imaging astrocyte activity. Science 320:1597–1599
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–457
Yang H, Downs JC, Sigal IA, Roberts MD, Thompson H, Burgoyne CF (2009) Deformation of the normal monkey optic nerve head connective tissue following acute iop elevation within 3-D histomorphometric reconstructions. Invest Ophthalmol Vis Sci (Epub 2009/2007/2025)
Yeh TH, Lee da Y, Gianino SM, Gutmann DH (2009) Microarray analyses reveal regional astrocyte heterogeneity with implications for neurofibromatosis type 1 (NF1)-regulated glial proliferation. Glia 57:1239–1249
Zador Z, Stiver S, Wang V, Manley GT (2009) Role of aquaporin-4 in cerebral edema and stroke. Handb Exp Pharmacol 190:159–170
Zhang M, Strnatka D, Donohue C, Hallows JL, Vincent I, Erickson RP (2008) Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1-/- mice. J Neurosci Res 86:2848–2856
Zhao L, Burt AD (2007) The diffuse stellate cell system. J Mol Histol 38:53–64
Acknowledgments
Work in the authors’ laboratories is supported by grants from NIH, RO1 NS057624 (MVS), P50 AG16570 (HVV) and P01 AG12435 (HVV), the Roman Reed Spinal Cord Injury Research Fund (MVS) and Wings for Life (MVS) and the Daljit S. and Elaine Sarkaria Chair in Diagnostic Medicine (HVV). Nicole Yin and Spencer Tung assisted with preparation of illustrations.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sofroniew, M.V., Vinters, H.V. Astrocytes: biology and pathology. Acta Neuropathol 119, 7–35 (2010). https://doi.org/10.1007/s00401-009-0619-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0619-8