
Abstract
The ability of dendritic cells (DCs) to initiate and regulate adaptive immune responses is fundamental for maintaining immune homeostasis upon exposure to self or foreign antigens. The immune regulatory function of DCs is strictly controlled by their distribution as well as by cytokines, chemokines, and transcriptional programming. These factors work in conjunction to determine whether DCs exert an immunosuppressive or immune-activating function. Therefore, understanding the molecular signals involved in DC-dependent immunoregulation is crucial in providing insight into the generation of organismal immunity and revealing potential clinical applications of DCs. Considering the many breakthroughs in DC research in recent years, in this review we focused on three basic lines of research directly related to the biological functions of DCs and summarized new immunotherapeutic strategies involving DCs. First, we reviewed recent findings on DC subsets and identified lineage-restricted transcription factors that guide the development of different DC subsets. Second, we discussed the recognition and processing of antigens by DCs through pattern recognition receptors, endogenous/exogenous pathways, and the presentation of antigens through peptide/major histocompatibility complexes. Third, we reviewed how interactions between DCs and T cells coordinate immune homeostasis in vivo via multiple pathways. Finally, we summarized the application of DC-based immunotherapy for autoimmune diseases and tumors and highlighted potential research prospects for immunotherapy that targets DCs. This review provides a useful resource to better understand the immunomodulatory signals involved in different subsets of DCs and the manipulation of these immune signals can facilitate DC-based immunotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dendritic cells (DCs), as key outposts of the immune response, are crucial for maintaining the central and peripheral tolerance mechanisms under steady-state conditions. Alternatively, under inflammatory conditions, danger signals from sites of infection or cancer promote the recruitment, activation, and maturation of DCs, ultimately leading to antigen-specific T cell responses [1, 2]. The migration of DCs towards lymph nodes and their upregulation of co-stimulatory molecules, such as CD40, CD80, and CD86, as well as secretion of multiple cytokines, including interferon (IFN)-α/β, interleukin (IL)-10, and IL-12, are induced by danger signals [3, 4]. These events determine the activation and polarization of T cells. Owing to their role in initiating T cell immunity, DCs have been identified as key regulators in guiding immunotherapy for autoimmune diseases and tumors [3, 5]. Existing evidence indicates that the presence of DCs or expression of DC-specific transcriptional signatures positively correlates with CD8+ T cell infiltration in tumors and improved prognosis, while selective depletion of DCs reduces the number of T cells in autoimmune diseases and attenuates disease severity [6,7,8,9].
The biological functions and markers of DCs may vary considerably in different microenvironments; this increases the difficulty of classifying DCs. Currently, a uniform classification system is to divide DCs into conventional DCs (cDCs), plasmacytoid DCs (pDCs), and monocyte-derived DCs (moDCs) based on their origin and differentiation pathways. Over the past two decades, research surrounding DCs has yielded extraordinary insights into various aspects of biological and medical applications. Nonetheless, these advances have highlighted many fundamental unanswered questions surrounding the function and mechanisms of DCs. Addressing these questions can accelerate the translation of this acquired knowledge on DCs into corresponding clinical applications. Therefore, the key aims of this review are to provide an overview of the molecular signals that control DC differentiation, antigen presentation, and immune regulation, and to highlight the potential applications of targeting DCs for immunotherapy. We also discuss the advancements in understanding how DCs crosstalk with other immune cells, as well as their role in shaping the tumor microenvironment. Overall, this review aims to comprehensively evaluate the signaling pathways that are involved in DC biology and their potential implications in disease treatment.
Developmental and biological functions of DC subsets
In 1973, Steinman and Cohn described a type of stellate cell structure in mouse spleens and lymph nodes, and termed them DCs [10]; prior to which macrophages and B cells were considered the primary antigen-presenting cells (APCs) [11]. Their experiments over the next few years confirmed that DCs are far more potent as APCs in stimulating T cell responses than other cell types that express the major histocompatibility complex (MHC) and co-stimulatory molecules [12]. DCs are present in almost all tissues throughout the body and exhibit a high degree of heterogeneity. Different DC subpopulations are defined based on their development, anatomical location, and immunological characteristics.
Development and transcriptional regulation of DC subsets
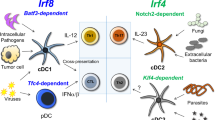
DCs originate from common myeloid progenitors (CMPs). Their main types include cDCs, pDCs, and moDCs (Fig. 1). In certain inflammatory conditions, the transcription factor Nur77 is responsible for promoting the differentiation of CMP into monocytes, which subsequently develop into moDCs [13, 14]. cDCs and pDCs are considered to have originated from a shared lineage of DC precursors known as common DC precursors (CDPs), which lack monocyte/macrophage differentiation potential and also develop from CMPs [15]. CDPs are typically characterized by a lack of lineage markers (LIN−) and are accompanied by the expression of Fms-like tyrosine kinase 3 (FLT3), macrophage colony-stimulating factor receptor (M-CSFR; also known as CD115), and receptor tyrosine kinase KIT (SCFR; also known as CD117) [16]. The transcription factor Zbtb46 is a specific marker for cDCs, which comprises two main subsets: the cDC1s and the more heterogeneous cDC2s [17].

Mouse dendritic cell development and lineage. DCs are derived from CDP, which is derived from LMPPs differentiated from HSC in the bone marrow. Branching CMPs of LMPPs are driven by the transcription factor Nuf77 to differentiate into monocytes and then further differentiate into M-CSF-dependent macrophages or GM-CSF-dependent moDCs. High expression of PU.1 is associated with the specification of these two cell types. Zbtb46 expression in pre-cDC1s and pre-cDC2s is a specific marker for the specification of cDCs. cDC2 subsets express high levels of IRF4, whereas pDCs and cDC1 express high levels of IRF8. Id2 is a suppressor of Tcf4, impairing Tcf4-driven pDCs specification and promoting cDC1s specification. HSC, hematopoietic stem cell; LMPP, lymphoid- primed multipotent progenitors; CLP, common lymphoid progenitors; GMP, granulocyte–macrophage progenitors; cMoP, common monocyte progenitors; IRF8, interferon regulatory factor 8; BATF, basic leucine zipper ATF-like transcription factor; SIRPα, signal regulatory protein alpha
cDC1 and cDC2 subsets
cDC1s and cDC2s play special roles in the immune activation of CD8+ and CD4+ T cells, respectively, through differential processing and presentation of antigens and the production of corresponding cytokines [18, 19]. In the bone marrow, upregulation of CD11c expression facilitates CDPs to further develop into pre-cDCs, which are excreted from the bone marrow via the blood and differentiated into cDC1 and cDC2 populations in tissues [11, 20]. cDC1s develop from pre-DCs in an IRF8-dependent manner, with cell surface expression of the chemokine receptor XCR1, C-type lectin–like receptor (CLEC9A), and CD205 [21,22,23]. In addition, Nfil3 and Id2 are essential for the differentiation of cDC1s. Selective deletion of cDC1s occurs in mice with Batf3, Nfil3, or Id2 knockout [4]. Interestingly, the effects of deletions in Batf3, Id2, or Nfil3 in mice can be repaired by ectopic expression of IRF8, suggesting that high expression of IRF8 is central to cDC1 development [24]. Similarly, the compensatory effects of other members of the BATF family maintain IRF8 expression under some inflammatory conditions, prompting to cDC1 production in Batf3−/− mice [25].
Compared to cDC1s, cDC2s have a different transcriptional profile and can be identified by the expression of SIRPα and CD11b, with the key transcription factor being IRF4 [11]. Although cDC2s are present in IRF4−/− mice, their numbers are substantially reduced and they lose the ability to migrate peripherally to the lymph nodes [26, 27]. In addition, the KLF4 transcription factor and Notch2 receptor are essential for the development of cDC2s [28, 29]. cDC2s are further divided into several subsets based on their phenotype and function owing to their high heterogeneity. High expression of the endothelial cell-selective adhesion molecule (ESAM) has been used to distinguish Notch2-dependent cDC2s (ESAMhi cDC2s) [30]. ESAMhi cDC2s have a high value-added rate and high expression of MHCII compared to the ESAMlo cDCs, which preferentially express monocyte-associated genes, including chemokine (Ccr2) receptors and lysozyme (Lyz1, Lyz2) [30]. Brown et al. defined the two subpopulations of cDCs: cDC2sA and cDC2sB, based on the mutually exclusive expression of the transcription factors T-bet or RORγT [31]. The cDC2sA population corresponds, at least in part, to the previously identified ESAMhi cDC2s. cDC2sB has a phenotype similar to that of ESAMlo cDC2s. T-bet+ cDC2sA are present throughout lymphoid and non-lymphoid tissues, in contrast to ESAMhi cells, which are found only in the spleen and mesenteric lymph nodes [32].
Human and mouse cDCs are thought to be relatively evolutionarily conserved. They are both derived from CDPs and comprise mainly two basic subtypes, cDC1s and cDC2s. Similar to mouse cDCs, the development of human cDC1s’ depends on Batf3 and IRF8 expression. Short hairpin RNA-mediated knockdown of Batf3 selectively inhibits the development of human cDC1s in vitro [33]. Patients with IRF8 mutations were found to have defective helper T cell function, likely due to reduced CD11c-expressing DCs, and had reduced resistance to infections, including Mycobacterium infections [34]. Human cDC1s were further identified by the expression of CD141 and CADM1 in addition to the expression of the mouse cDC1s-specific markers XCR1 and CLEC9A. In contrast, human cDC2s are commonly identified by the expression of CD1c (BDCA1) and CLEC10A (CD301a), in addition to SIRPα [35]. In a recent study, single-cell RNA-sequencing analysis was used to classify human cDC2s into DC2 and DC3 and was validated by parallel observations in their murine cDC2s homologs, including ESAMhi cDC2s and ESAMlo cDC2s [36].
pDC subsets
pDCs are derived from CDPs and are consistently produced in the bone marrow. They, then, mature and migrate to the periphery, where they lose their proliferative ability and only survive for a short time [37]. However, a portion of pDCs may originate from the lymphoid lineage, as evidenced by the discovery of a population of LIN−KIT+SCA1+CD34+FLT3+ cells that appear to be directly derived from lymphoid-primed multipotent progenitors; this population expresses high levels of Tcf4 with prominent pDC differentiation potential [38]. The pDCs possess elevated levels of Toll-like receptors (TLRs) that are capable of sensing endosomal nucleic acids, such as TLR7 and TLR9 [39, 40]. These receptors enable pDCs to recognize viral single-stranded RNA and unmethylated CpG motif-containing DNA [41]. FLT3 and its ligand (FLT3L) play crucial roles in the development of pDCs [42]. FLT3L drives spontaneous differentiation of pDCs and lineage bifurcation of pDCs and cDCs through the activation of signal transducer and transcription 3 (STAT3) and mammalian target of rapamycin (mTOR) [43]. Subsequent specification of pDCs requires the involvement of E protein transcription factor TCF4 (E2-2), which is a key factor in the identification of pCDs in mice and humans. [44]. Id2, a repressor of E2-2, may disrupt the E2-2-driven transcriptional program of pDCs, directing cell differentiation towards cDC1s [16]. Thus, Id2 is essentially absent in pDCs and relatively highly expressed in cDC1s. The numbers of pDCs are increased in Id2-/- mice. Overexpression of Id2 or de-repression of Id2 caused by the loss of the transcriptional repressor ZEB2 results in impaired development of pDCs and enhanced development of cDC1s [45,46,47]. In conclusion, these studies reveal a complex regulatory network for the development of pDCs and highlight the close relationship between the genetic mechanisms involved in the specification of cDCs and pDCs.
moDC subsets
In mice, two subsets of CMP-derived mononuclear phagocytes exist: CCR2−CX3Cr1hi LY6C− and CCR2+CX3Cr1loLY6C+CD62L+; the latter is a precursor to moDCs [48]. In contrast to the steady state, cDC2s express moDC-specific markers such as Ly6C, CD64, and MAR1 in the inflammatory microenvironment, which makes the identification of moDCs challenging [49]. To address this, Guilliams et al. compiled the available gene expression data for DCs and macrophage subsets. The findings showed that the expression of Fc receptors for IgG (FcγRs) is highly selective in humans and mice [50]. Specifically, moDCs highly express most activating and inhibitory FcγRs, while cDCs and pDCs predominantly express inhibitory FcγRIIB [50]. Of note, LY6C+ monocytes can differentiate into iNOS+ microbicidal macrophages in addition to CCR2-dependent moDCs [51, 52]. Menezes et al. suggested that PU.1 is critical in regulating the polarization of Ly6C+ monocytes and that high levels of PU.1 can selectively promote GM-CSF-dependent moDC production and downregulate the production of iNOS+ macrophages [53]. Furthermore, the aryl hydrocarbon receptor AHR serves as a molecular switch that plays a role in determining the fate of monocytes. In the presence of macrophage colony-stimulating factor (M-CSF), monocytes preferably differentiate into macrophages, whereas in the presence of IL-4 and TNF-α together with AHR ligands, they tend to differentiate into moDCs [54]. Overall, inflammatory monocytes have multiple fates, and the precise conditions in the microenvironment that drive the differentiation of monocytes towards moDCs rather than macrophages, as well as the specific molecular coordinators involved, remain to be further defined.
Antigen uptake and presentation by DC cells
In the previous section, the development and origin of the cDC, pDC, and moDC subsets were discussed. However, why these cells constitute the DC population is unclear. All of these cells bridge natural and adaptive immunity, including sensing the tissue environment, processing and presenting antigens, and promoting T cell responses, even though the pDC and moDC subsets are far less capable of initiating T cell responses compared to cDCs. Resting state DCs, are considered immature and must undergo an intricate and rigorous series of antigen-acquisition processes prior to activation and maturation[55]. Mature DCs exhibit reduced phagocytosis, increased antigen presentation and migration, and upregulated expression of various co-stimulatory molecules including CD40, CD70, and CD86, as well as MHC class I and class II molecules [40, 56].
Molecular players in antigen recognition and internalization
Interaction of pattern recognition receptors (PRRs) with pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns is a prerequisite for initiating immune responses in DCs. In mice and humans, the PRR families include TLRs, retinoic acid-inducible gene-I-like receptors (RLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), and range of intracellular DNA sensors [57,58,59]. High expression of TLR3, which senses viral dsRNA, is conserved in human and mouse cDC1s. TLR3 specifically initiates the efficient cDC1-mediated clearance of infected cells by CD8+ T cells [11, 60, 61]. Although the generation of a robust type I IFN response in cDC1s is biased towards TLR3, TLR9 activation is equally able to regulate Myd88-IRF7 signaling in cDC1s via NCoR1, inducing a moderate antiviral response [62, 63]. Additionally, mouse cDC1s express TLR11 and TLR13, whereas human cDC1s highly express TLR8[14, 64, 65]. Unlike cDC1s, cDC2s rapidly accumulate in mesenteric lymph nodes in a TLR5-dependent manner after soluble flagellin immunization [66]. cDC2s also express TLR2, TLR4, and TLR7 for sensing bacterial and viral associated PAMPs [36, 67]. The pDC-mediated antiviral response is largely dependent on the activation of TLR9 signaling and subsequent Myd88-IRF7 signaling[68]. In addition, TLR7 is responsible for the induction of inflammatory signaling following SARS-CoV-2 infection in a stem cell–based human pDC model [69]. Meanwhile, endosomal TLRs are responsible for initiating the initial steps in the generation of type I IFN in pDCs, whereas RLRs contribute to the latter stages of the IFN response [58, 70].
RLRs are constitutively expressed in cDCs and are the primary molecules used by cDCs to sense foreign nucleic acids [71]. As cytoplasmic sensors of viral RNA, RLRs interact with an adaptor protein (MAVS) upon activation, thereby triggering a signaling cascade that includes the secretion of inflammatory cytokines and chemokines [72, 73]. The calcium-dependent carbohydrate recognition domain in CLRs recognizes glycans in the fungal cell wall and is central to the body’s resistance to fungal infections [74]. CLR activation recruits Syk to initiate downstream signaling, leading to the production of ROS and activation of the NF-κB pathway mediated by the CARD9/Bcl-10/MALT1 complex [75, 76]. Expression of one CLR members, DNGR-1 (encoded by CLEC9A), is largely restricted to cDC1s, which promote phagosome rupture and cross-presentation of dead cell-associated antigens via the DNGR-1 pathway [77, 78]. When annexin A2, a protein commonly found in individuals with nasopharyngeal carcinoma, interacts with CLRs, an immunosuppressive response and release of IL-10 from DCs can be triggered [79].
Increasing evidence indicates that different members of the PRRs family can counteract pathogens by interacting with each other or attenuating excessive inflammation by mutual antagonism [80, 81]. Kim et al. showed that the collaboration between TLR2 and NOD2 is required for the induction of pro-IL-1β and NOD-like receptor pyrin domain-containing 3 (NLRP3) in Helicobacter pylori-infected murine-derived DCs [82]. In addition, NLRX1, a member of the NLR family, negatively regulates RLR-mediated type I IFN production in human pDCs [71]. In conclusion, the differences in intrinsic immune receptor expression between different subsets of DCs confer the corresponding ability of DCs to respond appropriately in response to different pathogens and stress signals (Fig. 2).

PRRs of DCs and their signaling pathways. The four major subgroups of DCs (cDC1s, cDC2s, pDCs, and moDCs), each have distinct PRRs. TLR2, TLR4, TLR5, TLR7, TLR8, TLR9, and TLR11 activate TRAF6 predominantly through Myd88; this then leads to the activation of IKKs and the release of NF-κB for nuclear translocation. In addition, TRAF6 can promote AP1 activity through MAPK signaling [58]. TLR3 signaling is primarily mediated by TRIF. TLR3 can promote the activation of TRAF6 and IRF3/IRF7. Activated RLRs interact with MVAS through their signaling structural domain, CARD, to activate TBK1, which then phosphorylates IRF3 and IRF7, translocating them from the cytoplasm to the nucleus [73]. In addition, the RLR signaling pathway activates NLRP3 inflammatory vesicles, which promote the formation of IL-1β. When NLR receptors are activated, they aggregate and activate downstream signaling molecules, including RIPK2, which in turn triggers the activation of NF-κB [58]. CLR activation recruits multiple kinases, including sky and the CARD9/Bcl10/MALT1 (CBM) complex, ultimately leading to activation of NF-κB [59]. TRAF6, TNF receptor associated factor 6; TRIF, TIR domain-containing adapter-inducing interferon-beta; ROS, reactive oxygen species; CARD, caspase recruitment domain; TBK1, TANK-binding kinase 1; RIPK2, receptor-interacting serine/threonine protein kinase 2
As tissue sentinels, DCs take up antigens through phagocytosis, micro- or macropinocytosis, and endocytosis [40]. After acquisition by DCs, the antigen is processed either by an endogenous or exogenous pathway. In general, antigens processed by the endogenous pathway are bound to MHC I molecules and presented to CD8+ cells, whereas antigens processed by the exogenous pathway are bound to MHC II molecules and presented to CD4+ cells [4, 40]. However, DCs are also capable of presenting peptides of exogenously internalized antigens via MHC I molecules, a process known as ‘cross-presentation’ [83]. Autophagy protein-mediated degradation of extracellular material provides substrates for MHC class II presentation to late endosomes and lysosomes [84, 85]. Conditional knockdown of the autophagy protein ATG5 in cDCs reportedly resulted in mice that were completely resistant to the development of experimental autoimmune encephalomyelitis (EAE) and had significantly reduced CD4+ T cell aggregation in the central nervous system [86]. DCs possess fewer proteases and are more susceptible to modulation by pH, unlike macrophages. Thus, they degrade internal antigens slowly but more efficiently recover immunogenic peptides and assemble peptide-MHC II complexes [87]. Reactive oxygen species (ROS), generated by the NADPH oxidase (NOX) complex or by electron leakage from mitochondrial aerobic respiration, promote alkalinization of phagosomal pH and inhibit rapid antigen degradation by acidic lysosomal proteases [88]. In contrast, activation of the transcription factor TFEB in mouse DCs induces a reduction in lysosomal pH and elevation in lysosomal protease expression, which results in downregulation of processing and presentation by MHC class II antigens and upregulation of processing and presentation by MHC class II antigens [89, 90]. In addition, the immunosuppressive function of the anti-inflammatory cytokine IL-10 is because of its ability to reduce the expression of MHC II molecules [91].
Antigen presentation
DCs are the predominant cell population responsible for presenting antigens in vivo and induce the activation of antigen-specific T cells through the interaction of the antigenic peptide-MHC complex with TCRs in combination with appropriate co-stimulatory signals. cDC1s highly express MHC class I molecules and possess a superior cross-presentation capability, which are necessary for CD8+ T cell activation. The binding of DNGR-1 to F-actin in cDCs exposed to dead cell debris facilitates cross-presentation of relevant antigens in tumors. Therefore, abundant DNGR-1 is positively correlated with the survival of cancer patients [92,93,94]. Secreted gelsolin blocks DNGR-1-dependent cross-presentation and inhibits antitumor CD8+ T-cell responses [92]. In contrast, WD Repeat- and FYVE domain-containing protein 4 (WDFY4)-regulated vesicular transport pathway is essential for DNGR-1 to bind dead cell antigens and facilitate cross-presentation [95]. TLR stimulation also upregulates the cross-presentation of cDC1s, which is associated with a reduction in Rab34-dependent phagolysosome fusion[96]. Earlier, pDCs were not considered cross-presenting DCs. However, a plethora of evidence indicates that pDCs can activate CD8+ cells by cross-presenting antigens in humans and mice [88, 97, 98]. Unlike cDC1s, the ability of pDCs to cross-present and activate CD8+ T cell responses are mainly dependent on mitochondrial ROS production rather than NOX1/2-mediated ROS production [88]. moDCs can also cross-present cell-associated antigens by using a different transcriptional program for cDCs [99].
DC-derived exosomes (Dex) are nanosized membrane vesicles that are initially formed by the inward budding of the endosomal membrane of DCs and subsequent release via the cell membran [90, 100]. Like DCs, Dex carries functional peptide-MHC complexes, co-stimulatory molecules (CD80 and CD86), and a variety of surface membrane proteins (integrins and ICAMs) that interact with immune cells [74, 100]. As a result, Dex can initiate antigen-specific CD4+ and CD8+ T cell responses directly or indirectly through antigen uptake and presentation. Exosomes derived from alpha-fetoprotein (AFP)-expressing DCs activate specific CD8 antitumor immune responses in tumor tissues [101]. Although Dex stimulate T cells in vitro and promotes increased secretion of IFN-γ and IL-2, these exosomes appear to be effective only for activated T cells, memory T cells, and T cell lines and are inefficient for the development of naïve T cells [101,102,103]. Compared to directly activating T cells with antigen-specific signals, the Dex antigen presentation pathway can stimulate T cell responses more efficiently by utilizing bystander DCs as intermediaries. One mechanism involves the direct transfer of peptide-MHC complexes to bystander DCs following the binding of Dex to the cell membrane of bystander DCs. Another possible mechanism is that Dex binds to bystander DCs and are reprocessed by the endosomal pathway, followed by the transfer of antigen epitopes of these Dex MHC molecules on bystander DCs for presentation to T-cells [102, 104].
DCs maintain immune homeostasis through multiple signaling pathways
Although DCs are classified as innate immune cells because of their ability to recognize danger signals via multiple PRRs, as APC, they also play an important role in initiating the subsequent adaptive immune response. DCs provide a constant supply of antigens to T cells under infectious/inflammatory and steady-state conditions [105, 106]. Under noninflammatory conditions, DCs can present self-antigens derived from local tissues to T cells without inducing active autoimmunity [107]. These findings strongly suggest that DCs play a role in inducing peripheral immune tolerance and maintaining immune homeostasis.
DC-mediated T cell homeostasis
The specification of an appropriate T cell fate is an important aspect of the effective adaptive immune function of T cells. For example, CD8+ cytotoxic T lymphocytes (CTL) kill infected or tumor cells, Th1 cells counteract intracellular bacterial and protozoal immune responses, Th2 cells respond to parasitic infections of the body, and Th17 cells counteract extracellular bacteria, and mycobacteria. DCs induce antigen-specific activation and expansion of T cells through peptide-MHC complexes and co-stimulatory signals, Expression of IRF4 by cDC2s is critical for the activation of CD4+ T cells; this may be attributed to the ability of IRF4 in enhancing the formation of peptide-MHC II complexes in cDC2s [4, 108]. DCs similarly instruct the fate of CD4+ cell polarization during antigen presentation (Fig. 2).
cDC1s are a major source of IL-12 in vivo and IL-12 signaling is essential for Th1 cell differentiation in mice and humans [109,110,111]. The absence of cDC1s in parasitic infection models such as Trichuris muris and Leishmania major infections is associated with impaired Th1 responses, suggesting that certain protective Th1 responses require the support of cDC1s in vivo [110, 112]. Unlike cDC1s, cDC2s adept at inducing Th2 and Th17 responses and are required for Th2 cell differentiation in the skin, lung, and intestine [113,114,115]. In particular, in the lungs of mice following house-dust extract inhalation, the partially mature Ly6C+ cDC2s lack CCR7 expression and promote Th17 differentiation, whereas the more mature CD200+ cDC2s expresse CCR7 and induce Th2 differentiation [116]. Tussiwand et al. suggested that differentiation of Th2 cells driven by cDC2s is dependent on the transcription factor KLF4 [28]. Conditional deletion of KLF4 in cDC2s impairs the Th2 cell response, but not Th1, Th17, or CTL responses [28]. The IL-13-induced signaling pathway promotes the STAT6-dependent differentiation of cDC2s and, subsequently, Th2 cells in the mouse dermis, while also reducing Th17 cell responses [117]. In humans, activation of formyl peptide receptor 3 (FPR3) enhances cDC2-mediated Th2 responses by decreasing IL-12 expression [118]. Interestingly, cDC1s have been recently found to similarly promote Th2 differentiation via p38α signaling [119]. TNFR2-cDC2s can promote the body’s Th1/17 immune response by activating moDCs after intranasal immunization with mucosal adjuvants [120]. Th17 cells play a key role in host defense and mucosal tissue homeostasis and are drivers of a variety of autoimmune diseases [121,122,123]. Furthermore, cDC2s provide many of the signals required for Th17 differentiation and promote the activation of latent transformation growth factor (TGF)-β via αvβ8 integrin [124,125,126]. Activation of TGF-β is an important requirement for non-pathogenic Th17 differentiation. Regulation of the C-type lectin dectin-1-mediated type I IFN response in human DCs allows TGF-β activation and promotes a nonpathogenic Th17 cellular immune response during fungal infection [127]. In mouse models of lung infection with Aspergillus fumigatus or in intestinal infection with Citrobacter rodentium, IL-23 that is specifically produced by cDC2s, can help establish an effective Th17 response [32, 115].
CD4+ T follicular helper (Tfh) cells support the germinal center response and maintain a prolonged humoral immune response [128,129,130]. A reduction in pre-Tfh cells was observed in Notch2−/−, irf4−/− and other cDC2s-specific deletion mouse models; these mice had impaired humoral immunity and reduced germinal center B cells[131, 132]. Therefore, cDC2s are necessary and sufficient for Tfh cell induction. In addition, DCs enhance Tfh cell differentiation via high expression of CD25 (IL-2 receptor alpha chain) and quenching of T cell-derived IL-2, a potent inhibitor of Tfh differentiation[133]. cDC2s and CD14+ macrophages in human tonsils synergistically induce Tfh cell polarization and effector molecule production[134]. Although the absence of DCs results in a slight reduction in Treg numbers, their presence is essential for maintaining the homeostatic proliferation of Tregs [135,136,137]. The dynamic equilibrium between Tregs and DCs is discussed in the next subsection. Activated CD4+ T cells optimize the cellular immune response by enhancing CD8+ T cell clonal expansion and differentiation, and DCs may act as a bridge in this process [138,139,140]. DCs that interact with CD4+ T cells can activate antigen-specific CD8+ T cells, even in the absence of CD4+ T cells after the interaction [141]. In contrast, recognition of cognate antigens on DCs by CD4+ T cells induces the production of CCL3/4, which attracts CD8+ T cells via CCR5 to recognize antigens on CD4+T-DC pairs [142]. The CCR5 ligand also attracts pDCs to the vicinity of CD8+T-DC pairs, with subsequent pDC-derived type I IFN production, further promoting the functions of cDC1s [143]. Takagi et al. determined that pDCs inhibit the induction of CD4+ T cell responses and are involved in the initiation of CD8+ T cell responses under antigenic stimulation and microbial infection [144]. In contrast, antigen-specific CD8+ T cell responses were reduced when pDCs were depleted, suggesting an active role for pDCs in cross-initiating CD8+ T cells [144]. Indeed, under certain conditions, pDCs can transfer antigens to cDC1s by producing pDC-derived exosomes(pDex), which enable cross-priming of CD8+ T cells [145]. Similarly, deficiency in cDC1s results in reduced numbers and function of memory CTL in immunized mice [146].
Altogether, these data suggest that multiple DC subpopulations orchestrate T cell differentiation in vivo and that infection, immunity, and location determine the ultimate polarized fate of T cells. Interestingly, the functional dichotomy between DC1s and DC2s is not invariant and can change in certain inflammatory settings wherein DC2s acquire the ability to activate CD8+ T cells. In a recent study, an IFN-I-induced activation state of novel cDC1s was identified, which, unlike the conventional cross-presentation of cDC1s, can acquire and present intact tumor-derived peptide-MHC I complexes, thereby promoting a specific CD8+ antitumor response [147].
DCs establish central and peripheral tolerance
Several studies have highlighted that DCs can induce negative selection of thymocytes in vivo, rather than positive selection, to maintain central tolerance [148, 149]. Medullary thymic epithelial cells (mTECs) express many tissue-specific antigens to avoid tissue-specific T-cell production. Thymic DCs can either directly present or cross-present autoantigens that are shed from mTECs and kill T cells if the peptide-MHC complex is strong [150]. Even peripheral DCs can migrate into the thymic medulla to induce self-reactive thymocytes and the formation of naturally occurring Tregs [151]. Circulating DCs can be recruited to the thymic medulla with the assistance of p-selectin, integrin VLA-4 interacting with its ligand VCAM-1, and pertussis toxin-sensitive chemoattractant signaling [149]. Husein et al. also demonstrated that peripheral pDCs can transport antigens to the thymus in the absence of TLR signaling in a CCR9-dependent manner to promote the central negative selection of T cells [152]. However, constitutive deletion of DCs did not affect the normal T cell pool of mice, and central tolerance was not disrupted under steady-state conditions, suggesting that DCs may not be critical for the establishment of central tolerance [135, 153].
Although central tolerance is closely coordinated by multiple mechanisms, it has certain dysregulations. For example, some auto-reactive T cells escape negative selection and some harmless antigens can not be expressed in the thymus [150]. To avoid the resulting immune homeostatic dysfunction, the second layer of peripheral tolerance in the organism is crucial. DCs are similar to sentinels that patrol the body and are widely distributed in peripheral tissues including the skin, kidney, lung, and respiratory mucosa, where they exhibit greater self-tolerance [154]. Relative to normal subsets of DCs, tolerogenic DCs (tolDCs) exhibit lower cross-presentation capacity and lower expression of co-stimulatory molecules [155]. A recent study reported that the Tim-3 bridging protein Bat3 acts as an endogenous regulator of tolDC function[156]. Mechanistically, the lack of Bat3 in DCs leads to enhanced steroidogenesis and suppresses Th1, Th17, and CTL cell responses in a paracrine fashion, thereby diminishing autoimmunity [156]. A tolerogenic subpopulation of DCs (DC-10) that stably express CD163, CD141, CD14, and CD16, produces IL-10 in the absence of IL-12 and enables the induction of T regulatory type 1 (Tr1) cell differentiation, was identified in human peripheral blood and spleen [157].
At steady state, tolDCs have a quiescent or semi-mature nature, in which they trap and process antigens to promote T cell anergy and Treg expansion rather than inducing T cell activation [154]. Several mechanisms underlying the induction of peripheral tolerance by DCs have been identified in mice and humans (Fig. 3). First, cytotoxic T lymphocyte-associated antigen 4 (CTLA4) expressed on T cells competitively binds to B7 molecules on DCs, inducing T-cell anergy [158]. Second, many tolDCs express programmed cell death-ligand 1 (PD-L1 and PD-L2), which induces the resting state of T cells upon binding to programmed cell death protein 1 (PD-1) expressed on T cells[159]. In addition to direct contact with T cells, DCs can induce peripheral tolerance through the secretion of cytokines and metabolites including TGF-β, IL-10, IL-27, indoleamine-2,3-dioxygenase (IDO), kynurenine, and retinoic acid[5]. The roles of the regulatory cytokines IL-10 and TGF-β in Treg proliferation and development have been widely demonstrated [160]. TolDCs express the IDO, rate-limiting enzyme that promotes T-cell apoptosis by blocking the cell cycle of T cells [161]. Notably, IDO is not constitutively expressed in DCs but requires induction by multiple mediators (e.g., TGF-β, endotoxin, TNF, IL-1, and IFN-γ) [55, 161, 162]. Moreover, IL-27 suppresses Th1, Th2, and Th17 cell-mediated immune responses and limit the development of central nervous system (CNS) autoimmunity [163, 164]. In addition, IL-27 regulates CD39 on DCs and Tregs and induces Tr1 cells capable of producing IL-10 [165]. The development of acute graft-versus-host disease is exacerbated by impaired Treg suppression and reduced Tr1 numbers in mice with a deficiency of the p28 subunit of IL-27 in DCs [166]. Furthermore, a clinical trial using human autologous tolDCs for kidney transplantation demonstrated that DCs can shape the T cell response to tolerance by producing high levels of lactate, which is a novel mechanism identified for the suppression of T cell immunity by tolDCs [167].

The role of DCs in the induction of immune tolerance and immune response. After activation of DCs by antigens from pathogens or with danger signals, DCs integrate multiple factors to induce T cell polarisation. As an important source of IL-12, DCs promote Th1 cell polarisation. The specific DCs-derived factors that promote Th2 differentiation are unclear, but the transcription factor KLF4 and the STAT6 and p32α signalling pathways play an important role in this pathway. DCs-derived IL-6 is involved in the differentiation of Tfh cells, and in the presence of DCs-derived IL-23 together with TGF-β induces Th17 differentiation [130]. DCs promote the proliferation and function of antigen-specific CTL through antigen cross-presentation, co-stimulatory signalling and derived cytokines such as IFN-I and IL-12 [4]. DCs promote the proliferation and function of antigen-specific CTL through antigen cross-presentation, co-stimulatory signalling and derived cytokines such as IFN-I and IL-12. tolDCs induce immune tolerance in the body by direct or indirect means. PDL1/2 is expressed on most tolDCs, and binding to PD1 on T cells induces T cell apoptosis. In addition, tolDCs secrete a variety of cytokines or metabolites with tolerance activity, such as TGF-β, IL-10, IL-27, Kynurenine, RA and lactate, which tend to promote differentiation of Treg and Breg, creating further suppression of Teff
Similar to Tregs, regulatory B cells (Bregs) are functionally involved in the production of tolerance, highly expression IL-10, TGF-β, and IL-35, and can stimulate T cell anergy as well as regulate invariant natural killer T cell responses [168, 169]. Although Bregs are normally triggered by inflammatory signals, they appear to be induced by DCs during the maintenance of peripheral tolerance [5]. DC-derived IFN-β and CD40 ligands induce the differentiation of mouse spleen B cells into CD19hiFcγIIbhi Bregs, which inhibit the CD4+ T cell response by secreting IL-10 [170]. In a group of EAE mouse models, antigen-specific vitamin D3 (vitD3) tolerogenic DC cell therapy halted disease progression by inducing multiple tolerance mechanisms, including increased Bregs, reduced natural killer (NK) cells, and immunomodulatory NKT activation [171]. These findings show that not only Tregs but also Bregs, play a role in the maintenance of peripheral tolerance by DCs.
Promotion of antitumor immunity by DCs
DCs are capable of initiating antigen-specific T cells to generate robust and durable T cell-driven immune responses; therefore, they are known as key regulators of antitumor responses. The tumor microenvironment (TME) can evolve a variety of immunosuppressive mechanisms that impede the effectiveness of DC maturation or DC-induced immune responses, enabling tumors to thrive under unfavorable conditions [172, 173]. Although most tumors contain only a small number of mature DCs, the presence of these DCs is positively associated with improved survival and prognosis in mouse tumor models as well as patients with cancer [94, 174]. The accumulation of DCs in the TME depends on the local production of growth factors and chemokines that promote the recruitment and expansion of DCs [1]. Subsequently, these DCs transport tumor antigens into draining lymph nodes (DLN) in a CCR7-dependent manner; this transport is critical for the initiation of anti-tumor T cells [175,176,177]. In the absence of CCR7, antitumor T cells in the DLN are no longer efficiently stimulated despite loading of DCs with tumor antigens [176]. NK cells produce XCL1, XCL2, and CCL5 chemokines, which contribute to the recruitment of DCs to the TME; in contrast, FLT3L expressed by NK cells can further promote the differentiation and expansion of DCs [178, 179]. PGE2-mediated tumor immune escape is partly because of the direct inhibition of NK cell production of CCL5 and XCL1 by PGE2, which impairs the localization and accumulation of cDC1s in the TME [178]. Several recent studies have highlighted the potential contribution of microbiota-derived PAMP in counteracting tumor immunity via the activation of DCs [1, 180, 181]. In addition, microbial-derived STING agonists (e.g., c-di-AMP) induce IFN-I production via monocytes in the TME to regulate crosstalk between NK cells and DCs, thereby enhancing the efficacy of immune checkpoint blockade [182]. In turn, the absence of these signals polarizes the differentiation of mononuclear phagocytes to tumor-promoting macrophages [182].
Given that CD8+ T cells are often considered the primary effector of antitumor immunity, promoting effective tumor-associated antigen presentation by DCs has been the focus of cancer immunotherapy. The number and stimulatory phenotype of cDC1s in draining lymph nodes has been reported to decrease with tumor progression, and restoration of the cDC1s axis with Flt3L/anti-CD40 treatment resulted in the expansion of tumor-specific CD8+ T cells and a reduction in tumor burden [183]. Consistent with this, Batf3−/− or Cd207creIrf8fl/fl mice have reduced numbers of tumor-infiltrating CD8+ T cells owing to the lack of cDC1s, thereby promoting the progression of lung adenocarcinoma and fibrosarcoma [21, 184]. The expression of β-linked proteins in cancer cells leads to a reduction in CCL4 concentration in tumors, which prevents CCR5-mediated recruitment of cDC1s and impedes the normal activation of CD8+ T cells [185]. Interestingly, selective deletion of MHC II in cDC1s can prevente early activation of CD4+ T cells by tumor-derived antigen [186]. This suggests that in the presence of tumor-derived antigens, CD40 signaling in cDC1s is critical not only for CD8+ T cell initiation but also for early activation of naïve CD4+ T cells.
In contrast to cDC1s, cDC2s present tumor-associated antigens directly to CD4+ T cells or transfer them to lymphoid tissue-resident DCs [29, 187]. In oropharyngeal squamous cell carcinoma models, cDC2s are required for Th1 cell polarization and the production of high levels of IL-12 and IL-18 [188]. Tumor infiltration by cDC2s is also positively correlated with infiltration by Tbet+ and tumor-specific T cells [188]. In a mouse model of degenerative fibrosarcoma, cDC2s expressing IFN-stimulated genes can acquire and present intact tumor-derived peptide/MHC class I complexes that activate antitumor CD8+ T cell responses [147]. Despite the ability of pDCs to produce large amounts of IFN-α/β in the activated state, their specific role in tumors remains controversial [189]. Activation of pDCs in tumor-draining lymph nodes or the presence of TGF-β results in the constitutive expression of the IDO immunosuppressive enzyme, which induces immune tolerance and facilitates tumor progression [190, 191]. moDCs can act as a source of IL-12 to induce a protective Th1 response mice; however, their ability to produce IL-12 is much lower than that of cDC1s [192]. In contrast, moDCs exert immunosuppressive functions in tumors through the production of nitric oxide, a potential T-cell suppressor molecule [193]. The heterogeneity of moDCs may explain their conflicting roles in tumors. Furthermore, different tumor models with different immune backgrounds may contribute to this discrepancy. In the TME, DCs communicate not only with immune cells but also with cancer cells through multiple pathways. For example, tumor-derived VEGF can inhibit FLT3L-mediated activation of NF-κB and negatively affect the production and function of cDCs in vivo [194]. Thus, different subsets of DCs coordinate the body’s antitumor immunity by interacting with multiple cell types (Fig. 3).
Immunotherapy based on DCs and their signaling pathways
The ability of DCs to induce immune tolerance and adaptive immune responses makes them an optimal candidate for research in cellular immunotherapy. Numerous attempts have been made to exploit the potential of DC-based immunotherapies, from suppressing autoimmune diseases and establishing transplant tolerance to inducing antitumor immunity[195,196,197,198].
The role of DCs in autoimmune diseases and therapeutic perspectives
As previously mentioned, DCs play a double-edged sword role in autoimmune diseases. Prolonged stimulation by autoantigens induces DC activation and maturation, which subsequently initiates self-reactive T and B cells, which disrupts immune tolerance and sustains tissue inflammation [199, 200]. An increase in the maturation phenotype of DCs, such as upregulated expression of co-stimulatory molecules, such as CD86, and increased secretion of pro-inflammatory cytokines, such as IL-12 and IFN-I, have been documented in systemic lupus erythematosus (SLE) patients [201, 202]. Additionally, pDCs isolated from patients with rheumatoid arthritis (RA) secrete higher levels of IFN-α than those from healthy donors [203]. The altered distribution or function of DCs are also part of the pathogenesis of autoimmune diseases [204, 205]. Compared to healthy individuals, patients with autoimmune diseases have a higher distribution of DCs at pathological sites and a lower distribution in the blood [206,207,208]. Specifically, pDC infiltration that is nearly completely absent in healthy tissue has been observed at significantly higher levels in various pathological tissues, such as the skin of patients with SLE, and muscle tissue of patients with idiopathic inflammatory myopathies [207, 209,210,211]. This may be attributed to the increased migration ability of DCs in the pathologic state. For example, an increased CCR7 expression in DCs of patients with SLE promotes enhanced migration of CCR7-dependent DCs to the skin [212, 213]. In addition, elevated myeloid growth factors, including IL-3 and GM-CSF, contribute to the inflammatory pathways and pathology of SLE [214, 215]. Activation of the IL-3- or GM-CSF-induced JAK2-STAT5 signaling pathway is necessary to establish mTORC1 activity, which is required for IFN-I production by pDCs at nephrotic sites in patients with SLE [216].
In EAE, DC signals induce pathogenic T cell development following the interaction between prostaglandin D2 (PGD2) and its receptor PTGDR [217]. Mitogen-activated protein kinase p38α regulates the expression of cytokines and co-stimulatory molecules in DCs and promotes pathogenic Th17 differentiation and progression of EAE by interacting with the IL-23 receptor (IL-23R) [218]. The intrinsic IFN-γ-JAK1-STAT1 signaling pathway in DCs promotes the expression of the co-inhibitory molecule PD-L1 and limits T cell proliferation, which is essential for iTreg production and peripheral tolerance during EAE [219]. Furthermore, a recent study showed that optineurin in DCs inhibits the downstream transcription of IL-10 by suppressing JAK2-STAT3 interactions, and inhibition of STAT3 phosphorylation reportedly facilitates the progression of EAE in mice [220]. A mouse model of psoriasiform dermatitis demonstrated that the production of IL-36 by skin cells drives the pathological IL-23/IL-17/IL-22 axis through DCs to promote disease progression, whereas deletion of CD11c+ DCs inhibits disease progression [221]. IL-17 A downregulates protein phosphatase 6 (PP6) expression in psoriatic keratinocytes, indirectly promoting TLR7-dependent RNA sensing and IL-6 production by DCs, which subsequently drives the hyperproliferation of keratinocytes [222]. Single-cell analysis of lesional and non-lesional skin from patients with psoriasis by Nakamizo et al. revealed a significant enrichment of CD14+ DC3s in damaged skin; these cells are one of the key cell types co-expressing IL-1B and IL-23 A, which are two cytokines critical for psoriasis pathogenesis [223]. In contrast, DCs with immunologic tolerance properties can suppress inflammation by inducing Treg or Breg production and, consequently, inhibit the activation of self-reactive T and B cells [200, 204]. In conclusion, prior data from mouse models and humans suggest that DCs, especially pro-inflammatory DCs, are important in maintaining the activation and polarization of effector T-cells in autoimmune diseases.
Currently, two main types of therapies for autoimmune diseases target DCs (Fig. 4). One approach is systemic immunomodulation by small-molecule drugs or monoclonal antibodies that selectively target a specific function of DCs, including cytokine secretion, antigen processing, antigen presentation, and migratory capacity, or deplete a subpopulation of pro-inflammatory DCs that are critical for the disease pathogenesis [208]. The second approach involves tolDCs with immune tolerance, which are induced in vivo or in vitro. These tolDCs usually exhibit an immature or semi-mature phenotype with low expression of co-stimulatory molecules, reduced secretion of inflammatory cytokines, and increased secretion of anti-inflammatory cytokines [224]. The first drug targeting the co-stimulatory signal between DCs and T cells was the fusion protein CTLA4-Ig, known as abatacept, which has been approved for the treatment of RA with successful results [225, 226]. Several clinical trials targeting pDC for the treatment of SLE have achieved favorable results [227]. VIB7734, a monoclonal antibody targeting the pDC-specific marker immunoglobulin-like transcript 7(ILT7), rapidly and efficiently depletes pDCs in vivo, and the reduction in pDCs in the skin correlates with reduced local IFN-I activity and alleviates clinical disease presentations [228]. B-cell lymphoma 2 (Bcl-2) is critical for pDC survival and IFN-I production in lupus mice or patients with SLE, and the use of the selective Bcl-2 inhibitor venetoclax (ABT-199) significantly reduces auto-reactive B cell and total lymphocyte counts and alleviates disease symptoms in female patients [229, 230]. Inflammatory cytokines secreted by DCs are involved in the hyperactivity of T and B cells as well as tissue damage and are positively associated with disease activity in a range of autoimmune diseases, including type 1 diabetes (T1D), SLE, and RA. Therefore, the inhibition of the action of these cytokines is an important strategy for treating the disease. Tocilizumab blocks the binding of IL-6 to the IL-6 receptor, and its safety and efficacy have been demonstrated in clinical trials of RA, juvenile idiopathic arthritis, and SLE [231, 232]. Etanercept has also been used as an TNF-α blocker in the treatment of many autoimmune diseases, including T1D, psoriasis, and ankylosing spondylitis [233]. In addition, treatment of humanized CCR7 mice with the anti-human CCR7 monoclonal antibody 8H3-16A12 produced favorable prophylactic and therapeutic effects against arthritis [234].

The role of DCs subsets in the tumor microenvironment. The antitumor effects of DCs start with the uptake of TAAs in the tumor microenvironment, followed by the activation and migration of DCs into the DLN. At the same time, tumors interfere with the maturation, recruitment and function of DCs through a series of signals such as secretion of VEGF, PEG2, β-catenin, TGF-β, and upregulation of Tim-3 expression of DCs. DCs migrating into the DLN deliver TAAs to naive T cells via cross-presentation and direct presentation, which promotes differentiation of tumor-specific T helper cells and CTL. There is also an interaction between DCs and NK cells, with IL-12 secreted by DCs promoting the activation and proliferation of NK cells, while NK cells further promote the recruitment and expansion of DCs via XCL1/5, CCL5 and FLT3L. pDCs can produce IFN-I and Granzyme to kill tumor cells, but can also inhibit the killing effect of effector T cells by secreting IDO. pDCs can produce IFN-I and Granzyme to kill tumor cells, but can also inhibit the killing effect of effector T cells by secreting IDO. moDCs exhibit dual effects on CD4+ T cells via NO and IL-12. TAAs. tumor- associated antigens; VEGF, vascular endothelial growth factor
Although immunosuppressive drugs used to treat autoimmune diseases are effective in improving disease outcomes, their use is often accompanied by the risk of chronic infection or cancer development [235]. In this context, tolDCS-based therapy is a promising alternative that specifically abrogates pathological autoimmune or inflammatory responses without compromising protective immune responses against other pathogens and malignancies [236]. Not only DCs themselves, but also various immunomodulatory Dex that are produced by DCs and modified with anti-inflammatory molecules, such as IL-10, IL-4, TGF-β, IDO and CTLA4, have shown promising results for the treatment of immune diseases and chronic inflammation [237,238,239,240,241]. Many small molecules such as dexamethasone, acetylsalicylic acid, minocycline, and vitamin D3, induce tolDC formation [242]. Moreover, dexamethasone and vitamin D3 similarly prevent the maturation of DCs in humans and mice in vitro, leading to a tolerogenic phenotype with downregulation of co-stimulatory molecules and reduced IL-12 production [243, 244]. Rapamycin, IL-10, and GM-CSF have been incorporated into protocols for the in vitro generation of human monocyte-derived tolDCs [5]. Many clinical trials have been performed to assess the feasibility of putative tolDC therapies for the treatment of autoimmune and other inflammatory diseases following the development of these protocols, and to increase understanding of DC-induced self-tolerance (Table 1). In a clinical trial evaluating the safety and tolerability of autologous tolDCs in Crohn’s disease (CD), intraperitoneal administration of tolDCs led to clinical improvement and good treatment tolerability in one-third of the patients [245]. Phase I clinical trials using tolDCs pulsed with proinsulin peptide that were administered intradermally to treat patients with T1D, have also confirmed the antigen-specific immunomodulation of tolDCs as a therapeutic option [246]. In addition, several clinical trials based on tolDCs in multiple sclerosis, RA, and organ transplantation have demonstrated encouraging results [197, 247]. However, significant challenges remain in the large-scale production of protocols for tolDCs with different phenotypes and tolerabilities, as well as in the dose and route of administration for different patients.
DCs in cancer therapy
Over the last two decades, clinical trials of DC-related immunotherapy have focused primarily on tumors, rather than autoimmune diseases, and have shown promising results (Table 1). Therapies targeting DCs focus on enhancing or restoring the function of DCs, increasing their number, or inducing de novo antitumor immunity. The therapies can be broadly categorized into DC vaccines and other DC-related trials (Fig. 5). WNT2 secreted by tumor cells or cancer-associated fibroblasts inhibits DC-mediated antitumor T cell responses via the SOCS3/p-JAK2/p-STAT3 signaling cascade, promoting the malignant progression of solid tumors including colorectal cancer, esophageal squamous cell carcinoma, gastric cancer, and breast cancer [248,249,250,251]. The use of an anti-WNT2 monoclonal antibody significantly restored antitumor T cell responses in mouse tumors by increasing active DCs [250]. Furthermore, the STAT3 signaling pathway is equally involved in the downstream transcription of IL-6, IL-10, and VEGF, which can impair the differentiation of DCs and inhibit the production of IL-12 in human moDCs [220, 252, 253]. Local injection of a STAT3 antisense oligonucleotide (CpG-STAT3ASO) activates human DCs and promotes CD8 + T-cell recruitment to tumor sites [254]. Many inhibitors of STAT3, including BP1-102, STX-0119, and HJC0123, have been identified clinically and shown to have pro-apoptotic and antitumor activities against STAT3-overactivated cancer cells [255].

DCs-based immunotherapy. Current strategies for DCs-based treatment of autoimmune diseases and tumors fall into two broad categories. One is the direct targeting of DCs in vivo. In the treatment of autoimmune diseases, the targets of various DCs (e.g., IL-12, Bal-2, ILT7, CCR7) are usually used to induce immune tolerance or suppress their immune response. In the treatment of tumors, DCs are usually targeted by various DCs (e.g., TLRs, CLRs, STING, CD40) to enhance or restore the function of DCs in order to achieve anti-tumor immunity. Another strategy is vaccination of DCs. Precursors of DCs (or mature cDCs and pDCs) are isolated from patient blood, induced to amplify and differentiate with different cocktails, and DCs are pulsed with relevant antigens to give them antigenic specificity. After a final safety and efficacy assessment, they are injected back to the patient
As knowledge of the DC signaling pathway accumulates, strategies to directly target DCs in vivo to enhance antitumor immunity are being increasingly developed. Activation of TLR on DCs induces their maturation and promotes the production of pro-inflammatory cytokines and chemokines; therefore, TLR agonists are also widely used to activate DCs [40]. The TLR7/8 ligand imiquimod has been used to treat non-melanoma skin cancers because of its ability to promote CCL2-dependent recruitment of pDCs and induce the secretion of tumor-killing TRAIL and granzyme B from pDCs [256]. Similarly, the TLR3-specific agonist ARNAX effectively initiates DCs to induce antitumor immunity without causing systemic inflammation in mice [257]. Activation of CD40 on DCs upregulates the secretion of co-stimulatory molecules and IL-12 [258]. A newly developed CD40 agonist antibody, 2141-V11b, has been shown to activate long-term systemic antitumor immunity in cDC1s-driven CD8+ T cells in orthotopic bladder cancer models and is now in phase I clinical studies [259]. However, the increased potency of Fc-engineered CD40 antibodies in clinical use is accompanied by increased toxicity, such as cytokine release syndrome and macrophage-dependent hepatotoxicity [260,261,262,263]. Therefore, Salomon et al. designed bispecific antibodies that preferentially target CD40 on DCs, increasing their specificity and safety while preserving their effective antitumor activity [264]. In addition, VitE can restore the function of DCs in TME by inhibiting intrinsic checkpoint protein tyrosine phosphatase SHP1 and enhancing tumor antigen cross-presentation by DCs and Dex [265]. Eliminating the ability of DCs to induce tolerance in the TME is another strategy for enhancing the antitumor immunity of DCs. In this regard, four IDO inhibitors are in clinical development, and preliminary findings indicate their antitumor activity [266]. Similarly, the anti-TIM-3 antibody upregulates CXCL9 on DCs and promotes cDC1-mediated antitumor immunity, which has been successfully combined with PD-1/PD-L1 blockade to inhibit tumor growth [267, 268].
DC vaccines aim to provide the immune system with in vitro-trained DCs, which are usually isolated from the peripheral blood of patients, properly activated, and loaded with tumor antigens or tumor lysates, to induce a strong antitumor response [196]. Treatment with personalized neoantigen peptide-pulsed autologous DC vaccine (Neo-DCVac) reportedly promotes the secretion of cytokines, such as IL-12, IFN-γ, and TNF-α in patients with metastatic lung cancer, with a disease control rate of 75% [269]. In a phase 2 study, the use of a DC vaccine electroporated with Wilms’ tumor 1 (WT1) mRNA in patients with acute myeloid leukemia was shown to be a safe and feasible regimen in promoting tumor infiltration of multi-epitope WT1-specific CD8+ T cells and effectively prevent or delay cancer recurrence after standard chemotherapy [270]. Owing to the rarity of cDCs or pre-DCs in the blood, moDCs are currently used in most clinical studies. The addition of autologous tumor lysate moDC vaccine (DCVax-L) to standard therapy in patients with glioblastoma (GBM) prolongs patient survival and has a favorable safety profile, with only 2.1% of patients experiencing treatment-related grade 3 or 4 adverse events [271]. However, endogenous DCs are required for the initiation of tumor-specific T cells after moDC vaccination [15]. Owing to their excellent antigen-presenting ability and high specificity, cDCs are considered promising antitumor vaccine candidates [272, 273]. HER2 peptide-pulsed cDC1 vaccines induce specific anti-HER2 CD4+ and CD8+ immune responses in the treatment of patients with HER2pos tumors, independent of the vaccination route [274]. It is important to note that cDC1s vaccines can directly engage in antigen presentation to drive tumor-specific CD8+ T cell responses in mice independent of host cDC1s [275].
Conclusion and outlook
Undoubtedly, the advances made in DCs research in recent years have led to increased knowledge of many key transcription factors and cytokines that are involved in the regulation of DCs differentiation and function. To capitalize on these discoveries, a growing number of clinical trials based on DCs therapies are being initiated. Some trials have shown promising safety and efficacy in the treatment of autoimmune diseases and cancers. However, considerable challenges remain in establishing effective and applicable DC vaccines. One challenge is ensuring that DC vaccines avoid the effects of immunosuppressive factors in TME, including IDO, IL-10, TGF-β, and PEG2. Moreover, most DC vaccines do not effectively migrate to the lymph nodes to mobilize the T cell immune response. Moreover, DC-based immunotherapies are often expensive and require complex manufacturing processes, which limit access to a wide range of patients.
The primary focus of research on DCs is to identify the precise conditions which determine whether DCs exert their immunosuppressive or immune-activating functions. The diversity of different DCs subpopulations in terms of origin, location, phenotype, and function makes this challenging. Future research should explore the biological properties of DCs in controlling T cell immunity to maximize their therapeutic potential. In addition, continuous advancement in technology has the potential to revolutionize the field of DC-based immunotherapy. The use of nanotechnology may enable the more effective delivery of DC-targeted drugs or vaccines. Advancements in gene editing technologies like CRISPR/CAS9 may enable the development of more effective and specific DC-based therapies. Overall, continued research in the field of DC biology and immunotherapy has the potential to revolutionize the treatment of a wide range of diseases and offers hope to patients with a wide range of disorders.
Data Availability
Not applicable.
References
Marciscano AE, Anandasabapathy N. The role of dendritic cells in cancer and anti-tumor immunity. Semin Immunol. 2021;52:101481. https://doi.org/10.1016/j.smim.2021.101481.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–77. https://doi.org/10.1038/nrc3258.
Verneau J, Sautes-Fridman C, Sun CM. Dendritic cells in the tumor microenvironment: prognostic and theranostic impact. Semin Immunol. 2020;48:101410. https://doi.org/10.1016/j.smim.2020.101410.
Hilligan KL, Ronchese F. Antigen presentation by dendritic cells and their instruction of CD4 + T helper cell responses. Cell Mol Immunol. 2020;17(6):587–99. https://doi.org/10.1038/s41423-020-0465-0.
Morante-Palacios O, Fondelli F, Ballestar E, Martinez-Caceres EM. Tolerogenic dendritic cells in autoimmunity and inflammatory Diseases. Trends Immunol. 2021;42(1):59–75. https://doi.org/10.1016/j.it.2020.11.001.
Fu C, Jiang A. Dendritic cells and CD8 T cell immunity in Tumor Microenvironment. Front Immunol. 2018;9:3059. https://doi.org/10.3389/fimmu.2018.03059.
Giles DA, Duncker PC, Wilkinson NM, Washnock-Schmid JM, Segal BM. CNS-resident classical DCs play a critical role in CNS autoimmune disease. J Clin Invest. 2018;128(12):5322–34. https://doi.org/10.1172/JCI123708.
Whartenby KA, Calabresi PA, McCadden E, Nguyen B, Kardian D, Wang T, et al. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc Natl Acad Sci U S A. 2005;102(46):16741–6. https://doi.org/10.1073/pnas.0506088102.
Gardner A, Ruffell B. Dendritic cells and Cancer Immunity. Trends Immunol. 2016;37(12):855–65. https://doi.org/10.1016/j.it.2016.09.006.
Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. morphology, quantitation, tissue distribution. J Exp Med. 1973;137(5):1142–62. https://doi.org/10.1084/jem.137.5.1142.
Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M. Reis e Sousa C. Dendritic cells revisited. Annu Rev Immunol. 2021;39:131–66. https://doi.org/10.1146/annurev-immunol-061020-053707.
Paul WE. Bridging innate and adaptive immunity. Cell. 2011;147(6):1212–5. https://doi.org/10.1016/j.cell.2011.11.036.
Leon B, Ardavin C. Monocyte-derived dendritic cells in innate and adaptive immunity. Immunol Cell Biol. 2008;86(4):320–4. https://doi.org/10.1038/icb.2008.14.
Schlitzer A, McGovern N, Ginhoux F. Dendritic cells and monocyte-derived cells: two complementary and integrated functional systems. Semin Cell Dev Biol. 2015;41:9–22. https://doi.org/10.1016/j.semcdb.2015.03.011.
Gardner A, de Mingo Pulido A, Ruffell B. Dendritic cells and their role in Immunotherapy. Front Immunol. 2020;11:924. https://doi.org/10.3389/fimmu.2020.00924.
Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015;15(8):471–85. https://doi.org/10.1038/nri3865.
Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med. 2012;209(6):1153–65. https://doi.org/10.1084/jem.20112675.
Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. https://doi.org/10.1146/annurev-immunol-020711-074950.
Gutierrez-Martinez E, Planes R, Anselmi G, Reynolds M, Menezes S, Adiko AC, et al. Cross-Presentation of Cell-Associated Antigens by MHC class I in dendritic cell subsets. Front Immunol. 2015;6:363. https://doi.org/10.3389/fimmu.2015.00363.
Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324(5925):392–7. https://doi.org/10.1126/science.1170540.
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha + dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–100. https://doi.org/10.1126/science.1164206.
Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, et al. IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells. Respectively Immun. 2016;45(3):626–40. https://doi.org/10.1016/j.immuni.2016.08.013.
Bachem A, Hartung E, Guttler S, Mora A, Zhou X, Hegemann A, et al. Expression of XCR1 characterizes the Batf3-Dependent lineage of dendritic cells capable of Antigen Cross-Presentation. Front Immunol. 2012;3:214. https://doi.org/10.3389/fimmu.2012.00214.
Seillet C, Jackson JT, Markey KA, Brady HJ, Hill GR, Macdonald KP et al. CD8alpha + DCs can be induced in the absence of transcription factors Id2, Nfil3, and Batf3. Blood 2013;121(9):1574–1583. doi:https://doi.org/10.1182/blood-2012-07-445650.
Tussiwand R, Lee WL, Murphy TL, Mashayekhi M, Kc W, Albring JC, et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature. 2012;490(7421):502–7. https://doi.org/10.1038/nature11531.
Bajana S, Turner S, Paul J, Ainsua-Enrich E, Kovats S. IRF4 and IRF8 act in CD11c + cells to regulate terminal differentiation of lung tissue dendritic cells. J Immunol. 2016;196(4):1666–77. https://doi.org/10.4049/jimmunol.1501870.
Durai V, Murphy KM. Functions of murine dendritic cells. Immunity. 2016;45(4):719–36. https://doi.org/10.1016/j.immuni.2016.10.010.
Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity. 2015;42(5):916–28. https://doi.org/10.1016/j.immuni.2015.04.017.
Saito Y, Komori S, Kotani T, Murata Y, Matozaki T. The role of Type-2 conventional dendritic cells in the regulation of Tumor Immunity. Cancers (Basel). 2022;14(8). https://doi.org/10.3390/cancers14081976.
Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity. 2011;35(5):780–91. https://doi.org/10.1016/j.immuni.2011.08.013.
Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallee VP, Mendoza A, et al. Transcriptional basis of mouse and human dendritic cell heterogeneity. Cell. 2019;179(4):846–863e824. https://doi.org/10.1016/j.cell.2019.09.035.
Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14(9):937–48. https://doi.org/10.1038/ni.2679.
Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, Murphy KM, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood. 2012;119(25):6052–62. https://doi.org/10.1182/blood-2012-01-406967.
Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365(2):127–38. https://doi.org/10.1056/NEJMoa1100066.
Collin M, Ginhoux F. Human dendritic cells. Semin Cell Dev Biol. 2019;86:1–2. https://doi.org/10.1016/j.semcdb.2018.04.015.
Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356(6335). https://doi.org/10.1126/science.aah4573.
Zhan Y, Chow KV, Soo P, Xu Z, Brady JL, Lawlor KE, et al. Plasmacytoid dendritic cells are short-lived: reappraising the influence of migration, genetic factors and activation on estimation of lifespan. Sci Rep. 2016;6:25060. https://doi.org/10.1038/srep25060.
Onai N, Kurabayashi K, Hosoi-Amaike M, Toyama-Sorimachi N, Matsushima K, Inaba K, et al. A clonogenic progenitor with prominent plasmacytoid dendritic cell developmental potential. Immunity. 2013;38(5):943–57. https://doi.org/10.1016/j.immuni.2013.04.006.
Lui G, Manches O, Angel J, Molens JP, Chaperot L, Plumas J. Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PLoS ONE. 2009;4(9):e7111. https://doi.org/10.1371/journal.pone.0007111.
Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27(1):74–95. https://doi.org/10.1038/cr.2016.157.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11(5):373–84. https://doi.org/10.1038/ni.1863.
Schmid MA, Kingston D, Boddupalli S, Manz MG. Instructive cytokine signals in dendritic cell lineage commitment. Immunol Rev. 2010;234(1):32–44. https://doi.org/10.1111/j.0105-2896.2009.00877.x.
Sathaliyawala T, O’Gorman WE, Greter M, Bogunovic M, Konjufca V, Hou ZE, et al. Mammalian target of rapamycin controls dendritic cell development downstream of Flt3 ligand signaling. Immunity. 2010;33(4):597–606. https://doi.org/10.1016/j.immuni.2010.09.012.
Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135(1):37–48. https://doi.org/10.1016/j.cell.2008.09.016.
Scott CL, Soen B, Martens L, Skrypek N, Saelens W, Taminau J, et al. The transcription factor Zeb2 regulates development of conventional and plasmacytoid DCs by repressing Id2. J Exp Med. 2016;213(6):897–911. https://doi.org/10.1084/jem.20151715.
Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol. 2003;4(4):380–6. https://doi.org/10.1038/ni903.
Reizis B. Plasmacytoid dendritic cells: Development, Regulation, and function. Immunity. 2019;50(1):37–50. https://doi.org/10.1016/j.immuni.2018.12.027.
Chow KV, Sutherland RM, Zhan Y, Lew AM. Heterogeneity, functional specialization and differentiation of monocyte-derived dendritic cells. Immunol Cell Biol. 2017;95(3):244–51. https://doi.org/10.1038/icb.2016.104.
Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity. 2020;52(6):1039–1056e1039. https://doi.org/10.1016/j.immuni.2020.04.005.
Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcgamma receptors in dendritic cells and macrophages. Nat Rev Immunol. 2014;14(2):94–108. https://doi.org/10.1038/nri3582.
Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6(3):498–510. https://doi.org/10.1038/mi.2012.89.
Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37(6):1076–90. https://doi.org/10.1016/j.immuni.2012.08.026.
Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, et al. The heterogeneity of Ly6C(hi) Monocytes Controls their differentiation into iNOS(+) macrophages or monocyte-derived dendritic cells. Immunity. 2016;45(6):1205–18. https://doi.org/10.1016/j.immuni.2016.12.001.
Goudot C, Coillard A, Villani AC, Gueguen P, Cros A, Sarkizova S, et al. Aryl Hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages. Immunity. 2017;47(3):582–596e586. https://doi.org/10.1016/j.immuni.2017.08.016.
Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6(6):476–83. https://doi.org/10.1038/nri1845.
Tiberio L, Del Prete A, Schioppa T, Sozio F, Bosisio D, Sozzani S. Chemokine and chemotactic signals in dendritic cell migration. Cell Mol Immunol. 2018;15(4):346–52. https://doi.org/10.1038/s41423-018-0005-3.
Kotsias F, Cebrian I, Alloatti A. Antigen processing and presentation. Int Rev Cell Mol Biol. 2019;348:69–121. https://doi.org/10.1016/bs.ircmb.2019.07.005.
Heaton SM, Borg NA, Dixit VM. Ubiquitin in the activation and attenuation of innate antiviral immunity. J Exp Med. 2016;213(1):1–13. https://doi.org/10.1084/jem.20151531.
Plato A, Hardison SE, Brown GD. Pattern recognition receptors in antifungal immunity. Semin Immunopathol. 2015;37(2):97–106. https://doi.org/10.1007/s00281-014-0462-4.
Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433(7028):887–92. https://doi.org/10.1038/nature03326.
Davey GM, Wojtasiak M, Proietto AI, Carbone FR, Heath WR, Bedoui S. Cutting edge: priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J Immunol. 2010;184(5):2243–6. https://doi.org/10.4049/jimmunol.0903013.
Arpaia N, Barton GM. Toll-like receptors: key players in antiviral immunity. Curr Opin Virol. 2011;1(6):447–54. https://doi.org/10.1016/j.coviro.2011.10.006.
Ahad A, Smita S, Mishra GP, Biswas VK, Sen K, Gupta B, et al. NCoR1 fine-tunes type-I IFN response in cDC1 dendritic cells by directly regulating Myd88-IRF7 axis under TLR9. Eur J Immunol. 2020;50(12):1959–75. https://doi.org/10.1002/eji.202048566.
Hemont C, Neel A, Heslan M, Braudeau C, Josien R. Human blood mDC subsets exhibit distinct TLR repertoire and responsiveness. J Leukoc Biol. 2013;93(4):599–609. https://doi.org/10.1189/jlb.0912452.
van der Aar AM, de Groot R, Sanchez-Hernandez M, Taanman EW, van Lier RA, Teunissen MB, et al. Cutting edge: virus selectively primes human langerhans cells for CD70 expression promoting CD8 + T cell responses. J Immunol. 2011;187(7):3488–92. https://doi.org/10.4049/jimmunol.1101105.
Flores-Langarica A, Cook C, Muller Luda K, Persson EK, Marshall JL, Beristain-Covarrubias N, et al. Intestinal CD103(+)CD11b(+) cDC2 conventional dendritic cells are required for primary CD4(+) T and B cell responses to Soluble Flagellin. Front Immunol. 2018;9:2409. https://doi.org/10.3389/fimmu.2018.02409.
Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32(2):279–89. https://doi.org/10.1016/j.immuni.2010.01.013.
Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434(7036):1035–40. https://doi.org/10.1038/nature03547.
van der Sluis RM, Cham LB, Gris-Oliver A, Gammelgaard KR, Pedersen JG, Idorn M, et al. TLR2 and TLR7 mediate distinct immunopathological and antiviral plasmacytoid dendritic cell responses to SARS-CoV-2 infection. EMBO J. 2022;41(10):e109622. https://doi.org/10.15252/embj.2021109622.
Agod Z, Fekete T, Budai MM, Varga A, Szabo A, Moon H, et al. Regulation of type I interferon responses by mitochondria-derived reactive oxygen species in plasmacytoid dendritic cells. Redox Biol. 2017;13:633–45. https://doi.org/10.1016/j.redox.2017.07.016.
Fekete T, Bencze D, Szabo A, Csoma E, Biro T, Bacsi A, et al. Regulatory NLRs Control the RLR-Mediated type I Interferon and inflammatory responses in human dendritic cells. Front Immunol. 2018;9:2314. https://doi.org/10.3389/fimmu.2018.02314.
Loo YM, Gale M. Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011;34(5):680–92. https://doi.org/10.1016/j.immuni.2011.05.003.
Thoresen D, Wang W, Galls D, Guo R, Xu L, Pyle AM. The molecular mechanism of RIG-I activation and signaling. Immunol Rev. 2021;304(1):154–68. https://doi.org/10.1111/imr.13022.
Wang Y, Xiang Y, Xin VW, Wang X-W, Peng X-C, Liu X-Q, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13(1). https://doi.org/10.1186/s13045-020-00939-6.
Tang J, Lin G, Langdon WY, Tao L, Zhang J. Regulation of C-Type lectin receptor-mediated antifungal immunity. Front Immunol. 2018;9:123. https://doi.org/10.3389/fimmu.2018.00123.
Liu CH, Liu H, Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol. 2017;14(12):963–75. https://doi.org/10.1038/cmi.2017.88.
Iborra S, Izquierdo HM, Martinez-Lopez M, Blanco-Menendez N, Reis e Sousa C, Sancho D. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. J Clin Invest. 2012;122(5):1628–43. https://doi.org/10.1172/JCI60660.
Iborra S, Martinez-Lopez M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, et al. Optimal generation of Tissue-Resident but not circulating memory T cells during viral infection requires crosspriming by DNGR-1(+) dendritic cells. Immunity. 2016;45(4):847–60. https://doi.org/10.1016/j.immuni.2016.08.019.
Chao PZ, Hsieh MS, Cheng CW, Hsu TJ, Lin YT, Lai CH, Oncotarget et al. 2015;6(1):159–170. doi:https://doi.org/10.18632/oncotarget.2700.
Oviedo-Boyso J, Bravo-Patiño A, Baizabal-Aguirre VM. Collaborative action of Toll-Like and Nod-Like receptors as modulators of the inflammatory response to pathogenic Bacteria. Mediat Inflamm. 2014;2014:1–16. https://doi.org/10.1155/2014/432785.
Kawai T, Akira S. Toll-like receptors and their crosstalk with other Innate receptors in infection and immunity. Immunity. 2011;34(5):637–50. https://doi.org/10.1016/j.immuni.2011.05.006.
Kim DJ, Park JH, Franchi L, Backert S, Nunez G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1beta production in Helicobacter pylori infected dendritic cells. Eur J Immunol. 2013;43(10):2650–8. https://doi.org/10.1002/eji.201243281.
Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12(8):557–69. https://doi.org/10.1038/nri3254.
Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26(1):79–92. https://doi.org/10.1016/j.immuni.2006.10.018.
Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108(42):17396–401. https://doi.org/10.1073/pnas.1113421108.
Keller CW, Sina C, Kotur MB, Ramelli G, Mundt S, Quast I, et al. ATG-dependent phagocytosis in dendritic cells drives myelin-specific CD4(+) T cell pathogenicity during CNS inflammation. Proc Natl Acad Sci U S A. 2017;114(52):E11228–37. https://doi.org/10.1073/pnas.1713664114.
Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307(5715):1630–4. https://doi.org/10.1126/science.1108003.
Oberkampf M, Guillerey C, Mouries J, Rosenbaum P, Fayolle C, Bobard A, et al. Mitochondrial reactive oxygen species regulate the induction of CD8(+) T cells by plasmacytoid dendritic cells. Nat Commun. 2018;9(1):2241. https://doi.org/10.1038/s41467-018-04686-8.
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–7. https://doi.org/10.1126/science.1174447.
Samie M, Cresswell P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nat Immunol. 2015;16(7):729–36. https://doi.org/10.1038/ni.3196.
Wang L, Gao T, Li Y, Xie Y, Zeng S, Tai C, et al. A long-term anti-inflammation markedly alleviated high-fat diet-induced obesity by repeated administrations of overexpressing IL10 human umbilical cord-derived mesenchymal stromal cells. Stem Cell Res Ther. 2022;13(1):259. https://doi.org/10.1186/s13287-022-02935-8.
Giampazolias E, Schulz O, Lim KHJ, Rogers NC, Chakravarty P, Srinivasan N, et al. Secreted gelsolin inhibits DNGR-1-dependent cross-presentation and cancer immunity. Cell. 2021;184(15):4016–4031e4022. https://doi.org/10.1016/j.cell.2021.05.021.
Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. 2018;24(8):1178–91. https://doi.org/10.1038/s41591-018-0085-8.
Michea P, Noel F, Zakine E, Czerwinska U, Sirven P, Abouzid O, et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol. 2018;19(8):885–97. https://doi.org/10.1038/s41590-018-0145-8.
Theisen DJ, Davidson JTt, Briseno CG, Gargaro M, Lauron EJ, Wang Q, et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science. 2018;362(6415):694–9. https://doi.org/10.1126/science.aat5030.
Alloatti A, Kotsias F, Pauwels AM, Carpier JM, Jouve M, Timmerman E, et al. Toll-like receptor 4 Engagement on dendritic cells restrains Phago-Lysosome Fusion and promotes Cross-Presentation of Antigens. Immunity. 2015;43(6):1087–100. https://doi.org/10.1016/j.immuni.2015.11.006.
Hoeffel G, Ripoche AC, Matheoud D, Nascimbeni M, Escriou N, Lebon P, et al. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity. 2007;27(3):481–92. https://doi.org/10.1016/j.immuni.2007.07.021.
Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity. 2008;29(3):352–61. https://doi.org/10.1016/j.immuni.2008.09.002.
Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, et al. Distinct Transcriptional Programs Control Cross-Priming in classical and monocyte-derived dendritic cells. Cell Rep. 2016;15(11):2462–74. https://doi.org/10.1016/j.celrep.2016.05.025.
Pitt JM, Andre F, Amigorena S, Soria JC, Eggermont A, Kroemer G, et al. Dendritic cell-derived exosomes for cancer therapy. J Clin Invest. 2016;126(4):1224–32. https://doi.org/10.1172/JCI81137.
Lu Z, Zuo B, Jing R, Gao X, Rao Q, Liu Z, et al. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J Hepatol. 2017;67(4):739–48. https://doi.org/10.1016/j.jhep.2017.05.019.
Yao Y, Fu C, Zhou L, Mi QS, Jiang A. DC-Derived Exosomes for Cancer Immunotherapy. Cancers (Basel). 2021;13(15). https://doi.org/10.3390/cancers13153667.
Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14(3):195–208. https://doi.org/10.1038/nri3622.
Samuel M, Gabrielsson S. Personalized medicine and back-allogeneic exosomes for cancer immunotherapy. J Intern Med. 2021;289(2):138–46. https://doi.org/10.1111/joim.12963.
Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2002;99(1):351–8. https://doi.org/10.1073/pnas.231606698.
Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. https://doi.org/10.1146/annurev.immunol.21.120601.141040.
Scheinecker C, McHugh R, Shevach EM, Germain RN. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J Exp Med. 2002;196(8):1079–90. https://doi.org/10.1084/jem.20020991.
Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol. 2014;15(2):161–7. https://doi.org/10.1038/ni.2795.
Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, et al. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells. Eur J Immunol. 1996;26(3):659–68. https://doi.org/10.1002/eji.1830260323.
Martinez-Lopez M, Iborra S, Conde-Garrosa R, Sancho D. Batf3-dependent CD103 + dendritic cells are major producers of IL-12 that drive local Th1 immunity against Leishmania major infection in mice. Eur J Immunol. 2015;45(1):119–29. https://doi.org/10.1002/eji.201444651.
Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. 2011;35(2):249–59. https://doi.org/10.1016/j.immuni.2011.08.008.
Luda KM, Joeris T, Persson EK, Rivollier A, Demiri M, Sitnik KM, et al. IRF8 transcription-factor-dependent classical dendritic cells are essential for intestinal T cell homeostasis. Immunity. 2016;44(4):860–74. https://doi.org/10.1016/j.immuni.2016.02.008.
Mattiuz R, Merad M. Innate IL-13 licenses dermal type 2 dendritic cells for efficient T helper 2 cell responses. Nat Rev Immunol. 2021;21(5):275. https://doi.org/10.1038/s41577-021-00552-9.
Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A. CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity. 2013;39(4):733–43. https://doi.org/10.1016/j.immuni.2013.08.029.
Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b + dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38(5):970–83. https://doi.org/10.1016/j.immuni.2013.04.011.
Izumi G, Nakano H, Nakano K, Whitehead GS, Grimm SA, Fessler MB, et al. CD11b(+) lung dendritic cells at different stages of maturation induce Th17 or Th2 differentiation. Nat Commun. 2021;12(1):5029. https://doi.org/10.1038/s41467-021-25307-x.
Mayer JU, Hilligan KL, Chandler JS, Eccles DA, Old SI, Domingues RG, et al. Homeostatic IL-13 in healthy skin directs dendritic cell differentiation to promote T(H)2 and inhibit T(H)17 cell polarization. Nat Immunol. 2021;22(12):1538–50. https://doi.org/10.1038/s41590-021-01067-0.
Klaver D, Posch B, Geisler A, Hermann M, Reider N, Heufler C. Peptides from allergenic lipocalins bind to formyl peptide receptor 3 in human dendritic cells to mediate T(H)2 immunity. J Allergy Clin Immunol. 2020;145(2):654–65. https://doi.org/10.1016/j.jaci.2019.07.008.
Han M, Ma J, Ouyang S, Wang Y, Zheng T, Lu P, et al. The kinase p38alpha functions in dendritic cells to regulate Th2-cell differentiation and allergic inflammation. Cell Mol Immunol. 2022;19(7):805–19. https://doi.org/10.1038/s41423-022-00873-2.
Mansouri S, Patel S, Katikaneni DS, Blaauboer SM, Wang W, Schattgen S, et al. Immature lung TNFR2(-) conventional DC 2 subpopulation activates moDCs to promote cyclic di-GMP mucosal adjuvant responses in vivo. Mucosal Immunol. 2019;12(1):277–89. https://doi.org/10.1038/s41385-018-0098-0.
McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of Cytokines in Health and Disease. Immunity. 2019;50(4):892–906. https://doi.org/10.1016/j.immuni.2019.03.021.
Zhang D, Jin W, Wu R, Li J, Park SA, Tu E, et al. High glucose intake exacerbates autoimmunity through reactive-oxygen-species-mediated TGF-beta cytokine activation. Immunity. 2019;51(4):671–681e675. https://doi.org/10.1016/j.immuni.2019.08.001.
Zhang M, Zhou L, Xu Y, Yang M, Xu Y, Komaniecki GP, et al. A STAT3 palmitoylation cycle promotes T(H)17 differentiation and colitis. Nature. 2020;586(7829):434–9. https://doi.org/10.1038/s41586-020-2799-2.
Linehan JL, Dileepan T, Kashem SW, Kaplan DH, Cleary P, Jenkins MK. Generation of Th17 cells in response to intranasal infection requires TGF-beta1 from dendritic cells and IL-6 from CD301b + dendritic cells. Proc Natl Acad Sci U S A. 2015;112(41):12782–7. https://doi.org/10.1073/pnas.1513532112.
Kim TG, Kim SH, Park J, Choi W, Sohn M, Na HY, et al. Skin-specific CD301b(+) dermal dendritic cells drive IL-17-Mediated Psoriasis-Like Immune response in mice. J Invest Dermatol. 2018;138(4):844–53. https://doi.org/10.1016/j.jid.2017.11.003.
Liu H, Chen F, Wu W, Cao AT, Xue X, Yao S, et al. TLR5 mediates CD172alpha(+) intestinal lamina propria dendritic cell induction of Th17 cells. Sci Rep. 2016;6:22040. https://doi.org/10.1038/srep22040.
Gringhuis SI, Kaptein TM, Remmerswaal EBM, Drewniak A, Wevers BA, Theelen B, et al. Fungal sensing by dectin-1 directs the non-pathogenic polarization of T(H)17 cells through balanced type I IFN responses in human DCs. Nat Immunol. 2022. https://doi.org/10.1038/s41590-022-01348-2.
Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–42. https://doi.org/10.1016/j.immuni.2014.10.004.
Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol. 2016;34:335–68. https://doi.org/10.1146/annurev-immunol-041015-055605.
Chakarov S, Fazilleau N. Monocyte-derived dendritic cells promote T follicular helper cell differentiation. EMBO Mol Med. 2014;6(5):590–603. https://doi.org/10.1002/emmm.201403841.
Briseno CG, Satpathy AT, Davidson JTt, Ferris ST, Durai V, Bagadia P, et al. Notch2-dependent DC2s mediate splenic germinal center responses. Proc Natl Acad Sci U S A. 2018;115(42):10726–31. https://doi.org/10.1073/pnas.1809925115.
Krishnaswamy JK, Gowthaman U, Zhang B, Mattsson J, Szeponik L, Liu D, et al. Migratory CD11b(+) conventional dendritic cells induce T follicular helper cell-dependent antibody responses. Sci Immunol. 2017;2(18). https://doi.org/10.1126/sciimmunol.aam9169.
Li J, Lu E, Yi T, Cyster JG. EBI2 augments tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature. 2016;533(7601):110–4. https://doi.org/10.1038/nature17947.
Durand M, Walter T, Pirnay T, Naessens T, Gueguen P, Goudot C, et al. Human lymphoid organ cDC2 and macrophages play complementary roles in T follicular helper responses. J Exp Med. 2019;216(7):1561–81. https://doi.org/10.1084/jem.20181994.
Birnberg T, Bar-On L, Sapoznikov A, Caton ML, Cervantes-Barragan L, Makia D, et al. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity. 2008;29(6):986–97. https://doi.org/10.1016/j.immuni.2008.10.012.
Suffner J, Hochweller K, Kuhnle MC, Li X, Kroczek RA, Garbi N, et al. Dendritic cells support homeostatic expansion of Foxp3 + regulatory T cells in Foxp3.LuciDTR mice. J Immunol. 2010;184(4):1810–20. https://doi.org/10.4049/jimmunol.0902420.
Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. 2009;206(9):1853–62. https://doi.org/10.1084/jem.20090746.
Castellino F, Germain RN. Cooperation between CD4 + and CD8 + T cells: when, where, and how. Annu Rev Immunol. 2006;24:519–40. https://doi.org/10.1146/annurev.immunol.23.021704.115825.
Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4 + and CD8 + T cell activation to localized viral infection. Immunity. 2015;43(3):554–65. https://doi.org/10.1016/j.immuni.2015.07.020.
Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. 2019;19(2):89–103. https://doi.org/10.1038/s41577-018-0088-1.
Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4 + T-helper and a T-killer cell. Nature. 1998;393(6684):474–8. https://doi.org/10.1038/30989.
Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8 + T cells to sites of CD4 + T cell-dendritic cell interaction. Nature. 2006;440(7086):890–5. https://doi.org/10.1038/nature04651.
Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, et al. CD8(+) T cells orchestrate pDC-XCR1(+) dendritic cell spatial and functional cooperativity to optimize priming. Immunity. 2017;46(2):205–19. https://doi.org/10.1016/j.immuni.2017.01.003.
Takagi H, Fukaya T, Eizumi K, Sato Y, Sato K, Shibazaki A, et al. Plasmacytoid dendritic cells are crucial for the initiation of inflammation and T cell immunity in vivo. Immunity. 2011;35(6):958–71. https://doi.org/10.1016/j.immuni.2011.10.014.
Fu C, Peng P, Loschko J, Feng L, Pham P, Cui W, et al. Plasmacytoid dendritic cells cross-prime naive CD8 T cells by transferring antigen to conventional dendritic cells through exosomes. Proc Natl Acad Sci U S A. 2020;117(38):23730–41. https://doi.org/10.1073/pnas.2002345117.
Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, et al. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell. 2015;162(6):1322–37. https://doi.org/10.1016/j.cell.2015.08.004.
Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, et al. Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity. 2022;55(2):308–323e309. https://doi.org/10.1016/j.immuni.2021.10.020.
Brocker T, Riedinger M, Karjalainen K. Targeted expression of major histocompatibility complex (MHC) class II molecules demonstrates that dendritic cells can induce negative but not positive selection of thymocytes in vivo. J Exp Med. 1997;185(3):541–50. https://doi.org/10.1084/jem.185.3.541.
Bonasio R, Scimone ML, Schaerli P, Grabie N, Lichtman AH, von Andrian UH. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat Immunol. 2006;7(10):1092–100. https://doi.org/10.1038/ni1385.
Waisman A, Lukas D, Clausen BE, Yogev N. Dendritic cells as gatekeepers of tolerance. Semin Immunopathol. 2017;39(2):153–63. https://doi.org/10.1007/s00281-016-0583-z.
Proietto AI, van Dommelen S, Zhou P, Rizzitelli A, D’Amico A, Steptoe RJ, et al. Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc Natl Acad Sci U S A. 2008;105(50):19869–74. https://doi.org/10.1073/pnas.0810268105.
Hadeiba H, Lahl K, Edalati A, Oderup C, Habtezion A, Pachynski R, et al. Plasmacytoid dendritic cells transport peripheral antigens to the thymus to promote central tolerance. Immunity. 2012;36(3):438–50. https://doi.org/10.1016/j.immuni.2012.01.017.
Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity. 2012;37(2):264–75. https://doi.org/10.1016/j.immuni.2012.05.025.
Devi KS, Anandasabapathy N. The origin of DCs and capacity for immunologic tolerance in central and peripheral tissues. Semin Immunopathol. 2017;39(2):137–52. https://doi.org/10.1007/s00281-016-0602-0.
Iberg CA, Jones A, Hawiger D. Dendritic cells as inducers of Peripheral Tolerance. Trends Immunol. 2017;38(11):793–804. https://doi.org/10.1016/j.it.2017.07.007.
Tang R, Acharya N, Subramanian A, Purohit V, Tabaka M, Hou Y, et al. Tim-3 adapter protein Bat3 acts as an endogenous regulator of tolerogenic dendritic cell function. Sci Immunol. 2022;7(69):eabm0631. https://doi.org/10.1126/sciimmunol.abm0631.
Comi M, Avancini D, Santoni de Sio F, Villa M, Uyeda MJ, Floris M, et al. Coexpression of CD163 and CD141 identifies human circulating IL-10-producing dendritic cells (DC-10). Cell Mol Immunol. 2020;17(1):95–107. https://doi.org/10.1038/s41423-019-0218-0.
Brunner-Weinzierl MC, Rudd CE. CTLA-4 and PD-1 control of T-Cell Motility and Migration: implications for Tumor Immunotherapy. Front Immunol. 2018;9:2737. https://doi.org/10.3389/fimmu.2018.02737.
Vander Lugt B, Riddell J, Khan AA, Hackney JA, Lesch J, DeVoss J, et al. Transcriptional determinants of tolerogenic and immunogenic states during dendritic cell maturation. J Cell Biol. 2017;216(3):779–92. https://doi.org/10.1083/jcb.201512012.
Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol. 2010;108:111–65. https://doi.org/10.1016/B978-0-12-380995-7.00004-5.
Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4(10):762–74. https://doi.org/10.1038/nri1457.
Jurgens B, Hainz U, Fuchs D, Felzmann T, Heitger A. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood. 2009;114(15):3235–43. https://doi.org/10.1182/blood-2008-12-195073.
Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–42. https://doi.org/10.1146/annurev.immunol.22.012703.104758.
Sweeney CM, Lonergan R, Basdeo SA, Kinsella K, Dungan LS, Higgins SC, et al. IL-27 mediates the response to IFN-beta therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav Immun. 2011;25(6):1170–81. https://doi.org/10.1016/j.bbi.2011.03.007.
Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. 2013;14(10):1054–63. https://doi.org/10.1038/ni.2695.
Gong H, Ma S, Chen J, Yang B, Liu S, Liu X, et al. Dendritic cell-derived IL-27 p28 regulates T cell program in pathogenicity and alleviates acute graft-versus-host disease. Signal Transduct Target Ther. 2022;7(1):319. https://doi.org/10.1038/s41392-022-01147-z.
Marin E, Bouchet-Delbos L, Renoult O, Louvet C, Nerriere-Daguin V, Managh AJ, et al. Human tolerogenic dendritic cells regulate Immune responses through Lactate Synthesis. Cell Metab. 2019;30(6):1075–1090e1078. https://doi.org/10.1016/j.cmet.2019.11.011.
Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16(2):219–30. https://doi.org/10.1016/s1074-7613(02)00274-1.
Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366–70. https://doi.org/10.1038/nature12979.
Qian L, Qian C, Chen Y, Bai Y, Bao Y, Lu L, et al. Regulatory dendritic cells program B cells to differentiate into CD19hiFcgammaIIbhi regulatory B cells through IFN-beta and CD40L. Blood. 2012;120(3):581–91. https://doi.org/10.1182/blood-2011-08-377242.
Mansilla MJ, Contreras-Cardone R, Navarro-Barriuso J, Cools N, Berneman Z, Ramo-Tello C, et al. Cryopreserved vitamin D3-tolerogenic dendritic cells pulsed with autoantigens as a potential therapy for multiple sclerosis patients. J Neuroinflammation. 2016;13(1):113. https://doi.org/10.1186/s12974-016-0584-9.
Giovanelli P, Sandoval TA, Cubillos-Ruiz JR. Dendritic cell metabolism and function in tumors. Trends Immunol. 2019;40(8):699–718. https://doi.org/10.1016/j.it.2019.06.004.
Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26(5):638–52. https://doi.org/10.1016/j.ccell.2014.09.007.
Truxova I, Kasikova L, Hensler M, Skapa P, Laco J, Pecen L, et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J Immunother Cancer. 2018;6(1):139. https://doi.org/10.1186/s40425-018-0446-3.
Brandum EP, Jorgensen AS, Rosenkilde MM, Hjorto GM. Dendritic cells and CCR7 expression: an important factor for Autoimmune Diseases, chronic inflammation, and Cancer. Int J Mol Sci. 2021;22(15). https://doi.org/10.3390/ijms22158340.
Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for Tumor Antigen trafficking and priming of T cell immunity in Melanoma. Cancer Cell. 2016;30(2):324–36. https://doi.org/10.1016/j.ccell.2016.06.003.
Liu J, Zhang X, Cheng Y, Cao X. Dendritic cell migration in inflammation and immunity. Cell Mol Immunol. 2021;18(11):2461–71. https://doi.org/10.1038/s41423-021-00726-4.
Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the Tumor Microenvironment promoting Cancer Immune Control. Cell. 2018;172(5):1022–1037e1014. https://doi.org/10.1016/j.cell.2018.01.004.
Kyrysyuk O, Wucherpfennig KW. Designing Cancer Immunotherapies that Engage T cells and NK cells. Annu Rev Immunol. 2022. https://doi.org/10.1146/annurev-immunol-101921-044122.
Shi Y, Zheng W, Yang K, Harris KG, Ni K, Xue L, et al. Intratumoral accumulation of gut microbiota facilitates CD47-based immunotherapy via STING signaling. J Exp Med. 2020;217(5). https://doi.org/10.1084/jem.20192282.
Xavier JB, Young VB, Skufca J, Ginty F, Testerman T, Pearson AT, et al. The Cancer Microbiome: distinguishing Direct and Indirect Effects requires a systemic view. Trends Cancer. 2020;6(3):192–204. https://doi.org/10.1016/j.trecan.2020.01.004.
Lam KC, Araya RE, Huang A, Chen Q, Di Modica M, Rodrigues RR, et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell. 2021;184(21):5338–5356e5321. https://doi.org/10.1016/j.cell.2021.09.019.
Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan SL, et al. Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1(+) CD8(+) T cells in tumor-draining lymph nodes. Immunity. 2021;54(10):2338–2353e2336. https://doi.org/10.1016/j.immuni.2021.08.026.
Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature. 2020;580(7802):257–62. https://doi.org/10.1038/s41586-020-2134-y.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5. https://doi.org/10.1038/nature14404.
Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature. 2020;584(7822):624–9. https://doi.org/10.1038/s41586-020-2611-3.
Ruhland MK, Roberts EW, Cai E, Mujal AM, Marchuk K, Beppler C, et al. Visualizing synaptic transfer of Tumor Antigens among dendritic cells. Cancer Cell. 2020;37(6):786–799e785. https://doi.org/10.1016/j.ccell.2020.05.002.
Santegoets SJ, Duurland CL, Jordanova EJ, van Ham VJ, Ehsan I, Loof NM, et al. CD163(+) cytokine-producing cDC2 stimulate intratumoral type 1 T cell responses in HPV16-induced oropharyngeal cancer. J Immunother Cancer. 2020;8(2). https://doi.org/10.1136/jitc-2020-001053.
Murphy TL, Murphy KM. Dendritic cells in cancer immunology. Cell Mol Immunol. 2022;19(1):3–13. https://doi.org/10.1038/s41423-021-00741-5.
Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114(2):280–90. https://doi.org/10.1172/JCI21583.
Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12(9):870–8. https://doi.org/10.1038/ni.2077.
Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity. 2007;26(4):519–31. https://doi.org/10.1016/j.immuni.2007.01.017.
Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. 2016;7:13720. https://doi.org/10.1038/ncomms13720.
Ohm JE, Shurin MR, Esche C, Lotze MT, Carbone DP, Gabrilovich DI. Effect of vascular endothelial growth factor and FLT3 ligand on dendritic cell generation in vivo. J Immunol. 1999;163(6):3260–8.
Banchereau J, Klechevsky E, Schmitt N, Morita R, Palucka K, Ueno H. Harnessing human dendritic cell subsets to design novel vaccines. Ann N Y Acad Sci. 2009;1174:24–32. https://doi.org/10.1111/j.1749-6632.2009.04999.x.
Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the Art and Beyond. Clin Cancer Res. 2016;22(8):1897–906. https://doi.org/10.1158/1078-0432.CCR-15-1399.
Passeri L, Marta F, Bassi V, Gregori S. Tolerogenic dendritic cell-based approaches in autoimmunity. Int J Mol Sci. 2021;22(16). https://doi.org/10.3390/ijms22168415.
Thomson AW, Robbins PD. Tolerogenic dendritic cells for autoimmune disease and transplantation. Ann Rheum Dis. 2008;67(Suppl 3):iii90–96. https://doi.org/10.1136/ard.2008.099176.
Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113(15):3418–27. https://doi.org/10.1182/blood-2008-12-180646.
Liu J, Zhang X, Cao X. Dendritic cells in systemic lupus erythematosus: from pathogenesis to therapeutic applications. J Autoimmun. 2022;132:102856. https://doi.org/10.1016/j.jaut.2022.102856.
Crispin JC, Vargas-Rojas MI, Monsivais-Urenda A, Alcocer-Varela J. Phenotype and function of dendritic cells of patients with systemic lupus erythematosus. Clin Immunol. 2012;143(1):45–50. https://doi.org/10.1016/j.clim.2011.12.004.
Ding D, Mehta H, McCune WJ, Kaplan MJ. Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J Immunol. 2006;177(9):5878–89. https://doi.org/10.4049/jimmunol.177.9.5878.
Nehmar R, Mariotte A, de Cauwer A, Sibilia J, Bahram S, Georgel P. Therapeutic perspectives for interferons and plasmacytoid dendritic cells in rheumatoid arthritis. Trends Mol Med. 2018;24(4):338–47. https://doi.org/10.1016/j.molmed.2018.02.001.
Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol. 2013;13(8):566–77. https://doi.org/10.1038/nri3477.
Wehr P, Purvis H, Law SC, Thomas R. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clin Exp Immunol. 2019;196(1):12–27. https://doi.org/10.1111/cei.13256.
Cederblad B, Blomberg S, Vallin H, Perers A, Alm GV, Ronnblom L. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon-alpha- producing cells. J Autoimmun. 1998;11(5):465–70. https://doi.org/10.1006/jaut.1998.0215.
Jin O, Kavikondala S, Sun L, Fu R, Mok MY, Chan A, et al. Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient CD83 expression. Lupus. 2008;17(7):654–62. https://doi.org/10.1177/0961203308089410.
Coutant F, Miossec P. Altered dendritic cell functions in autoimmune diseases: distinct and overlapping profiles. Nat Rev Rheumatol. 2016;12(12):703–15. https://doi.org/10.1038/nrrheum.2016.147.
Fiore N, Castellano G, Blasi A, Capobianco C, Loverre A, Montinaro V, et al. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol Immunol. 2008;45(1):259–65. https://doi.org/10.1016/j.molimm.2007.04.029.
Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, Silvestris F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. 2008;58(1):251–62. https://doi.org/10.1002/art.23186.
Sozzani S, Vermi W, Del Prete A, Facchetti F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. 2010;31(7):270–7. https://doi.org/10.1016/j.it.2010.05.004.
Nie YJ, Mok MY, Chan GC, Chan AW, Jin OU, Kavikondala S, et al. Phenotypic and functional abnormalities of bone marrow-derived dendritic cells in systemic lupus erythematosus. Arthritis Res Ther. 2010;12(3):R91. https://doi.org/10.1186/ar3018.
Gerl V, Lischka A, Panne D, Grossmann P, Berthold R, Hoyer BF, et al. Blood dendritic cells in systemic lupus erythematosus exhibit altered activation state and chemokine receptor function. Ann Rheum Dis. 2010;69(7):1370–7. https://doi.org/10.1136/ard.2009.111021.
Fishman P, Kamashta M, Ehrenfeld M, Vianna J, Hughes GR, Sredni D, et al. Interleukin-3 immunoassay in systemic lupus erythematosus patients: preliminary data. Int Arch Allergy Immunol. 1993;100(3):215–8. https://doi.org/10.1159/000236414.
Gottschalk TA, Tsantikos E, Hibbs ML. Pathogenic inflammation and its therapeutic targeting in systemic Lupus Erythematosus. Front Immunol. 2015;6:550. https://doi.org/10.3389/fimmu.2015.00550.
Grzes KM, Sanin DE, Kabat AM, Stanczak MA, Edwards-Hicks J, Matsushita M, et al. Plasmacytoid dendritic cell activation is dependent on coordinated expression of distinct amino acid transporters. Immunity. 2021;54(11):2514–2530e2517. https://doi.org/10.1016/j.immuni.2021.10.009.
Zheng J, Sariol A, Meyerholz D, Zhang Q, Abrahante Llorens JE, Narumiya S, et al. Prostaglandin D2 signaling in dendritic cells is critical for the development of EAE. J Autoimmun. 2020;114:102508. https://doi.org/10.1016/j.jaut.2020.102508.
Huang G, Wang Y, Vogel P, Kanneganti TD, Otsu K, Chi H. Signaling via the kinase p38alpha programs dendritic cells to drive TH17 differentiation and autoimmune inflammation. Nat Immunol. 2012;13(2):152–61. https://doi.org/10.1038/ni.2207.
Vogel A, Martin K, Soukup K, Halfmann A, Kerndl M, Brunner JS, et al. JAK1 signaling in dendritic cells promotes peripheral tolerance in autoimmunity through PD-L1-mediated regulatory T cell induction. Cell Rep. 2022;38(8):110420. https://doi.org/10.1016/j.celrep.2022.110420.
Wang J, Wang J, Hong W, Zhang L, Song L, Shi Q, et al. Optineurin modulates the maturation of dendritic cells to regulate autoimmunity through JAK2-STAT3 signaling. Nat Commun. 2021;12(1):6198. https://doi.org/10.1038/s41467-021-26477-4.
Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012;122(11):3965–76. https://doi.org/10.1172/JCI63451.
Lou F, Sun Y, Xu Z, Niu L, Wang Z, Deng S, et al. Excessive polyamine generation in Keratinocytes promotes Self-RNA sensing by dendritic cells in Psoriasis. Immunity. 2020;53(1):204–216e210. https://doi.org/10.1016/j.immuni.2020.06.004.
Nakamizo S, Dutertre CA, Khalilnezhad A, Zhang XM, Lim S, Lum J, et al. Single-cell analysis of human skin identifies CD14 + type 3 dendritic cells co-producing IL1B and IL23A in psoriasis. J Exp Med. 2021;218(9). https://doi.org/10.1084/jem.20202345.
Khan FU, Khongorzul P, Raki AA, Rajasekaran A, Gris D, Amrani A. Dendritic cells and their immunotherapeutic potential for treating type 1 diabetes. Int J Mol Sci. 2022;23(9). https://doi.org/10.3390/ijms23094885.
Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, et al. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science. 1992;257(5071):792–5. https://doi.org/10.1126/science.1496399.
Cagnotto G, Willim M, Nilsson JA, Compagno M, Jacobsson LTH, Saevarsdottir S, et al. Abatacept in rheumatoid arthritis: survival on drug, clinical outcomes, and their predictors-data from a large national quality register. Arthritis Res Ther. 2020;22(1):15. https://doi.org/10.1186/s13075-020-2100-y.
Durcan L, O’Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet. 2019;393(10188):2332–43. https://doi.org/10.1016/S0140-6736(19)30237-5.
Karnell JL, Wu Y, Mittereder N, Smith MA, Gunsior M, Yan L, et al. Depleting plasmacytoid dendritic cells reduces local type I interferon responses and disease activity in patients with cutaneous lupus. Sci Transl Med. 2021;13(595). https://doi.org/10.1126/scitranslmed.abf8442.
Lu P, Fleischmann R, Curtis C, Ignatenko S, Clarke SH, Desai M, et al. Safety and pharmacodynamics of venetoclax (ABT-199) in a randomized single and multiple ascending dose study in women with systemic lupus erythematosus. Lupus. 2018;27(2):290–302. https://doi.org/10.1177/0961203317719334.
Zhan Y, Carrington EM, Ko HJ, Vikstrom IB, Oon S, Zhang JG, et al. Bcl-2 antagonists kill plasmacytoid dendritic cells from lupus-prone mice and dampen interferon-alpha production. Arthritis Rheumatol. 2015;67(3):797–808. https://doi.org/10.1002/art.38966.
Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010;62(2):542–52. https://doi.org/10.1002/art.27221.
Straub RH, Harle P, Yamana S, Matsuda T, Takasugi K, Kishimoto T, et al. Anti-interleukin-6 receptor antibody therapy favors adrenal androgen secretion in patients with rheumatoid arthritis: a randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2006;54(6):1778–85. https://doi.org/10.1002/art.21826.
Mastrandrea L, Yu J, Behrens T, Buchlis J, Albini C, Fourtner S, et al. Etanercept treatment in children with new-onset type 1 diabetes: pilot randomized, placebo-controlled, double-blind study. Diabetes Care. 2009;32(7):1244–9. https://doi.org/10.2337/dc09-0054.
Moschovakis GL, Bubke A, Friedrichsen M, Ristenpart J, Back JW, Falk CS, et al. The chemokine receptor CCR7 is a promising target for rheumatoid arthritis therapy. Cell Mol Immunol. 2019;16(10):791–9. https://doi.org/10.1038/s41423-018-0056-5.
Scotta C, Fanelli G, Hoong SJ, Romano M, Lamperti EN, Sukthankar M, et al. Impact of immunosuppressive drugs on the therapeutic efficacy of ex vivo expanded human regulatory T cells. Haematologica. 2016;101(1):91–100. https://doi.org/10.3324/haematol.2015.128934.
Wraith DC. The future of Immunotherapy: a 20-Year perspective. Front Immunol. 2017;8:1668. https://doi.org/10.3389/fimmu.2017.01668.
Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD. Effective treatment of inflammatory disease models with exosomes derived from dendritic cells genetically modified to express IL-4. J Immunol. 2007;179(4):2242–9. https://doi.org/10.4049/jimmunol.179.4.2242.
Bianco NR, Kim SH, Ruffner MA, Robbins PD. Therapeutic effect of exosomes from indoleamine 2,3-dioxygenase-positive dendritic cells in collagen-induced arthritis and delayed-type hypersensitivity disease models. Arthritis Rheum. 2009;60(2):380–9. https://doi.org/10.1002/art.24229.
Peche H, Heslan M, Usal C, Amigorena S, Cuturi MC. Presentation of donor major histocompatibility complex antigens by bone marrow dendritic cell-derived exosomes modulates allograft rejection. Transplantation. 2003;76(10):1503–10. https://doi.org/10.1097/01.TP.0000092494.75313.38.
Elashiry M, Elsayed R, Cutler CW. Exogenous and endogenous dendritic cell-derived Exosomes: Lessons learned for Immunotherapy and Disease Pathogenesis. Cells. 2021;11(1). https://doi.org/10.3390/cells11010115.
Cai Z, Zhang W, Yang F, Yu L, Yu Z, Pan J, et al. Immunosuppressive exosomes from TGF-beta1 gene-modified dendritic cells attenuate Th17-mediated inflammatory autoimmune disease by inducing regulatory T cells. Cell Res. 2012;22(3):607–10. https://doi.org/10.1038/cr.2011.196.
Cauwels A, Tavernier J. Tolerizing strategies for the treatment of Autoimmune Diseases: from ex vivo to in vivo strategies. Front Immunol. 2020;11:674. https://doi.org/10.3389/fimmu.2020.00674.
Piemonti L, Monti P, Allavena P, Sironi M, Soldini L, Leone BE, et al. Glucocorticoids affect human dendritic cell differentiation and maturation. J Immunol. 1999;162(11):6473–81.
Ferreira GB, Vanherwegen AS, Eelen G, Gutierrez ACF, Van Lommel L, Marchal K, et al. Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell Rep. 2015;10(5):711–25. https://doi.org/10.1016/j.celrep.2015.01.013.
Jauregui-Amezaga A, Cabezon R, Ramirez-Morros A, Espana C, Rimola J, Bru C, et al. Intraperitoneal administration of Autologous Tolerogenic dendritic cells for refractory Crohn’s Disease: a phase I study. J Crohns Colitis. 2015;9(12):1071–8. https://doi.org/10.1093/ecco-jcc/jjv144.
Nikolic T, Zwaginga JJ, Uitbeijerse BS, Woittiez NJ, de Koning EJ, Aanstoot HJ, et al. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide-for type 1 diabetes. Lancet Diabetes Endocrinol. 2020;8(6):470–2. https://doi.org/10.1016/S2213-8587(20)30104-2.
Willekens B, Presas-Rodriguez S, Mansilla MJ, Derdelinckx J, Lee WP, Nijs G, et al. Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): a harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ Open. 2019;9(9):e030309. https://doi.org/10.1136/bmjopen-2019-030309.
Kramer N, Schmollerl J, Unger C, Nivarthi H, Rudisch A, Unterleuthner D, et al. Autocrine WNT2 signaling in fibroblasts promotes colorectal cancer progression. Oncogene. 2017;36(39):5460–72. https://doi.org/10.1038/onc.2017.144.
Xiu DH, Liu GF, Yu SN, Li LY, Zhao GQ, Liu L, et al. Long non-coding RNA LINC00968 attenuates drug resistance of breast cancer cells through inhibiting the Wnt2/beta-catenin signaling pathway by regulating WNT2. J Exp Clin Cancer Res. 2019;38(1):94. https://doi.org/10.1186/s13046-019-1100-8.
Huang TX, Tan XY, Huang HS, Li YT, Liu BL, Liu KS, et al. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut. 2022;71(2):333–44. https://doi.org/10.1136/gutjnl-2020-322924.
Zhang Z, Wang J, Dong X. Wnt2 contributes to the progression of gastric cancer by promoting cell migration and invasion. Oncol Lett. 2018;16(3):2857–64. https://doi.org/10.3892/ol.2018.9050.
Ott PA, Adams S. Small-molecule protein kinase inhibitors and their effects on the immune system: implications for cancer treatment. Immunotherapy. 2011;3(2):213–27. https://doi.org/10.2217/imt.10.99.
Nefedova Y, Cheng P, Gilkes D, Blaskovich M, Beg AA, Sebti SM, et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol. 2005;175(7):4338–46. https://doi.org/10.4049/jimmunol.175.7.4338.
Moreira D, Sampath S, Won H, White SV, Su YL, Alcantara M, et al. Myeloid cell-targeted STAT3 inhibition sensitizes head and neck cancers to radiotherapy and T cell-mediated immunity. J Clin Invest. 2021;131(2). https://doi.org/10.1172/JCI137001.
Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–48. https://doi.org/10.1038/nrclinonc.2018.8.
Drobits B, Holcmann M, Amberg N, Swiecki M, Grundtner R, Hammer M, et al. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J Clin Invest. 2012;122(2):575–85. https://doi.org/10.1172/JCI61034.
Matsumoto M, Takeda Y, Seya T. Targeting toll-like receptor 3 in dendritic cells for cancer immunotherapy. Expert Opin Biol Ther. 2020;20(8):937–46. https://doi.org/10.1080/14712598.2020.1749260.
Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV. Therapeutic activity of Agonistic, Human Anti-CD40 monoclonal antibodies requires selective FcgammaR Engagement. Cancer Cell. 2016;29(6):820–31. https://doi.org/10.1016/j.ccell.2016.05.001.
Garris CS, Wong JL, Ravetch JV, Knorr DA. Dendritic cell targeting with Fc-enhanced CD40 antibody agonists induces durable antitumor immunity in humanized mouse models of bladder cancer. Sci Transl Med. 2021;13(594). https://doi.org/10.1126/scitranslmed.abd1346.
Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, et al. Clinical and biological effects of an agonist anti-CD40 antibody: a Cancer Research UK phase I study. Clin Cancer Res. 2015;21(6):1321–8. https://doi.org/10.1158/1078-0432.CCR-14-2355.
Ruter J, Antonia SJ, Burris HA, Huhn RD, Vonderheide RH. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10(10):983–93. https://doi.org/10.4161/cbt.10.10.13251.
Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25(7):876–83. https://doi.org/10.1200/JCO.2006.08.3311.
Byrne KT, Leisenring NH, Bajor DL, Vonderheide RH. CSF-1R-Dependent Lethal Hepatotoxicity when Agonistic CD40 antibody is given before but not after Chemotherapy. J Immunol. 2016;197(1):179–87. https://doi.org/10.4049/jimmunol.1600146.
Salomon R, Rotem H, Katzenelenbogen Y, Weiner A, Cohen Saban N, Feferman T, et al. Bispecific antibodies increase the therapeutic window of CD40 agonists through selective dendritic cell targeting. Nat Cancer. 2022;3(3):287–302. https://doi.org/10.1038/s43018-022-00329-6.
Yuan X, Duan Y, Xiao Y, Sun K, Qi Y, Zhang Y, et al. Vitamin E enhances Cancer Immunotherapy by reinvigorating dendritic cells via Targeting Checkpoint SHP1. Cancer Discov. 2022;12(7):1742–59. https://doi.org/10.1158/2159-8290.CD-21-0900.
Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer. 2015;3:51. https://doi.org/10.1186/s40425-015-0094-9.
de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 regulates CD103(+) dendritic cell function and response to chemotherapy in breast Cancer. Cancer Cell. 2018;33(1):60–74e66. https://doi.org/10.1016/j.ccell.2017.11.019.
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94. https://doi.org/10.1084/jem.20100643.
Ding Z, Li Q, Zhang R, Xie L, Shu Y, Gao S, et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct Target Ther. 2021;6(1):26. https://doi.org/10.1038/s41392-020-00448-5.
Anguille S, Van de Velde AL, Smits EL, Van Tendeloo VF, Juliusson G, Cools N, et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood. 2017;130(15):1713–21. https://doi.org/10.1182/blood-2017-04-780155.
Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16(1):142. https://doi.org/10.1186/s12967-018-1507-6.
Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. https://doi.org/10.1146/annurev.immunol.18.1.767.
Saxena M, Bhardwaj N. Re-emergence of dendritic cell vaccines for Cancer Treatment. Trends Cancer. 2018;4(2):119–37. https://doi.org/10.1016/j.trecan.2017.12.007.
Lowenfeld L, Mick R, Datta J, Xu S, Fitzpatrick E, Fisher CS, et al. Dendritic cell vaccination enhances Immune responses and induces regression of HER2(pos) DCIS Independent of Route: results of Randomized Selection Design Trial. Clin Cancer Res. 2017;23(12):2961–71. https://doi.org/10.1158/1078-0432.CCR-16-1924.
Ferris ST, Ohara RA, Ou F, Wu R, Huang X, Kim S, et al. cDC1 vaccines drive Tumor rejection by Direct Presentation independently of host cDC1. Cancer Immunol Res. 2022;10(8):920–31. https://doi.org/10.1158/2326-6066.CIR-21-0865.
Acknowledgements
We are grateful to the Sichuan Provincial Department of Science and Technology, West China Hospital, Sichuan University for their financial support. We are grateful to Servier Medical ART (SMART) for part of the material and Biorender for some of the inspiration.
Funding
This work was supported by the Key Project of the Science and Technology Department of Sichuan Province (NO. 2022YFH0100), the National Natural Science Foundation of China (NO. 82171829), the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (NO. ZYYC21012), and the Fundamental Research Funds for the Central Universities (20822041E4084).
Author information
Authors and Affiliations
Contributions
HC drafted the manuscript. WC, YL, JZ and XS edited the manuscript. DZ supervised the work and edited the manuscript. All authors contributed to the article and approved it for publication.
Corresponding author
Ethics declarations
Ethics approval
(with approval No.)
Not applicable.
of interest.
The authors declare that the work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, H., Chen, W., Lin, Y. et al. Signaling pathways involved in the biological functions of dendritic cells and their implications for disease treatment. Mol Biomed 4, 15 (2023). https://doi.org/10.1186/s43556-023-00125-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-023-00125-3