
Abstract
Dendritic cells (DCs) are specialized immune sentinels that play key role in maintaining immune homeostasis by efficiently regulating the delicate balance between protective immunity and tolerance to self. Although DCs respond to maturation signals present in the surrounding milieu, multiple layers of suppression also co-exist that reduce the infringement of tolerance against self-antigens. These tolerance inducing properties of DCs are governed by their origin and a range of other factors including distribution, cytokines, growth factors, and transcriptional programing, that collectively impart suppressive functions to these cells. DCs directing tolerance secrete anti-inflammatory cytokines and induce naïve T cells or B cells to differentiate into regulatory T cells (Tregs) or B cells. In this review, we provide a detailed outlook on the molecular mechanisms that induce functional specialization to govern central or peripheral tolerance. The tolerance-inducing nature of DCs can be exploited to overcome autoimmunity and rejection in graft transplantation.
Similar content being viewed by others

Introduction
Since the discovery of dendritic cells (DCs) in 1973 by Dr. Ralph Steinman and Dr. Zanvil A. Cohn [1], numerous studies have been conducted to understand the complex biology of these cells. Based on their functional responses, DCs have been described as “gatekeepers of the immune system”, able to orchestrate the components of the immune system for a favorable effect in an organism [1, 2]. As gatekeepers, DC maintain immune homeostasis by both activating adaptive immunity or contributing to tolerance. DCs that stimulate adaptive immunity have been referred to as stimulatory DCs (sDCs) while those that suppress immune response have been termed tolerogenic DCs [3].These terms describe DC by their functional state, and are not intended to confer that a single subset is solely dedicated to one state or another at all times.
How are tolerance and immunity simultaneous achieved?
Under steady-state conditions, naïve or immature DCs (iDCs) are poorly immunogenic. However, once these cells sense “danger” signals in the periphery (such as by pattern recognition receptors such as Toll-like-receptors), they differentiate into mature DCs (mDCs) with an upregulated expression of MHC, co-stimulatory molecules and chemokine receptors (eg. CCR7). This enables DC migration to the nearest lymph nodes for antigen presentation to T cells [4–8]. In translating these innate cues such as TLR mediated danger signals, into an adaptive outcome, DCs provide a bridge to link these innate and adaptive immune arms. The concept that immature DCs tolerize, and mature DC’s prime, however is a likely oversimplification [9]. iDCs that have matured in the absence of inflammatory signal acquire the “semi-mature” phenotype and express CCR7 receptors like mDCs but exhibit regulatory or tolerogenic functions [10]. Furthermore it remains unknown how during an ongoing immune response in vivo tolerance and immunity are simultaneously orchestrated.
In addition to maturation, localization is a key variable in DC activity. In their capacity as sentinels, DC distribute broadly in peripheral tissues (eg. skin, lung, meninges/choroid, mucosa) where they exhibit a high turn-over rate, patrol and migrate from peripheral tissues to draining lymphoid organs. These DC may be pre-conditioned towards greater self-tolerance or upon tissue entry acquire phenotypic and functional changes in response to environmental stimuli to achieve tolerogenic vs. immunogenic function. DC in tissues encounter a variety of foreign antigens and maintain tolerance in response to both sterile and non-sterile injury, while existing in tissues with variable rates of turn over. It remains unknown if and how these cells have evolved conserved mechanisms of maintaining self tolerance. Homeostatic maturation linked to migration from tissues and leading to tolerance [11, 12] has been distinguished from danger signal based licensing leading to adaptive immunity [13].
DCs achieve their regulatory function by inducing apoptosis of inflammatory T-cells, restoring immune homeostasis (regulating pro- and anti-inflammatory reactions), and/or by expansion of regulatory T cells (Tregs) [14–16]. Inhibiting the destruction of self-reactive T cells that have escaped thymic selection is also mediated through peripheral tolerance of DCs, thus limiting chances of autoimmunity [17]. DCs have a critical role in maintaining peripheral tissue homeostasis in the steady state, permitting self tolerance [18]. Conditioning DCs to impart tolerance has clinical utility in diseases such as graft-versus-host-disease (GvHD) [19–21]. In this chapter, we will provide background on the origin and differentiation of DCs, known factors that influence their tolerogenic properties, processes regulating DC mediated Treg function, and therapeutic opportunities associated with their tolerogenic face.
Overview of DC subsets and functional specialization
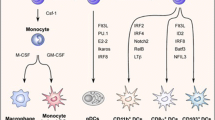
As sentinels, DC are functionally, programmatically, and phenotypically diversified into several cellular subsets (or subtypes) in lymphoid organs, circulation and tissues. DCs may be categorized primarily into four classes based on their phenotypic and functional maturation: myeloid or classical (also called conventional) DCs (cDCs), plasmacytoid DCs (pDCs), monocyte-derived DCs (moDCs), and Langerhans cells (LCs) [22, 23]. pDCs and cDCs participate in inducing both adaptive immunity and tolerance while moDCs develop mainly under conditions of inflammation. Langerhans cells (LC), are present in the steady state and are distinct members of the monophagocyte lineage, which like Kuppfer cells of the liver or microglia of the brain are tissue specific [24, 25].While sharing common cell surface markers and functionality with myeloid or classical DCs, LC develop from precursors and progenitors in the macrophages and monocytes lineage and thus have undergone recent reclassification. Like other DC, LC induce either tolerance or active immunity depending on the environmental stimuli [22].
DCs in mice express the integrin CD11c and MHC-II in varying amounts, and can be further classified phenotypically based on the expression level of markers such as CD8α, CD4, CD11b, PDCA-1, Langerin along with a continuous growing list of other markers. Differential expression of these markers defines sub-populations or contributes to the mixed DC subsets that reside in specific organs, particularly the secondary lymphoid organs (SLOs) [5, 26]. Phenotypically, classical DCs (cDCs) exhibit high levels of CD11c and MHC-II, while plasmacytoid cells (pDCs) that are specialized type I interferon (IFN) producing cells show low expression of the same markers [5, 27]. Conventional DCs may be further categorized by location and considered as tissue resident in peripheral tissue sites such as the skin, lung, or gut and which have migratory capacity (migDCs, MHCII high, CD11c intermediate), or, lymphoid-resident classical DCs (LN or spleen cDC, MHCII intermediate, CD11c high). LCs of the epidermis do not develop in the same manner as other classical DCs but like other tissue DCs constitutively migrate to the draining LN.
Central lymphoid
Classical DCs reside in secondary lymphoid organs (SLOs) like local lymph nodes, spleen and thymus, and are either CD11b + (CD4+ CD8α−) DCs, or CD8α +(CD11b- CD4−)DCs, In some cases a third group of CD4− CD8α− DCs [28] has been described in mice. This nomenclature was recently simplified based on DC development and can now be described as classical type 1 or 2 DCs (cDC1s or cDC2s), in which cDC1s comprise of CD8α + and CD103+ while cDC2s represent CD11b + and CD172a + DC populations [29].Use of CD8α+expression as a distinguishing feature on DCs is specific to mice [19] as in humans these populations are decribed as BDCA1+ (which corresponds to CD11b +CD4+ CD8α− DCs) or BDCA3+ DC (which corresponds to CD11b− CD4− CD8α +DCs). Pre-DCs that mark their entry into lymph nodes through high endothelial venules (HEV) is shown to express high levels of CD62L and inhibition of this receptor prevented the accumulation of cDCs in the LNs, but not in the spleen where entry of pre-cDCs occur through marginal sinuses [30].
Peripheral tissue
Migratory DCs (migDCs) in contrast patrol peripheral tissues like skin and mucosa where they are exposed to continuous environmental signals. In the steady state 5–7 % have been estimated to navigate to nearest lymph node for antigen presentation to T cells [12]( while under local inflammatory conditions their migratory rate is significantly increased [6, 8]. Transendothelial migration of pre-DCs to non-lymphoid tissues such as skin is mediated by receptor-ligand interactions, such as CCR4-CCL22/CCL17, CCR6-CCL20, CCR2-CCL2, CCR5-CCL5, and CXCR3-CXCL9/CXCL10/CXCL11. On attaining maturation, DCs primarily express the chemokine receptors CCR7 (ligands CCL19 and CCL21) and CXCR4 (ligand CXCL12) [5, 8]. These chemokines are secreted to the microenvironment by the endothelial cells of the lymphatic vessels or by the stroma cells of the lymph node [31, 32]. All DC of the skin are capable of migration and include LC of the epidermis as well as dermal or interstitial DC.
Epidermis
Langerhans cells (LCs) were initially considered nerve cells of the skin due to their long dendrites when identified by Paul Langerhans in 1868 [33]. LC are found within the epidermis and mucosae of the skin while dermal DCs (dDCs) majorly reside in the dermis of the skin. LCs are characterized by the presence of an intracellular organelle called Birbeck granules (BG) and expresses high levels of its associated protein Langerin/CD207, a C-type lectin receptor that is responsible for the development of these granules [34]. While in some contexts LC have been ascribed with a suppressive role during contact sensitivity [35], dose and model dependent difference have also implicated LC in mediating inflammation [36] and LC have been implicated in transporting a model antigen like OVA to the skin draining LN for priming as well during tape stripping [37]. Functional specialization of LC in mediating T cell priming in response to Candida albicans [38, 39] has been found to be specific only for T helper 17 (Th17) and not for CTLs (cytotoxic lymphocytes). Mouse and human LCs that constitute about 1–5 % of the epidermal cell population also express E-cadherin (keratinocyte adhering molecule), CD205 and MHC-II molecules. Human LCs, in addition express high amounts of CD1a, a molecule that mediates non-peptide, cell-wall mycobacterial antigen components [40] and lipid antigen [41] presentation to T cells .
Dermis
Skin (and lung and gut) DC may also be dermal or interstitial DCs and the majority of dermal DC depend on the growth factor Flt3L. In mice 3 major populations of dermal DC that depend on Flt3L exist [42, 43] though some dermal macrophage or monocyte derived populations have been appreciated [24]. Of Flt3L dependent populations that are Langerin low, CD11b+ or CD11b− cells [43, 44], relate more closely and share KLF4 (Kruppel-like factor 4) dependence [45]. Functionally CD11b-(or CD103+) has been associated with TH2 allergy while CD11b + DCs have been linked to Th17 responses [46, 47] A third population of Langerin + cDC are present in the dermis of mice (and in lung and gut).Though langerin+, these cells express lower levels of langerin than dermal DCs [34, 48]. In contrast to LCs, dermal langerin + DCs co-express the αE integrin CD103, instead of EpCAM (Epithelial Cell Adhesion Molecule) which is predominant in LCs [36, 49, 50]. Functionally these Langerin + CD103+ cells are of critical importance as they are known to mediate tumor/viral cross presentation. Thus in addition to LC of the epidermis, 3 dermal/interstitial DC populations exist in skin lung and gut. Recently a revised classification has been proposed to simplify the two main populations in tissues as cDC1 and cDC2 (for Langerin + CD103+ DC vs. Langerin-CD11b + DC in mouse or BDCA3 vs. BDCA1 in human) [29], by ontogeny. This classification system leaves out the CD11b- subset involved in TH2 responses which comprise a larger fraction in skin and lung than in gut (unpublished data). Based on markers, dDCs may be sub-divided into CD11bloCD207+, CD11bloCD207− and CD11bhiCD207− cells [44]. Among dermal DCs, Langerin + CD103+ DC express high amounts of XCR1(XC Chemokine Receptor 1) [51]. XCR1+ expression is highly specific for CD8α cDCs [13]. In human skin, counterparts are BDCA1+ (akin to CD11b+), BDCA3+ (Langerin + CD103+) or may be double positive [52] No clear counterpart to CD11b- DC has been definitively identified.
moDCs (Monocyte-derived DCs) are distinct from conventional DCs. They originate from Ly6C+ monocytes following inflammation and express phenotypic markers such as CD11b (in high amounts), CD11c (intermediate expression), MHCII and MAC3 (a glycoprotein expressed on the surface of macrophages), but remain CD4− and CD8−[28, 53–56]. moDCs are prevalent in both central as well as peripheral lymphoid and non-lymphoid organs.
Overview of DC origin and differentiation
Early precursors
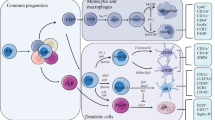
The multipotent CD34+hematopoietic stem cells (HSCs) in the bone marrow acts as the precursor to multi-lineage progenitors which gives further rise to the common lymphoid progenitor (CLP) as well as the common myeloid progenitor (CMP) populations. These CLPs and CMPs have the potential to generate into any DC subtypes (cDCs and pDCs), suggesting developmental flexibility in DC formation [26, 57, 58]. Adoptive transfer of these early precursors into irradiated animals has shown high potential in cDC and pDC generation [57–60]. Although CMPs and CLPs have almost similar efficiency to produce DCs at single cell level, CMPs exceed in cell population and therefore serve as the major DC precursor. CMPs generate LN and splenic DCs efficiently while CLPs contribute highly towards production of thymic DCs and meager level of splenic DCs [58, 61]. Thymic cDCs have also been shown to originate from thymic precursors residing locally in thymus [58, 59]. However, the ability of CLPs or CMPs to differentiate into DCs is confined to only those subsets expressing Flt3 (fms-related tyrosine kinase 3) receptor (also known as CD135), a kinase with strong homology to c-fms and c-Kit [62, 63]. Interaction of Flt3 with Flt3 ligand (Flt3L, a cytokine) induces the generation of cDCs (or pre-cDCs) and pDCs under steady-state conditions [63–65]. Pre-cDCs migrate to lymphoid and other non-lymphoid tissues from the bone marrow to ultimately differentiate into mature cDCs [28]. Mice lacking expression of the Flt3 receptor (in some cases also referred as Flk-2) or production of Flt3L DC-poetin show severe decreases in pDC and cDC subsets [66, 67]. On the other hand, Flt3− progenitors lacking the ability to generate DCs were enforced to produce both plasmacytoid and conventional DCs on transfection with Flt3 [68].The cDCs and pDCs residing in blood, peripheral organs or lymphoid organs represent the “immature” stage of the cell whose primary function is to maintain immune homeostasis until they encounter an antigen to finally differentiate into mature DCs with immunogenic nature. This model of development suggests that all DCs may be tolerogenic at initial differentiation from their precursors, unless further triggered by an environmental insult. DC functional plasticity is then further determined during their differentiation process, which governs their final outcome depending on the surrounding factors. Central to this premise is that tolerogenic programing should be shared as part of a core signature expressed in all immature DC irrespective of origin. How such programing is governed or maintained is unknown.
Intermediate precursors
Commitment to specific DC subtypes is further controlled by intermediate precursors that are devoid of lineage specific markers (lin−) but retain the early HSC surface markers like CD117 and act as common precursors for both macrophages and DCs in the bone marrow [69]. This defines the nature of myeloid precursors to differentiate into a macrophage dendritic cell progenitor (MDP) lineage with Lin−c-kit+CX3CR1+phenotype [69, 70]. These MDPs could generate lymphoid and non-lymphoid resident cDCs, some pDCs and macrophages, but were not observed to produce any granulocytes. This may be further supported by two other studies in which cell populations in the marrow having a CD31+CD11c+Ly6C+or Lin− Csf-1R+phenotype were found to show restricted developmental potential and to serve as the precursors of cDCs, pDCs and macrophages [66, 71].
Common dendritic progenitors (CDPs), identified as giving rise to cDCs and pDCs were initially isolated from bone marrow with the phenotype Lin−Flt3+c-kitintCSF-1R+. These CDPs on being transferred to lethally irradiated, sublethally irradiated or unirradiated animals generated splenic and LN cDCs and pDCs, but no macrophages [63, 64]. MDPs have slightly higher expression of the stem cell factor c-kit (or CD117) and sustain its cellular proliferation in the presence of M-CSF in contrast to CDPs which are more Flt3 dependent [63]. This finding draws a lineage that CDPs are the immediate downstream progeny of MDPs along with monocytes.
Downstream precursors
Downstream precursors which further give rise to only a defined group of DCs were identified in the case of mouse splenic DCs with CD11bloCD11cintCD43intCD45RAloSIRPαintMHCII−CD8−CD4−phenotype that produced CD11b+ and CD8+cDCs, but no pDCs [28].Based on the persistent expression of the receptor Flt3 in all the precursors of cDCs, it has been revealed that these pre-classical DCs (pre-cDCs) might have their origin from CDPs in the bone marrow where they acquire the last stage precursor phenotype and later on home to lymphoid and non-lymphoid tissues through the blood circulation [30, 70]. Final differentiation of pre-cDCs into the immature conventional DCs occurs in the local migrated tissues. Even in non-lymphoid tissues such as liver, kidney and intestine, pre-cDCs have been shown to develop into cDCs with CD103 and some CD11b expression [72, 73]. The pDC in contrast is thought to have developed completely in the bone marrow and probably thereafter migrate to other tissues through the blood stream [26].
Although monocytes usually contribute to DC formation only during inflammatory conditions, few recent reports have highlighted their involvement in the production of CD103−CD11b+ DCs in the intestine, FcγRI+ DCs in the muscular tissues and CD11b+Esamlo DCs in the spleen during steady-state conditions [70, 72, 74, 75]. One caveat to these studies is that they are based on phenotypic population sorting and adoptive transfer to define lineages. While these markers help to enrich populations and may do so sequentially they may fail to appropriately discriminate one progenitor cell from another. Now with the advent of single cell sequencing it is likely a variety of committed intermediates may be rapidly identified suggesting commitment to a fate may happen earlier in immune development from the bone marrow. As such recently preDC subsets giving rise to classical DC were identified [25, 26].
Peripheral DC precursors
Langerhans cell precursors
Langerhans cells (LCs) constitute the major migratory DC population in the peripheral tissues (both dermis and epidermis) and undergo transit to the neighboring lymph nodes at a basal level in the steady state. Mouse skin contains a reservoir of LC precursors, which have their origin in Ly6C expressing mouse monocytes. The Ly6C+inflammatory monocytes differentiate into MHCII+ Langerin+ expressing LC cells within few days in the skin and CSF-1R serve as one of the critical factors in the migration and generation of skin LCs from these precursors [76]. This portends that the population of LCs in steady state may be derived from non-inflammatory Ly6C+ monocytes during the early developmental stage as mice deficient in CSF-1R (or M-CSFR) monocytes has been shown to lack Langerhans cells [26, 76]. However, another study has emphasized the role of monocytes derived from fetal liver to be the immediate precursor of the major LC pool with yolk-sac myeloid progenitors contributing to only 10 % of the total pool [77]. LC regeneration in skin could also be attained from Flt3+ hematopoietic precursors which have been transferred to these peripheral tissues, although mice lacking M-CSFR expression showed no Flt3+ differentiation into LCs signifying the key role of this receptor in LC development [26, 78]. Dependence on M-CSFR and evidence of monocytic origin suggests that LCs possess a distinct developmental pathway in comparison to their splenic DC counterpart. Thus, LCs of the skin and some other tissue-specific DCs follow a different pathway of development unlike conventional DCs which are Flt3 dependent. Mice deficient in M-CSFR fail to generate LCs, thereby identifying the M-CSF/M-CSFR axis as a critical factor in this unique DC subset generation [76].
Dermal DC precursors
The dermal DCs have a short life span and a minor subset of these DCs also expresses the endocytic receptor langerin. In the steady state these cells are largely derived from precursors such as pre-cDCs (Langerin + CD103+, CD11b+, CD11b-) or are monocyte-derived. The generation of dermal cDCs from pre-cDCs is dependent upon Flt3 ligand [43] while monocyte-derived dermal DCs originate from Ly6C+ expressing monocytes that require CCR2 signaling [25, 51, 79].
Hematopoetic factors influencing the capacity for DCs to maintain homeostasis
Dendritic cell function is not only governed by late differentiation cues such as danger signals that define their final phenotypic or functional state but also those factors that regulate DC development, to permit inflammatory or tolerogenic activity. Signals that drive the fate of DCs include growth factors, transcription factors and cytokines. DC development from the HSC precursor requires Flt3L-STAT3 signaling pathway. Mice deficient in signal transducer and activator of transcription 3 (STAT3, a transcription factor operating downstream of Flt3) show low levels of DC numbers indicating that STAT3 contributes to Flt3L induced DC differentiation in steady-state [80]. Although Flt3L signaling has been known to be the major contributing factor in DC development, GM-CSF and STAT5 signaling also contribute to immature DC generation [2, 81]. Transgenic mice that overexpress GM-CSF have shown moderate increase in the DC population [82]. GM-CSF, which is generated by activated NK cells, T cells and by tissue stromal cells are however detected in higher quantities during inflammation than in the steady state. This indicates that GM-CSF remains dispensable in steady state conditions, but serves to bean important differentiation factor for DCs in the inflammatory situations [83]. On the other hand, Flt3L expressed by activated T cells and tissue stroma cells in the soluble or membrane bound form is indispensable for development of pDCs and DCs residing in lymphoid organs [84].
FLT3L and transcriptional programing
Flt3L drives the development of immature DCs [85, 86]. Therefore Flt3L has been used therapeutically to treat autoimmune disease [87, 88]). On the other hand when Flt3L is used to expand DC numbers and is given with a TLR adjuvant to mature DC, Flt3L can enhance DC priming [42, 89]. In some contexts, Flt3L treatment of DCs also contributes to entry of T cells into tumors [90, 91] but close attention must be paid to contexts in which TLR adjuvant was given to mature DC.
Other transcription factors like E2–2 have been documented to induce pDC development [92], while RelB (NF-κβ/Rel family member) and Batf3 have been linked to CD8α−cDC and CD8α+cDC development respectively [93, 94]. These transcription factors may act either in early DC development or are involved in the differentiation of late DC subsets.
PU.1 (encoded by Sfpi1) is a transcription factor belonging to ETS group of DNA binding proteins that lies upstream of GM-CSF and Flt3, and induces DC commitment through both pathways in both steady and inflammatory conditions [68, 95]. Mice deficient in PU.1 hematopoietic cells have abrogated DC differentiation from myeloid progenitors [95]. Gfi-1 (a transcriptional repressor) has also been documented to antagonize PU.1 function by repressing the Sfpi1 gene which then displaces PU.1 from positive autoregulatory elements in multipotential progenitors thereby resulting in DC generation rather than the predefined macrophage fate [96]. Deletion of the zinc finger Gfi-1 which regulates STAT3 activation also leads to deficiencies in lymphoid and myeloid DCs in all lymphoid organs, but increases LC numbers instead [97] .
Zbtb46 transcription factor (from the zinc finger family, also known as Btbd4 or zDC) is an evolutionary conserved protein and its expression is highly restricted to cDC lineage among immune cells, apart from its presence in erythroid progenitors and endothelial cells. Expression of Zbtb46 starts at the pre-cDC stage of development and continues to be expressed on CD8+ and CD11b+cDCs in spleen, CD103+ and some CD11b+cDCs in non-lymphoid tissues, whereas remain absent in pDCs and macrophages [70, 98, 99]. Development of migDCs which are Langerin−CD11b−and are Flt3 responsive have been found to be depleted in Zbtb46 deficient mice, thereby relating them to cDC type [43]. Zbtb46 has been recently identified as a marker of the classical DC lineage and may provide exciting prospects in DC targeted vaccine development. Indeed, Zbtb46 and Flt3 dependent cDCs have been reported to induce T cell and humoral immune response following HIV-derived gag p24 peptide and adjuvant immunization [42].
Transcription factors such as interferon regulatory factors (IRFs), E2–2, E2A, Spi-B, Id2, Batf3and Runx3 act downstream in DC development and guide the differentiation of specific DC subsets. IRFs 2, 4 and 8 have been regarded as the key transcription factors that are linked to a defined DC diversification. Irf-2 deficient mice showed reduced levels of CD8α−DCs and partly decreased LC population [100], while mice lacking Irf-4 displayed low levels of CD8α−DCs and somewhat reduced pDCs [101, 102]. Irf-8 knockout mice lack major DC subsets such as CD8α+ DCs, LCs and pDCs [103, 104][Fig.1]. DC differentiation by GM-CSF is dependent on IRF-4 signaling while Flt3L preferentially require IRF-8 (also known as ICSBP, interferon consensus sequence-binding protein) [102].

IRF2, IRF-4 and IRF-8 play major roles in maintaining different DC subsets
Transcription factors that belong to the basic helix-loop-helix (HLH) family of E proteins (E12, E47, HEB, E2a, E2–2) bind on the sequence CACCTG E-box as homodimers or heterodimers. These E proteins are activators of transcription and their function can be inhibited by another HLH family of proteins called the inhibitors of DNA binding (Id) proteins which sequesters E proteins and prevent their binding to target sites [23, 105]. E2–2 deletion in mice has led to ablation of pDC development and haploinsufficiency of E2–2 (Pitt-Hopkins syndrome) in rare human patients showed impaired pDC formation [92]. E2–2 may be regarded as a master regulator in pDC development as they bind to signature genes such as Irf7,Irf8, SpiB and BDCA-2 [23]. Id2−/− mice on the other hand show increased levels of pDCs, but lack LCs and have severely reduced CD8α+cDCs [106]. Transforming growth factor beta (TGF-β1) has been shown to induce Id2 expression, thereby revealing the association of TGF-β deficient mice with LC deficiency that was observed in Id2−/− mice [106]. Unlike PU.1, deficiency of Spi-B transcription factor only affects pDC development. Over expression of Spi-B on the other hand impairs Id2 expression and enhances E2–2 activity, indicating that Id2 acts as a regulator of the pDC/cDC balance [107]. Ablation of Spi-B expression abrogated the ability of E2–2 to induce pDC differentiation showing that E2–2 and Spi-B act jointly towards the development of pDCs from its precursors in humans [107].
The bZIP transcription factor Batf3, also known as Jun dimerization protein p21SNFT represses NFAT-AP1 activity by dimerizing with Jun and inhibiting Jun-Fos heterodimer formation. Batf3 is more highly expressed in DCs than other immune cells and plays a pivotal role in the development of migratory CD103+CD11b−cross-presenting DCs in peripheral tissues such as skin [108]. Although mice with Batf3 deletion have shown reduced CD8α+ DCs in the spleen, there are now reports that absence of Batf3 does not decrease the CD8α+ DC population but affects their cross-presentation of antigens to CD8+ T cells [94]. Lack of another transcription factor called Runx3 (runt domain family) impairs TGF-β mediated inhibition of DC maturation and leads to defect in LC development [109].A list of the various factors guiding DC subset development in various tissues has been described in Table 1.
The life span of steady state DCs also has an influential role in maintaining immune homeostasis and its disruption may lead to autoimmune disorders. Deficiency in apoptotic inducing genes, overexpression of anti-apoptotic genes and inactivation of Fas-induced death signaling disrupts the normal DC function and causes disorders [2].CD11bhiregulatory DCs show high Fas expression and TGF-β produced by the surrounding stromal cells could induce high Fas expression on these regulatory DCs through ERK activation [121].There thus exists a multitude of fators that control DC maturation in homeostatic and inflammatory conditions.
DCs dendritic cells, cDCs conventional DCs, pDCsplasmacytoid DCs, LCs langerhans cells, Id inhibitor of DNA binding, GM-CSF granulocyte macrophage colony-stimulating factor, M-CSF macrophage colony-stimulating factor, MDPs macrophage dendritic cell progenitor, CDPs common dendritic progenitors.
Central tolerance
DCs maintain central tolerance by mediating negative selection of self antigens in the thymus resulting in either removal of autoreactive T cells or are rendered innocuous by tolerance induction [122]. Circulating DCs migrate to the medulla region of thymus through a three-step mechanism that involves P-selectin adhesion, interaction of VLA-4 integrin with VCAM-1 ligand and signaling by chemoattractant sensitive to pertussis toxin [122]. Thymic dendritic cells (TDCs) contribute to Treg induction in vivo and more interestingly, even peripheral DCs can migrate to the medullary region of thymus to efficiently induce Treg development and deletion of self reactive thymocytes [123]. IL-7 like thymic stromal lymphoietin (TSLP) expressed by Hassall’s corpuscles in the thymus medulla induces the tolerogenic phenotype on DCs rendering them the ability to convert naïve T cells into CD4+CD25+Foxp3+Treg cells [124, 125]. Plasmacytoid DCs in the human thymus can also induce the development of Tregs which produce IL-10 more efficiently than Tregs generated by TDCs [126]. However, in the extra-thymic sites such as the skin and lung, TSLP promotes Th2 response apart from Treg induction suggesting that other unknown factors also contribute to the DC function in the thymus [127]. One answer to the debate that whether DC maturation in the homeostatic state is a stochastic or a defined process comes from a recent finding by Ardouin et al., where it was shown that there is a functional convergence of the transcriptomic signals during maturation of the homeostatic XCR1+ thymic DCs and DCs induced by PRRs (pattern recognition receptors) [128]. XCR1+ thymic DCs also expressed unique interferon-stimulated gene (ISG) signature than XCR1+ DCs of peripheral origin [128].
Peripheral tolerance
Self-antigens presented on MHC to autoreactive T cells that have escaped thymic selection pose a potential threat to the immune balance. To avert the breach of immune homeostasis by these autoreactive T cells, a second tier of peripheral tolerance exist in which DCs likely provide a major contribution in the tolerance induction. Tolerogenic DCs may be naïve immature cells or alternatively activated cells that exhibit resistance to maturation even in the presence of an inducing signal. Immature DCs (iDCs) express high levels of CCR1, CCR2, CCR6, CCR5, CCR7, CXCR1, CXCR2, FcγRII, DEC-205, TLRs, PD-L1 molecules and have lower expression of MHC-II and other co-stimulatory molecules such as CD40, CD80, CD86, CD52 and PD-L2 [129–131].
On encountering danger signals, splenic CD8α−DCs migrate to the T cell zone of the spleen and as with CD8α+DCs, they express high levels of MHC, co-stimulatory molecules and cytokines such as IL12p70 to induce naïve and memory T cell activation. CD40 present on the cell surface of these DCs interact with CD40L expressed by activated T cells and induces further maturation of both the DC subtypes. Matured CD8α−DCs induces Foxp3+CD4+CD25+regulatory T cell (Treg) expansion. This provides an inhibitory loop for excessive T cell response and a means to maintain peripheral tolerance against self antigens which are presented by the same DCs that also capture foreign antigens [19]. Contrastingly in the steady state, DCs have a quiescent or semi-mature nature wherein they capture and process exogenous antigens, and fail to induce naïve T cell activation. Instead they promote T cell anergy and regulatory T cell expansion. Known mechanisms by which DCs induce peripheral tolerance include by (i) enhancing the synthesis of indoleamine 2,3-dioxygenase (IDO), (ii) inducing and maintaining T cell anergy through the expression of programmed cell death ligand 1 (PDL-1) and (iii) promoting deletion of T cells that are antigen specific through upregulated expression of CD95L (FasL) or TRAIL (TNF-related apoptosis-inducing ligand) [132]. Tolerance induction by IDO requires the ligation of B7–1/B7–2 (CD80/CD86) present on DC subsets with soluble or membrane bound form of CTLA4 (cytotoxic T-lymphocyte antigen 4, also known as CD28) expressed by T cells [133, 134]. IDO is a rate-limiting enzyme that catalyzes the degradation of the essential amino acid tryptophan into various tryptophan-derived metabolites which then inhibits T cell proliferation by impairing the cell cycle machinery and promotes T cell apoptosis [135, 136].However, IDO is not constitutively expressed in DCs and requires induction by multiple mediators. In addition to IFN-γ, IDO can be induced by TGF-β, endotoxin, TNF, IL-1 and may be inhibited by cytokines such as IL-6 that downregulates expression of CD119 (IFN-γ receptor chain) [3, 19, 135]. Some reports indicate induction of IDO during inflammation is largely secondary to IFN-γ dependent effects [137]. Depletion of tryptophan increases the inhibitory receptors ILT3 and ILT4 (Immunoglobulin like transcripts) expression on DCs which then favor the upregulation of CD4+CD25+Foxp3+ Tcell suppressor function [138]. This tolerogenic property induced upon DCs as a result of tryptophan deficiency is associated with GCN2 (general control non depressing 2) kinase mediated stress response pathway [138]. Although low tryptophan level may seem to hamper the normal inflammatory functions, its presence in autoimmunity balances the peripheral tolerance. Apart from IDO, DCs can also produce the enzyme heme oxygenase-1 (HO-1) that degrades heme and produces carbon monoxide (CO) which inhibits DC mediated inflammation [139]. The anti-inflammatory effect of HO-1 may be induced through Treg activation [140], although the exact mechanism still needs to be further studied. It was previously observed that PD-L1, IDO, and Fas expression is particularly enriched in tissue derived DCs as compared to their LN counterparts [42, 70]. Thus regulation of DC function in vivo may relate to tissue specific programing at sites of active self antigen release.
Cross-presentation ability of DCs
DCs are the main cross-presenting cells to present exogenous antigens on MHC Class I molecules to initiate cytotoxic T cell immune responses. However, cross-presentation not only induces an inflammatory response, but can also generate T cell tolerance. Under non-inflammatory or steady state conditions, constitutive cross presentation of self-antigens or tissue-associated antigens by immature CD8α+ DCs induce inactivation and/or deletion of proliferating CD8+ T cells [141–143]. As such cross presentation of self antigens limits the chances of autoimmunity. Various studies have documented that Batf3 dependent DC subsets are better cross-presenters than other subsets [144]. In tissues such as lungs and skin, cross-presentation is limited to DC subpopulations that express Langerin and CD103 molecules [145, 146]. These CD103 expressing DCs are also present in lymph nodes and their exact mechanism of cross presentation still remains unclear [147].The proposed mechanism of efficient cross-presentation by DCs has been associated with the recruitment of lower levels of lysosomal proteases in their phagosomes [148]. As a result, there is substantial decrease in the rate of degradation of internalized proteins. Another series demonstrated that DCs have a lower acidic pH in their endosomes and phagosomes than macrophages. This is due to both deficiency of V-ATPase assembly in lysosomes [149] as well as high recruitment of NADPH NOX2 in phagosomes and endosomes of immature DCs [150]. NOX2 generates high level of reactive oxygen species (ROS), which consumes the protons, and makes the endocytic compartment of DCs alkaline. This alkalinity results in degradation of internalized proteins at a lower rate than other phagocytes and facilitates peptide loading on MHC-I [147]. Indeed, NOX2 deficient DCs have observed fall in pH, increase in proteolysis and decrease in the effectiveness of cross-presentation. The role of DCs in inducing tolerance through cross-presentation however needs to be more precisely evaluated to develop therapeutic targets for autoimmune diseases. However Flt3L which selectively skews DC development towards an increase in cross-presenting DCs in spleen [85] or in LN [42] may be useful in the absence of an adjuvant to induce tolerance.
Interaction of DCs with other immune cells
Treg cells and DCs
Tregs that induce tolerance or immunosuppressive functions are broadly categorized based on origin. Natural Tregs (nTregs) that develop in the thymus during fetal development while adaptive Tregs (aTregs) originate in non lymphoid organs such as SLOs [3, 151]. Foxp3 transcription factor which guides the suppressive programming of T cells gets upregulated in naïve T cells upon self antigen recognition in the thymus during development [152]. These self-antigens either appear during development, appear in the post-natal period, or are transported to the thymus from the periphery by migrating DCs [122, 153]. Tregs express a plethora of anti-inflammatory cytokines such as TGF-b, IL-10, IL-35 and/or inhibitory receptors like CTLA4 (cytotoxic T-lymphocyte antigen 4), GITR (Glucocorticoid induced tumor necrosis factor receptor), LAG-3 (lymphocyte activation gene 3), CD73, CD39 among several others [154, 155].
The role of DCs in regulating the tolerogenic or inflammatory function of T cells is dependent on their maturation and antigen presenting capability. Immature DCs in the absence of any maturation signal induces naïve T cells to differentiate into the fork head box P3 (Foxp3) expressing regulatory T cell (Treg) or regulates the function of already differentiated Treg cells [156, 157]. In such cases, even the presence of antigens in the lymphoid organs did not alter the regulatory functions of Treg cells and led to antigen specific tolerance. To induce T cell effector responses, DCs provide three concomitant input signals which includes antigen display by MHC-II molecules, co-stimulatory signals such as CD40, CD80, CD86, CD275, OX40L and cytokines (IL-2, IL-1β, IL-12, IL-6, IL-8, IL-18) generated by DCs or other cells in the surrounding milieu [3, 158]. A major mechanism by which immature DCs induce their tolerogenic function is by presenting antigen to T cells without additional costimulation or cytokine environment. iDCs usually express low levels of MHC-II and costimulatory ligands and receptors. However, immature dendritic cells when exposed to high levels of TNF-α, IFN-γ or manipulated to cause E-cadherin inhibition or CCR7 upregulation develop into mature DCs phenotypically and not functionally. Such treated DCs induce naïve T cells to differentiate into adaptive Treg cells (aTregs) [3, 159]. Although CCR7 upregulation is predominant in matured DCs, it has been seen that few iDCs residing in the peripheral tissues such as skin also express CCR7 which allows them to migrate to nearby lymph nodes and favor Treg development [8, 160, 161]. In fact CCR7 deficient iDCs show impaired migration that leads to compromise in tolerance induction [7, 162]. Thus, tolerance induction is not only due to lack of costimulatory or cytokine signaling, but is also dependent on prior exposure of iDCs to tolerance inducing maturation signals. A different type of DCs called the exhausted DCs (exDCs) have also been observed in vitro after extended exposure to LPS signals. These exDCs unlike the matured DCs have lost their ability to induce T cell maturation, and instead produce anti-inflammatory cytokines such as IL-10 which elicitsTH2 and non-polarized memory T cell responses [163, 164]. These exDCs thus induces a state of endotoxin or cross-tolerance. However, the role of exDCs in inducing Treg responses in vivo still needs to be elucidated.
The anti-inflammatory cytokine IL-10 that is generated in the surrounding microenvironment in tolerogenic conditions by leukocytes and other structural cells such as intestinal epithelial cells (IECs) can induce immature DCs to develop into tolerance inducing DCs in the peripheral tissues. These DCs themselves acquire the ability to generate IL-10 and migrate to neighboring lymphoid organs where IL-10 produced by the DCs regulates the development and proliferation of Treg cells [3]. IL-10 induces the expression of specific genes such as SLAM (signaling lymphocytic activation molecule) and SOCS3 (suppressor of cytokine signaling 3). SOCS3 deficiency prolongs STAT1 and STAT3 activation after IL-6 stimulation but have normal activation of STAT1 after stimulation with interferon-gamma (IFN-gamma). Conversely, cells deficient in SOCS1 exhibit prolonged IFN-γ induced STAT1 activation suggesting that SOCS1 and SOCS3 display reciprocal functions [165]. IL-6 inhibits the suppression induced by Treg cells [166], thus highlighting that SOCS3 which negatively regulate IL-6 prolongs Treg function. IL-10R induced expression of SLAM activates src homology 2 domain containing protein tyrosine phosphatase 1 (SHP-1) that dephosphorylates the cytoplasmic tail of costimulatory receptors (CD28, CD2, ICOS) and deactivates their function [167, 168].
Severe cases of autoimmunity have been documented in animals lacking functional iDCs, possibly due to reduction in Treg function [3, 169]. Deficiency in TGF-β activating αvβ8 integrin or inactivation of TGF-β receptor signaling disrupts the tolerance inducing nature of DCs and causes autoimmune diseases [170, 171]. DCs and Tregs have also been shown to display a regulatory feedback loop in maintaining immune homeostasis. Loss of DCs either decreased Treg cells or reduced the expression of Foxp3 in Tregs, which in turn led to secretion of IFN-γ and IL-17 by Treg cells so as to restore the dying DC population [172].
B regulatory cells (Bregs) and DCs
Apart from inducing proliferation and activation of naïve and memory B cells, DCs have also been reported to induce regulatory B cell function and induce tolerance [131, 173]. Under inflammatory conditions, cDCs normally produce IL-12 that induces follicular B cells to develop into IL-12 and IFN-γ producing effector cells [174]. However in the process of maintaining peripheral tolerance, regulatory DCs can induce the differentiation of splenic B cells into IL-10 producing CD19hiFcγIIbhiBreg cells which inhibit CD4 T cell response [175]. Both bone-marrow and primary lung derived DCs have also been able to suppress IgE production in B cells by class switch recombination and hence play significant role in inducing B cell regulatory function [176].
NK-T cells and DCs
iNKT (invariant natural killer T) cells can also induce peripheral tolerance and dampen immune responses to prevent autoimmune disorders such as rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis and type 1 diabetes [18]. The contact of iNKT cells with immature DCs through CD1d receptor triggers phenotypic maturation of DCs towards tolerogenic type that promote Treg differentiation by secreting high levels of IL-10 and prevent autoimmunity [177].
Mast cells and DCs
Mast cells (MCs) that induce allergic reactions also contribute to immune homeostasis either through their mechanism of inducing Treg immunosuppressive functions or by conditioning immature DCs to migrate to regional lymph nodes where they suppress T cell inflammatory responses [178–180]. Local production of GM-CSF and TNF-α by MCs causes increased migration of tolerogenic DCs to the peripheral lymph nodes [178]. However, the direct role of MCs in inducing DC differentiation into the tolerogenic type is mediated after direct contact of MCs with iDCs. Interaction of PD-1 expressing MCs with PD-L1 and PD-L2 expressing iDCs led to increased synthesis of IDO in immature DCs which further activated the non-canonical NF-κβ and STAT-3 pathway in iDCs that are associated with induction of regulatory T cell functions [181].
DC targeted vaccines
Due to the inherent tolerogenic nature of DCs, they are often considered as therapeutic targets or vaccines in autoimmune and transplantation related diseases (GvHD). Exposure of DCs to pharmacological agents such as vitamin A, vitamin D3, rapamycin, cyclosporine A, dexamethasone or growth factors and cytokines like TGF-β, TNF, IL-10 induces the semi-mature type of DCs that become phenotypically matured but exhibit immunosuppressive functions [131]. Such tolerogenic DCs show marked prospects in reducing autoimmune disorders, increasing allograft survival and inhibit GvHD following stem cell transplantation.
Immunosuppressants such as glucocorticoids bind to glucocorticoid receptors (GR) in DCs and instruct glucocorticoid response elements (GRE) to negatively regulate the activity of the canonical NF-κβ family and the inflammatory cytokines [182]. DCs on acquiring maturation signals normally induce the phosphorylation of Iκβ (inhibitor of NF-κβ) by inhibitor kinase β (IKKβ) and releases p65 (or Rel-A) for nuclear translocation. However during tolerogenic induction, NIK (NFκB-inducing kinase) and IKKα are activated towards Rel-B dimer formation thereby resulting in the inhibition of the canonical NFκβ pathway [183, 184]. Glucocorticoids induce naïve DCs to convert into the tolerogenic type by operating through this NF-κβ pathway. Small molecules that act as antagonists of NF-κβ and IKKβ function also exhibit a pivotal role in inducing Treg suppressor functions through tolerance inducing DCs and thus assist in reducing autoimmune encephalomyelitis, allograft rejections and other autoimmune diseases [3, 185, 186]. In parallel to this view, deletion of IKKβ (an activator of NF-κβ) prevented the accumulation of migratory non-lymphoid tissue DCs (NLT-DCs) in lymph nodes and attenuated the formation of regulatory T cells in vivo, summarizing that a NF-κβ dependent pathway exists for the development of tolerance inducing DCs [11].
However, administration of immunosuppressants following transplantation is critical to attain therapeutic benefit as few suppressive agents can limit the total population of peripheral DC cells along with T cells. Use of costimulatory signaling inhibitors in this respect might serve as better option as they neither impair nor promote the DC mediated Treg activation. For example, patients treated with Belatacept or CTLA4-Ig (a B7-CD28 co-stimulaion blocking molecule) increased HLA-G plasma level in renal transplant patients than calcineurin inhibitor treated individuals. DCs served as the source of HLA-G in these patients and interaction of ILT-4 with HLA-G suppresses T cell stimulation [187]. Tolerogenic DCs that are antigen specific can also be generated in vitro and administered to patients with immune dysfunction. However, issues like timing, dose, route of administration and type of DC subsets to be used for tolerance induction needs to be carefully examined before designing such immunosuppressive regimens [21, 188]. CDX-301, a recombinant human Flt3L type molecule with similar amino acid sequence and activity has shown effective increase in peripheral monocytes, hematopoietic stem cells as well as in myeloid DCs and pDCs in healthy volunteers [189]. The potential of Flt3L as a therapy in transplantation has already been documented in murine models and with human trials still to be performed. [190]. While DCs are exploited for their inherent ability to induce tolerance in autoimmunity and graft transplantation, this activity remains a barrier to cancer therapy. In such cases, inhibitory molecules that will alleviate development of tolerance inducing DC and favor inflammation inducing mDC differentiation may prove beneficial. The role of tolerogenic DCs and immunogenic DCs as targeted vaccines in allograft survival and in cancer immunotherapy are summarized in Fig. 2.

Dendritic cells (DCs) as targeted vaccines in organ transplantation and in cancer immunotherapy. Differentiation of tolerogenic DCs from naïve DCs using pharmacological mediators, specific growth factors and cytokines may lead to anergy or deletion of alloreactive T cells while Tregs (regulatory T cells) get expanded and allows allograft survival through immune suppression. Conversely for cancer immunotherapy, mature DCs can be formed after exposure to stimulants like microbial products or DC antibodies (with tumor antigens) thereby allowing expansion of CD4+ T and CD8+ T cells, which then initiate anti-tumoral immunity
Future perspectives
Homeostasis encompasses the notion that the immune system must both activate a response and restore and preserve tolerance. Tolerogenic activity by DCs regulates dysfunction and likely prevents allergies, asthma, autoimmunity, and transplant rejection. The stable tolerogenic disposition of DCs must be maintained even under the influence of maturation signals. These DCs achieve suppressive functions by either inhibiting T cell based inflammation or by activating regulatory T cells. However, the existing knowledge on DC induced suppression of immune response and the factors regulating DC differentiation are still incomplete. Studies on the effect of cytokines in DC development may address key questions related to how the immune balance is maintained. In depth studies on the cross-presenting ability of DCs and tolerance induction might provide another facet of DC biology. Understanding the spatiotemporal regulation of DC populations during an in vivo response will also help to understand how these divergent roles are orchestrated. Modulating DC functions by cellular therapy or by novel drugs may serve as a better means of inducing immune suppression rather than the use of traditional immunosuppressants that elicit detrimental effects on immune cells and compromises immune defense against cancer or pathogenic attack. Tolerance inducing DC based therapy may thereby provide a safer approach of achieving immune suppression. Although this aim of achieving clinical translation is promising, undefined gaps in our knowledge of DC tolerance, require exploration before we can derive full benefit in therapies.
References
Steinman RM, Cohn ZA (1973) Identification of a novel cell type in peripheral lymphoid organs of mice. J Exp Med 137:1142–1162
Hammer GE, Ma A (2013) Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu Rev Immunol 31:743–791
Maldonado RA, von Andrian UH (2010) How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol 108:111–165
Banchereau J, Briere F, Caux C et al (2000) Immunobiology of dendritic cells. Annu Rev Immunol 18:767–811
Alvarez D, Vollmann EH, von Andrian UH (2008) Mechanisms and consequences of dendritic cell migration. Immunity 29:325–342
Forster R, Schubel A, Breitfeld D et al (1999) CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99:23–33
Martín-Fontecha A, Sebastiani S, Höpken UE et al (2003) Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med 198:615–621
Ohl L, Mohaupt M, Czeloth N et al (2004) CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 21:279–288
Reis e Sousa C (2006) Dendritic cells in a mature age. Nat Rev Immunol 6:476–483
Lutz MB, Schuler G (2002) Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol 23:445–449
Baratin M, Foray C, Demaria O et al (2015) Homeostatic NF-κβ signaling in steady-state migratory dendritic cells regulates immune homeostasis and tolerance. Immunity 42:627–639
Nirschl CJ, Anandasabapathy N (2016) Duality at the gate: skin dendritic cells as mediators of vaccine immunity and tolerance. Hum Vaccines Immunother 12:104–116
Dalod M, Chelbi R, Malissen B, Lawrence T (2014) Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J 33:1104–1116
Steinman RM, Banchereau J (2007) Taking dendritic cells into medicine. Nature 449:419–426
Cools N, Ponsaerts P, Van Tendeloo VFI, Berneman ZN (2007) Balancing between immunity and tolerance: an interplay between dendritic cells, regulatory T cells, and effector T cells. J Leukoc Biol 82:1365–1374
Hill M, Cuturi MC (2010) Negative vaccination by tolerogenic dendritic cells in organ transplantation. Curr Opin Organ Transplant 15:738–743
Steinman RM, Hawiger D, Nussenzweig MC (2003) Tolerogenic dendritic cells. Annu Rev Immunol 21:685–711
Manicassamy S, Pulendran B (2011) Dendritic cell control of tolerogenic responses. Immunol Rev 241:206–227
Morelli AE, Thomson AW (2007) Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol 7:610–621
Mendieta-Zerón H (2011) Developing immunologic tolerance for transplantation at the fetal stage. Immunotherapy 3:1499–1512
Ezzelarab M, Thomson AW (2011) Tolerogenic dendritic cells and their role in transplantation. Semin Immunol 23:252–263
Chopin M, Nutt SL (2014) Establishing and maintaining the Langerhans cell network. Semin Cell Dev Biol 41:1–7
Chopin M, Allan RS, Belz GT (2012) Transcriptional regulation of dendritic cell diversity. Front Immunol 3:26
Tamoutounour S, Guilliams M, MontananaSanchis F et al (2013) Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity 39:925–938
Merad M, Ginhoux F, Collin M (2008) Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol 8:935–947
Shortman K, Naik SH (2007) Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol 7:19–30
Cella M, Jarrossay D, Facchetti F et al (1999) Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 5:919–923
Naik SH, Metcalf D, van Nieuwenhuijze A et al (2006) Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol 7:663–671
Guilliams M, Ginhoux F, Jakubzick C et al (2014) Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14:571–578
Liu K, Victora G, Schwickert T et al (2009) In vivo analysis of dendritic cell development and homeostasis. Science 324:392–397
MartIn-Fontecha A, Sebastiani S, Höpken UE et al (2003) Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med 198:615–621
Saeki H, Moore AM, Brown MJ, Hwang ST (1999) Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol 162:2472–2475
Wolff K (1967) The fine structure of the Langerhans cell granule. J Cell Biol 35:468–473
Valladeau J, Ravel O, Dezutter-Dambuyant C et al (2000) Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 12:71–81
Kaplan DH, Igyártó BZ, Gaspari AA (2012) Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol 12:114–124
Bursch LS, Wang L, Igyarto B et al (2007) Identification of a novel population of Langerin + dendritic cells. J Exp Med 204:3147–3156
Nizza ST, Campbell JJ (2014) CD11b + Migratory dendritic cells mediate CD8 T cell cross-priming and cutaneous imprinting after topical immunization. PLoS One 9:e91054
Clausen BE, Stoitzner P (2015) Functional specialization of skin dendritic cell subsets in regulating T cell responses. Front Immunol 6:1–19
Igyártá BZ, Haley K, Ortner D et al (2011) Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper responses. Immunity 35:260–272
Hunger RE, Sieling PA, Ochoa MT et al (2004) Langerhans cells utilize CD1a and langerin to efficiently present nonpeptide antigens to T cells. J Clin Invest 113:701–708
De Jong A, Cheng T, Huang S et al (2014) CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Imunol 15:177–185
Anandasabapathy N, Feder R, Mollah S et al (2014) Classical Flt3L-dependent dendritic cells control immunity to protein vaccine. J Exp Med 211:1875–1891
Mollah SA, Dobrin JS, Feder RE et al (2014) Flt3L dependence helps define an uncharacterized subset of murine cutaneous dendritic cells. J Invest Dermatol 134:1265–1275
Henri S, Poulin LF, Tamoutounour S et al (2010) CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med 207:189–206
Tussiwand R, Everts B, Grajales-Reyes GE et al (2015) Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 42:916–928
Schlitzer A, McGovern N, Teo P et al (2013) IRF4 transcription factor-dependent CD11b + dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 38:970–983
Nakano H, Free ME, Whitehead GS et al (2012) Pulmonary CD103+ dendritic cells prime Th2 responses to inhaled allergens. Mucosal Immunol 5:53–65
Henri S, Vremec D, Kamath A et al (2001) The dendritic cell populations of mouse lymph nodes. J Immunol 167:741–748
Borkowski TA, Nelson AJ, Farr AG, Udey MC (1996) Expression of gp40, the murine homologue of human epithelial cell adhesion molecule (ep-CAM), by murine dendritic cells. Eur J Immunol 26:110–114
Nagao K, Ginhoux F, Leitner WW et al (2009) Murine epidermal Langerhans cells and langerin-expressing dermal dendritic cells are unrelated and exhibit distinct functions. Proc Natl Acad Sci U S A 106:3312–3317
Malissen B, Tamoutounour S, Henri S (2014) The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol 14:417–428
Haniffa M, Shin A, Bigley V et al (2012) Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103 + Nonlymphoid dendritic cells. Immunity 37:60–73
Nakano H, Lin KL, Yanagita M et al (2009) Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute TH1 immune responses. Nat Immunol 10:394–402
Leon B, Martinez del Hoyo G, Parrillas V et al (2004) Dendritic cell differentiation potential of mouse monocytes: monocytes represent immediate precursors of CD8- and CD8+ splenic dendritic cells. Blood 103:2668–2676
Hohl TM, Rivera A, Lipuma L et al (2009) Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 6:470–481
Cheong C, Matos I, Choi J et al (2010) Microbial stimulation fully differentiates monocytes to DC-SIGN / CD209+ dendritic cells for immune T cell areas. Cell 143:416–429
Chicha L (2004) Clonal type I interferon-producing and dendritic cell precursors are contained in both human lymphoid and myeloid progenitor populations. J Exp Med 200:1519–1524
Manz MG, Traver D, Miyamoto T et al (2001) Dendritic cell potentials of early lymphoid and myeloid progenitors. Blood 97:3333–3341
Wu L, D’Amico A, Hochrein H et al (2001) Development of thymic and splenic dendritic cell populations from different hemopoietic precursors. Blood 98:3376–3382
Traver D, Akashi K, Manz M et al (2000) Development of CD8α-positive dendritic cells from a common myeloid progenitor. Science 290:2152–2154
Manz MG, Traver D, Akashi K et al (2001) Dendritic cell development from common myeloid progenitors. Ann N Y Acad Sci 938:167–173
Lyman SD, Jacobsen SE (1998) C-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood 91:1101–1134
Onai N, Obata-Onai A, Schmid MA et al (2007) Identification of clonogenic common Flt3 + M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol 8:1207–1216
Naik SH, Sathe P, Park H-Y et al (2007) Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 8:1217–1226
Karsunky H, Merad M, Cozzio A et al (2003) Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med 198:305–313
Waskow C, Liu K, Darrasse-jèze G et al (2008) FMS-like tyrosine kinase 3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol 9:676–683
McKenna HJ (2001) Role of hematopoietic growth factors/flt3 ligand in expansion and regulation of dendritic cells. Curr Opin Hematol 8:149–154
Onai N, Obata-Onai A, Tussiwand R et al (2006) Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon-producing and dendritic cell development. J Exp Med 203:227–238
Fogg DK, Sibon C, Miled C et al (2006) A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311:83–87
Merad M, Sathe P, Helft J et al (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31:563–604
Bruno L, Seidl T, Lanzavecchia A (2001) Mouse pre-immunocytes as non-proliferating multipotent precursors of macrophages, interferon-producing cells, CD8α + and CD8α- dendritic cells. Eur J Immunol 31:3403–3412
Ginhoux F, Liu K, Helft J et al (2009) The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med 206:3115–3130
Bogunovic M, Ginhoux F, Helft J et al (2009) Origin of the lamina propria dendritic cell network. Immunity 31:513–525
Langlet C, Tamoutounour S, Henri S et al (2012) CD64 expression distinguishes monocyte-derived and conventional dendritic cells and reveals their distinct role during intramuscular immunization. J Immunol 188:1751–1760
Lewis KL, Caton ML, Bogunovic M et al (2011) Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35:780–791
Ginhoux F, Tacke F, Angeli V et al (2006) Langerhans cells arise from monocytes in vivo. Nat Immunol 7:265–273
Hoeffel G, Wang Y, Greter M et al (2012) Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 209:1167–1181
Mende I, Karsunky H, Weissman IL et al (2006) Flk2+ myeloid progenitors are the main source of Langerhans cells. Blood 107:1383–1390
Ginhoux F, Collin MP, Bogunovic M et al (2007) Blood-derived dermal langerin + dendritic cells survey the skin in the steady state. J Exp Med 204:3133–3146
Laouar Y, Welte T, Fu XY, Flavell RA (2003) STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity 19:903–912
Lutz MB, Suri RM, Niimi M et al (2000) Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur J Immunol 30:1813–1822
Vremec D, J. Lieschke G, R. Dunn A, et al. (1997) The influence of granulocyte/macrophage colony-stimulating factor on dendritic cell levels in mouse lymphoid organs. Eur J Immunol 27:40–44.
Hamilton JA (2008) Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 8:533–544
Lyman SD, Jacobsen SEW (1998) C-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood 91:1101–1134
Bozzacco L, Trumpfheller C, Huang Y et al (2010) HIV gag protein is efficiently cross-presented when targeted with an antibody towards the DEC-205 receptor in Flt3 ligand-mobilized murine DC. Eur J Immunol 40:36–46
Dudziak D, Kampfhorst AO, Heidkamp G et al (2007) Differential antigen processing by dendritic cell subsets in vivo. Science 315:107–111
Tarbell KV, Petit L, Zuo X et al (2007) Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med 204:191–201
Whartenby KA, Small D, Calabresi PA (2008) FLT3 inhibitors for the treatment of autoimmune disease. Expert Opin Investig drugs 17:1685–1692
Dhodapkar MV, Sznol M, Zhao B et al (2014) Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med 6:232ra51
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523:231–235
Salmon H, Idoyaga J, Rahman A et al (2016) Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44:924–938
Cisse B, Caton ML, Lehner M et al (2008) Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 135:37–48
Wu L, D’Amico A, Winkel KD et al (1998) RelB is essential for the development of myeloid-related CD8α- dendritic cells but not of lymphoid-related CD8α + dendritic cells. Immunity 9:839–847
Hildner K, Edelson BT, Purtha WE et al (2008) Batf3 deficiency reveals a critical role for CD8α + dendritic cells in cytotoxic T cell immunity. Science 322:1097–1100
Anderson KL, Perkin H, Surh CD et al (2000) Transcription factor PU.1 is necessary for development of thymic and myeloid progenitor-derived dendritic cells. J Immunol 164:1855–1861
Spooner CJ, Cheng JX, Pujadas E et al (2009) A recurrent network involving the transcription factors PU.1 and Gfi1 orchestrates innate and adaptive immune cell fates. Immunity 31:576–586
Rathinam C, Geffers R, Yucel R et al (2005) The transcriptional repressor Gfi1 controls STAT3-dependent dendritic cell development and function. Immunity 22:717–728
Satpathy AT, Kc W, Albring JC et al (2012) Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med 209:1135–1152
Meredith MM, Liu K, Darrasse-Jeze G et al (2012) Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med 209:1153–1165
Ichikawa E, Hida S, Omatsu Y et al (2004) Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor-2. Proc Natl Acad Sci U S A 101:3909–3914
Suzuki S, Honma K, Matsuyama T et al (2004) Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc Natl Acad Sci U S A 101:8981–8986
Tamura T, Tailor P, Yamaoka K et al (2005) IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol 174:2573–2581
Schiavoni G, Mattei F, Borghi P et al (2004) ICSBP is critically involved in the normal development and trafficking of Langerhans cells and dermal dendritic cells. Blood 103:2221–2228
Aliberti J, Schulz O, Pennington DJ et al (2003) Essential role for ICSBP in the in vivo development of murine CD8α + dendritic cells. Blood 101:305–310
Merad M, Manz MG (2009) Dendritic cell homeostasis. Blood 113:3418–3427
Hacker C, Kirsch RD, Ju X-S et al (2003) Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol 4:380–386
Nagasawa M, Schmidlin H, Hazekamp MG et al (2008) Development of human plasmacytoid dendritic cells depends on the combined action of the basic helix-loop-helix factor E2-2 and the Ets factor Spi-B. Eur J Immunol 38:2389–2400
Edelson BT, Kc W, Juang R et al (2010) Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha + conventional dendritic cells. J Exp Med 207:823–836
Fainaru O, Woolf E, Lotem J et al (2004) Runx3 regulates mouse TGF-beta-mediated dendritic cell function and its absence results in airway inflammation. EMBO J 23:969–979
Esashi E, Wang YH, Perng O et al (2008) The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity 28:509–520
Tussiwand R, Lee W-L, Murphy TL et al (2012) Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490:502–507
Wu L, Nichogiannopoulou A, Shortman K, Georgopoulos K (1997) Cell-autonomous defects in dendritic cell populations of Ikaros mutant mice point to a developmental relationship with the lymphoid lineage. Immunity 7:483–492
Allman D, Dalod M, Asselin-Paturel C et al (2006) Ikaros is required for plasmacytoid dendritic cell differentiation. Blood 108:4025–4034
Jackson JT, Hu Y, Liu R et al (2011) Id2 expression delineates differential checkpoints in the genetic program of CD8alpha + and CD103+ dendritic cell lineages. EMBO J 30:2690–2704
Platzer B, Jörgl A, Taschner S et al (2004) RelB regulates human dendritic cell subset development by promoting monocyte intermediates. Blood 104:3655–3663
Kingston D, Schmid MA, Onai N et al (2009) The concerted action of GM-CSF and Flt3-ligand on in vivo dendritic cell homeostasis. Blood 114:835–843
Greter M, Helft J, Chow A et al (2012) GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 36:1031–1046
Kel JM, Girard-Madoux MJH, Reizis B, Clausen BE (2010) TGF-β is required to maintain the pool of immature langerhans cells in the epidermis. J Immunol 185:3248–3255
Wang Y-G, Kim KD, Wang J et al (2005) Stimulating lymphotoxin beta receptor on the dendritic cells is critical for their homeostasis and expansion. J Immunol 175:6997–7002
Wu Q, Wang Y, Wang J et al (1999) The requirement of membrane lymphotoxin for the presence of dendritic cells in lymphoid tissues. J Exp Med 190:629–638
Qian C, Qian L, Yu Y et al (2013) Fas signal promotes the immunosuppressive function of regulatory dendritic cells via the ERK/β-catenin pathway. J Biol Chem 288:27825–27835
Bonasio R, Scimone ML, Schaerli P et al (2006) Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat Immunol 7:1092–1100
Proietto AI, van Dommelen S, Zhou P et al (2008) Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc Natl Acad Sci U S A 105:19869–19874
Watanabe N, Wang Y-H, Lee HK et al (2005) Hassall’s corpuscles instruct dendritic cells to induce CD4 + CD25+ regulatory T cells in human thymus. Nature 436:1181–1185
Besin G, Gaudreau S, Menard M et al (2008) Thymic stromal lymphopoietin and thymic stromal lymphopoietin-conditioned dendritic cells induce regulatory T-cell differentiation and protection of NOD mice against diabetes. Diabetes 57:2107–2117
Martin-Gayo E, Sierra-Filardi E, Corbi AL, Toribio ML (2010) Plasmacytoid dendritic cells resident in human thymus drive natural Treg cell development. Blood 115:5366–5375
Ziegler SF, Artis D (2010) Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol 11:289–293
Ardouin L, Luche H, Chelbi R et al (2016) Broad and largely concordant molecular changes characterize tolerogenic and immunogenic dendritic cell maturation in thymus and periphery. Immunity 45:305–318
McGrath MM, Najafian N (2012) The role of coinhibitory signaling pathways in transplantation and tolerance. Front Immunol 3:1–17
Yang J, Riella LV, Chock S et al (2011) The novel costimulatory programmed death ligand 1/B7.1 pathway is functional in inhibiting alloimmune responses in vivo. J Immunol 187:1113–1119
Li H, Shi B (2015) Tolerogenic dendritic cells and their applications in transplantation. Cell Mol Immunol 12:24–30
Morelli AE, Thomson AW (2003) Dendritic cells: regulators of alloimmunity and opportunities for tolerance induction. Immunol Rev 196:125–146
Munn DH, Sharma MD, Mellor AL (2004) Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J Immunol 172:4100–4110
Grohmann U, Orabona C, Fallarino F et al (2002) CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol 3:1097–1101
Mellor AL, Munn DH (2004) IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 4:762–774
Jürgens B, Hainz U, Fuchs D et al (2009) Interferon-γ-triggered indoleamine2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood 114:3235–3243
Grohmann U, Puccetti P (2002) The immunosuppressive activity of proinflammatory cytokines in experimental models : potential for therapeutic intervention in autoimmunity. Curr Drug Targets Inflamm Allergy 1:77–87
Brenk M, Scheler M, Koch S et al (2009) Tryptophan deprivation induces inhibitory receptors ILT3 and ILT4 on dendritic cells favoring the induction of human CD4 + CD25+ Foxp3+ T regulatory cells. J Immunol 183:145–154
Chauveau C, Rémy S, Royer PJ et al (2005) Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood 106:1694–1702
Yamashita K, Ollinger R, McDaid J et al (2006) Heme oxygenase-1 is essential for and promotes tolerance to transplanted organs. FASEB J 20:776–778
Luckashenak N, Schroeder S, Endt K et al (2008) Constitutive crosspresentation of tissue antigens by dendritic cells controls CD8+ T cell tolerance in vivo. Immunity 28:521–532
Bonifaz L, Bonnyay D, Mahnke K et al (2002) Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med 196:1627–1638
Belz GT, Behrens GMN, Smith CM et al (2002) The CD8 + dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J Exp Med 196:1099–1104
Segura E, Amigorena S (2015) Cross-Presentation in Mouse and Human Dendritic Cells, 1st ed. Adv Immunol
Bedoui S, Whitney PG, Waithman J et al (2009) Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol 10:488–495
del Rio M-L, Rodriguez-Barbosa J-I, Kremmer E, Förster R (2007) CD103− and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J Immunol 178:6861–6866
Amigorena S, Savina A (2010) Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol 22:109–117
Lennon-Duménil A-M, Bakker AH, Maehr R et al (2002) Analysis of protease activity in live antigen-presenting cells shows regulation of the phagosomal proteolytic contents during dendritic cell activation. J Exp Med 196:529–540
Trombetta ES, Ebersold M, Garrett MP, Mellman I (2003) Activation of lysosomal function during dendritic cell maturation. Science 299:1400–1403
Savina A, Jancic C, Hugues S et al (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126:205–218
Lio CWJ, Hsieh CS (2008) A two-step process for thymic regulatory T cell development. Immunity 28:100–111
Kim JM, Kim JM, Rudensky A, Rudensky A (2006) The role of the transcription factor Foxp3 in the development of regulatory T cells. Immunol Rev 212:86–98
Vigouroux S, Yvon E, Biagi E, Brenner MK (2004) Antigen-induced regulatory T cells. Blood 104:26–33
Vignali DAA, Collison LW, Workman CJ (2008) How regulatory T cells work. Nat Rev Immunol 8:523–532
Tang Q, Bluestone JA (2008) The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol 9:239–244
Ostroukhova M, Seguin-devaux C, Oriss TB et al (2004) Tolerance induced by inhaled antigen involves CD4 + T cells expressing membrane-bound TGF-β and FOXP3. J Clin Invest 114:28–38
Tsuji NM, Kosaka A (2008) Oral tolerance: intestinal homeostasis and antigen-specific regulatory T cells. Trends Immunol 29:532–540
Cronin SJF, Penninger JM (2007) From T-cell activation signals to signaling control of anti-cancer immunity. Immunol Rev 220:151–168
Tisch R (2010) Immunogenic versus tolerogenic dendritic cells: a matter of maturation. Int Rev Immunol 29:111–118
Hintzen G, Ohl L, del Rio M-L et al (2006) Induction of tolerance to innocuous inhaled antigen relies on a CCR7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J Immunol 177:7346–7354
Worbs T, Bode U, Yan S et al (2006) Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J Exp Med 203:519–527
Förster R, Davalos-Misslitz AC, Rot A (2008) CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol 8:362–371
Langenkamp A, Messi M, Lanzavecchia A, Sallusto F (2000) Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol 1:311–316
Langenkamp A, Casorati G, Garavaglia C et al (2002) T cell priming by dendritic cells: thresholds for proliferation, differentiation and death and intraclonal functional diversification. Eur J Immunol 32:2046–2054
Croker BA, Krebs DL, Zhang J-G et al (2003) SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 4:540–545
Pasare C, Medzhitov R (2003) Toll pathway-dependent blockade of CD4 + CD25+ T cell-mediated suppression by dendritic cells. Science 299:1033–1036
Veillette A, Latour S (2003) The SLAM family of immune-cell receptors. Curr Opin Immunol 15:277–285
Dennis KL, Blatner NR, Gounari F, Khazaie K (2013) Current status of interleukin-10 and regulatory T-cells in cancer. Curr Opin Oncol 25:637–645
Ohnmacht C, Pullner A, King SBS et al (2009) Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med 206:549–559
Laouar Y, Town T, Jeng D et al (2008) TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 105:10865–10870
Travis MA, Reizis B, Melton AC et al (2007) Loss of integrin αvβ8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449:361–365
Darrasse-Jeze G, Deroubaix S, Mouquet H et al (2009) Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med 206:1853–1862
Volchenkov R, Karlsen M, Jonsson R, Appel S (2013) Type 1 regulatory T cells and regulatory B cells induced by tolerogenic dendritic cells. Scand J Immunol 77:246–254
Lund FE (2008) Cytokine-producing B lymphocytes - key regulators of immunity. Curr Opin Immunol 20:332–338
Qian L, Qian C, Chen Y et al (2012) Regulatory dendritic cells program B cells to differentiate into CD19hiFcγIIbhi regulatory B cells through IFN-β and CD40L. Blood 120:581–591
Obayashi K, Doi T, Koyasu S (2007) Dendritic cells suppress IgE production in B cells. Int Immunol 19:217–226
Caielli S, Conforti-Andreoni C, Di Pietro C et al (2010) On/off TLR signaling decides proinflammatory or tolerogenic dendritic cell maturation upon CD1d-mediated interaction with invariant NKT cells. J Immunol 185:7317–7329
de Vries VC, Pino-Lagos K, Nowak EC et al (2011) Mast cells condition dendritic cells to mediate allograft tolerance. Immunity 35:550–561
Leveson-Gower DB, Sega EI, Kalesnikoff J et al (2013) Mast cells suppress murine GVHD in a mechanism independent of CD4 + CD25+ regulatory T cells. Blood 122:3659–3665
Lu L-F, Lind EF, Gondek DC et al (2006) Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 442:997–1002
Rodrigues CP, Ferreira ACF, Pinho MP et al (2016) Tolerogenic IDO(+) dendritic cells are induced by PD-1-expressing mast cells. Front Immunol 7:9
Ito K, Chung KF, Adcock IM (2006) Update on glucocorticoid action and resistance. J Allergy Clin Immunol 117:522–543
Puccetti P, Grohmann U (2007) IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol 7:817–823
Bonizzi G, Karin M (2004) The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25:280–288
Zhang X, Li M, Lian D et al (2008) Generation of therapeutic dendritic cells and regulatory T cells for preventing allogeneic cardiac graft rejection. Clin Immunol 127:313–321
Iruretagoyena MI, Lezana JP, Hermoso M et al (2006) Inhibition of nuclear factor-kappa B enhances the capacity of immature dendritic cells to induce antigen-specific tolerance in experimental autoimmune encephalomyelitis. J Pharmacol Exp Ther 318:59–67
Bahri R, Naji A, Menier C et al (2009) Dendritic cells secrete the immunosuppressive HLA-G molecule upon CTLA4-Ig treatment: implication in human renal transplant acceptance. J Immunol 183:7054–7062
Hu J, Wan Y (2010) Tolerogenic dendritic cells and their potential applications. Immunology 132:307–314
Anandasabapathy N, Breton G, Hurley A et al (2015) Efficacy and safety of CDX-301, recombinant human Flt3L, at expanding dendritic cells and hematopoietic stem cells in healthy human volunteers. Bone Marrow Transplant 50:924–930
He S, Chu J, Vasu S et al (2014) FLT3L and plerixafor combination increases hematopoietic stem cell mobilization and leads to improved transplantation outcome. Biol Blood Marrow Transplant 20:309–313
Acknowledgements
This work was supported by the Melanoma Research Alliance-Brigham and Women’s Hospital (BWH) combined grant (to N.A.) and by the BWH Department of Dermatology. This work was also supported by a Clinic and Laboratory Integration Program (CLIP) award from the Cancer Research Institute (C.L.S), National Institute of Arthritis and Musculoskeletal and Skin Disease K23 AR063461-01A1, and AR070234-01A1 (to N.A.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare financial grant support for research from Sanofi Pharmaceuticals for pre-clinical project development.
Additional information
This article is a contribution to the special issue on Dendritic Cell Subsets and Immune-mediated Diseases - Guest Editor: Francisco Quintana
Rights and permissions
About this article

Cite this article
Devi, K...S.P., Anandasabapathy, N. The origin of DCs and capacity for immunologic tolerance in central and peripheral tissues. Semin Immunopathol 39, 137–152 (2017). https://doi.org/10.1007/s00281-016-0602-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-016-0602-0