
Abstract
Ibrutinib (IBR) is a biopharmaceutical classification system (BCS) class II drug and an irreversible Bruton’s tyrosine kinase (BTK) inhibitor. IBR has an extremely low oral bioavailability due to the activity of the CYP3A4 enzyme. The current intention of the research was to enhance solubility followed by oral bioavailability of IBR using the hot melt extrusion (HME) technique by formulating drug-drug cocrystals (DDCs). Ketoconazole (KET) is an active CYP3A4 inhibitor and was selected based on computational studies and solubility parameter prediction. Differential scanning calorimetry (DSC), Fourier transform infrared spectroscopy (FT-IR), powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA), proton nuclear magnetic resonance (1H NMR), and scanning electron microscopy (SEM) evaluations were employed for estimating the formation of IBR-KET DDCs. The IBR-KET DDC system was discovered to have a hydrogen bond (H-bond) and π-π-stacking interactions, in accordance with the computational results. Further, IBR-KET DDCs showed enhanced solubility, stability, powder dissolution, in vitro release, and flow properties. Furthermore, IBR-KET-DDCs were associated with enhanced cytotoxic activity in K562-CCL-243 cancer cell lines when compared with IBR and KET alone. In vivo pharmacokinetic studies have shown an enhanced oral bioavailability of up to 4.30 folds of IBR and 2.31 folds of KET through IBR-KET-DDCs compared to that of the IBR and KET suspension alone. Thus, the prepared IBR-KET-DDCs using the HME technique stand as a favorable drug delivery system that augments the solubility and oral bioavailability of IBR along with KET.
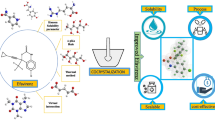
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Many active pharmaceutical ingredients (APIs) do not exhibit the expected physicochemical characteristics that make them suitable for formulation development. Poor oral bioavailability caused by low water solubility affects a large number of APIs [1]. According to estimates, new chemical entities (NCEs) which make up to 75% of anticancer drugs are mostly water-insoluble [2]. Their aqueous solubility is a major obstacle to the development of formulations and clinical application of these anticancer medicines [3]. Over the past few decades, numerous initiatives have been launched to enhance the properties of anticancer drugs and address the lower solubility issues through a variety of novel formulations like amorphous solid dispersions (ASDs) [4], complexation [5], lipid-based nanoparticles [6], nanoparticles [7], and crystal engineering [8]. From them all, co-crystallization has gained a lot of interest in the last few years. “Pharmaceutical cocrystals are defined as homogeneous crystalline materials which contain APIs and biologically safe cocrystal formers (CCFs) at a fixed ratio in the same crystal lattice via noncovalent interactions” [9]. Cocrystal formation can change a given API’s physicochemical characteristics, such as solubility, dissolution, bioavailability, stability, and tabletability, without affecting the molecular structure [10].
Drug-drug cocrystals (DDCs) are emerging subsets of cocrystals made up of two different API molecules and offer many advantages when compared with APIs [11]. DDCs are competent solid forms for dual drug administration during any therapy. Compared to any other alternative methods for dual drug therapy, DDCs may be capable of enhancing the physical and chemical properties of pertinent drugs, like refining the dissolution rate of either of the drugs, augmenting the solubility and bioavailability, extending the stability of molecules which lack stability, and hence improving the management of existing products [12].
The DDCs no doubt have several advantages but they also have a few disadvantages which are discussed here. DDCs may not necessarily be able to show their synergistic effects and positive effects during the therapy. The selection of suitable codrugs may stand to be a greater task since they may not show definite impact as required. And also sometimes there might be challenges with respect to fixed-dose drug combinations in attaining the same ratio as available.
Well-known approaches for the formulation of DDCs are solvent evaporation, liquid-assisted grinding, ultrasonic method, cooling crystallization, and hot melt extrusion (HME) [13]. HME, on the other hand, is one of the latest approaches for co-crystallization. HME technique is solvent free which leads to endless manufacturing of the product with minimal to no contamination and can stand as a green synthesis technique.
In this current study, we have approached several in silico and screening techniques to select a suitable coformer or drug to prepare DDCs including the prediction of synthons using the supramolecular synthon approach, calculating the difference in the ΔpKa values, calculating solubility parameter via Hansen solubility parameter approach, and molecular dynamics simulation studies. In silico studies were particularly selected for screening a suitable coformer or codrug, this helps in the faster screening of the suitable codrugs by decreasing the use of multiple chemicals and also eliminating the key time period occupied for the coformer/codrug screening.
Ibrutinib (IBR), 1-[(3R)-3-[4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]piperidin-1-yl]prop-2-en-1-one, has been permitted as an effective first-line treatment for numerous lymphoproliferative illnesses, such as small lymphocytic lymphoma, chronic lymphocytic leukemia, and mantle cell lymphoma. Ibrutinib functions by permanently blocking the BTK [14]. IBR is a BCS class II drug that is insoluble in water and has a low oral bioavailability, necessitating higher oral doses [15]. IBR also suffers from high first-pass metabolism through the CYP3A4 enzyme leading to its very low oral bioavailability of around 3% [16]. Recently, cocrystals of IBR with a number of carboxylic acids have been designed to enhance the oral bioavailability and solubility of IBR [17]. This encouraged us to explore DDCs of IBR with a CYP3A4 inhibitor by oral administration.
Ketoconazole (KET), cis-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-imidazole-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazine, is an antifungal drug that is highly efficient in treating mycoses both superficially and systemically [18]. Due to its weak basicity and being a BCS class II drug, KET faces a low water solubility [19]. KET is administered topically and orally; however, its lower solubility at pH greater than 3 is a significant drawback to its effectiveness when administered orally. Solubility and bioavailability enhancements would reduce the dosage required to achieve the desired therapeutic effect which can potentially decrease the chance of hepatotoxicity [20]. Several studies have proved the solubility improvement of KET using co-crystallization using several phenolic and carboxylic acids [21, 22].
KET is also a well-known CYP3A4 inhibitor, and IBR suffers diminished oral bioavailability due to the same enzyme activity [23]. In this paper, we addressed the formulation of IBR-KET-DDCs aiming to improve IBR physicochemical properties by using KET as a CYP3A4 enzyme inhibitor. IBR-KET-DDCs may decrease the first-pass effect on IBR and may simultaneously increase oral bioavailability. The obtained IBR-KET-DDCs were subjected to several characterization studies, solubility, stability, and dissolution studies were also studied to assess their application pharmaceutically.
Materials and Methods
Materials
IBR was received as a gift sample from Hetero Drugs Ltd., Hyderabad, India. KET was received from Gloss Pharmaceuticals Pvt. Ltd., Ahmedabad, India. All other chemicals and solvents were purchased from various companies. The purity of all the chemicals and solvents used in this study has been confirmed analytically.
Suitability of KET as a Coformer/Codrug to Prepare DDCs
The selection of a suitable coformer or codrug for IBR was initially screened using hydrogen bonding which stands as a key in the development of cocrystals. Molecules that possess thermodynamic hydrogen-bonding properties increase the chance of forming a stable cocrystal. KET was further tested to confirm its efficiency to form DDCs with IBR through multiple in silico modeling.
Molecular Dynamics (MD) Simulation Study
MD was done by utilizing Schrodinger Material Science Suite licensed by Schrodinger, LLC, New York, NY.
Structure Construction
An inbuilt 2D sketcher of Schrodinger Maestro 2020-3 was used to represent the chemical structure of IBR, KET, and the complex of IBR-KET. Further 3D rendering of the molecular structures was done.
Disordered System and MD Simulation
An equimolar ratio with 100 molecules each was selected, and the structures were optimized at 100 ns with constant temperature and pressure.
Cluster Extraction
The dimers of IBR-KET were selected, and cluster extraction was set to a maximum intermolecular distance of 4Å.
Gas Phase Energy Calculations
Dimers of IBR-KET were selected to perform jaguar optimization studies through the molecular minimization module. These dimers were further optimized using B3LYP-D3 theory, at a basis set of 6-31G** using density function theory (DFT). The solvation was run at the gas phase since there is no solvent used for IBR-KET DDC formulation. The least interaction energy ring conformations were generated using the OPLS3e module [24, 25].
This data processing computed the interaction energies of the IBR, KET, and IBR-KET complex. Thus, Eq. 1 was used to determine the cocrystal’s interaction energy.
Solubility Parameter Calculation
Hildebrand Solubility Parameter Using a Computational Approach
Cocrystals are non-ionic in nature consisting of drug and coformer in their supramolecular crystal lattices that display miscibility at a molecular stage with non-ionic interactions [26]. From this perspective, a computational miscibility estimation was achieved by the Hildebrand solubility parameter (δ), and “δ” numbers of IBR and KET were calculated through the Schrodinger maestro suit.
Both the molar volume (Vm) and the energy of vaporization (ΔEV) are taken into account when calculating (Eq. 2).
Trajectory analysis using the molecular dynamics (MD) model was executed for the dimer clusters of the IBR-KET complex to obtain the solubility parameter of each molecule. The selection of the suitable coformer to form DDCs depends on the closer “δ” values of the molecules.
Hansen Solubility Parameter Approach
Determining the miscibility of drugs with polymers is carried out commonly using the Hansen solubility parameter (HSP) [27]. Additionally, HSP values can forecast how drugs will interact with coformers. The Hofytzer van Krevelen approach, which includes the functional groups in the molecular structure, was evaluated for the present DDCs among the numerous ways used to determine HSP value [28].
The solubility parameter δ is used to measure the cohesive energy or the attraction among molecules in a material. The δ is the sum of dispersion (δd), polar (δp), and cohesive (δh) forces.
Equation 3 below helps in computing the total solubility parameter (δt).
The 4, 5, and 6 equations provided below can be used to determine the values of δd, δp, and δh.
in which
i is the functional groups within the molecule,
fdi is the role of functional groups to the dispersion forces,
fpi is the role of functional groups to the polar forces,
fhi is the role of functional groups to the hydrogen bonding energy, and
Vi is the role of functional groups to the molar volume.
Formulation of IBR-KET DDCs
The formation of IBR-KET DDCs was further explored by liquid assisted grinding (LAG) technique for initial confirmation, and the optimization of IBR-KET DDCs was performed using HME. KET was selected as coformer/codrug for IBR based on a number of assessments involving the hetero synthon approach, in silico modeling, difference in pKa values, and biological impact as mentioned previously.
Preparation of IBR-KET DDCs by LAG Technique
First, the practicality of IBR-KET DDCs was explored using the LAG technique. IBR and KET were taken precisely in molar ratios of 1:1 and 1:2, and the mixture was mixed for 30 minutes at ambient temperature by adding 100 μL of methanol in a mortar and pestle. To get rid of any remaining solvent, a hot air oven was set to 60°C for 30 minutes to dry the final product [29]. By analyzing the obtained samples using DSC and FTIR, DDC formation was validated.
Preparation of DDCs by HME Technique
The IBR-KET DDCs were engineered via an HME (Omicron 10P model, Steer Engineering Pvt Ltd., India) that has a screw size of 1.71. Optimization of IBR-KET DDCs was made using the OFAT method where a single factor was considered at a time. Based on past trials by the LAG technique, IBR and KET were accurately weighed in the equimolar ratio (1:1). 5 g of both IBR and KET was accurately weighed, mixed for five minutes, and then fed into the screw via the feeder. The barrel’s screw speed has been adjusted to 75 rpm, and the barrel temperatures were set to B1, 30, 110, 154, and 105 degrees Celsius. Figure 1 provides an overview of the manufacturing of DDCs utilizing a one-step HME technique to make this concept easier to understand. Zone B1’s temperature was automatically adjusted to 30°C in order to allow the solid feed to be delivered to zone B2. To make sure that the final product was in a solid state and not in a liquid or a melt state, the temperature in zone B4 was regulated at 105°C. Further, milling and sieving of the IBR-KET DDCs were done using mesh sizes 40 and 60. The substance that passed through 40 and remained on 60 was used for additional analysis [30].

Illustration of HME to acquire IBR-KET-DDCs. The barrel heating zones are represented by B1, B2, B3, and B4
Flow Property Analysis
Using the below-provided Eqs. 7 and 8, IBR, KET, and IBR-KET DDCs angle of repose, bulk density, tap density, Hausner’s ratio (HR), and Carr’s index (CI) were calculated. In order to illustrate that an acceptable dosage form for the DDCs could be created, the DDCs that were retained on mesh 60 after passing through a sieve with a mesh size of 40 were used in the study.
where ρtap stands for tap density, and ρbulk stands for bulk density [31].
Characterization of Cocrystals
Differential Scanning Calorimetry (DSC)
The DSC for IBR, KET, and IBR-KET DDCs was carried out utilizing (Shimadzu-TA-60 WS, Kyoto, Japan) to assess the samples endotherm-onset and endotherm-transition temperatures. IBR, KET, and IBR-KET DDCs were heated from 25°C to 350°C at a rate of 10°C per minute while sealed in an empty aluminum pan. At a flow rate of 40 mL/min, nitrogen purge was performed to create an inert environment. A blank aluminum pan was placed as a blank reference before performing the analysis. For each sample, a thermogram was plotted against heat flow and temperature [32].
Fourier Transform Infrared Spectroscopy (FTIR)
The individual samples of IBR, KET, and IBR-KET DDCs were subjected to FTIR analysis, and the spectra were scanned at a wave number from 400–4000 cm−1 [33]. FTIR analysis was performed using Bruker’s Alpha II compact attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectrometer to measure infrared wavelengths. Initially, a blank correction was performed. The ATR crystal was then covered with a sample (15–20 mg) using a micro spatula. To achieve thorough contact between the sample and the crystal, the anvil arm was rotated.
Powder X-Ray Diffraction (PXRD)
To obtain the PXRD patterns of IBR, KET, and DDCs, an X-ray diffractometer (Rigaku Co., Tokyo, Japan) was employed. The study was conducted at room temperature, with a 2θ range between 5 and 80, with 0.02θ scans per minute [34]. The X-ray beam was diffracted and recorded by a standard scintillation counter.
Proton Nuclear Magnetic Resonance (1H NMR)
The potential molecular interaction between IBR and KET was investigated using the NMR analyzer Bruker ASCEND TM 400 (Billerica, Massachusetts, USA). Deuterated chloroform was chosen to form a solution of IBR, KET, and IBR-KET DDCs [35].
Thermogravimetric Analysis for Thermal Stability (TGA)
The TGA for IBR, KET, and IBR-KET DDCs was subjected to analysis (TGA; STA7200RV, Hitachi High-Tech Science Corporation, Tokyo, Japan) to estimate thermostability. A 10 mg sample was put in a platinum pan and heated to 500°C at a rate of 10°C/min, and the weight changes were measured [36].
Morphological Studies
Scanning Electron Microscopy (SEM)
IBR, KET, and IBR-KET DDCs were subjected to surface morphology through SEM analysis (EVO MA18 Oxford EDS (Zeiss, Oberkochen, Germany). Individually, IBR, KET, and IBR-KET DDCs were placed on an Al stub that was conducted using gold sputtering, and then a ten-torr vacuum was used. The samples were then subjected to an electron beam scan, and the Smart SEM program was used to collect SEM photomicrograph pictures [37].
Simultaneous Estimation of IBR and KET Using High-Performance Liquid Chromatography (HPLC)
IBR and KET were measured with the use of the HPLC method in both analytical and rat plasma samples [38]. An automatic sampler, an Ultraviolet-visible detector, and twin-piston pumps were equipped in the HPLC instrument (Shimadzu Company, Japan). Chromatograms were examined using LC solution software. A reverse phase C18 column (Kromasil C18) was used to carry out the chromatographic separation. Acetonitrile (ACN) and 10 milli molar phosphate buffer pH 6.8 were used as the mobile phase at a ratio of 56:44 v/v and a flow rate of 1.2 mL/min, respectively. With a 20-minute runtime, the analysis was performed at 260 nm. There was good linearity in the linearity curve for the analyzed samples, from a range of 50 to 10,000 ng/mL, r2 value of 0.999, and equation (y = 24.519x + 182) for IBR and an r2 value of 0.999 and equation (y = 17.71x - 106.27) for KET; (“x” is the conc. of IBR and KET and “y” is the peak area). The IBR, KET, and internal standard dasatinib (DAS) were eluted in the biological sample with no interference. The linearity curve for the biological samples was performed in the concentration range from 25 to 10,000 ng/mL, with an r2 value of 0.998 and straight-line equation (y = 0.0021x - 0.2389) for IBR and an r2 value of 0.999 and equation (y = 0.0045x + 0.2513) for KET, representing the linearity.
Saturated Solubility Studies
Saturated solubility experiments on IBR, KET, and IBR-KET DDCs were performed via respective pH buffers with different values, such as pH 1.2 (HCl buffer), pH 4.5 (sodium acetate buffer), pH 6.8, pH 7.4, (phosphate buffer), and water [39]. To a 1.5-mL microcentrifuge tube filled with water and various buffer solutions, excess amounts of plain IBR, KET, and IBR-KET DDCs were added. Utilizing a tube rotator from Maas Instruments in Mumbai, India, the tubes were rotated for 2 days (48 hrs) at 50 rpm. At 10,000 rpm, the sample was centrifuged for 15 minutes, and a 0.22-micron filter was used to filter the supernatant and subjected to HPLC analysis for determining the content of IBR, KET, and IBR-KET DDCs by the procedure explained in "Simultaneous Estimation of IBR and KET Using High-Performance Liquid Chromatography (HPLC)" section.
Powder Dissolution Experiments
Powder dissolution studies for IBR, KET, and IBR-KET DDCs were achieved by using a type II USP dissolution apparatus from Mumbai, India’s Electrolab India Pvt. Ltd. The phosphate buffer pH 6.8 and pH 1.2 0.1N HCl buffer were utilized as a dissolving media for this purpose. The powdered samples of IBR, KET, and IBR-KET DDCs of an appropriate amount (5 mg equivalent of IBR, KET, and IBR-KET DDCs) were added to a flask containing 600 mL of pH 1.2 and pH 6.8 buffers and maintained at 150 rpm and 37 ± 5°C. At intervals of 5, 10, 15, 30, 45, 60, 90, 120, and 180 minutes, an aliquot of the sample was withdrawn and filtered through a 0.22-micron filter [40]. The filtrate was accurately diluted, and the IBR and KET concentrations were determined using the HPLC procedure depicted in "Simultaneous Estimation of IBR and KET Using High-Performance Liquid Chromatography (HPLC)" section.
In Vitro Drug Release Profile
A dialysis bag approach was employed to assess in vitro drug release. IBR, KET, and IBR-KET DDCs (10 mg) suspensions of 1 mL were prepared using 0.25% Na-CMC (sodium carboxymethyl cellulose) and were added into the previously soaked dialysis membrane, 12000 Daltons in molecular weight. About 100 milliliters of pH 6.8 buffer was used as the release media using a beaker. The release study was executed at a temperature of 37°C ± 0.5°C and at a rotation rate of 150 rpm. At intervals of 0, 0.5, 1, 2, 3, 4, 6, 12, 24, and 48 hours, 1 milliliter of the aliquots was withdrawn and restored with the constant volume of media. As described in "Simultaneous Estimation of IBR and KET Using High-Performance Liquid Chromatography (HPLC)" section, aliquots were filtered, diluted, and subjected to HPLC analysis [41]. The studies were all carried out in triplicates (n = 3), and the mean SD of the data was reported.
Accelerated Stability Studies
IBR, KET, and IBR-KET DDC samples were put through an accelerated stability test at a temperature of 40°C and relative humidity (RH) of 75%. A sealed desiccator holding an excessive amount of calcium carbonate powder was used to maintain the 75% RH condition. Samples were collected at 1, 3, and 6 months and were subjected to analysis. PXRD, drug content, DSC, and moisture uptake studies were conducted to detect possible phase changes [42].
Moisture Uptake Studies
The Coulometric Karl Fischer (KF) titration method was used to estimate the presence of water in the formulation by using Lab Junction’s Microprocessor KF Moisture Titrator LJ-1760-1 (India). After being dissolved in methanol, the weighed samples were moved into the titration chamber for evaluation. About 10 mg of the sample was analyzed, and each titration test was carried out in triplicate [43].
Drug Content Estimation
The drug content of IBR-KET DDCs was calculated by dissolving the DDCs (equivalent to 5 mg of pure IBR and KET individually) in 5 mL methanol and 15 minutes of sonication. The solution of the samples was centrifuged at 10,000 rpm for 10 min. The supernatant was filtered and diluted as needed and then subjected to HPLC analysis as outlined in "Simultaneous Estimation of IBR and KET Using High-Performance Liquid Chromatography (HPLC)" section to determine its drug concentration. The stock solutions of IBR and KET were made in the same concentration of 1 mg/mL using methanol [44].
In Vitro Cell Viability Assay
The K562-CCL-243 leukemia cell line and the human keratinocyte (HK) cell line have been procured from the American Type Culture Collection (ATCC). The following items were acquired from HiMedia Laboratories (Mumbai, India): penicillin/streptomycin solution, Dulbecco’s modified Eagle’s medium (DMEM), and fetal bovine serum (FBS). For sub-culturing HK and K562 cells, fetal bovine serum (ten percent) and penicillin/streptomycin (one hundred IU/mL) were added to DMEM media. The cells were grown in a sterilized, humidified atmosphere at 37°C with 5% CO2.
Cell Viability by MTT Assay
According to the literature, the MTT assay (3-(4,5-dimethylthiazol2-y1)-2,5-diphenyl tetrazolium bromide) was employed to estimate the in vitro cytotoxicity of HK and K562 cells [45]. HK/K562 cells were seeded in 96-well plates at a density of 2–104 cells per well, and the cells were then incubated for 24 hours at 37°C. The free drug and formulations (100 μL) of different concentrations have been added to the wells incubating for 24 h. The HK and K562 control cells received no treatment. Supernatant media was discarded after 24 h. Further, 100 microliters of MTT solution (5 mg/mL) was included and was incubated for 4 hours followed by substituting the MTT solution with 150 microliters of DMSO. Using a microplate reader, the absorbance of the well plate was evaluated with 30 minutes of incubation (SpectraMax™, Molecular Devices, USA) at 590 nm (reference wavelength 620 nm). The % cell viability was estimated using Eq. 9:
In which Abs (sample) is the absorbance of the cells treated with either IBR, KET, or IBR-KET DDCs solution. Abs (control) is the average absorbance of the control cells. The study was conducted in triplicates, and the results were presented as mean SD.
The half-maximal inhibitory concentrations (IC50) of IBR, KET, and IBR-KET DDCs which can indirectly reflect the cytotoxicity were calculated.
In Vivo Pharmacokinetic Study
Wistar rats weighing about 250 ± 50 grams were used to study the pharmacokinetics of IBR, KET, and IBR-KET DDCs. Before the study commenced, approval from the Institutional Animal Ethics Committee (IAEC), Manipal Academy of Higher Education, Manipal (IAEC/KMC/99/2021) was obtained. The usage and care of animals were handled in accordance with institutional and national regulations. The Wistar rats were separated into 3 groups (n = 3) (IBR suspension, KET suspension, and IBR-KET DDCs suspension). The Wistar rats were administered at 50 mg/kg of IBR and KET via oral gavage with relevant formulations. Following oral administration, blood samples (0.3 mL) were collected from the retro-orbital venous plexus at intervals of 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 10, 12, 24, and 48 hours. To separate the plasma, the blood samples were immediately centrifuged for 10 minutes at 20,000×g rpm in a centrifuge at 4°C [46]. HPLC-UV was used to quantify the IBR and KET concentrations in rat plasma simultaneously. IBR and KET were extracted from the rat plasma using the protein precipitation technique, as described in "Extraction Procedure of IBR and KET from Plasma" section. As an internal standard (IS), DAS was used at a concentration of 20 μg/mL. For analysis, 80 microliters of the sample was injected into the HPLC. The process outlined in "Simultaneous Estimation of IBR and KET Using High-Performance Liquid Chromatography (HPLC)" section was used to elute the samples. PK solver was used to calculate the pharmacokinetic parameters. The pharmacokinetic parameters taken into consideration for evaluation were maximum plasma concentration (Cmax), area under the plasma concentration-time curve (AUC), elimination half-life (Kel), absorption half-life (t1/2), and mean residence time (MRT).
Extraction Procedure of IBR and KET from Plasma
IBR and KET were extracted from the plasma samples using the protein precipitation method (PP) [47]. About 90 microliters of thawed plasma was taken, and 10 microliters of DAS (IS) was added to the plasma. The sample was thoroughly mixed and a further 400 microliters of chilled methanol was added. Again, the sample was subjected to mixing and later centrifuged at 20,000×g rpm for 10 min. After the samples were separated, the supernatant was collected and the HPLC conditions explained in "Simultaneous Estimation of IBR and KET Using High-Performance Liquid Chromatography (HPLC)" section were used to elute the drugs.
Statistics
A one-way ANOVA was considered for the statistical analysis (GraphPad Prism 6.0). Using Tukey’s post-hoc test, individual variations between the treatment groups were found; P < 0.05 indicated statistical significance.
Results and Discussion
Coformer Selection
IBR, as seen in Fig. 2a, contains the π-π-interaction site in addition to one H-bond donor and seven H-bond acceptors. According to studies, coformers with the greatest propensity for hydrogen bond formation have a better chance of acting as co-crystallizing agents. There is a potential that IBR might form a hydrogen bond with its donor site since KET in Fig. 2b also contains a significant number of hydrogen bond acceptor sites.

Chemical structure of a Ibrutinib and b Ketoconazole
Molecular Dynamics (MD) Simulation Study
Gas phase energy computations were conducted on the basis of density functional theory (DFT) via B3LYP-D3 molecular set of quantum mechanics basis of 6–31G**.
The prophesied properties of IBR, KET, and the complex of IBR-KET have been listed in Table I. The gas phase energy for IBR and IBR-KET complex was predicted at an equimolar ratio as shown in Fig. 3a, and the hydrogen bonding interaction is illustrated in Fig. 3b. It was discovered that there was an approximately 3 times increment in the projected gas-phase energy of the IBR-KET complex. KET was therefore considered as a possible coformer/codrug to perform further experimental studies. Also, the interaction energy of the IBR-KET complex was observed to be lower which may help in the stability of the complex. A molecule with lower interaction energy depicts the presence of attractive forces within the complex which helps in decreasing the potential energy of the bonds. In short, the less the potential energy more the stability of the molecules since their mobility is restricted. As shown in Fig. 4, the MD simulation π-π stacking interaction was observed between IBR and KET which can play a prominent role in the formulation of IBR-KET DDCs in addition to the intermolecular hydrogen bond formation.

a Molecular dynamic simulation of IBR and KET at equimolar ratio and b H bond interaction between IBR and KET

π-π stacking interaction between IBR and KET
Solubility Parameter Calculation
Hildebrand Solubility Parameter Using a Computational Approach
Hildebrand solubility parameter (δ) analysis data of IBR and KET is mentioned in Table II. Studies have shown that the IBR has a δ value of 25.85 MPa0.5. Theoretically, “δ” is “an indicator of solvency behavior depending on the molecular cohesive energy and relies on the principle that those materials with similar cohesive energy density result in solvation and/or miscibility.” In order to attain solid-state molecular miscibility, a coformer should exhibit a δ value similar to that of the IBR. According to the given statement, KET was considered appropriate as it showed a definite δ value of 21.84 MPa0.5, respectively.
Hansen Solubility Parameter
The Hofytzer van Krevelen technique has been used to estimate the drug carrier solubility parameter. The extent of the miscibility is the “difference in solubility parameters between the drug and the coformer.” The coformer whose solubility parameter (SP) difference is less than 7 to 10 MPa0.5 will likely show good miscibility. The difference between the SP of IBR and KET was discovered to be less than 7MPa0.5, as indicated in Table III. Therefore, it is possible that KET and IBR are miscible.
From Table III, “V” depicts the molar volume of the compound.
Flow Property Study
From the visual observation, it was understood that IBR-KET DDCs of molar ratio 1:1 were successfully extruded through HME. From Table IV, it appears that the overall micromeritic properties proved to be good as Hausner’s ratio was within the allowable range for HME extruded IBR-KET DDCs. With regard to flow properties, HME produced a better product. The IBR-KET DDCs via HME improved flowability versus IBR and KET alone.
Characterization of Cocrystals
Differential Scanning Calorimetry (DSC)
From Fig. 5, plain IBR, KET, and the IBR-KET-HME DDCs samples were analyzed by DSC. Plain IBR has shown its specific melting endotherm peak initiating at 155.61°C and the melting point appearing at 158.09°C. This was discovered to be in accordance with the value mentioned in the literature [48, 49]. IBR further did not show any degradation up to 350°C which ensures that extrusion at greater temperatures is feasible. KET displayed an endotherm onset at 148.58°C and a melting point at 152.21°C. IBR-KET DDCs via HME displayed the onset of endotherms at 124.28°C and the melting point peak at 131.57°C indicating the formation of a new complex. Endotherms of either IBR or KET alone were absent in the IBR-KET DDCs representing a successful molar interaction.

DSC thermogram of Ibrutinib, Ketoconazole, and IBR-KET-DDCs
Fourier Transform Infrared Spectroscopy (FTIR)
Figure 6 shows the characteristic absorption frequency bands in the IBR’s FTIR spectra, which were associated with the symmetric and anti-symmetric N-H(NH2) stretching vibrations at 3469 cm−1 and 3436 cm−1, respectively. Additionally, the C=O stretching vibration absorption peak for the ketone group was at 1652 cm−1, and the C=C stretching vibration absorption peak for the aromatic group was at 1613 cm−1 and 1520 cm−1, respectively. The pure KET shows intense absorption peaks of the C=O stretching vibration at 1637 cm−1, absorption peaks of C=C aromatic stretching at 1505 cm−1, absorption peaks of the tertiary amine at 1242 cm−1 and 1207 cm−1, and absorption peaks of C-Cl stretching at 850 cm−1, respectively. The FTIR spectra of IBR-KET DDCs showed several modifications as compared to the IBR, which may have resulted from intermolecular interaction. One evident distinction was the peak shape of the IBR-KET DDCs at 3400 cm−1 in the IR spectrum, which was substantially wider than that of IBR. In addition, the IBR’s multiple peaks created by the N-H stretching vibration had unified, and the wavenumber of the N-H stretching vibration had changed. For instance, in IBR-KET DDCs, the N-H stretching vibration of IBR shifted from 3469 cm−1 to 3431 cm−1. The stretching vibration of C=O of IBR in DDCs moved from 1652 cm−1 to 1642 cm−1. Also, there was a shift in the C=C aromatic stretching of both IBR and KET in IBR-KET DDCs, i.e., 1613 cm−1 to 1574 cm−1 in the case of IBR and 1520 cm−1 to 1517 cm−1 in case of KET. As seen, a significant broad peak covered the initial prominent absorption peaks of the N-H stretching vibration in the case of IBR-KET DDCs. As a result, it was hypothesized that the -NH2 of IBR and the C=O bond of KET may create the H-bond interactions. Additionally, compared to IBR and KET, peak changes for the aromatic skeleton stretching vibration had also been seen in IBR-KET DDCs. For instance, compared to IBR and KET, the aromatic skeleton stretching vibration of IBR-KET DDCs displayed a lower wavenumber. This could be inferred from the computational study outcomes, which were the evidence for the existence of the π-π stacking interaction.

FTIR spectra of Ibrutinib, Ketoconazole, and IBR-KET-DDCs
Powder X-Ray Diffraction (PXRD)
The PXRD patterns of IBR, KET, and the IBR-KET DDCs are illustrated in Fig. 7. The IBR diffractogram has prominent 2θ values at 5.60°, 10.72°, 13.68°, 18.88°, 21.24°, 22.34°, 23.48°, and 28.88°; the KET diffractogram has their 2θ values at 6.8°, 10.22°, 15.46°, 16.96°, 18.82°, 20.74°, 23.22°, 23.73°, and 27.03°; and the crystalline character of the IBR and KET is responsible for these diffraction peaks. The IBR-KET DDCs produced characteristic peaks at 2θ values of 15.63°, 19.44°, 20.94°, 23.25°, and 27.24°, which were absent in both IBR and KET. This was due to interactions between the two molecules, which led to the formation of new crystalline material.

PXRD data of Ibrutinib, Ketoconazole, and IBR-KET-DDCs
Proton Nuclear Magnetic Resonance (1H NMR)
IBR, KET, and IBR-KET-DDCs have been characterized using 1H NMR spectral technique. The 1H NMR spectrum of IBR consists of R-CH shifts from 1-benzene was observed at δ6.3–7.66 ppm; R-CH from piperidine and 1 alpha -N-R from methine was observed at δ3.78 ppm. The aromatic C-NH signal was observed at δ4.5 ppm. The CH signal from 4-pyrimidine was observed at δ8.37 ppm. In the case of KET, CH2-methylene signal was observed at δ2.50 ppm, and CH2 from 1,3-dioxolan signal was observed at δ3.65 ppm. CH from imidazole was observed at δ7.49 ppm, and CH from 1-benzene signal was observed at δ6.9–7.02 ppm. The 1H NMR spectra of the IBR-KET DDCs in Fig. 8c illustrate the presence of proton signals of IBR and KET in Fig. 8a and b, respectively. All the 1H NMR signals were identical to that of the pure forms of IBR and KET, and a slight broadening in a few peaks of DDCs was observed when compared to that of the pure IBR and KET 1H NMR signals. These show that the IBR and KET in the produced IBR-KET DDCs formed intermolecular hydrogen bonds.

1H NMR spectra of a Ibrutinib, b Ketoconazole, and c IBR-KET-DDCs
Thermogravimetric Analysis for Thermal Stability (TGA)
To investigate the IBR-KET DDC thermal stability in comparison to pure IBR and KET, TGA was used to investigate weight variations of all the samples with changes in temperature (Fig. 9 and Table V). As demonstrated in the TGA graph, the decompositions of IBR and KET were initiated at 353°C and 345°C and were accomplished in a single step. Yet, DDCs showed an extended decomposition rate when compared to IBR and KET individually. The decomposition initiation temperature of DDCs was 397°C, which represents the enhanced thermal stability of the IBR-KET DDCs than that of the pure IBR and KET. This implies that developed IBR-KET DDCs improved the thermal stabilities of the pure drugs.

TGA data of Ibrutinib, Ketoconazole, and IBR-KET-DDCs
Morphological Studies
Scanning Electron Microscopy (SEM)
Figure 10a and b shows SEM images of IBR and KET. IBR and KET display broken, irregularly shaped crystals, while IBR-KET DDCs in Fig. 10c show crystals with smooth surfaces. The smooth surfaces and clustered IBR-KET DDCs in Fig. 10c indicate that the DDCs processed by HME were packed closely together, which might be attributable to effective mixing inside the barrel. These variations in the DDC shape and size are indicative of molecular interaction between the IBR-KET. As a result, the DDC morphology was wholly distinct from that of the IBR and KET, indicating the development of novel crystalline material. When developing pharmaceutical solid dosage forms, the surface morphology of IBR-KET DDCs may have a positive impact on physicochemical and mechanochemical parameters such as solubility, dissolution, flowability, and compressibility properties.

SEM analysis of a ibrutinib, b ketoconazole, and c IBR-KET-DDCs
Saturation Solubility Studies
The saturation solubility of IBR, KET, and IBR-KET-DDCs was tested in water and multiple buffers of variable pH values, as illustrated in Table VI. IBR is a weak, poorly soluble base that has limited water solubility and displays a pH-dependent solubility [50]. Comparing the solubility of the IBR-KET DDCs to that of pure IBR and KET, DDCs were found to be significantly more soluble at pH 1.2 (35.88 times (IBR) and 12.28 times (KET)), pH 4.5 (21.15 times (IBR) and 9.46 times (KET)), pH 6.8 (5.45 times (IBR) and 7.88 times (KET)), pH 7.4 (6.29 times (IBR) and 7.15 times (KET)), and water (6.94 times (IBR) and 10.98 times (KET)). This may be due to the fact that the drugs individually dissociated from the DDC complex into the medium, causing the IBR and KET to become supersaturated and form metastable polymorphs or nanocrystalline clusters, which resulted in a relatively high solubility, this was possible due to the spring and parachute mechanism observed by the DDCs [51,52,53].
Powder Dissolution Experiments
The powder dissolution studies of IBR, KET, and DDCs formulations were conducted in both pH 1.2 and pH 6.8 to simulate the stomach and intestinal pH conditions, and the outcomes of the powder dissolution study are given in Figs. 11 and 12. The dissolution profile for IBR and KET indicated a release rate in pH 1.2 with 46.50% and 51.66% being dissolved after 180 min. Only 7.57% and 12.60% of IBR and KET were dissolved after 180 minutes, indicating a slow rate of dissolution under pH 6.8. IBR and KET both had notable increase in their rate of dissolution during the 180-minute cumulative drug release (IBR-KET-DDCs), reaching 62.86% and 67.52%, respectively. The IBR-KET-DDC dissolution rate was superior at pH 6.8, suggesting that the IBR and KET can quickly release under intestinal pH conditions, when IBR-KET DDCs are administered orally, thus creating the premise for its rapid and sufficient absorption. This rapid dissolution of DDC could be explained by the intermolecular interaction that occurs when IBR and KET form IBR-KET DDCs. This can also be supported by the fact that the IBR-KET DDCs have a higher solubility than the IBR and KET, as supported by the fact that DDCs melting point is lower than that of IBR and KET. Additionally, the effective intermolecular complexation of the two components may account for the increased solubility of IBR-KET DDCs compared to IBR and KET. As a result, the novel IBR-KET DDC composition created by the HME methodology can be envisioned as an effective way to accelerate the dissolution of both IBR and KET.

Powder dissolution studies of ibrutinib, ketoconazole, and IBR-KET-DDCs in pH 1.2

Powder dissolution studies of ibrutinib, ketoconazole, and IBR-KET-DDCs in pH 6.8
In Vitro Drug Release Profile
Figure 13 shows the results of comparative studies on IBR, KET, and IBR-KET DDCs release behavior that was conducted at 37°C in pH 6.8 buffer solution. It is demonstrated that after 10 hours, the IBR-KET-DDC release concentration approaches equilibrium and a cumulative release amount with IBR about 26.57% and KET about 59.25%, respectively. While pure IBR and KET were only up to 4.77% and 10.35% at this time, proving the supersaturation state with the augmented physicochemical properties. Results suggest that high amounts of IBR-KET DDCs are released and retained more in the intestinal region before making it to the intended tissues, resulting in localized releases and also helping to reduce toxicity in healthy tissues. To lay the groundwork for the long-term and effective enrichment of IBR with more significant effects on target cells, the IBR-KET-DDCs can perform a dual duty of stabilizing and regulating the release behavior through electrostatic interaction. IBR-KET DDCs with superior release performance will help to increase oral bioavailability, retention time, and half-life, which will help create better conditions for drugs to prove their efficacy. Further, oral bioavailability results are proven via in vivo pharmacokinetic studies in the coming sections.

In vitro release studies of ibrutinib, ketoconazole, and IBR-KET-DDCs in pH 6.8
Accelerated Stability Studies
One of the fundamental principles of the pharmaceutical industry is the stability of a solid drug dosage form across temperature and relative humidity. As a result, IBR-KET DDCs must require accelerated stability testing. Under accelerated conditions of 40°C/75% RH, the physical phase stabilities of IBR-KET DDCs were implemented for 180 days, and samples were collected at every 1, 3, and 6 months. Figures 14 and 15 depict the outcomes of the accelerated stability studies (PXRD and DSC representation). The IBR-KET DDC PXRD patterns showed no apparent shift when compared with fresh IBR-KET DDCs, demonstrating that they remained stable followed by accelerated storage. Similar to this, the DSC thermograms of the IBR-KET DDCs remained unchanged throughout the accelerated storage indicating stability. Also, the moisture uptake studies were performed for IBR-KET-DDCs indicated ∼2% moisture content close to initial values (Table VII), proving their phase stability and humidity stability. According to the aforementioned findings, the IBR-KET DDC strong physical stability encouraged us to investigate further. The chemical stability experiments of IBR-KET-DDCs at 40°C/75% RH were assessed, and Table VIII displayed the outcomes of drug content analysis. The drug content barely changed in IBR-KET-DDCs, indicating that the individual drugs had strong chemical stability in the DDCs (Table VIII).

DSC thermogram of IBR-KET-DDCs at 40°C/75% RH for 6 months

PXRD spectra of IBR-KET-DDCs at 40°C/75% RH for 6 months
In Vitro Cell Viability Assay
By using the MTT assay, the cytotoxicity of IBR, KET, and IBR-KET-DDCs on HK and K562-CCL-243 leukemia cell lines was investigated (Figs. 16 and 17). IBR, KET, and IBR-KET-DDCs showed concentration-dependent cytotoxicity to K562-CCL-243 cancer cell lines and increased cell viability was observed in HK cell lines. The half maximal inhibitory concentration (IC50) of IBR and KET for K562-CCL-243 cell lines was 62.95 ± 8.382 μM and 63.94 ± 9.873 μM, respectively. Meanwhile, the IC50 value of IBR-KET-DDCs was 29.51 ± 2.909 μM, demonstrating that it has much improved anticancer activity than IBR alone (Fig. 18). These findings provide further evidence for the enhanced anticancer effect of IBR-KET-DDCs, which may reduce the effective dosage of IBR and reduce IBR’s dose-related side effects.

In vitro cell viability assay of ibrutinib, ketoconazole, and IBR-KET-DDCs in HK cell lines

In vitro cell viability assay of ibrutinib, ketoconazole, and IBR-KET-DDCs in K562-CCL-243 leukemia cell lines

IC50 values of ibrutinib, ketoconazole, and IBR-KET-DDCs in K562-CCL-243 leukemia cell lines
In Vivo Pharmacokinetic Study
An important factor influencing a drug’s application clinically is its improved oral bioavailability. Wistar rats were used to investigate the pharmacokinetic properties of IBR, KET, and IBR-KET-DDC suspension. Following oral administration of IBR, KET, and IBR-KET-DDCs suspension, the relevant plasma IBR and KET concentration curves are depicted in Figs. 19 and 20, and the related pharmacokinetic characteristics are included in Table IX. The drug-time curve suggests that the plasma displayed a maximum plasma concentration (452.97 ± 0.079 ng/mL) at 2.5 h after oral administration of IBR suspension and maximum plasma concentration (216.78 ± 1.31 ng/mL) at 1 h after oral administration of KET suspension. However, the plasma showed a significantly higher maximum plasma concentration (6590.21 ± 0.22 ng/mL) of IBR at 2 h and (638.95 ± 0.39 ng/mL) of KET at 2 h after oral administration of IBR-KET-DDCs. The AUC (0–48 h) for the IBR-KET-DDCs set was a 4.30 folds enhancement when compared to that for the IBR suspension group and 2.31 folds enhancement when compared to that for the KET suspension group. In addition, tmax (IBR) was lower in the IBR-KET-DDCs group compared to the IBR suspension group. This finding showed that the IBR-KET DDCs significantly increased the rate of drug absorption, which led to a quicker onset of action.

In vivo pharmacokinetic log plasma concentration vs. time profile of ibrutinib vs. ibrutinib in IBR-KET-DDCs

In vivo pharmacokinetic log plasma concentration vs. time profile of ketoconazole vs. ketoconazole in IBR-KET-DDCs
Following oral administration of IBR-KET DDC suspension, IBR oral bioavailability is increased, which can be summed up as follows:
-
(1)
Due to the interaction with KET and improved hydrogen bonding, the hydrophilicity of the weakly soluble IBR may have significantly increased
-
(2)
Also, the research work primary objective about using KET was to inhibit the CYP3A4 enzyme activity which leads to the first pass metabolism of the IBR affecting the oral bioavailability. From the pharmacokinetic data presented in Table IX, it was absolute that the oral bioavailability has increased when compared with the pure IBR suspension proving some evident CYP3A4 inhibitory activity
In a nutshell, the findings showed that IBR-KET-DDCs enhanced the oral bioavailability of both IBR and KET, which helped to improve the drug’s clinical efficacy.
Conclusions
In the current study, we successfully prepared IBR-KET-DDCs employing the HME technology without any organic solvents. The IBR-KET DDCs were evaluated by FTIR, DSC, PXRD, TGA, SEM, and NMR studies, and IBR and KET have a non-covalent interaction, according to the FTIR and NMR data. Molecular dynamics and simulation studies for screening suitable codrug/coformer for formulating DDCs were successfully employed. IBR-KET-DDCs did not undergo any physical or chemical alteration during their six-month storage. At various pH levels, the solubility of DDCs increased substantially in comparison to that of the IBR and KET. When compared to IBR and KET, IBR-KET-DDCs exhibit improved dissolution profiles and release rates, which significantly increases the oral bioavailability of the individual drugs. Additionally, investigations in in vitro cytotoxicity of IBR-KET-DDCs in K562-CCL-243 cancer cells illustrated that IBR-KET-DDCs displayed increased cytotoxicity than the pure IBR and KET. Additionally, the improved oral bioavailability demonstrates that the formation of IBR-KET DDCs is a potential strategy to increase the solubility and oral bioavailability of IBR. Due to its uninterrupted manufacturing method, HME technology will be not only helpful for scaling up but also for large-scale manufacturing.
References
Guo M, Sun X, Chen J, Cai T. Pharmaceutical cocrystals: A review of preparations, physicochemical properties and applications. Acta Pharmaceutica Sinica B. 2021;11:2537–64.
Gala UH, Miller DA, Williams RO. Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions. Biochimica et Biophysica Acta (BBA) - Reviews on. Cancer. 2020;1873:188319.
Kara DD, Rathnanand M. Cocrystals and drug–drug cocrystals of anticancer drugs: A perception towards screening techniques, preparation, and enhancement of drug properties. Crystals. 2022;12:1337.
Al-Obaidi H, Granger A, Hibbard T, Opesanwo S. Pulmonary drug delivery of antimicrobials and anticancer drugs using solid dispersions. Pharmaceutics. 2021;13:1056.
Khatun B, Baishya P, Ramteke A, Maji TK. Study of the complexation of structurally modified curcumin with hydroxypropyl beta cyclodextrin and its effect on anticancer activity. New J Chem. 2020;44:4887–97.
Lerata MS, D’Souza S, Sibuyi NRS, Dube A, Meyer M, Samaai T, et al. Encapsulation of variabilin in stearic acid solid lipid nanoparticles enhances its anticancer activity in vitro. Molecules. 2020;25:830.
Gomes HIO, Martins CSM, Prior JAV. Silver nanoparticles as carriers of anticancer drugs for efficient target treatment of cancer cells. Nanomaterials. 2021;11:964.
Sathisaran I, Dalvi SV. Engineering cocrystals of poorly water-soluble drugs to enhance dissolution in aqueous medium. Pharmaceutics. 2018;10:108.
Liu L, Yu Y-M, Bu F-Z, Li Y-T, Yan C-W, Wu Z-Y. The first cocrystallization of milrinone with nutraceuticals: The adjusting effects of hydrophilicity/hydrophobicity in cavities on the in vitro/in vivo properties of the cocrystals. Crystal Growth Des. 2022;22:1623–37.
Kumar S, Nanda A. Approaches to design of pharmaceutical cocrystals: A review. Mol Cryst Liq Cryst. 2018;667:54–77.
Banerjee M, Nimkar K, Naik S, Patravale V. Unlocking the potential of drug-drug cocrystals – A comprehensive review. J Controlled Release. 2022;348:456–69.
Wang X, Du S, Zhang R, Jia X, Yang T, Zhang X. Drug-drug cocrystals: Opportunities and challenges. Asian J Pharmaceutical Sci. 2021;16:307–17.
Douroumis D, Ross SA, Nokhodchi A. Advanced methodologies for cocrystal synthesis. Adv Drug Deliv Rev. 2017;117:178–95.
Alshetaili AS, Ansari MJ, Anwer MK, Ganaie MA, Iqbal M, Alshahrani SM, et al. Enhanced oral bioavailability of ibrutinib encapsulated poly (lactic-co- glycolic acid) nanoparticles: Pharmacokinetic evaluation in rats. CPA. 2019;15:661–8.
Song P, Tian Y, Hao G, Xu L, Sun Y, Sun Y. Preparation and evaluation of ibrutinib lipid-based formulations. J Drug Deliv Sci Technol. 2022;77:103912.
Tapaninen T, Olkkola AM, Tornio A, Neuvonen M, Elonen E, Neuvonen PJ, et al. Itraconazole increases ibrutinib exposure 10-fold and reduces interindividual variation—A potentially beneficial drug-drug interaction. Clin Transl Sci. 2020;13:345–51.
Shi X, Wang C, Chen Q, Shen S, Song S, Zhou X. Improving physicochemical properties of ibrutinib with cocrystal strategy based on structures and natures of the carboxylic acid co-formers. J Drug Deliv Sci Technol. 2021;63:102554.
Hossain Mithu MS, Ross SA, Hurt AP, Douroumis D. Effect of mechanochemical grinding conditions on the formation of pharmaceutical cocrystals and co-amorphous solid forms of ketoconazole – Dicarboxylic acid. J Drug Deliv Sci Technol. 2021;63:102508.
Kamble RN, Bothiraja C, Mehta PP, Varghese V. Synthesis, solid state characterization and antifungal activity of ketoconazole cocrystals. J Pharm Investig. 2018;48:541–9.
Martin FA, Pop MM, Borodi G, Filip X, Kacso I. Ketoconazole salt and co-crystals with enhanced aqueous solubility. Cryst Growth Des. 2013;13:4295–304.
Martin F, Pop M, Kacso I, Grosu IG, Miclăuş M, Vodnar D, et al. Ketoconazole-p-aminobenzoic acid cocrystal: Revival of an old drug by crystal engineering. Mol Pharm. 2020;17:919–32.
Chen YM, Rodríguez-Hornedo N. Cocrystals mitigate negative effects of high pH on solubility and dissolution of a basic drug. Cryst Growth Des. 2018;18:1358–66.
Weiss J, Foerster KI, Weber M, Burhenne J, Mikus G, Lehr T, et al. Does the circulating ketoconazole metabolite N-deacetyl ketoconazole contribute to the drug-drug interaction potential of the parent compound? Eur J Pharmaceutical Sci. 2022;169:106076.
Mehta CH, Narayan R, Aithal G, Pandiyan S, Bhat P, Dengale S, et al. Molecular simulation driven experiment for formulation of fixed dose combination of darunavir and ritonavir as anti-HIV nanosuspension. J Mol Liq. 2019;293:111469.
Desai PP, Patravale VB. Curcumin cocrystal micelles—Multifunctional nanocomposites for management of neurodegenerative ailments. J Pharm Sci. 2018;107:1143–56.
Das T, Mehta CH, Nayak UY. Multiple approaches for achieving drug solubility: An in silico perspective. Drug Discov Today. 2020;25:1206–12.
Pisay M, Bhaskar KV, Mehta CH, Nayak UY, Koteshwara KB, Mutalik S. Drug-carrier miscibility in solid dispersions of glibenclamide and a novel approach to enhance its solubility using an effervescent agent. AAPS PharmSciTech. 2022;23:284.
Kshirsagar SM, Chatale BC, Amin PD. Comparative evaluation of ibuprofen co-crystals prepared by solvent evaporation and hot melt extrusion technology. Journal of Drug Delivery Science and Technology. 2022;67:103003.
Chaudhari KR, Savjani JK, Savjani KT, Shah H. Improved pharmaceutical properties of ritonavir through co-crystallization approach with liquid-assisted grinding method. Drug Dev Ind Pharm. 2021;47:1633–42.
Narala S, Nyavanandi D, Alzahrani A, Bandari S, Zhang F, Repka MA. Creation of hydrochlorothiazide pharmaceutical cocrystals via hot-melt extrusion for enhanced solubility and permeability. AAPS PharmSciTech. 2022;23:56.
Butreddy A, Sarabu S, Bandari S, Dumpa N, Zhang F, Repka MA. Polymer-assisted aripiprazole–adipic acid cocrystals produced by hot melt extrusion techniques. Cryst Growth Des. 2020;20:4335–45.
Baptista JA, Castro RAE, Rosado MTS, Maria TMR, Silva MR, Canotilho J, et al. Polymorphic cocrystals of the antimalarial drug pyrimethamine: Two case studies. Cryst Growth Des. 2021;21:3699–713.
Nikam VJ, Patil SB. Pharmaceutical cocrystals of nebivolol hydrochloride with enhanced solubility. J Cryst Growth. 2020;534:125488.
Pantwalawalkar J, More H, Bhange D, Patil U, Jadhav N. Novel curcumin ascorbic acid cocrystal for improved solubility. J Drug Deliv Sci Technol. 2021;61:102233.
Cui W, He Z, Zhang Y, Fan Q, Feng N. Naringenin cocrystals prepared by solution crystallization method for improving bioavailability and anti-hyperlipidemia effects. AAPS PharmSciTech. 2019;20:115.
Liu H, Tong HHY, Zhou Z. Feasibility of thermal methods on screening, characterization and physicochemical evaluation of pharmaceutical cocrystals. J Therm Anal Calorim. 2022;147:12947–63.
Gowda BHJ, Nechipadappu SK, Shankar SJ, Chavali M, Paul K, Ahmed MG, et al. Pharmaceutical cocrystals of efavirenz: Towards the improvement of solubility, dissolution rate and stability. Mater Today: Proceed. 2022;51:394–402.
Li X, Liu X, Song J, Wang C, Li J, Liu L, et al. Drug–drug cocrystallization simultaneously improves pharmaceutical properties of genistein and ligustrazine. Cryst Growth Des. 2021;21:3461–8.
Hong M, Li S, Ji W, Qi M-H, Ren G. Cocrystals of lenvatinib with sulfamerazine and salicylic acid: Crystal structure, equilibrium solubility, stability study, and anti-hepatoma activity. Cryst Growth Des. 2021;21:3714–27.
Salas-Zúñiga R, Rodríguez-Ruiz C, Höpfl H, Morales-Rojas H, Sánchez-Guadarrama O, Rodríguez-Cuamatzi P, et al. Dissolution advantage of nitazoxanide cocrystals in the presence of cellulosic polymers. Pharmaceutics. 2020;12:23.
Yu Y-M, Liu L, Bu F-Z, Li Y-T, Yan C-W, Wu Z-Y. A novice cocrystal nanomicelle formulation of 5-fluorouracil with proline: The design, self-assembly and in vitro/vivo biopharmaceutical characteristics. Int J Pharma. 2022;617:121635.
Wang L, Yan Y, Zhang X, Zhou X. Novel pharmaceutical cocrystal of lenalidomide with nicotinamide: Structural design, evaluation, and thermal phase transition study. Int J Pharma. 2022;613:121394.
Hao H, Zhang Y, Hu X, Guo W, Yang C, Wang J. Cocrystallization of 5-fluorouracil with gallic acid: A novel 5-fluorouracil cocrystal displaying synergistic anti-tumor activity both in oral and intraperitoneal injection administration. Eur J Pharm Biopharm. 2023;187:12–23.
Vasilev NA, Surov AO, Voronin AP, Drozd KV, Perlovich GL. Novel cocrystals of itraconazole: Insights from phase diagrams, formation thermodynamics and solubility. Int J Pharm. 2021;599:120441.
Ch S, Padaga SG, Ghosh B, Roy S, Biswas S. Chitosan-poly(lactide-co-glycolide)/poloxamer mixed micelles as a mucoadhesive thermo-responsive moxifloxacin eye drop to improve treatment efficacy in bacterial keratitis. Carbohydrate Polymers. 2023;312:120822.
Liu Y, Yang F, Zhao X, Wang S, Yang Q, Zhang X. Crystal structure, solubility, and pharmacokinetic study on a hesperetin cocrystal with piperine as coformer. Pharmaceutics. 2022;14:94.
Écsiová D, Šimkovič M, Uher M, Hroch M. High-throughput salting-out assisted liquid-liquid extraction using a 3D printed device and its application in the quantification of ibrutinib and its metabolite PCI-45227 in human serum. J Pharm Biomed Anal. 2022;219:114923.
Dain IA, Zolotov SA, Demina NB, Zolotova AS, Buzanov GA, Retivov VM, et al. Complex dispersions of poloxamers and mesoporous carriers with ibrutinib. OpenNano. 2022;8:100073.
Fathi M, Sodeifian G, Sajadian SA. Experimental study of ketoconazole impregnation into polyvinyl pyrrolidone and hydroxyl propyl methyl cellulose using supercritical carbon dioxide: Process optimization. The J Supercrit Fluids. 2022;188:105674.
Ashar F, Hani U, Osmani RAM, Kazim SM, Selvamuthukumar S. Preparation and optimization of ibrutinib-loaded nanoliposomes using response surface methodology. Polymers. 2022;14:3886.
Zhang J, Liu M, Zeng Z. The antisolvent coprecipitation method for enhanced bioavailability of poorly water-soluble drugs. Int J Pharm. 2022;626:122043.
Bavishi DD, Borkhataria CH. Spring and parachute: How cocrystals enhance solubility. Progress Cryst Growth Charact Mater. 2016;62:1–8.
Babu NJ, Nangia A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst Growth Des. 2011;11:2662–79.
Acknowledgements
The authors are thankful to (i) Manipal College of Pharmaceutical Sciences, Manipal Academy of Higher Education, Manipal, India, for providing the TMA Pai scholarship to Divya Dhatri Kara; Steer Life Private Limited, Bangalore and Manipal-Government of Karnataka Bio incubator, Manipal Academy of Higher Education, Manipal for providing the necessary facilities, (ii) Department of Pharmaceutics, Manipal College of Pharmaceutical Sciences for facilities of Schrodinger’s Suit computer simulations, and (iii) SP Accure Labs Private Limited, Hyderabad.
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal The research work was supported by an IMF grant from Manipal Academy of Higher Education, Manipal.
Author information
Authors and Affiliations
Contributions
Conceptualization: Divya Dhatri Kara and Mahalaxmi Rathnanand; methodology: Divya Dhatri Kara, Pragathi Devanand Bangera, and Mahalaxmi Rathnanand; software: Chetan Hasmukh Mehta and Divya Dhatri Kara; validation: Divya Dhatri Kara and Mahalaxmi Rathnanand; formal analysis: Divya Dhatri Kara, Pragathi Devanand Bangera, and Katikala Tanvi; investigation: Divya Dhatri Kara and Pragathi Devanand Bangera; resources: Mahalaxmi Rathnanand; data curation: Divya Dhatri Kara and Pragathi Devanand Bangera; writing—original draft preparation, Divya Dhatri Kara; writing—review and editing, Mahalaxmi Rathnanand; supervision and funding acquisition: Mahalaxmi Rathnanand.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kara, D.D., Bangera, P.D., Mehta, C.H. et al. In Silico Screening as a Tool to Prepare Drug-Drug Cocrystals of Ibrutinib-Ketoconazole: a Strategy to Enhance Their Solubility Profiles and Oral Bioavailability. AAPS PharmSciTech 24, 164 (2023). https://doi.org/10.1208/s12249-023-02621-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-023-02621-9