
Abstract
Background
Coronavirus disease 2019 (COVID-19) is caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2, previously named 2019-nCov), a novel coronavirus that emerged in China in December 2019 and was declared a global pandemic by World Health Organization by March 11th, 2020. Severe manifestations of COVID-19 are caused by a combination of direct tissue injury by viral replication and associated cytokine storm resulting in progressive organ damage.
Discussion
We reviewed published literature between January 1st, 2000 and June 30th, 2020, excluding articles focusing on pediatric or obstetric population, with a focus on virus-host interactions and immunological mechanisms responsible for virus associated cytokine release syndrome (CRS). COVID-19 illness encompasses three main phases. In phase 1, SARS-CoV-2 binds with angiotensin converting enzyme (ACE)2 receptor on alveolar macrophages and epithelial cells, triggering toll like receptor (TLR) mediated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ƙB) signaling. It effectively blunts an early (IFN) response allowing unchecked viral replication. Phase 2 is characterized by hypoxia and innate immunity mediated pneumocyte damage as well as capillary leak. Some patients further progress to phase 3 characterized by cytokine storm with worsening respiratory symptoms, persistent fever, and hemodynamic instability. Important cytokines involved in this phase are interleukin (IL)-6, IL-1β, and tumor necrosis factor (TNF)-α. This is typically followed by a recovery phase with production of antibodies against the virus. We summarize published data regarding virus-host interactions, key immunological mechanisms responsible for virus-associated CRS, and potential opportunities for therapeutic interventions.
Conclusion
Evidence regarding SARS-CoV-2 epidemiology and pathogenesis is rapidly evolving. A better understanding of the pathophysiology and immune system dysregulation associated with CRS and acute respiratory distress syndrome in severe COVID-19 is imperative to identify novel drug targets and other therapeutic interventions.
Similar content being viewed by others


Introduction
Since it was first reported from Wuhan, China in December 2019, Coronavirus Disease 2019 (COVID-19) has rapidly spread across the globe and was declared a global pandemic by the WHO on March 11th, 2020 [1]. As of July 18th, 2020, 188 countries have been affected with more than 14 million confirmed cases and over 600,000 fatalities [2]. Being a novel virus, there has been a steep learning curve about its microbiology, host interactions, mechanism of immune dysregulation in humans, and tissue injury. Multi-modality therapeutic options are being explored on an emergent basis with limited evidence of efficacy [3].
We provide a focused review of the published literature regarding the pathophysiology of COVID-19 with an emphasis on the anti-viral and immunomodulatory therapies.
Methodology
We conducted searches on PubMed and Google Scholar for any articles between January 1st, 2000 and June 30th, 2020, with the search terms “Coronavirus or COVID-19” in conjunction with the search terms “transmission”, “pathogenesis”, “immune response”, “cytokines”, “interleukin (IL) inhibitor”, “antiviral therapy”. Due to limited published literature related to COVID-19 in the pediatric and obstetric population and their unique aspects, we exclude articles pertaining to that population. We also reviewed information published on the World Health Organization (WHO), Centers for Disease Control and Prevention (CDC), and John Hopkins University Center for Systems Science and Engineering (CSSE) websites.
Epidemiology
The Huanan Seafood Wholesale Market in Wuhan, China, the purported origin site of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), was the epicenter of new cases of COVID-19 from December 2019 to January 2020. In February 2020, the epicenter shifted initially to Italy and Spain, and subsequently to the United States of America (USA) in March 2020 [4, 5]. Estimated case fatality rate with COVID-19 ranges from 0.5 to 3% [6, 7]. However, mortality is higher in males, patients with comorbidities including diabetes mellitus, heart disease or hypertension, and those over age 60 years [8, 9].
Wu et al. reviewed the epidemiology of 72,314 COVID-19 patients in China and noted that the predominant age distribution was 30–79 years of age, but observed increasing case fatality rate (CFR) in older patients (> 80) [10]. Less than 2% of identified patients were less than 18 years of age. While similar patterns have been reported in Europe and the United States, the interpretation of epidemiological data is limited by the testing characteristics of the specific community, with likely under-representation of asymptomatic patients [2]. Further, variable transmission rates are also observed based upon characteristics of the local community (e.g. urban vs rural, age distribution, etc.) and any public health policies in place for containment or mitigation such as quarantines, shelter-in-place orders, mask-wearing, or contact tracing.
Clinical presentation
The spectrum of clinical manifestations ranges from asymptomatic to life-threatening. However, more than 80% of patients have mild symptoms or are asymptomatic [11, 12]. The most frequently reported symptoms include fever (80–90%) and dry cough (50–70%). There may be associated severe fatigue and dyspnea. Loss of taste and smell have also been reported. Gastrointestinal symptoms (nausea, vomiting, diarrhea) are present in less than 5% of patients. Symptoms typically resolve within 5–10 days. However, approximately 14% of patients have severe disease requiring hospitalization and 5% may have critical illness evidenced by adult respiratory distress syndrome (ARDS), respiratory failure, shock and/or multi-organ dysfunction [13, 14].
Predictors of severe disease
Clinical predictors of poor outcome include advanced age, male gender, hypertension, diabetes mellitus and coronary artery disease [15, 16]. Laboratory predictors of critical disease include lymphopenia, elevated levels of D-Dimer, pro Brain-type Natriuretic Peptide (pro-BNP), troponin I, and creatinine [9, 15, 16]. High levels of inflammatory markers such as IL-6, C-reactive protein (CRP), and ferritin are also associated with more severe disease [17]. Qin et al. described that patients with severe COVID-19 infection had significantly lower circulating B cells, T cells, and Natural killer (NK) cells on flow cytometry as compared to non-severe cases, endorsing the hypothesis that immune dysregulation plays a role in disease severity [18].
Mode of transmission
SARS-CoV-2 is a member of the betacoronavirus (β-CoV) family. In the last 20 years, the most lethal strains of the β-CoVs causing epidemics include Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) in 2002, Middle Eastern Respiratory Disease coronavirus (MERS-CoV) in 2011 and SARS-CoV-2 in 2020. SARS-CoV-2 is purportedly spread to humans from bats via intermediate hosts such as turtles and pangolins, however this is currently controversial [19,20,21,22,23]. The main mechanism of spread for both SARS-CoV and SARS-CoV-2 seems to be human-to-human transmission [3, 20]. COVID-19 is predominantly thought to be spread via droplets and fomites [24], with very limited aerosolization, and recent data indicates possible fecal–oral spread as well [11, 24, 25]. Patients can be contagious for 24–48 h before symptom onset [6, 10]. The incubation period is 2–15 days, with a mean of 5.1 days. Most (97.5%) patients develop symptoms within 11.5 days [11]. The virus can survive up to 1–2 days on glass and metal surfaces, and up to 4–5 days on plastic surfaces [26]. It is unclear if a significant amount of SARS-CoV-2 is present in breast milk, urine, or semen for transmission. Vertical transmission from pregnant mothers to infants remains a controversial topic, but there are emerging reports of damage to the placenta from COVID-19 [24], and SARS-CoV-2 RNA has been detected on the fetal side of the placenta [27].
Testing
Microbiologic diagnosis of SARS-CoV-2 is made by real-time reverse transcriptase-polymerase chain reaction (rt RT-PCR), serology, and rapid antigen detecting kits [28]. True sensitivity of PCRs from nasopharyngeal swabs varies from 30 to 70% depending on the phase of illness [29]. Since PCR has a significant false-negative rate, a negative PCR should be interpreted in context with the clinical manifestation, disease phase, and radiological findings. Virus-specific immunoglobulins (Ig)—IgG and IgM antibodies can be detected beyond day 5 of infection and can be detected in those who have active disease or recovered, although delays in seroconversion beyond 14 days have been reported [30]. Further, there are some reports that antibodies produced against SARS-CoV-2 are short-lived and may not be fully protective [31]. The sensitivity and specificity of serologic assays differ based on the specific methodology utilized (e.g. ELISA, agglutination, or complement-fixation). Numerous serology kits with variable false negative and false positive rates are currently in the process of being developed, and appropriate implementation requires validation.
Serological testing as a diagnostic tool for COVID-19 is limited by the fact that seroconversion may be significantly delayed after the onset of illness, although it may have increasing utility during later phases of disease when viral loads are lower [32]. A clearer understanding of the kinetics of antibody production during infection is critical for understanding the specific role of serological testing as a diagnostic tool, as well as an instrument for seroepidemiological and vaccine evaluation studies [33].
SARS-COV-2 structure
Coronaviruses are spherical, positive-sense, single-stranded, non-segmented ribonucleic acid (RNA) surrounded by a lipid capsule derived from the host cell membrane, which has a characteristic surface spike glycoprotein [34]. The general structure comprises of four essential proteins: the spike (S) protein responsible for attachment to host cell receptors, the membrane (M) protein which promotes membrane curvature and binds to the nucleocapsid), the envelop (E) protein which helps with viral assembly and release, and the nucleocapsid N protein (helps with viral replication) [35, 36]. In vitro studies demonstrate that viral non-structural proteins and E2 glycoprotein have a high affinity binding to the porphyrin portion of heme of infected cells [36]. The replication of SARS-CoV-2 is shown in Fig. 1.

Viral replication pathway of Covid-19. The virus first attaches to the ACE2 receptor and internalizes into the respiratory epithelial cell and causes the release of its genome. The S protein (spikes on the viral surface responsible for attachment to host cell receptors), M protein (shapes the virion, promotes membrane curvature and binds to the nucleocapsid), E protein (helps with viral assembly and release)
SARS-CoV-2 shares structural similarity to both Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) (approximately 80% similarity) and Middle Eastern Respiratory Disease Coronavirus (MERS-CoV); thereby studies on SARS and MERS are extrapolated and applied to COVID-19 [37]. There is a possibility of emerging strains within SARS-CoV-2, including the so-called S-type and L-type. At present, there is no clear evidence that one strain is more virulent than another, and the two strains do not represent distinct targets for drugs and vaccine development [38, 39].
SARS-COV-2 invasion of host cell using angiotensin converting enzyme-2 (ACE2) receptor
As shown above, SARS-CoV-2 enters the host by binding to the host cell ACE2 receptor, a membrane-bound protein found on the surface of type 2 pneumocytes, epithelial cells, and enterocytes [34]. In addition to ACE2 mediated endocytosis, direct entry into cells may also occur from cell surface. Transmembrane protease serine 2 (TMPRSS2) is another host cellular protein which has been described as a co-factor for SARS-CoV-2 entry into cells, and Camostat is a clinically relevant drug has been observed to be inhibitory towards SARS-CoV-2 particles in cell culture systems [40].
ACE2 exists in a soluble form in the alveolar fluid where it potentially plays an important role in protection from ARDS [30, 41]. ACE2 receptor expression on epithelial cells increases with age and may partly explain why children are less prone to infection by SARS-CoV-2 [42]. Further, children have higher levels of soluble ACE2 activity within alveolae during ARDS, which has been hypothesized to cause improved lung repair mechanisms compared to adults and be protective against developing COVID-19 [42].
There is controversy regarding the role of ACE inhibitors or ARBs in potentially increasing the virulence of COVID-19 via the upregulation of ACE2 [43]. Although there is currently no convincing evidence to suggest that ACE inhibitors or ARBs have a beneficial or harmful role in COVID-19, further studies are needed to resolve this question. Current consensus by several societies such as the European Society of Cardiology is that there should be no change in the utilization of these agents in patients infected with COVID-19 [44, 45].
Since ACE2 is vital for viral entry to a host cell, a novel treatment strategy utilizing recombinant human ACE2 protein is being studied in a randomized trial in China, looking at its use as a competitive inhibitor as well as a mediator to promote lung repair. Meanwhile, the University of Minnesota has launched a phase 2 clinical trial (NCT04312009) to evaluate the efficacy of losartan, which is an ARB, in COVID-19 pneumonia.
SARS COV-2 replication and evasion of the immune system
Virus-infected cells typically activate the immune system via cytotoxic cells, interferons or antibodies. The MHC1 (Major Histocompatibility Complex 1) is an antigen presenting cell that causes resultant autophagy by lysosomal degradation. The ORF-2 protein of SARS-CoV-2, which is expressed by both L-type and S-subtypes, down-regulates MHC1 by decreasing total protein and beta-2 microglobulin expression in a dose-dependent and incubation time dependent manner [46]. This causes a decrease in cell surface expression, and hence decreases lysosomal degradation by autophagy, and subsequently decreases cell elimination. This is similar to viruses that cause chronic infection such as HIV (Human Immunodeficiency Virus), which lead to maladaptive immune system while maintaining active replication.
Another mechanism that aids with the elimination of virus-infected cells is increased expression of cytokines. IFN alpha and IFN beta are produced systemically by epithelial cells, monocytes and alveolar macrophages and aggravate lung pathology during respiratory infections. Human intestinal epithelial cells (HIEC) are known to be infected by SARS-CoV-2 and can assist by generating an IFN-mediated intrinsic immune response [47]. IFN gamma is produced locally by lung resident dendritic cells and has been shown to inhibit lung epithelial repair after viral recognition [48, 49]. Non-Structural Protein 1 (NSP1) is another protein that decreases IFN production by preventing translation of IFNs and pro-inflammatory cytokines and IFN-stimulated anti-viral components by binding mRNA translation machinery and enucleated cleavage and degradation of lost mRNA [50].
TLR3 is activated by sensing viral replication from dying cells. It causes increase in IFN gamma, which results in compromise by epithelial cells and predisposes to secondary bacterial infections.
Emerging anti-viral therapeutic options
Our knowledge of clinical manifestations and pathogenesis of COVID-19 is rapidly evolving as is our understanding of how best to manage this illness and associated complications. Given the rapidly changing data, several therapies are being used solely based on anecdotal evidence. While immediate access to new data is critical in a pandemic such as COVID-19, it can also propagate misinformation as the data is not verified or peer-reviewed in many instances.
Several anti-viral therapies are under investigation for the treatment of COVID-19, either as monotherapies or in combination with other agents [51]. The only medication for which FDA has issued emergency use authorization (EUA) is Remdesivir, which was approved on May 1st, 2020. Table 1 summarizes the key anti-viral therapies currently being investigated in clinical trials. We will discuss some of the promising therapies based on mechanisms of action.
Viral mRNA synthesis inhibitors
Remdesivir (GS-5734) is a viral nucleotide (adenosine) analog, which incorporates into nascent viral RNA chains and results in premature termination [51,52,53]. In vitro studies suggest that the combination of remdesivir and CQ could inhibit viral replication even at low concentrations [51]. The multi-center trial, commonly known as SOLIDARITY (NCT04321616), was one of the first large-scale trials aiming to compare HCQ, remdesivir, and the combination of HCQ/remdesivir in hospitalized COVID-19 patients. This trial was started as a five-arm adaptive design trial aimed at studying the primary outcome of in-house mortality at 3 weeks. Other outcomes measures included comparison of mechanical ventilation occurrence, viral clearance, and markers of inflammation [54]. On March 24th, 2020, the Czech Republic approved the use of remdesivir in critically ill patients [39]. Some adverse effects of remdesivir are transaminitis, nausea, and vomiting—further studies are underway to evaluate for side effects.
Based on the finding of the Adaptive COVID-19 treatment trial (ACTT), Remdesivir may be most beneficial if given to patients with severe COVID-19 lung involvement before mechanical ventilation [55]. Remdesivir use was associated with a reduced median time to recovery (11d v 15 days). A mortality benefit was observed (8% v 11.6%) but was not statistically significant.
Favipiravir (T-705), a synthetic nucleoside analog, functions as a chain terminator at the site of incorporation of the viral RNA, thereby inhibiting the RNA-dependent RNA polymerase. Umifenovir is a membrane fusion inhibitor originally developed for influenza. A multicenter trial comparing favipiravir to umifenovir in COVID-19 patients with moderate symptoms and chest imaging abnormalities showed a 71% recovery at day 7 for those receiving favipiravir, compared to 55% in the group receiving umifenovir [43]. Another trial in China on 80 patients infected with SARS-CoV-2 compared response to Interferon (IFN)-α combination with either favipiravir or lopinavir (LPV)/ritonavir (RTV). The combination with favipiravir was shown to expedite viral clearance (4 days versus 11 days, shorten recovery time (statistically significant at 92% versus 62%) and cause improvement in chest imaging [56]. Based on these studies, Favipiravir received marketing approval in China on February 17th, 2020. Common side effects of favipiravir include nausea, headache, and diarrhea. Favipiravir may cause embryotoxicity [57].
Endosomal function inhibitors
CQ and its derivative, HCQ have historically been used for malaria and amebiasis. They are also widely utilized in the treatment of auto-immune conditions such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), and in recent years have been seen to have activity against a wide range of viruses in-vitro such as Ebola virus and SARS-CoV-1 [58, 59].
Their anti-viral effects are achieved by multiple mechanisms, primarily by alkalinizing the normally acidic endosomal pH of the infected cells, limiting virus-cell fusion, and modifying glycosylation of receptors [60]. They also affect cell signaling and have an immunomodulatory effect by blocking proinflammatory cytokines, particularly tumor necrosis factor-alpha (TNFα) and IL-6, IL-1β production by activated alveolar macrophages and downregulation of TNF receptors on monocytes resulting in decreased monocyte activation. They also reduce the severity of ARDS by decreasing TNF-α mediated opening of tight junctions of epithelial cells, as well as upregulation of leukocyte adhesion molecules (LAM) and hence decreasing leukocyte extravasation into damaged alveoli [61, 62]. Apart from these, another mechanism by which CQ/HCQ is postulated to interfere with SARS-CoV-2 relates to the virus’s ability to block hemoglobin synthesis. CQ/HCQ competes with the porphyrin to bind to the E2 portion of the virus, thus freeing the porphyrin to incorporate into hemoglobin [36].
Based on these observations, these agents were thought to be promising prophylactic and therapeutic options for COVID-19. HCQ was investigated both as monotherapy as well as in combination with Azithromycin (an antibiotic added to prevent bacterial super-infection) for COVID-19 [63]. Preliminary data from the open-label non-randomized French trial on six patients of this combination showed viral load reduction [63]. On March 28, 2020, Chloroquine and Hydroxychloroquine sulfate were issued EUA to treat hospitalized adults and adolescents who were unable to be enrolled in a clinical trial. However, based on continuous review of the available scientific evidence, the EUA was revoked on June 15, 2020, as there was no clear demonstratable effect [64].
Adverse effects of CQ and HCQ include prolonged QTc interval, arrhythmias, and dilated cardiomyopathy which may be of concern given the high incidence of cardiac arrhythmias and sudden cardiac death being noted in critically ill COVID patients [65,66,67]. Photosensitivity and retinal damage can also occur, and rare cases of Stevens-Johnson syndrome have been reported [68]. They should be avoided in patients with glucose-6-phosphate-dehydrogenase (G-6-PD) deficiency or porphyrias [67].
Other therapies to inhibit viral host attachment and viral replication
LPV and RTV are both antiviral agents used in combination to treat human immunodeficiency virus (HIV). LPV is a protease inhibitor that acts by preventing maturation of the HIV-1 virus, while RTV is a cytochrome P3A4 (CYP3A4) inhibitor which increases plasma levels of LPV. In a randomized study, involving 199 patients with severe COVID-19, there was some anecdotal success, but the combination did not demonstrate significant improvement in death rates, rates of oxygen desaturation, and rates of intubation compared to the standard of care treatment in matched controls [36]. In SARS, LPV and RTV were most effective when used in combination with ribavirin [69]. There is some evidence based on viral-host cell proteomic mapping demonstrating SARs-CoV-2 binds directly to a protein that is targeted by ribavirin [70]. Adverse effects may include diarrhea, nausea, rash, and asthenia.
Other anti-viral therapies currently in clinical trials are summarized in Table 1.
COVID associated cytokine storm and lung injury
Some patients with COVID-19 undergo progressive clinical decline which is characterized by the following phases: early phase, pulmonary phase, and hyper inflammation phase. We describe the phases of COVID-19 infection, discuss immune dysregulation resulting in cytokine storm, and review immunomodulatory options being studied in this disease. Therapeutic options are summarized in Table 1.
Early phase: phase one
The first phase of COVID-19 infection generally presents as fever and cough triggered by robust viral replication within the respiratory epithelium. The innate immune system is the primary mediator of inflammation during this phase. Viral particles are recognized by Toll-like receptors (TLR) on macrophages, neutrophils and dendritic cells (DC), which typically activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ƙβ) pathways leading to transcription of several cytokines including IL-6 and IFNƴ [71,72,73]. IFNs induce Janus Kinase (JAK) and activator of transcriptor (STAT) pathways, which promote expression IFN-stimulation genes [74]. SARS-CoV-2 appears to inhibit the NF-ƙβ -TLR4 pathway and thereby delays IFN production, allowing unchecked viral replication [75]. The N protein of SARS-CoV-2 appears to antagonize the host response and the IFN response. This provides a potential therapeutic target, as utilizing IFN gamma earlier could help modulate the immune response and potentially decrease disease severity.
Pulmonary phase: phase two
Progression to the second phase of SARS-CoV-2 infection is characterized by the development of hypoxia. The direct virus-induced cytopathic effect on type two pneumocytes via ACE2 receptors activates the innate immune system, resulting in a large influx of monocytes, macrophages, and heavy infiltration of neutrophils [76]. They produce nitric oxide, growth factors, and transforming growth factor-beta (TGF-β) which contribute to oxidative injury, leading to capillary leak and alveolar basement membrane damage utilizing the TLR-4- NF-ƙβ pathway [77]. Loss of the type two pneumocytes also impairs endogenous lung repair mechanisms, leading to the cascading progression of injury.
Hyperinflammation phase: phase three
As COVID-19 disease progresses to Phase 3, which appears approximately 9–12 days after the onset of illness, there may be development of ARDS, CRS, septic shock and cardiac complications [9, 18].
SARS-CoV-2 drives a lower anti-viral transcriptional response compared to other respiratory viruses, resulting in low IFN-I and IFN-III levels and elevated chemokine levels [78]. T lymphocytes, activated macrophages, and neutrophils migrate towards and infiltrate the alveolar microenvironment, releasing pro-inflammatory chemokines and cytokines including IL-1, IL-6, and IL-8, IL-17 and TNF [79, 80].
Cytokines implicated in COVID-19-associated lung injury and CRS include IL-1β, TNF-α and IL-6, which activate other proinflammatory pathways via the JAK-STAT pathway and activation of Th cells [17, 77, 81, 82]. IL-6 also recruits macrophages at sites of injury and promotes inflammation and ARDS. The SARS E protein causes IL-1β secretion, leading to lung inflammation and injury [83]. IL-1β also accelerates the production of TNF-α, which in turn promotes the apoptosis of lung epithelial and endothelial cells [84]. IL-1β, TNF-α and IL-6 provide an integrated, amplified inflammatory response, leading to a breach in alveolar basement membrane integrity. This subsequently increases vascular permeability, leading to pulmonary edema, which is associated with pulmonary deterioration even with ventilator support. This is schematically represented in Fig. 2.

Alveolar micro-environment showing pathophysiology of acute respiratory distress syndrome (ARDS). The Th response causes the release of IL-17 which activates TNF alpha which enhances epithelial injury and activates neutrophils to cause degranulation. IL-6 is produced by alveolar macrophages which also stimulates neutrophils. Once the epithelial integrity of alveolus is breached epithelial sodium channel (Enac) channels and Na/K channels fail to maintain homeostasis eventually leading to an increase in permeability of capillaries causing exudation of fluids. T reg cells also trigger TGF beta which causes fibrosis to the damaged epithelial membrane. Most of the COVID-19 patients present with ground-glass opacities and fibrosis of their lungs
Secondary hemophagocytic syndrome (sHLH) may also develop during this phase, and is characterized by fever, cytopenias, hyperferritinemia, and an increase in proinflammatory cytokines including IL-6 and IL-18 [85, 86]. This could be useful to consider when treating a patient with COVID-19—white blood cell count, CRP, and D-dimer may be helpful to monitor and to distinguish between CRS versus secondary bacterial infections.
Another aspect that warrants further study is the consequence of elevated D-dimer, as it is unclear at present if it is reflective of an acute phase only, or potentially a disseminated intravascular coagulopathy (DIC) phenomena occurring in the lungs secondary to the sHLH.
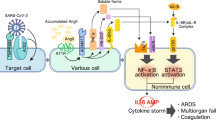
An overview of the immune dysregulation in COVID-19 is shown in Fig. 3.

Immune dysregulation in cytokine release syndrome. The involvement of the immune system in COVID-19 is in 3 phases: initial inflammation which is characterized by delayed interferon response and robust viral replication, the pulmonary inflammation phase which is characterized by sub-optimal T-cell and antibody response, leading to increased vascular leakage and permeability and impaired viral clearance, and the hyperinflammation phase which is characterized by very severe infiltration of monocytes, macrophages and neutrophils—this leads to progressive lung injury and ARDS as well as hemodynamic instability and shock
Recovery phase
This phase can occur at any time during the disease and is divided into the early recovery stage (ERS) and the late recovery stage (LRS). IL-6 production and circulating inflammatory monocytes were noted in ERS, with the potential for ongoing lung injury. In LRS, patients’ serum contains an abundance of antibodies. It is speculated that dendritic cells (DC) produce IL-18 which promotes proliferation of B cells as well as IL-7 which promotes T cell proliferation, IL-2 secretion and B cell proliferation, and antibody production [87].
Non-selective immunomodulators
Corticosteroids
The use of corticosteroids was much debated for COVID-19 as steroids are immunosuppressive, potentially delaying viral clearance and increased risk of secondary infection. An observational study on intensive care unit (ICU) patients with MERS reported that high doses of corticosteroids were associated with more severe disease but did not increase ninety-day mortality [88]. Other meta-analyses in SARS showed no benefit of steroid use [89, 90]. An observational trial by Yuan et al. showed no benefit from Methylprednisone. However, preliminary data from the UK RECOVERY trial showed that low dose dexamethasone (6 mg PO or IV daily for 10 days) reduced mortality by 35% in intubated patients and by 20% in hospitalized patients requiring oxygen supplementation compared to patients receiving standard of care, but had no effect in patients who did not require oxygen supplementation [91]. Based on these data, Dexamethasone is now recommended for patients with severe COVID-19 (requiring oxygen) including those on mechanical ventilation by the NIH and IDSA [92, 93].
Non-steroidal anti-inflammatory drugs (NSAIDs)
There is controversy regarding NSAID use for symptom relief with COVID-19. The French National Agency for Medicines and Health Products Safety suggested that COVID-19 viral clearance may be delayed by NSAIDs. The European Medical Association (EMA) did not support this statement, due to lack of supporting evidence [94].
Thalidomide
Thalidomide blocks the NF-ƙB binding to gene promotors, reducing the production of IL-6, TNF-α and chemokines [95]. It increases circulating NK cells and increases IFN-ƴ production by T cells. It is FDA approved to treat multiple myeloma in combination with low dose dexamethasone and trials suggest activity in influenza-associated lung injury. Trials are evaluating the role of Thalidomide in COVID-19 (NCT04273529). This drug has well-known teratogenic effects [96].
Cytokine inhibitors
IL-6 inhibitors
IL-6 is one of the key players in accelerating a cytokine storm. Several IL-6 antagonists are being studied for safety and efficacy in COVID-19. Tocilizumab, a prototype IL-6 receptor antagonist, is the most studied and is currently used in managing cytokine storm in chimeric antigen receptor Antibody (CAR-T) therapy. It was approved in China for the treatment of severe COVID-19 in March 2020. An initial trial in China where Tocilizumab was administered to 20 patients with severe COVID-19, targeting cytokine storm demonstrated promising results, with 19 patients discharged in stable condition 2 weeks after administration. Chest imaging showed significant improvement on day four to five [97]. Anecdotal reports from other large centers suggest rapid improvement in some patients with improved oxygenation often within 24 to 48 h of administration. Also, treatment may be more effective earlier in the disease course than when ARDS fully develops. Typically, a single 8 mg/kg dose is administered. Notable adverse effects of Tocilizumab include increased risk of secondary infection, liver dysfunction, and cytopenias [98].
A similar anti-IL-6 agent, Sarilumab, is being investigated in clinical trials for COVID-19 (e.g. NCT04315298). Siltuximab is a chimeric anti-IL-6 monoclonal antibody that binds to soluble and membrane-bound forms of IL-6, preventing binding to soluble and membrane-bound receptors. It is used in the treatment of CAR-T induced CRS not responding to tocilizumab, and hence may play a role in COVID-19 induced CRS as well [99, 100]. Side effects include cytopenias, edema, hypotension, and increased risk of secondary infections.
IL-1 inhibitors
IL-1 is another pro-inflammatory cytokine that feeds the cytokine storm. It mediates inflammation in the lungs, leading to fever, ARDS and fibrosis. Anakinra, an IL-1 blocker, is used to treat RA in adults and neonatal-onset multisystem dysfunction (NOMID), as well as used off-label for neurotoxicity complications of CAR-T therapy. This is currently being investigated for COVID-19 induced CRS [85, 101]. Adverse effects include hypersensitivity, neutropenia, and infections [102].
JAK inhibitors
The regulation of the JAK-STAT pathway is essential for cross interaction between various cytokine signaling pathways leading to an uncontrolled pro-inflammatory state. JAK inhibitors such as Ruxolitinib and Fedratinib target the pro-inflammatory JAK/STAT pathway and are approved for myeloproliferative disorders [17, 103, 104]. Ruxolitinib is also approved for steroid-refractory graft versus host disease (GVHD) which are JAK/STAT-driven diseases. Baricitinib is another JAK inhibitor currently used in RA which is being investigated for efficacy in COVID-19 (NCT04320277). It also may have anti-viral activity by reducing clathrin-mediated endocytosis [105]. Common dose-limiting side effects of JAK inhibitors include cytopenias, hyperlipidemia and increased risk of secondary infection.
Immune effector cell therapy
NK cell therapy
NK cells are recruited to site of infection by chemokines, activated by cytokines produced from infected cells, like IL-12, IL-15, IL-18, and IFN. Activated NK cells counter the virus by increased IFN-ƴ production and NK cell-mediated cytolysis of infected cells. Possible blunting of NK responses by SARS-CoV-2 may allow disease progression [21, 106, 107]. Given its anti-viral properties, allogeneic, “off the shelf”, NK cell infusions, derived from healthy donors, are being evaluated for efficacy in COVID-19 associated pneumonia. NK infusions are generally well tolerated.
Mesenchymal cells (MSC)
Cell-based therapy, especially mesenchymal stem cell therapy, is considered to be one of the most promising therapeutic approaches aiming to provide opportunities to treat several diseases. MSC have diverse immunomodulatory and regenerative properties [54]. Previous trials have shown evidence of stabilized and improved lung function in patients with ARDS who received MSC without any treatment-related adverse effects. Given the hypothesis that MSC therapy might prevent the triggering of cytokine storm and promote endogenous repair, several clinical trials are Looking at the safety and therapeutic potential of MSC from various sources in SARS-CoV-2 (e.g. NCT04313322) [108, 109]. Since infusions may carry the risk of microcirculation injury, MSC derived exosomes, which can be delivered by aerosol inhalation, are also being evaluated for safety and efficacy in severe COVID-19 pneumonia (NCT04276987). Availability and large-scale manufacturing are potential issues.
Complement inhibitors
In addition to DIC, the complement pathway contributes to lung injury in SARS, and it may contribute to the high incidence of fatal microvascular and macrovascular thrombosis associated with COVID-19 [110]. Eculizumab, which is approved to treat rare complement-mediated disorders like paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), neuromyelitis optica spectrum disorder and myasthenia gravis, is being evaluated for safety and efficacy in COVID-19 (NCT04288713). Immunosuppression is a major side effect. In general, patients should be vaccinated against meningococcus and pneumococcus prior to use [111]. However, this may not be possible in COVID-19 patients being considered for this therapy.
Programmed cell death (PD)-1 inhibitors
These likely function by delaying T cell exhaustion. Camrelizumab, a fully humanized PD-1 monoclonal antibody, is currently approved to treat lymphoma in China and is now being investigated as an immunoregulatory therapeutic option for COVID-19. Clinical efficacy of camrelizumab plus thymosin in patients with COVID-19 will be evaluated in clinical trial NCT04268537 [112]. Adverse effects of immunotherapy are generally related to breakthrough autoimmunity and may include rash, diarrhea, colitis, and thyroid dysfunction [112].
Therapies utilizing passive immunity
Convalescent plasma exchange, which utilizes passive immunity, may be an effective treatment strategy. Serum rich in anti-SARS-CoV-2 Ab can be obtained from recovered donors and transfused to infected patients. Shen et al. reported transfusing hyperimmune plasma on 5 critically ill patients infected with COVID-19, who had severe pneumonia, rapid progression, and persistently high viral load despite treatment, as well as severe ARDS mechanical ventilation. These patients received transfusion with convalescent plasma with SARS-CoV-2v-2 specific antibody with the resultant resolution of ARDS within two weeks, and 3 of these patients were extubated within 2 weeks. All patients clinically improved around a week later [113]. This appears promising and is being investigated in several countries for critically ill patients [114].
A summary of all the therapies discussed above is listed in Table 1.
Conclusion
Given the worsening trajectory of the COVID-19 pandemic, there is a global race to develop effective therapeutic interventions. Since SARS-CoV-2 is a novel virus, our understanding regarding its host interaction and resultant inflammatory responses is still evolving. Most therapeutic agents currently under investigation are based on prior observations with SARS or experience in immune dysregulation. Moreover, with rapid publication pace and immediate access to data before formal peer-review, there are emerging challenges in ensuring the accuracy of published information for clinical use. The first medication to receive EUA was HCQ/chloroquine. However, based on subsequent data, this EUA was revoked. Currently, the only direct anti-viral agent with EUA for COVID-19 is Remdesivir.
A key mechanism driving COVID-19 associated mortality may be the cytokine storm augmenting lung injury. While the precise pathways driving CRS and ARDS are yet to fully understood, high levels of pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α characterize the cytokine storm. There is encouraging preliminary data in CRS and ARDS with the immunomodulators like Tocilizumab, an IL-6 inhibitor. These agents may be used alone or in conjunction with other treatments, such as dexamethasone, in severe disease.
Cellular therapy may also have a role in treating and reducing lung injury in COVID-19. Based on their application as cancer treatments, NK cells are known to exert direct cytotoxic effects on virally infected cells and produce IFN-ƴ and TNFα to boost the host immune response. MSCs, with prior use in the treatment of GVHD, fibrotic liver, and lung diseases, may also improve COVID-19 associated lung damage. There is a potential role for the development of agents aimed at enhancing immune surveillance by specifically targeting ORF8 or NSP1 to impair MHC1 antigen presentation.
Convalescent plasma from recovered patients is also an attractive treatment option for critically ill or rapidly deteriorating patients. Ultimately, the hope is to develop vaccinations effective in prevention, but this may take several months or years to develop.
Most of the evidence on current therapeutic agents are based on small observational studies and need to be validated by larger studies and RCTs. Given the paucity of information regarding therapeutic agents and their administration, there is an urgent need for studies to evaluate all aspects of therapy, including the timing of administration, potential synergism between treatments, and potential toxicities. It is also crucial to balance the need to expedite the utilization of potentially helpful medications with the need to ensure patient safety.
Availability of data and materials
Not applicable.
Abbreviations
- SARS-CoV-2:
-
Severe Acute Respiratory Syndrome Coronavirus 2
- COVID-19:
-
Coronavirus disease 2019
- ARDS:
-
Adult respiratory distress syndrome
- CRS:
-
Cytokine release syndrome
- IL:
-
Interleukin
- WHO:
-
World Health organization
- CDC:
-
Centers for disease control
- CSSE:
-
Center for Systems Science and Engineering
- ACE2:
-
Angiotensin converting enzyme
- TLR:
-
Toll like receptors
- NF-ƙB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- IFN:
-
Interferon
- TNF:
-
Tumor necrosis factor
- SARS:
-
Severe Acute Respiratory Syndrome
- MERS:
-
Middle East Respiratory Syndrome
- USA:
-
United States of America
- pro-BNP:
-
Pro brain-type natriuretic peptide
- CRP:
-
C-reactive protein
- T cells:
-
T lymphocytes
- B cells:
-
B lymphocytes
- NK:
-
Natural killer
- β-CoV:
-
Beta coronavirus
- SARS-CoV:
-
Severe Acute Respiratory Syndrome Coronavirus
- MERS-CoV:
-
Middle Eastern Respiratory Disease coronavirus
- rt RT-PCR:
-
Real time reverse transcription polymerase chain reaction
- BAL:
-
Bronchoalveolar lavage
- CT:
-
Computed tomography
- mRNA:
-
Messenger ribonucleic acid
- NSP1:
-
Nonstructural Protein 1
- TMPRSS2:
-
Transmembrane protease, serine 2
- ARB:
-
Angiotensin II type I receptor blockers
- NHC:
-
National Health Commission
- CQ:
-
Chloroquine phosphate
- HCQ:
-
Hydroxychloroquine
- IFN:
-
Interferon
- LPV:
-
Lopinavir
- RTV:
-
Ritonavir
- RA:
-
Rheumatoid arthritis
- SLE:
-
Systemic lupus erythematosus
- E:
-
Envelop protein
- LAM:
-
Leukocyte adhesion molecules
- G6PD:
-
Glucose-6-phosphate-dehydrogenase
- CYP3A4:
-
Cytochrome P450 3A4
- HIV:
-
Human immunodeficiency virus
- JAK:
-
Janus kinase
- STAT:
-
Activator of transcriptor pathway
- N protein:
-
Nucleocapsid protein
- M:
-
Membrane protein
- S:
-
Spike protein
- sHLH:
-
Secondary hemophagocytic syndrome
- ERS:
-
Early recovery stage
- LRS:
-
Late recovery stage
- DC:
-
Dendritic cells
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- EMA:
-
European Medical Association
- FDA:
-
U.S. Food and Drug Administration
- CAR-T:
-
Chimeric antigen receptor T-cells
- MSC:
-
Mesenchymal cells
- PNH::
-
Paroxysmal nocturnal hemoglobinuria
- aHUS::
-
Atypical hemolytic uremic syndrome
- PD-1::
-
Programmed cell death-1
- Ig::
-
Immunoglobulins
- DC::
-
Dendritic cells
- ICU::
-
Intensive care unit
- COPD: :
-
Chronic obstructive pulmonary disease
- NOMID::
-
Neonatal onset multisystem dysfunction
References
Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424–32.
John Hopkins Coronavirus Dashboard [Web]. Website2020. https://coronavirus.jhu.edu/map.html.
Ashour HM, Elkhatib WF, Rahman MM, Elshabrawy HA. Insights into the recent 2019 novel coronavirus (SARS-CoV-2) in light of past human coronavirus outbreaks. Pathogens (Basel, Switzerland). 2020;9(3):186.
organization Wh. Coronavirus disease 2019 (COVID-19) Situation Report-60.
Schiffmann A. COVID 19 Live Dashboard. In: Schiffmann A, editor. 2019.
Wilson N, Kvalsvig A, Barnard LT, Baker MG. Case-fatality risk estimates for COVID-19 calculated by using a lag time for fatality. Emerg Infect Dis. 2020. https://doi.org/10.3201/eid2606.200320.
Porcheddu R, Serra C, Kelvin D, Kelvin N, Rubino S. Similarity in case fatality rates (CFR) of COVID-19/SARS-COV-2 in Italy and China. J Infect Dev Ctries. 2020;14(2):125–8.
Weiss P, Murdoch DR. Clinical course and mortality risk of severe COVID-19. Lancet. 2020;395:1014–5.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) Outbreak in China: summary of a report of 72314 cases from the chinese center for disease control and prevention. JAMA. 2020;323:1239.
Linton NM, Kobayashi T, Yang Y, Hayashi K, Akhmetzhanov AR, Jung SM, et al. Incubation period and other epidemiological characteristics of 2019 novel coronavirus infections with right truncation: a statistical analysis of publicly available case data. J Clin Med. 2020;9(2):538.
Nishiura H, Kobayashi T, Suzuki A, Jung SM, Hayashi K, Kinoshita R, et al. Estimation of the asymptomatic ratio of novel coronavirus infections (COVID-19). Int J Infect Dis. 2020;94:154–5.
Bouadma L, Lescure FX, Lucet JC, Yazdanpanah Y, Timsit JF. Severe SARS-CoV-2 infections: practical considerations and management strategy for intensivists. Intensive Care Med. 2020;46:579–82.
Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992–1000.
Lippi G, Lavie CJ, Sanchis-Gomar F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): evidence from a meta-analysis. Progress Cardiovas Dis. 2020;63:390–1.
B. M. COVID-19 clinical guidance for the cardiovascular care team. Published online March 6, 2020. https://www.acc.org/~/media/665AFA1E710B4B3293138D14BE8D1213.pdf. Accessed 16 Mar 2020.
Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan China. Intensive Care Med. 2020;46:1294–7.
Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, Xie C, Ma K, Shang K, Wang W, Tian D-S. Dysregulation of immune response in patients with COVID-19 in Wuhan, China (February 17, 2020). Available at SSRN: https://ssrn.com/abstract=3541136. Lancet (pre-print). 2020.
Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microb Infect. 2020;9(1):221–36.
[The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China]. Zhonghua liu xing bing xue za zhi = Zhonghua liuxingbingxue zazhi. 2020;41(2):145–51.
Wong MC, Javornik Cregeen SJ, Ajami NJ, Petrosino JF. Evidence of recombination in coronaviruses implicating pangolin origins of nCoV-2019. bioRxiv. 2020;11:979.
Zhang T, Wu Q, Zhang Z. Probable pangolin origin of 2019-nCoV associated with outbreak of COVID-19. SSRN J. 2020. https://doi.org/10.2139/ssrn.3542586.
Li X, Zai J, Zhao Q, Nie Q, Li Y, Foley BT, et al. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J Med Virol. 2020;92:602–11.
Ong SWX, Tan YK, Chia PY, Lee TH, Ng OT, Wong MSY, et al. Air, surface environmental, and personal protective equipment contamination by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from a symptomatic patient. JAMA. 2020;323:1610.
Patane L, Morotti D, Giunta MR, Sigismondi C, Piccoli MG, Frigerio L, et al. Vertical transmission of COVID-19: SARS-CoV-2 RNA on the fetal side of the placenta in pregnancies with COVID-19 positive mothers and neonates at birth. Am J Obstet Gynecol MFM. 2020;2:100145.
Loeffelholz MJ, Tang Y-W. Laboratory diagnosis of emerging human coronavirus infections—the state of the art. Emerg Microbes Infect. 2020;9:747–56.
Wang W, Xu Y, Gao R, Lu R, Han K, Wu G, et al. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. 2020. https://doi.org/10.1001/jama.2020.3786.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46:586–90.
Wu F WA, Liu M, et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. [Pre-print]. In press 2020.
Perera RA, Mok CK, Tsang OT, Lv H, Ko RL, Wu NC, et al. Serological assays for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), March 2020. Euro Surveill. 2020. https://doi.org/10.2807/1560-7917.ES.2020.25.16.2000421.
Okba NMA, Muller MA, Li W, Wang C, GeurtsvanKessel CH, Corman VM, et al. Severe acute respiratory syndrome coronavirus 2-specific antibody responses in coronavirus disease patients. Emerg Infect Dis. 2020;26(7):1478–88.
Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pöhlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293–307.
Bárcena M, Oostergetel GT, Bartelink W, Faas FG, Verkleij A, Rottier PJ, et al. Cryo-electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc Natl Acad Sci U S A. 2009;106(2):582–7.
Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. 2020;92:667–74.
Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, et al. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 2020;7:1012–23.
Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS-CoV-2. Nat Sci Rev. 2020;7:1012–23.
Press Release:Coronavirus: Are there two strains and is one more deadly?: https://www.newscientist.com/article/2236544-coronavirus-are-there-two-strains-and-is-one-more-deadly/#ixzz6HFuOyP8y. 2020.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80.
Zhang H, Baker A. Recombinant human ACE2: acing out angiotensin II in ARDS therapy. Crit Care. 2017;21(1):305.
Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181(4):905–13.
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3.
Press Release: https://www.escardio.org/Councils/Council-on-Hypertension-(CHT)/News/position-statement-of-the-esc-council-on-hypertension-on-ace-inhibitors-and-ang. 2020.
Fosbol EL, Butt JH, Ostergaard L, Andersson C, Selmer C, Kragholm K, et al. Association of angiotensin-converting enzyme inhibitor or angiotensin receptor blocker use with COVID-19 diagnosis and mortality. JAMA. 2020;324:168.
Zhang Y. The ORF8 protein of SARS-CoV-2 mediates immune evasion through potently downregulating MHC-I. bioRxiv. 2020;162:5049.
Stanifer ML, Kee C, Cortese M, Zumaran CM, Triana S, Mukenhirn M, et al. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep. 2020;32(1):107863.
Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369(6504):712–7.
Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A, et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369(6504):706–12.
Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369(6508):1249–55.
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–71.
Shiraki K, Daikoku T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol Ther. 2020;209:107512.
Al-Tawfiq JA, Al-Homoud AH, Memish ZA. Remdesivir as a possible therapeutic option for the COVID-19. Travel Med Infect Dis. 2020;34:101615.
Xu Y, Li X, Zhu B, Liang H, Fang C, Gong Y, et al. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat Med. 2020;26:502–5.
Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of Covid-19—preliminary report. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2007764.
Cai Q, Yang M, Liu D, Chen J, Shu D, Xia J, et al. Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering. 2020. https://doi.org/10.1016/j.eng.2020.03.007.
Furuta Y, Komeno T, Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(7):449–63.
Barnard DL, Day CW, Bailey K, Heiner M, Montgomery R, Lauridsen L, et al. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antivir Chem Chemother. 2006;17(5):275–84.
Akpovwa H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem Funct. 2016;34(4):191–6.
Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005;2(1):69.
Weber SM, Levitz SM. Chloroquine interferes with lipopolysaccharide-induced TNF-α gene expression by a Nonlysosomotropic mechanism. J Immunol. 2000;165(3):1534–40.
Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. Effects of chloroquine on viral infections: an old drug against today's diseases. Lancet Infect Dis. 2003;3(11):722–7.
Gautret P, Lagier J-C, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;56:105949.
Colson P, Rolain J-M, Lagier J-C, Brouqui P, Raoult D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int J Antimicrob Agents. 2020;55:105932.
Farrell DF. Retinal toxicity to antimalarial drugs: chloroquine and hydroxychloroquine: a neurophysiologic study. Clin Ophthalmol. 2012;6:377–83.
Stokkermans TJ, Trichonas G. Chloroquine and hydroxychloroquine toxicity. StatPearls. Treasure Island (FL) 2020.
Ortel B, Sivayathorn A, Hönigsmann H. An unusual combination of phototoxicity and Stevens–Johnson syndrome due to antimalarial therapy. Dermatologica. 1989;178(1):39–42.
Cheng VCC, Tang BSF, Wu AKL, Chu CM, Yuen KY. Medical treatment of viral pneumonia including SARS in immunocompetent adult. J Infect. 2004;49(4):262–73.
Bojkova D, Klann K, Koch B, Widera M, Krause D, Ciesek S, et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583(7816):469–72.
Totura AL, Whitmore A, Agnihothram S, Schäfer A, Katze MG, Heise MT, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. 2015;6(3):e00638.
Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan China. Clin Infect Dis. 2020;71:762–8.
Woodland D. Toll-like receptors and viral immunity. Viral Immunol. 2012;25(5):347.
Kumar H, Koyama S, Ishii KJ, Kawai T, Akira S. Cutting edge: cooperation of IPS-1- and TRIF-dependent pathways in poly IC-enhanced antibody production and cytotoxic T cell responses. J Immunol. 2008;180(2):683–7.
Hirano T, Murakami M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity. 2020;52(5):731–3.
Ali RM, Al-Shorbagy MY, Helmy MW, El-Abhar HS. Role of Wnt4/beta-catenin, Ang II/TGFbeta, ACE2, NF-kappaB, and IL-18 in attenuating renal ischemia/reperfusion-induced injury in rats treated with Vit D and pioglitazone. Eur J Pharmacol. 2018;831:68–766.
Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–49.
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–45.
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202(3):415–24.
Baseler LJ, Falzarano D, Scott DP, Rosenke R, Thomas T, Munster VJ, et al. An acute immune response to middle east respiratory syndrome coronavirus replication contributes to viral pathogenicity. Am J Pathol. 2016;186(3):630–8.
Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv. 2020:2020.02.23.20026690.
Bunte K, Beikler T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int J Mol Sci. 2019;20(14):3394.
Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Castano-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–9.
Lu HL, Huang XY, Luo YF, Tan WP, Chen PF, Guo YB. Activation of M1 macrophages plays a critical role in the initiation of acute lung injury. Biosci Rep. 2018. https://doi.org/10.1042/BSR20171555.
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. The Lancet. 2020;395(10229):1033.
Karakike E, Giamarellos-Bourboulis EJ. Macrophage activation-like syndrome: a distinct entity leading to early death in sepsis. Front Immunol. 2019;10:55.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet. 2020;395(10223):497–506.
Arabi YM, Mandourah Y, Al-Hameed F, Sindi AA, Almekhlafi GA, Hussein MA, et al. Corticosteroid therapy for critically ill patients with middle east respiratory syndrome. Am J Respir Crit Care Med. 2018;197(6):757–67.
Stockman LJ, Bellamy R, Garner P. SARS: systematic review of treatment effects. PLoS Med. 2006;3(9):e343.
Russell CD, Millar JE, Baillie JK. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet. 2020;395(10223):473–5.
Peter Horby WSL, Jonathan Emberson, Marion Mafham, Jennifer Bell, Louise Linsell, Natalie Staplin, Christopher Brightling, Andrew Ustianowski, Einas Elmahi, Benjamin Prudon, Christopher Green, Timothy Felton, David Chadwick, Kanchan Rege, Christopher Fegan, Lucy C Chappell, Saul N Faust, Thomas Jaki, Katie Jeffery, Alan Montgomery, Kathryn Rowan, Edmund Juszczak, J Kenneth Baillie, Richard Haynes, Martin J Landray, RECOVERY Collaborative Group. Effect of Dexamethasone in Hospitalized Patients with COVID-19: Preliminary Report. MedRxIV. 2020.
Hanson KE, Caliendo AM, Arias CA, Englund JA, Lee MJ, Loeb M, et al. Infectious Diseases Society of America Guidelines on the Diagnosis of COVID-19. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa760.
Bhimraj A ea. Guidelines on the treatment and management of patients with COVID-19 Webpage2020. https://www.idsociety.org/practice-guideline/covid-19-guideline-treatment-and-management/#toc-5.
European Medical Society Press Release. https://www.ema.europa.eu/en/news/ema-gives-advice-use-non-steroidal-anti-inflammatories-covid-19. 2020.
Zhu H, Shi X, Ju D, Huang H, Wei W, Dong X. Anti-inflammatory effect of thalidomide on H1N1 influenza virus-induced pulmonary injury in mice. Inflammation. 2014;37(6):2091–8.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/021430lbl.pdf: Thalidomide package insert. 2006.
Xu X. Effective Treatment of Severe COVID-19 Patients with Tocilizumab. chinaXiv:20200300026v1: ahead of publication. 2020.
Touret F, de Lamballerie X. Of chloroquine and COVID-19. Antiviral Res. 2020;177:104762.
Khadka RH, Sakemura R, Kenderian SS, Johnson AJ. Management of cytokine release syndrome: an update on emerging antigen-specific T cell engaging immunotherapies. Immunotherapy. 2019;11(10):851–7.
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62.
Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. 2016;44(2):275–81.
Anakinra packet insert: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/103950s5136lbl.pdf.
Stebbing J, Phelan A, Griffin I, Tucker C, Oechsle O, Smith D, et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis.
Singer JW, Al-Fayoumi S, Taylor J, Velichko S, O'Mahony A. Comparative phenotypic profiling of the JAK2 inhibitors ruxolitinib, fedratinib, momelotinib, and pacritinib reveals distinct mechanistic signatures. PLoS ONE. 2019;14(9):e0222944.
Stebbing J, Phelan A, Griffin I, Tucker C, Oechsle O, Smith D, et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis. 2020;20(4):400–2.
The involvement of natural killer cells in the pathogenesis of severe acute respiratory syndrome. Am J Clin Pathol. 2004;121(4):507–11.
Paust S, Blish CA, Reeves RK. Redefining memory: building the case for adaptive NK cells. J Virol. 2017;91(20):e00169–e217.
Golchin A, Seyedjafari E, Ardeshirylajimi A. Mesenchymal stem cell therapy for COVID-19: present or future. Stem Cell Rev Rep. 2020;16(3):427–33.
Rajarshi K, Chatterjee A, Ray S. Combating COVID-19 with mesenchymal stem cell therapy. Biotechnol Rep (Amst). 2020;26:e00467.
Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio. 2018. https://doi.org/10.1128/mBio.01753-18.
Markham A, Keam SJ. Camrelizumab: first global approval. Drugs. 2019;79(12):1355–61.
Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA. 2020;323:1582.
Cunningham AC, Goh HP, Koh D. Treatment of COVID-19: old tricks for new challenges. Crit Care. 2020;24(1):91.
Acknowledgements
Authors are thankful to Joanna King MSMI, Creative Director Medical Illustration and Animation at Mayo Clinic Rochester, Minnesota for her help with illustrations in this review.
Funding
Illustrations for this review article were made using discretionary research funds to Dr. Sohail from Mayo Clinic College of Medicine and Science.
Author information
Authors and Affiliations
Contributions
All authors have contributed significantly to the writing of this manuscript and critical revisions. Additionally, SK, NA, and NAh drafted the figures and table, which the other authors subsequently edited. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors have declared they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Khadke, S., Ahmed, N., Ahmed, N. et al. Harnessing the immune system to overcome cytokine storm and reduce viral load in COVID-19: a review of the phases of illness and therapeutic agents. Virol J 17, 154 (2020). https://doi.org/10.1186/s12985-020-01415-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-020-01415-w