
Abstract
Background
The current coronavirus disease 2019 (COVID-19) outbreak caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has emerged in Wuhan, China, and has rapidly become a global challenge, creating major challenges to health systems in almost every country in the world it has turned into a pandemic. COVID-19 poses a risky clinical situation that can range from mild illness to severe respiratory failure, requiring admission to intensive care.
Main body
It is known that SARS-CoV-2 infection causes a cytokine storm in some critically ill patients. However, more and more evidence showed that there is a dramatic increase in cytokine levels in patients diagnosed with COVID-19. Midkine (MK) is involved in various physiological and pathological processes, which some of them are desired and beneficial such as controlling tissue repair and antimicrobial effects, but some others are harmful such as promoting inflammation, carcinogenesis, and chemoresistance. Also, MK is expressed in inflammatory cells and released by endothelial cells under hypoxic conditions.
Conclusions
Considering all this information, there are strong data that midkine, an important cytokine known to increase in inflammatory diseases, may be overexpressed in patients who are positive for COVID-19. The overexpression of MK reveals a picture leading to fibrosis and damage in the lung. Therefore, questions arise about how the expression of MK changes in COVID-19 patients and can we use it as an inflammation biomarker or in the treatment protocol in the future.
Similar content being viewed by others

Background
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is responsible for coronavirus disease 2019 (COVID-19), not only created an extremely important worldwide problem in health management, but also generated disturbances in societies’ economy and social and cultural structure [1]. Although, the majority of COVID-19-infected patients are asymptomatic or have “mild” symptoms, such as fever, fatigue, and dry cough, some patients can progress to severe disease manifestations, including pneumonia, pulmonary edema, vascular hyper-permeability, and acute respiratory distress syndrome (ARDS) [2]. In addition, very serious conditions such as respiratory failure, septic shock, multiple organ failure, and even death occur in approximately 5% of patients [3]. In such seriously ill COVID-19 patients, the production of pro-inflammatory mediators and cytokines, including tumor necrosis factor alpha (TNF-α), interleukin (IL) 1 beta (IL-1β), IL-6, IL-18, and interferon gamma (IFNϒ), is abnormally increased that result in a “cytokine storm,” causing a diffuse alveolar damage [4, 5]. Some symptoms attributed to these cytokines are fever, chills, headaches, dizziness, and fatigue. Additionally, these cytokines can contribute to severe pathologies such as cardiomyopathy, lung injury, and septic shock.
Midkine (MK), a low molecular-weight growth factor (a heparin-binding cytokine), is strongly expressed during embryogenesis, whereas it is downregulated to relatively low levels in healthy adults [6]. However, MK is overexpressed back in various pathologies, including inflammatory diseases and many malignancies [7, 8]. It is involved in several physiological and/or pathological cell functions, including survival, reproduction, repair, and growth, and has chemotactic activity as well as proinflammatory actions [9, 10]. Since the overproduction of pro-inflammatory mediators and cytokines plays important role in the pathogenesis of COVID-19, we propose that besides other cytokines, MK may be also overexpressed in SARS-CoV-2 infection and participate in or modulate the course of COVID-19. MK has chemotactic actions, resulting in the accumulation of inflammatory cells, such as macrophages and neutrophils, which in turn aggravate the inflammatory response [11]. Additionally, in the context of COVID-19, which uses angiotensin-converting enzyme 2 (ACE-2) receptor for its clinical manifestations, MK activates angiotensin-converting enzyme (ACE), leading to higher concentrations of Ang II, leading to impaired functions of the alveoli [12]. These and other pathophysiological functions of MK and the possible relationship to SARS-CoV-2 infection are discussed in this review.
Main text
Inflammation and cytokine storm in SARS-CoV-2-infected patients
Actually, cytokines attend to the normal immune response to infectious agents in healthy individuals. However, several pathogenic infections, including SARS-CoV-2, are often associated with an excessive cytokine release called cytokine storm, which result in tissue damage [13]. At the time by the binding of SARS-CoV-2 to ACE-2, which serves as a functional receptor for SARS-CoV-2, the immune system of patients becomes activated [14, 15]. This, in turn, induces the accumulation of inflammatory cells with subsequent production of pro-inflammatory cytokines and chemokines at the infection area, which can also spread to many extra-pulmonary organs [2].
Association and interaction between RAS and COVID-19
In physiological conditions, after the biotransformation of angiotensinogen to angiotensin I (Ang I) by renin, the ACE converts Ang I to angiotensin II (Ang II), which may contribute to inflammation, fibrosis, tissue damage, and edema in the lungs. On the other hand, Ang II is converted by ACE-2 to angiotensin (1–7), which has anti-inflammatory and vasodilatory properties that balanced the effects of Ang II. However, in COVID-19, the SARS-CoV-2 interacts with the RAS through ACE-2, which is necessary for the entry of the virus into pneumocytes as well as its replication [14]. Consequently, the expression of ACE-2 is downregulated by SARS-CoV-2, causing overactivation of renin-angiotensin-system (RAS) that results in increased pulmonary vasoconstriction, edema, hypoxia, and lung damage in COVID-19 patients [16, 17].
The effect of hypoxia and activation of hypoxia-inducible factor 1α (HIF-1α) in COVID-19
The lung, an organ exposed to a high amount of oxygen, is subject to various infections, including SARS-CoV-2. Due to fluid accumulation in the alveoli, as a result of SARS-CoV-2 invasion, the effectiveness of air exchange decreases dramatically, which then results in hypoxemia and subsequently ARDS [18, 19]. This hypoxic effect of viral occupation contributes to several pathophysiological changes in the lung and is also involved in all stages of COVID-19. Hypoxia, a powerful inflammatory stimulant, is also induced in inflammatory conditions [20–24]. Besides leading to the high amount of pro-inflammatory cytokines and the creation of cytokine storm on the infection region, hypoxia triggered simultaneously several pathophysiological processes, including induction of hypoxia-inducible factor-1α (HIF-1α). HIF-1α is expressed in certain cell types, including immune cells, and regulates cell metabolism and inflammation [25, 26]. Under normal pressure of oxygen in the bloodstream, the expression of HIF-1α caused by phagocytic cells, such as neutrophils and macrophages, is low. However, in infection sites, they increase HIF-1α expression, which in turn stimulates the expression of several pro-inflammatory cytokines [27, 28]. Owing to its proinflammatory properties, it has been suggested that inhibition of HIF-1α activity can reduce the SARS-CoV-2-related inflammation and relieve the severity of COVID-19 [29].
NETosis, oxidative stress, ROS, and COVID-19
Several pathophysiological mechanisms occur simultaneously when SARS-CoV-2 binds to ACE-2 in the lung. Primarily, monocytes recruited into the alveolar space secrete pro-inflammatory cytokines which are responsible for cytokine storm. Additionally, recruited macrophages release also cytokines and chemokines that augmented capillary permeability, pulmonary edema, and followed by neutrophil recruitment. Increased neutrophil invasion leads to the release of neutrophil extracellular traps (NETs) that are intracellular contents such as DNA, histones, and proteins. Several studies showed that this process, called NETosis, is associated closely with COVID-19 [30–32]. The excessive neutrophil degranulation precipitates lung injury and damages the alveolar-capillary barrier. In addition, NETosis is associated with increased levels of intracellular reactive oxygen species (ROS) of neutrophils [33]. But on the other hand, ROS can destroy pathogens directly by causing oxidative damage as well as indirectly, by inducing pathogen elimination via NETs formation in neutrophils [34].
Possible relationship between COVID-19 pathogenesis and MK
MK is involved in various physiological and pathological processes, which some of them are desired and beneficial such as controlling tissue repair and antimicrobial effects, but some others are harmful such as promoting inflammation, carcinogenesis, and chemo-resistance [35–37]. Although animal models of myocardial infarction have shown a protective role of MK for the injured cardiac tissue by its anti-apoptotic effect and its role in angiogenesis [38, 39], most studies showed that MK is harmful under chronic inflammatory conditions [40]. MK is a cytokine with strong pro-inflammatory characteristics, causing macrophage and neutrophil recruitment to the inflamed region and interact with other growth factors and cytokines, particularly TNFα. Furthermore, MK mediates and exhibits enhancement of fibrinolytic activity, which are important processes in the initial stage of inflammatory responses of various pathologies [11, 41, 42]. The expression of MK is induced by TNFα, an important component of cytokine storm in COVID-19, and vice versa [43, 44]. Therefore, it is highly possible that MK is contributed to the cytokine invasion and interacts with other cytokines and chemokines in SARS-CoV-2 infections. This pro-inflammatory effect and subsequently occurring several pathophysiological processes, in that MK is involved, could be detrimental rather than protective. In an animal study, it has been found that the expression of MK was induced in the lung endothelium of micro-vessels and alveolar-capillary endothelial cells by oxidative stress and upregulated by ACE, which hydrolyses Ang I to form Ang II. Ang II induces NADPH oxidase (Nox) expression, and the increased expression of Nox, which initiated ROS production, induced further oxidative stress, and subsequently accelerated MK and ACE generation [44]. Hypoxia, resulting from cytokine storm, inflammation, and other molecular mechanisms, including induction of hypoxia-inducible factor 1-α (HIF1-α), is an important component of SARS-CoV-2 infection [28, 45]. It has been shown that MK expression increased during hypoxia by the binding of HIF1-α to hypoxia-responsive elements located in the MK promoter [12, 46]. These findings suggest that a close relationship exists between MK, hypoxia, and HIF1-α, by enhancing each other’s pathophysiological effects. An important issue of COVID-19 is the development of ARDS, a result that may cause severe pulmonary injury and even death. Several studies demonstrated that MK is closely involved in the pathogenesis of ARDS. To clarify the possible role of the MK signaling pathway in ARDS, Zhang et al. showed that exposure to a mechanical stretch of lung epithelial cells led to an epithelial-mesenchymal transition profile associated with increased expression of ACE which was attenuated by silencing MK. Furthermore, they found out that the plasma levels of MK were higher in patients with ARDS than in healthy subjects [47]. Similarly, in idiopathic pulmonary fibrosis patients, the serum MK level was also higher compared to healthy subjects, supporting the role of MK in the development of ARDS [48]. Moreover, in midkine-deficient mice, low expression of collagen and α-smooth muscle actin, as well as a low value for the pathological lung fibrosis score, was detected. Thus, MK participates to the progression of pulmonary fibrosis, mainly by regulating inflammatory cell migration into the lung and augmenting TNF-α and tumor growth factor β (TGF-β) expression [48].
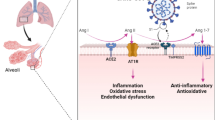
Table 1 and Fig. 1 summarizes the possible pathophysiological mechanisms of MK in relation to COVID-19.

Schematic representation of hypothetic actions of midkine in relation to SARS-CoV-2 infection. SARS CoV-2, severe acute respiratory syndrome coronavirus-2; RAS, renin-angiotensin system; Ang, angiotensin; ROS, reactive oxygen species; NETs, neutrophil extracellular traps; HIF-1, hypoxia-inducible factor-1
Conclusions
Current clinical observations indicate that SARS-CoV-2 infection can range from an unapparent non-symptomatic infection to severe pulmonary damage and multiorgan failure. The excessive secretion of several cytokines is closely related to the development of clinical symptoms in COVID-19 patients. This abnormal and uncontrolled production of cytokines has been observed in most of the patients with SARS-CoV-2-related pneumonia, increasing the progression of COVID-19 and mortality. Furthermore, the overproduction of inflammatory cytokines contributes to acute lung injury and ARDS. Therefore, to improve the outcome and reduce mortality of SARS-CoV-2, cytokine monitoring is recommended for the diagnosis and treatment of COVID-19. MK, which is a cytokine and growth factor and is significantly upregulated upon exposure to various harmful stimuli, including inflammation, is likely to accompany the cytokine attack that occurs in SARS-CoV-2 infections. Drugs targeting MK, such as antibodies and RNA aptamers that suppressed the generation and/or action of MK, may be a useful part of therapeutic modalities applied in the management of COVID-19.
Availability of data and materials
Not applicable to this manuscript.
Abbreviations
- COVID-19:
-
Coronavirus disease 2019
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- MK:
-
Midkine
- ARDS:
-
Acute respiratory distress syndrome
- TNF-α:
-
Tumor necrosis factor alpha
- IL:
-
Interleukin
- ACE-2:
-
Angiotensin-converting enzyme 2
- ACE:
-
Angiotensin-converting enzyme
- Ang I:
-
Angiotensin I
- Ang II:
-
Angiotensin II
- HIF-1α:
-
Hypoxia-inducible factor 1α
- NETs:
-
Neutrophil extracellular traps
- ROS:
-
Reactive oxygen species
- Nox:
-
NADPH oxidase
- TGF-β:
-
Tumor growth factor β
References
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 382(8):727–733. https://doi.org/10.1056/NEJMoa2001017
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China [published correction appears in Lancet. 2020 Jan 30;:]. Lancet. 2020;395(10223):497-506. doi:https://doi.org/10.1016/S0140-6736(20)30183-5
Williamson EJ, Walker AJ, Bhaskaran K, Bacon S, Bates C, Morton CE, Curtis HJ, Mehrkar A, Evans D, Inglesby P, Cockburn J, McDonald HI, MacKenna B, Tomlinson L, Douglas IJ, Rentsch CT, Mathur R, Wong AYS, Grieve R, Harrison D, Forbes H, Schultze A, Croker R, Parry J, Hester F, Harper S, Perera R, Evans SJW, Smeeth L, Goldacre B (2020) Factors associated with COVID-19-related death using OpenSAFELY. Nature. 584(7821):430–436. https://doi.org/10.1038/s41586-020-2521-4
Gao YM, Xu G, Wang B, Liu BC (2021) Cytokine storm syndrome in coronavirus disease 2019: a narrative review. J Intern Med. 289(2):147–161. https://doi.org/10.1111/joim.13144
Ye Q, Wang B, Mao J (2020) The pathogenesis and treatment of the ‘cytokine storm’ in COVID-19. J Infect. 80(6):607–613. https://doi.org/10.1016/j.jinf.2020.03.037
Kadomatsu K, Tomomura M, Muramatsu T (1988) cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem Biophys Res Commun. 151(3):1312–1318. https://doi.org/10.1016/s0006-291x(88)80505-9
Weckbach LT, Preissner KT, Deindl E (2018) The role of midkine in arteriogenesis, involving mechanosensing, endothelial cell proliferation, and vasodilation. Int J Mol Sci. 19(9):2559. https://doi.org/10.3390/ijms19092559
Filippou PS, Karagiannis GS, Constantinidou A (2020) Midkine (MDK) growth factor: a key player in cancer progression and a promising therapeutic target. Oncogene. 39(10):2040–2054. https://doi.org/10.1038/s41388-019-1124-8
Muramatsu T (2002) Midkine and pleiotrophin: two related proteins involved in development, survival, inflammation and tumorigenesis. J Biochem. 132(3):359–371. https://doi.org/10.1093/oxfordjournals.jbchem.a003231
Muramatsu T (2010) Midkine, a heparin-binding cytokine with multiple roles in development, repair and diseases. Proc Jpn Acad Ser B Phys Biol Sci. 86(4):410–425. https://doi.org/10.2183/pjab.86.410
Takada T, Toriyama K, Muramatsu H, Song XJ, Torii S, Muramatsu T (1997) Midkine, a retinoic acid-inducible heparin-binding cytokine in inflammatory responses: chemotactic activity to neutrophils and association with inflammatory synovitis. J Biochem. 122(2):453–458. https://doi.org/10.1093/oxfordjournals.jbchem.a021773
Hobo A, Yuzawa Y, Kosugi T, Kato N, Asai N, Sato W, Maruyama S, Ito Y, Kobori H, Ikematsu S, Nishiyama A, Matsuo S, Kadomatsu K (2009) The growth factor midkine regulates the renin-angiotensin system in mice. J Clin Invest. 119(6):1616–1625. https://doi.org/10.1172/JCI37249
Channappanavar R, Perlman S (2017) Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 39(5):529–539. https://doi.org/10.1007/s00281-017-0629-x
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 426(6965):450–454. https://doi.org/10.1038/nature02145
Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, Herridge M, Randolph AG, Calfee CS (2019) Acute respiratory distress syndrome. Nat Rev Dis Primers. 5(1):18. https://doi.org/10.1038/s41572-019-0069-0
Wan Y, Shang J, Graham R, Baric RS, Li F (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 94(7):e00127–e00120. https://doi.org/10.1128/JVI.00127-20
Hoffmann M, Kleine-Weber H, Schroeder S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271–280.e8. https://doi.org/10.1016/j.cell.2020.02.052
Lee KY (2017) Pneumonia, acute respiratory distress syndrome, and early immune-modulator therapy. Int J Mol Sci. 18(2):388. https://doi.org/10.3390/ijms18020388
Sarkar M, Niranjan N, Banyal PK (2017) Mechanisms of hypoxemia [published correction appears in Lung India. 2017;34(2):220]. Lung India. 34(1):47–60. https://doi.org/10.4103/0970-2113.197116
Gonzalez NC, Wood JG (2010) Alveolar hypoxia-induced systemic inflammation: what low PO(2) does and does not do. Adv Exp Med Biol. 662:27–32. https://doi.org/10.1007/978-1-4419-1241-1_3
Eltzschig HK, Carmeliet P (2011) Hypoxia and inflammation. N Engl J Med. 364(7):656–665. https://doi.org/10.1056/NEJMra0910283
Fröhlich S, Boylan J, McLoughlin P (2013) Hypoxia-induced inflammation in the lung: a potential therapeutic target in acute lung injury? Am J Respir Cell Mol Biol. 48(3):271–279. https://doi.org/10.1165/rcmb.2012-0137TR
Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA, Kourembanas S (2001) Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci U S A. 98(15):8798–8803. https://doi.org/10.1073/pnas.161272598
Watts ER, Walmsley SR (2019) Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol Med. 25(1):33–46. https://doi.org/10.1016/j.molmed.2018.10.006
Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation [published correction appears in Cell. 2003 May 2;113(3):419]. Cell. 112(5):645–657. https://doi.org/10.1016/s0092-8674(03)00154-5
Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 201(1):105–115. https://doi.org/10.1084/jem.20040624
Nizet V, Johnson RS (2009) Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 9(9):609–617. https://doi.org/10.1038/nri2607
Jahani M, Dokaneheifard S, Mansouri K (2020) Hypoxia: a key feature of COVID-19 launching activation of HIF-1 and cytokine storm. J Inflamm (Lond). 17(1):33. https://doi.org/10.1186/s12950-020-00263-3
Serebrovska ZO, Chong EY, Serebrovska TV, Tumanovska LV, Xi L (2020) Hypoxia, HIF-1α, and COVID-19: from pathogenic factors to potential therapeutic targets. Acta Pharmacol Sin. 41(12):1539–1546. https://doi.org/10.1038/s41401-020-00554-8
Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair CN, Weber A, Barnes BJ, Egeblad M, Woods RJ, Kanthi Y, Knight JS (2020) Neutrophil extracellular traps in COVID-19. JCI Insight. 5(11):e138999. https://doi.org/10.1172/jci.insight.138999
Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, Schneider AH, Caetité D, Tavares LA, Paiva IM, Rosales R, Colón D, Martins R, Castro IA, Almeida GM, Lopes MIF, Benatti MN, Bonjorno LP, Giannini MC, Luppino-Assad R, Almeida SL, Vilar F, Santana R, Bollela VR, Auxiliadora-Martins M, Borges M, Miranda CH, Pazin-Filho A, da Silva LLP, Cunha LD, Zamboni DS, Dal-Pizzol F, Leiria LO, Siyuan L, Batah S, Fabro A, Mauad T, Dolhnikoff M, Duarte-Neto A, Saldiva P, Cunha TM, Alves-Filho JC, Arruda E, Louzada-Junior P, Oliveira RD, Cunha FQ (2020) SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. 217(12):e20201129. https://doi.org/10.1084/jem.20201129
Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, dos Reis MC, de Castro GMM, da Silva Fontes Y, Todeschini AR, Freire-de-Lima L, Decoté-Ricardo D, Ferreira-Pereira A, Freire-de-Lima CG, Barroso SPC, Takiya C, Conceição-Silva F, Savino W, Morrot A (2020) The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep. 10(1):19630. https://doi.org/10.1038/s41598-020-76781-0
Reshi ML, Su YC, Hong JR (2014) RNA viruses: ROS-mediated cell death. Int J Cell Biol. 2014:467452–467416. https://doi.org/10.1155/2014/467452
Nguyen GT, Green ER, Mecsas J (2017) Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol. 7:373. https://doi.org/10.3389/fcimb.2017.00373
Ikutomo M, Sakakima H, Matsuda F, Yoshida Y (2014) Midkine-deficient mice delayed degeneration and regeneration after skeletal muscle injury. Acta Histochem. 116(2):319–326. https://doi.org/10.1016/j.acthis.2013.08.009
Svensson SL, Pasupuleti M, Walse B, Malmsten M, Mörgelin M, Sjögren C, Olin AI, Collin M, Schmidtchen A, Palmer R, Egesten A (2010) Midkine and pleiotrophin have bactericidal properties: preserved antibacterial activity in a family of heparin-binding growth factors during evolution. J Biol Chem. 285(21):16105–16115. https://doi.org/10.1074/jbc.M109.081232
Kang HC, Kim IJ, Park HW, Jang SG, Ahn SA, Yoon SN, Chang HJ, Yoo BC, Park JG (2007) Regulation of MDK expression in human cancer cells modulates sensitivities to various anticancer drugs: MDK overexpression confers to a multi-drug resistance. Cancer Lett. 247(1):40–47. https://doi.org/10.1016/j.canlet.2006.03.017
Takenaka H, Horiba M, Ishiguro H, Sumida A, Hojo M, Usui A, Akita T, Sakuma S, Ueda Y, Kodama I, Kadomatsu K (2009) Midkine prevents ventricular remodeling and improves long-term survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 296(2):H462–H469. https://doi.org/10.1152/ajpheart.00733.2008
Sumida A, Horiba M, Ishiguro H, Takenaka H, Ueda N, Ooboshi H, Opthof T, Kadomatsu K, Kodama I (2010) Midkine gene transfer after myocardial infarction in rats prevents remodelling and ameliorates cardiac dysfunction. Cardiovasc Res. 86(1):113–121. https://doi.org/10.1093/cvr/cvp386
Maruyama K, Muramatsu H, Ishiguro N, Muramatsu T (2004) Midkine, a heparin-binding growth factor, is fundamentally involved in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 50(5):1420–1429. https://doi.org/10.1002/art.20175
Wang J, Takeuchi H, Sonobe Y, Jin S, Mizuno T, Miyakawa S, Fujiwara M, Nakamura Y, Kato T, Muramatsu H, Muramatsu T, Suzumura A (2008) Inhibition of midkine alleviates experimental autoimmune encephalomyelitis through the expansion of regulatory T cell population. Proc Natl Acad Sci U S A. 105(10):3915–3920. https://doi.org/10.1073/pnas.0709592105
Sato W, Kadomatsu K, Yuzawa Y, Muramatsu H, Hotta N, Matsuo S, Muramatsu T (2001) Midkine is involved in neutrophil infiltration into the tubulointerstitium in ischemic renal injury. J Immunol. 167(6):3463–3469. https://doi.org/10.4049/jimmunol.167.6.3463
Shindo E, Nanki T, Kusunoki N, Shikano K, Kawazoe M, Sato H, Kaneko K, Muraoka S, Kaburaki M, Akasaka Y, Shimada H, Hasunuma T, Kawai S (2017) The growth factor midkine may play a pathophysiological role in rheumatoid arthritis. Mod Rheumatol. 27(1):54–59. https://doi.org/10.1080/14397595.2016.1179860
You Z, Dong Y, Kong X, Beckett LA, Gandour-Edwards R, Melamed J (2008) Midkine is a NF-kappaB-inducible gene that supports prostate cancer cell survival. BMC Med Genomics. 1(1):6. https://doi.org/10.1186/1755-8794-1-6
Taniguchi-Ponciano K, Vadillo E, Mayani H, Gonzalez-Bonilla CR, Torres J, Majluf A, Flores-Padilla G, Wacher-Rodarte N, Galan JC, Ferat-Osorio E, Blanco-Favela F, Lopez-Macias C, Ferreira-Hermosillo A, Ramirez-Renteria C, Peña-Martínez E, Silva-Román G, Vela-Patiño S, Mata-Lozano C, Carvente-Garcia R, Basurto-Acevedo L, Saucedo R, Piña-Sanchez P, Chavez-Gonzalez A, Marrero-Rodríguez D, Mercado M (2021) Increased expression of hypoxia-induced factor 1α mRNA and its related genes in myeloid blood cells from critically ill COVID-19 patients. Ann Med. 53(1):197–207. https://doi.org/10.1080/07853890.2020.1858234
Reynolds PR, Mucenski ML, Le Cras TD, Nichols WC, Whitsett JA (2004) Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J Biol Chem. 279(35):37124–37132. https://doi.org/10.1074/jbc.M405254200
Zhang R, Pan Y, Fanelli V, Wu S, Luo AA, Islam D, Han B, Mao P, Ghazarian M, Zeng W, Spieth PM, Wang D, Khang J, Mo H, Liu X, Uhlig S, Liu M, Laffey J, Slutsky AS, Li Y, Zhang H (2015) Mechanical stress and the induction of lung fibrosis via the midkine signaling pathway. Am J Respir Crit Care Med. 192(3):315–323. https://doi.org/10.1164/rccm.201412-2326OC
Misa K, Tanino Y, Wang X et al (2017) Involvement of midkine in the development of pulmonary fibrosis. Physiol Rep 5(16):e13383. https://doi.org/10.14814/phy2.13383
Acknowledgements
No acknowledgements.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
Both authors (SK and AŞA) equally contributed to the preparation of this review article. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable to this manuscript.
Consent for publication
Not applicable to this manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ketenci, S., Aynacıoğlu, A.Ş. The growth factor/cytokine midkine may participate in cytokine storm and contribute to the pathogenesis of severe acute respiratory syndrome coronavirus 2-infected patients. Egypt J Bronchol 15, 42 (2021). https://doi.org/10.1186/s43168-021-00087-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43168-021-00087-6