
Abstract
Background
Incontinentia pigmenti (IP) is a rare X-linked dominant disorder affecting the skin and other ectodermal tissues that is caused by a mutation of the IKBKG/NEMO gene.
Case presentation
We describe a Turkish family with IP. Sanger sequencing was performed in our patient with IP, and we identified DNA variant c.172_173delAA (p.Asn58SerfsTer79) in IKBKG. We found the same mutation in the patient's mother and grandmother.
Conclusion
Our report expands the mutation spectrum in this disorder and provides valuable information on the importance of the IKBKG. Our study shows that confirmation of the mutation analysis of IP in the suspected cases is necessary for future planning pregnancies.
Similar content being viewed by others


Background
Incontinentia pigmenti (IP; Bloch-Sulzberger syndrome) is a rare X-linked dominant multisystemic genodermatosis that primarily affects the ectodermal tissues such as skin, hair, nails, teeth, eyes, and central nervous system [1]. Clinical expression of IP differs between males and females, typically resulting in an in-utero exitus in males. It is caused by mutations in the Inhibitor of kappa B kinase gamma (IKBKG), previously NEMO gene. IKBKG, a 23 kb long gene that consists of 10 exons, is essential for the activation of the Nuclear factor-kappa B (NF- κB) transcription factor, which is involved in the prevention of Tumor Necrosis Factor-alpha (TNF-α)-induced apoptosis and the regulation of immune and inflammatory responses [2,3,4,5]. Although IP can be mainly diagnosed with clinical features, mutation analysis of IKBKG thus confirmation of the clinical diagnose are critical for the prenatal screening of future pregnancies.
Herein we present a case of IP confirmed with molecular genetic analysis.
Case presentation
A 40-day-old female patient was referred to our clinic for the complaint of vesiculobullous rashes on her arms and legs. Upon the patient’s history, it was noted that the rashes had appeared right after birth, and the patient had been taken to an intensive care unit (ICU). During that period, varicella and TORCH infections have been investigated, and no infection was reported. The patient’s mother stated that she had had similar lesions in her infancy, which receded later.
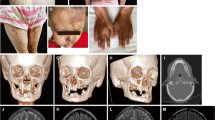
Upon physical examination, it was noted that the patient’s vital signs were stable, and her overall condition was good. She had vesiculobullous rashes with an erythematous base on her arms and verrucous, hyperkeratotic papules and plaques on her legs, all consistent with the course of Blaschko’s lines. Upon the mother’s physical examination, hypopigmented lesions on the skin were noted (Fig. 1).

Pictures of the patient and patients’ mother. A vesiculobullous rashes with an erythematous base on the arm of the patient, B verrucous, hyperkeratotic papules and plaques on the leg of the patient, C hypopigmented skin lesions of the mother
Based on the patient’s clinical features, IP was suspected and blood samples from the patient, the mother, the father, and the maternal grandmother were taken for molecular genetic testing. An informed consent was received from the family. DNA analysis of the patient’s sample revealed an AA deletion at 172–173 in IKBKG, which caused an Asn > Ser change. The same mutation was also detected in the mother and the maternal grandmother (Fig. 2).

Representation of electropherograms and pedigree of the family S318. A Pedigree and segregation of the IKBKG variant in the family. B Shows electropherograms of the p. Q59Rfs*78 variant. Het: heterozygous mutant, WT: wild type
After the diagnosis of IP was confirmed, the patient was followed up for additional systemic involvements. During her ocular examination at age 12 months, bilateral peripheric avascular areas and a fibrotic membrane in the right optic nerve were observed. Laser therapy was done to the avascular areas. The patient was later operated on at age 26 months, due to the growth of the fibrotic membrane in her right eye. Hairs were sparse. Nail dystrophia and hyperpigmented lesions on the skin developed later (Fig. 3). Dentition occurred at age 11 months and 15 teeth came out till age 4 (Fig. 3). Neuroimaging studies [Magnetic resonance imaging (MRI)] were conducted for possible central nervous system anomalies; none were reported nor have developed later.

Picture of the patient. A. Hyperpigmented lesions on legs, B. Cone-shaped teeth and partial anodontia
Discussion
IP, with an estimated birth prevalence of 1–9/1,000,000 (http://www.orpha.net/orphacom), is an X-linked dominant disease [6]. Because males lack a compensatory healthy X chromosome, the common deletion of IKBKG is lethal in males and the resulting disease form is mainly seen in females.
IP is caused by mutations in the IKBKG (inhibitor of kappa B kinase gamma) gene, also known as NEMO (nuclear factor-kappa-B essential modulator), located on Xq28. The encoded protein I-kappa-B kinase-gamma is involved in the activation of NF-κB, thus the regulation of apoptosis, inflammation, and immunity. Most of the cases have “de novo” deletions of exons 4–10, resulting in complete loss of encoded protein function [5, 6].
The phenotypic expression can range from mild skin involvements to severe neuro-ophthalmological symptoms, thus clinical variations, even in the same family, can be observed. This variability of expressivity is mainly caused by lyonization (X-inactivation) that leads to functional mosaicism [6, 7,8,9,10].
Dermatological findings develop in the first weeks of life and are generally the first to be observed, and as our patient, most patients present them.
80% of the cases have dental anomalies, making dental findings are the most seen after dermatological ones. On the contrary to dermatological, dental anomalies last for life and have a different diagnostic significance [9,10,11].
Ocular involvement is not common, around 35% of the cases are thought to have it. Strabismus, optic nerve atrophy, conjunctival pigmentation, iris hypoplasia, nystagmus, and uveitis are important non-retinal findings. Foveal hypoplasia, avascular retina, neovascularization, vitreous hemorrhage, and fibrovascular proliferation are important retinal findings, and they are usually associated with vascular abnormalities. Retinal detachment and blindness can be observed in advanced cases.
Retinal lesions are most usually observed in the first 12 months, thus making ophthalmological follow-ups during this period highly important. Non-retinal lesions tend to occur later, within age 2 years [12, 13].
Differential diagnosis of IP varies within different cutaneous stages of the disease. During the first vesicular stage, newborn vesiculobullous diseases should be suspected, infections such as bullous impetigo, herpes simplex, varicella, or immunological diseases such as dermatitis herpetiformis, epidermolysis bullosa acquisita, bullous pemphigus, neonatal pemphigus vulgaris should be investigated. Langerhans cell histiocytosis can also manifest as papules and vesicles during the newborn period. Additionally, bullous mastocytosis can manifest as localized erythema and bullae in the first year of life [14].
This variant that we detected in our study has not been reported. c.172_173delAA (p.Asn58SerfsTer79) variant was not found in healthy population, but its locus is covered in gnomAD exomes and genomes databases. This variant is located in exon 2 in IKBKG and causes a terminated protein. Therefore, we evaluated this deletion pathogenic according to the ACMG guideline [15].
Conclusion
In our case, the presence of vesicular and verrucous lesions during the newborn period and a similar history accompanying existing hyperpigmented and atrophic skin lesions in the mother made the clinical diagnosis of IP possible. The diagnosis was later confirmed with genetic testing.
Mutation analysis of IP suspected cases is essential for future planning pregnancies, along with the confirmation of the disease itself.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- IP:
-
Incontinentia pigmenti
- IKBKG:
-
Inhibitor of kappa B kinase gamma
- NF- κB:
-
Nuclear factor-kappa B
- TNF-α:
-
Tumor Necrosis Factor-alpha
- ICU:
-
Intensive care unit
- MRI:
-
Magnetic resonance imaging
- NEMO:
-
Nuclear factor-kappa-B essential modulator
References
Landy SJ, Donnai D (1993) Incontinentia Pigmenti (Bloch-Sulzberger Syndrome). J Med Genet 30:53–59
Sefiani A, Abel L, Heuertz S et al (1989) The gene for incontinentia pigmenti is assigned to Xq28. Genomics 4(3):427–429. https://doi.org/10.1016/0888-7543(89)90350-9
Smahl A, Hyden-granskog C, Peterlin B et al (1994) The gene for the familial form of incontinentia pigmenti (IP2) maps to the distal part of Xq28. Hum Mol Genet 3(2):273–278
Jin DY, Jeang KT (1999) Isolation of full-length cDNA and chromosomal localization of human NF-kappaB modulator NEMO to Xq28. J Biomed Sci 6(2):115–120
Smahi A, Courtois G, Vabres P et al (2000) Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 405(6785):466–472
Fusco F, Valente V, Fergola D, Pescatore A, Lioi MB, Ursini MV (2019) The Incontinentia Pigmenti Genetic Biobank: study design and cohort profile to facilitate research into a rare disease worldwide. Eur J Hum Genet 27(10):1509–1518
Levy M. Incontinentia pigmenti. In: Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com/contents/incontinentia-pigmenti. Accessed [24.10.2020].
Kenwrick S, Woffendin H, Jakins T et al (2001) Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am J Hum Genet 69(6):1210–1217
Berlin AL, Paller AS, Chan LS (2002) Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol 47(2):169–190
Happle R (1985) Lyonization and the lines of Blaschko. Hum Genet 70(3):200–206
Minić S, Trpinac D, Gabriel H, Gencik M, Obradović M (2013) Dental and oral anomalies in incontinentia pigmenti: a systematic review. Clin Oral Investig 17(1):1–8
Rosenfeld SI, Smith ME (1985) Ocular findings in incontinentia pigmenti. Ophthalmology 92(4):543–546
Goldberg MF, Custis PH (1993) Retinal and other manifestations of incontinentia pigmenti (Bloch-Sulzberger syndrome). Ophthalmology 100(11):1645–1654
Shah K. Incontinentia Pigmenti Differential Diagnoses. Medscape. https://emedicine.medscape.com/article/1114205-differential. Updated: [05.03.2019]. Accessed [24.10.2020].
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Acknowledgements
Not applicable.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
MG, BG, SB, NA followed up the patients. FBCE, MT performed the genetic analysis of the patient. FBCE, BG, NA drafted the manuscript. MT, NA critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Miami Institutional Review (USA), and the Ankara University Medical School Ethics Committee (Turkey).
Consent for publication
Informed consent was obtained from the patient for publication of this case report and accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ergin, F.B.C., Tekin, M., Güneş, M. et al. A Turkish case of incontinentia pigmenti with a deletion mutation at Inhibitor of kappa B kinase gamma gene. Egypt J Med Hum Genet 23, 6 (2022). https://doi.org/10.1186/s43042-022-00215-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00215-x