
Abstract
Background
In vitro plant regeneration via androgenesis or somatic embryogenesis is capable of inducing (epi)mutations that may affect sexual progenies. While epimutations are associated with DNA methylation, mutations could be due to the movement of transposons. The common notion is that both processes are linked. It is being assumed that demethylation activates transposable elements (TEs). Analysis of methylation changes and their relation with TEs activation in tissue cultures requires uniquely derived donor plants (Ds), their regenerants (Rs) and respective progeny (Ps) that would allow discrimination of processes not related to changes introduced via in vitro cultures. Moreover, a set of methods (RP-HPLC, SSAP, and MSTD) is needed to study whether different TEs families are being activated during in vitro tissue culture plant regeneration and whether their activity could be linked to DNA methylation changes or alternative explanations should be considered.
Results
The in vitro tissue culture plant regeneration in barley was responsible for the induction of DNA methylation in regenerants and conservation of the methylation level in the progeny as shown by the RP-HPLC approach. No difference between andro- and embryo-derived Rs and Ps was observed. The SSAP and MSTD approach revealed that Ds and Rs were more polymorphic than Ps. Moreover, Rs individuals exhibited more polymorphisms with the MSTD than SSAP approach. The differences between Ds, Rs and Ps were also evaluated via ANOVA and AMOVA.
Conclusions
Stressful conditions during plant regeneration via in vitro tissue cultures affect regenerants and their sexual progeny leading to an increase in global DNA methylation of Rs and Ps compared to Ds in barley. The increased methylation level noted among regenerants remains unchanged in the Ps as indicated via RP-HPLC data. Marker-based experiments suggest that TEs are activated via in vitro tissue cultures and that, independently of the increased methylation, their activity in Rs is greater than in Ps. Thus, the increased methylation level may not correspond to the stabilization of TEs movement at least at the level of regenerants. The presence of TEs variation among Ds that were genetically and epigenetically uniform may suggest that at least some mobile elements may be active, and they may mask variation related to tissue cultures. Thus, tissue cultures may activate some TEs whereas the others remain intact, or their level of movement is changed. Finally, we suggest that sexual reproduction may be responsible for the stabilization of TEs.
Similar content being viewed by others

Background
There is a growing body of evidence that even morphologically identical plants regenerated via tissue culture may not be uniform at the (epi)genetic level [1], and the regenerants may differ from their donors [2, 3]. These differences can be due to changes in DNA sequence and methylation patterns. Plant genomes are usually highly methylated [4]. At the DNA level, methylation may be responsible for the regulation of gene expression [5], plant development [6] or responses to abiotic stresses [7]. Thus, alterations in DNA methylation patterns may result in either morphological [8], physiological [9] or biochemical changes [10]. They may be exhibited either among regenerants and/or their generative progenies [11, 12]. Such epimutations may arise “spontaneously” and do not appear to follow the Mendelian rules of inheritance [13, 14].
In tissue culture, reprogramming of cells (via demethylation and de novo methylation) [15] is required to force plant regeneration [16]. The mode of plant regeneration via andro- and embryogenesis is affected by the ploidy level of the source tissue [17]. The absence or duration of the callus phase may modify the reprogramming processes [18] and regeneration via tissue culture is often triggered by abiotic stresses [19]. These may alter the DNA methylation pattern [20] and activate mobile elements [21]. Finally, sequence variation may be expected [22]. Thus, studies on DNA methylation in tissue culture may generate information on epigenetic processes induced during plant regeneration and could be used as an indicator of TEs activity [23, 24].
One of the approaches to such studies is the employment of the RP-HPLC [25]. This technique delivers information on global DNA methylation in plant genomes [26]. Experiments in oil palm (Elaeis guineensis) demonstrated that DNA methylation of ortet (parent plant) vs. regenerants was 17.26 vs. 16.88 %, respectively [27]. The corresponding data for the in vitro derived banana (Musa AAA) compared to conventionally propagated plants equaled to 17.7 and 22.5 %, respectively [28]. Sianipar et al. [8] found a 2.72 % drop of global methylation between mother plants of oil palm and embryogenesis-derived progeny. In cedar (Cedrus sp.), a 5.6 % decrease in DNA methylation was noted among regenerants during in vitro culture about the donor plants [29]. The same trend was seen in triticale (xTritico secale Wittm. ex A. Camus), where tissue culture induced a decrease in DNA methylation of the regenerants compare to donor plants [30]. The RFLP analysis based on the HpaII and MspI digests are in good agreement with presented data in the case of maize regenerants derived from embryos and their sexual progeny [31]. Evidently, tissue culture may induce epigenetic changes that influence not only regenerants but also their progeny.
Changes in DNA methylation are supposed to be closely related to the movement of the mobile elements [32]. On one side, it was demonstrated that a decrease in DNA methylation resulted in activation of TEs during tissue culture plant regeneration [33]. On the other hand, it has been suggested that either transposon movement is not related to tissue cultures [34], or that some transposons could be activated [35], whereas others are not, by in vitro tissue culture treatment [36]. Possibly, some transposons may be activated in response to the given stresses [37] whereas others are activated in other cases [38]. Although retrotransposon activity is considered to be one of the causes of variability induced in tissue cultures, it should be emphasized that they can also be responsible for pre-existing variation [39]. Among TEs with the activity that can be studied in tissue culture manipulations in cereals, a suitable candidate seems to be the group with the long terminal repeats (LTR) and the non-LTR retrotransposons—both of them are present in monocot and dicot angiosperms [40, 41]. One of the members of these retrotransposons, BARE-1, has homologues in different species e.g. barley, oat, wheat or rice [42, 43]. It has proven useful for detecting polymorphism in cereals [44].
To study TEs movement, one may apply techniques directed towards retrotransposon sequences e.g. inter retrotransposons amplification polymorphism (IRAP) [22], retrotransposons microsatellite amplified polymorphism (REMAP) [45], sequence specific amplified polymorphism (SSAP) [46] or methyl-sensitive transposon display (MSTD) [32] techniques. The IRAP technique was used to study sequence variation between parental plants and regenerants in three barley cultivars [44]. It was demonstrated that 29 % of cv. Golden Promise, 53 % of Tallon and 96 % of Mackay regenerants obtained via somatic embryogenesis differed from their parental forms. Similar studies with SSAP resulted in 19.66 % polymorphism between the donor plants, two callus-pools and eight regenerants developed from young inflorescence-derived calli of barley plants (H. brevisbulatum) [46]. In extension to IRAP and SSAP that can assess sequence changes, the MSTD based on the metAFLP approach [1, 30, 47] seems to be useful in similar studies.
Besides techniques suitable to study TEs movement in tissue culture conditions, the need of suitably derived plant material to test the putative impact of in vitro conditions for the epigenetic status of regenerated plants and their sexual progeny with the reference to TE activation and DNA methylation changes still remains. Evidently, the source of explants and donor plants, should not be affected (or such an influence should be minimized) by tissue culture itself. Possibly, this could be accomplished via using generative progeny of the DH plants [48]. However, to our best knowledge, there is no information available how many generative cycles are needed to stabilize/eliminate (if possible at all) the effects induced in tissue cultures. Nevertheless, it was shown that in triticale [30] regenerants derived via anther cultures started to reestablish their methylation status after one/two cycles. Similar data are not available in barley. However, it was shown [1] that donors derived from the progeny of DH plants were uniform both at the DNA and DNA methylation levels. Thus, to stabilize DNA methylation changes induced in regenerants during in vitro tissue cultures one should consider to use as a source of explants the tissues from the progeny of the regenerants. Obviously, control of the TEs movement is hardly possible; however, it was suggested that their movement could be controlled via generative cycle [48] giving the opportunity to study the linkage between TEs activity and DNA methylation.
The linkage between DNA methylation change due to in vitro tissue culture plant regeneration and the activation of transposons (e.g. Ty-1 Copia LTR family) is not entirely understood. It is not also clear whether the level TEs in Ds, Rs and Ps would be at the comparable level or not. The aim of the study was to verify whether different TEs families are being activated during in vitro tissue culture plant regeneration; whether their activity is transmitted to sexual progeny and whether DNA methylation is linked to TEs.
Results
Barley regenerants obtained via andro- and embryogenesis and used in the analysis were fully self-fertile. They did not exhibit any apparent morphological changes relative to the donor plants. Similarly, no visible changes in plant morphology or growth patterns were observed for sexual progenies of the regenerants.
RP-HPLC
RP-HPLC allowed the identification of peaks related to dC and 5mdC with retention times equal to 6.83 and 9.68 min, respectively. The average total amount of cytosine (dC + 5mdC) in the barley genome of Ds, Rs and Ps amounted to 23.38 %. The lowest mean value of the global methylation was observed in the donor plants (17.86 %), whereas the mean value in the regenerants and their progenies was 20 and 20.13 %, respectively (Table 1).
The RP-HPLC analysis of the regenerants from any of the two regeneration approaches showed that the level of the cytosine methylation of the embryogenic (RE) and androgenic (RA) derived regenerants was 20.1 and 19.88 %, whereas DNA methylation of their progenies was 20.17 and 20.09 % for PE and PA, respectively.
ANOVA showed the increase of global DNA methylation among the regenerants relative to the donor plants (F = 36.69; p < 0.0001, α = 0.01), whereas DNA methylation of the generative progeny was at the same level as that for the regenerants (F = 0.28; p = 0.60, α = 0.05) and varied from the donors (F = 26.46; p < 0.0001, α = 0.01). There were no differences between the two modes of plant regeneration (androgenesis vs. somatic embryogenesis) in the global DNA methylation levels (F = 0.61; p = 0.44, α = 0.05). Also, the difference between progenies derived from the two types of regenerants was insignificant (F = 0.04; p = 0.85, α = 0.05). There were no significant alterations among individual regenerants (F = 1.73; p = 0.15, α = 0.05) and the progenies (F = 0.66; p = 0.58, α = 0.05).
metAFLP
The metAFLP profiling resulted in stable and highly reproducible banding patterns amplified with metAFLP selective primer pairs as illustrated in the case of donors (Figs. 1, 2).

An example of the distinct SSAP profile generated with BARE1 EO377 directed and MGT selective primers. The Acc65I/MseI (left) and KpnI/MseI (right) metAFLP platforms. Lines 6–10, 11–17 and 18–24 represent profiles of the donors, regenerants and progenies, respectively. The metAFLP pattern using CpXpG-AGC/MCGT primer pair, (line 1–5) representing donor plants, is included
SSAP and MSTD markers
The primers used in SSAP and MSTD approach generated polymorphic (Fig. 1) as well as monomorphic profiles (Fig. 2). While the primers NIKITA and BARE1 EO377, resulted in highly polymorphic and hardly readable banding patterns (Fig. 1), the primers BARE LO45C, BARE LTR, SUKKULA 9900 and SUKKULA EO299 generated entirely or mostly monomorphic profiles detected for Ds, Rs and Ps samples (Fig. 2).

An example of the monomorphic SSAP profile generated with BARE LTR-directed and MCGT selective primers. The Acc65I/MseI (left) and KpnI/MseI (right) metAFLP platforms. Lines 6–10, 11–17 and 18–24 represent profiles of the donors, regenerants and progenies, respectively. The metAFLP pattern using the using CpXpG-AGC/MCGT primer pair (line 2–5 for Acc65I/MseI and 1–5 for KpnI/MseI) representing donor plants is included
The SSAP and MSTD approach using finally 12 selective primer pairs amplified 513 markers with 210 and 293 being polymorphic, respectively. There were 41, 39 and ten markers amplified via the SSAP method and shared exclusively among Ds, Rs and Ps, respectively. In the MSTD, these amounts were 75, 24 and 9, respectively. The SSAP markers were less polymorphic than the MSTD ones (Table 2). Moreover, markers in the progeny were less polymorphic than those in regenerants and donors. Shannon’s information indices followed the very similar pattern of changes in both approaches with the highest values for the donors and the lowest for the progenies.
Analysis of Molecular Variance demonstrated that the difference between Ds, Rs and Ps evaluated based on both approaches were significant (Table 3). The difference between Ds, Rs and Ps was also assessed for SSAP profiles (Φ PT = 0.289, p = 0.001) and MSTD data (Φ PT = 0.199, p = 0.001). Comparison of the SSAP data pointed at R–P as the one with the highest value of the variance (Table 3). The same comparison (R–P) for MSTD did not reach as high value as for SSAP. The highest Φ PT value contrasted D-P for MSTD markers (Table 3). Φ PT values demonstrated that explained variance between Rs and Ps was greater than that between Ds and Ps and between Ds and Rs based on SSAP markers. The explained variance was the highest between Ds and Ps and the lowest between Ds and Rs in the case of the MSTD markers.
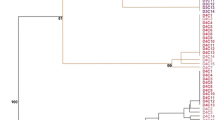
Ward’s method of clustering based on SSAP and MSTD markers divided the data into two independent parts. The first one reflected the results assessed via the SSAP and the second via MSTD approach. The SSAP method classified Ds, Rs, and Ps into three distinct groups (Fig. 3). The presented analysis pointed at donors and regenerants exhibiting the highest diversity compared to the progeny. However, a difference in variation between Rs and Ds was either negligible or slightly increased in Rs. The MSTD method resulted in one cluster encompassing of progeny and the other represented by donors and regenerants. The difference among regenerants was greater than among donors. Independently of the marker system, the variation level among sexual progeny was lower than that among donor plants and their regenerants. Moreover, the SSAP revealed smaller variation level among Ds, Rs, and Ps than the MSTD.

Clustering of Ds, Rs, and Ps based on SSAP and MSTD markers using Ward’s method with bootstrap values at the nodes. D, R, and P reflect donors, regenerants and their sexual progeny; “K” states for the SSAP whereas “M” for the MSTD results
Discussion
Visual inspection of Rs and Ps failed to identify any morphological differences among analyzed plants and all of them were in donor plant type suggesting the lack of the tissue culture induced and somaclonal variation. However, this result is in contrast to the data evaluated based on RP-HPLC approach indicating the increase in DNA methylation of the regenerants conserved in the progeny comparing to the donors. A similar change in the global DNA methylation related to in vitro regeneration, detected by the RP-HPLC method, was observed in oil palm (Elaeis guineensis) regenerants [49] as well as in Gentiana pannonica [50]. On the other hand, a decrease was found in triticale (xTritico secale Wittm. ex A. Camus) [30]. Thus, at least two alternative pathways of DNA methylation under tissue culture condition appear possible. In the first scenario, tissue culture induces an increase of DNA methylation of regenerants whereas in the second one demethylation is observed. It could be speculated that there might be some differences in demethylation and de novo methylation of species under tissue culture conditions. Alternatively, some differences could be related to the ploidy levels. It appears that at least some diploids (e.g. barley) would tend to increase their DNA methylation levels while polyploids (e.g. triticale) decrease their levels under tissue culture conditions. However, the difference between barley and triticale may also reflect the general instability of triticale [51]. The instability may be related to delayed replication of rye chromosomes [52]. However, available data [53] seems to indicate that the pathway is more species rather than the ploidy level specific. Such a notion appears to be true as the tested species at the same ploidy level (barley, oil palm) may either be affected by prevailing de novo methylation or demethylation of genomic DNA of the regenerated plants [29, 46, 49]. Interestingly, as the RP-HPLC delivers averaged information on DNA methylation change it may not reflect subtle alterations identified by marker-based approaches. Thus, in barley, the MSAP method [46] showed a 3 % drop in global DNA methylation of the restriction sites observed in Rs compared to Ds whereas metAFLP [1] demonstrated that both site DNA de novo methylation and demethylation changes nearly equally affected regenerants in comparison to donors. On the other hand, the RP-HPLC and metAFLP results assessed in Gentiana pannonica clearly demonstrated the increase in genomic methylation of the regenerants [50]. The differences between molecular approaches may suggest varying distribution of the sites or their presence in genome regions distinctly affected by DNA methylation changes because the MSAP approach is based on HpaII and MspI whereas metAFLP on Acc65I and MspI isoschizomers. Such a notion seems to be supported by the uneven distribution of AFLPs on genetic maps of wheat [54] and rye [55] which could also be the case in barley. Thus, molecular marker approaches could be valuable tools to study fluctuations in site DNA methylation pattern changes or those bind to specific genomic elements (i.e. transposons, genes).
It has been suggested that demethylation due to abiotic stresses may be responsible for the activation of mobile elements [56]. As metAFLP proved to be useful for the analysis of methylation changes, a slight modification that could utilize selective primers towards varied classes of TEs could be a method of choice. The most common retrotransposons shared among cereals are those classified as LTR [57, 58] and solo LTR families [59]. The former are represented by BARE-1, whereas the later by NIKITA and SUKKULA [59]. It was expected that utilization of the selective primers directed towards BARE-1, NIKITA and SUKKULA TE sequences in the SSAP and MSTD method would help in linking methylation changes among Ds, Rs and Ps and alterations in DNA sequences. Hierarchical clustering demonstrated that whereas Ds, Rs and Ps formed separate groups based on the SSAP approach, the level of genetic variation in Rs was slightly higher or nearly identical to that in Ds whereas in Ps was lower than in former cases. Similarly, the MSTD analysis confirmed more moderate variation among Ps than that among Rs and Ds individuals that formed the separate cluster. However, Rs exhibited higher variation than Ds. The differences among Rs identified by the MSTD are even greater than Rs produced by SSAP approach. The contrast between the three groups of plants is also evidenced by AMOVA and confirms that TEs activity exhibited among Rs could be related to DNA methylation changes induced in tissue cultures. Moreover, higher variation level among Rs assessed by the MSTD method than by the SSAP one is in agreement with our results in barley demonstrating that in vitro tissue cultures induce increased site DNA methylation compared to sequence changes [1] which could be linked to TEs activation in tissue cultures. The observed phenomenon is congruent with the data presented by the others [60, 61].
Our former study [1], as well as current analysis of donor plants based on the metAFLP approach, demonstrated that Ds were highly uniform at the (epi)genetic level. Moreover, independently of whether the SSAP or MSTD approach was applied the variation among Ds is evident. The level of DNA sequence polymorphisms among Ds (evaluated by the SSAP) was nearly identical to that observed among regenerants of the randomly chosen donor (as indicated e.g. by cluster analysis). It should be stressed that donors were the progeny of the tissue culture regenerated plants. Obviously, the donors could be affected by TEs movement (illustrated by SSAP and MSTD approaches) influencing our estimation of TE related variation. Alternatively, the difference between donors could be explained by the so-called pre-existing variation [39] that resulted in TE-related variation not revealed by the metAFLP approach. It is being suggested, however, that generative cycle should stabilize putative TE activity [48]. Thus, individual donor plants should be treated as uniform materials.
As individual donor plants were assumed to be a homozygous generative progeny of DH regenerants (see “Methods” section) they are adequate material for studying TEs movement due to in vitro tissue plant regeneration. Thus, most—if not all—of the TE-related variation observed in Rs (and transmitted to Ps) comparing to the given D plant was the result of the tissue culture plant regeneration. Alternatively, the polymorphic TE-related profiles revealed for Rs could be interpreted regarding background activity of some mobile elements, which to some extent may be responsible for the pre-existing variation not related to the in vitro tissue culture plant regeneration. If so, then, at least, some retrotransposons may generate variation that is not related to in vitro tissue culture.
Interestingly, some primers directed towards BARE-1and NIKITA TEs failed to amplify whereas those towards i.e. SUKKULA and the other BARE-1 amplified hardly polymorphic profiles. Thus, we tend to think that some TEs could be active whereas the others not under tissue culture plant regeneration. Such results could be explained by the different activity of various mobile elements [62] or even altered action of the same TE in distinct species due to e.g. stresses like tissue culture [35, 36]. Our data is in agreement with previous reports, indicating that distinct TEs could be activated due to in vitro tissue culture plant regeneration [35, 63].
Although we cannot entirely exclude that a decrease in genetic variation in Ps compared to Ds and Rs is due to the progeny that originated from a single regenerant, we suggest that TEs movement detected among Ps is reduced regarding sexual reproduction. This notion is supported by polymorphisms revealed via the MSTD and SSAP that decreased in Ps in parallel to a nearly identical level of DNA methylation of Ps and Rs as assessed by RP-HPLC approach. Thus, our results favor the hypothesis of TE stabilization due to sexual reproduction [48] indicating that reproductive cycle of in vitro regenerated DH plants may somewhat limit the level of variation related to retrotransposon activity (making such materials suitable for studies on TE related changes originating due to tissue culture plant regeneration methods). However, independent studies are needed to verify how many reproductive cycles are needed to eliminate/minimize the TEs activation due to tissue culture manipulations and whether this is dependent on species and mode of reproduction.
According to a common notion, plant regeneration via androgenesis should be less error prone than via embryogenesis due to the lack or hardly observed callus phase [64, 65], which is responsible for the release of cellular control over mutagenesis generated i.e. by the motion of transposons agents possibly induced as the result of genomic DNA demethylation [66]. The RP-HPLC approach, however, failed to assess differences in DNA methylation due to the mode of the in vitro tissue culture plant regeneration which is congruent with our previous studies in barley [1] based on the metAFLP approach, and those in triticale [47]. Thus, the presence of putative callus phase during embryogenesis is not the source of the tissue culture-induced variation. Alternatively, other factors (i.e. haploid and the diploid fabrics) diminished the level of variation in the case of embryogenesis derived regenerants [1]. Finally, we cannot exclude that, general metAFLP characteristics failed to detect subtle changes that were assessed based on the advanced ones evaluated in the case of triticale [47] demonstrating that the mode of plant regeneration could influence tissue culture-induced variation.
Conclusion
Stressful conditions during plant regeneration via in vitro tissue cultures affect regenerants and their sexual progeny leading to an increase in global DNA methylation of Rs and Ds compared to Ds in barley. The increased methylation level revealed among regenerants remains unchanged in the Ps as indicated via RP-HPLC data. It is usually assumed that such a change in methylation is related to stabilization of the TEs activity. Our marker based experiments showed that TEs seem to be activated via in vitro tissue cultures and that independently of the increased methylation their activity in Rs is greater than in Ps. Thus, the increased methylation level may not correspond to the stabilization of TEs movement at least at the level of regenerants. We have also shown the presence of TEs variation among Ds that were genetically and epigenetically uniform as indicated by the metAFLP approach. TEs variation assessed among Ds may suggest that at least some mobile elements may be active, and they may mask variation related to tissue cultures. It should be stressed however, that we cannot exclude that the donor plants being the progeny of DH regenerants failed to stabilize TEs activity most probably induced during tissue culture manipulations. Thus, tissue cultures may activate some TEs whereas the others remain intact, or their level of movement is changed. Moreover, sexual reproduction may be responsible for the stabilization of TEs (possibly due to stabilized methylation of the genome). Nevertheless, their movement is not eliminated.
Methods
Plant materials for RP-HPLC
The starting materials were full-sib progenies (Ds, donor plants; 22 plants) of individual doubled haploid (DH) barley (Hordeum vulgare L.; cv. Scarlett) plants. The DH plants originated via androgenesis in isolated microspore culture [67]. Ds served as a source of explants to obtain regenerants (Rs) either by androgenesis in anther cultures (labeled RA, 35 plants) or by somatic embryogenesis using immature zygotic embryo cultures (labeled RE, 37 plants). Randomly chosen regenerants (four from androgenesis and four from embryogenesis) were self-pollinated to obtain sexual progenies (labeled Ps, PE, 24 plants, PA, 22 plants) (Fig. 4).

A schematic representation of the plant materials. D stands for donor plants, R for regenerants and P for progeny. RE and RA refer to regenerants obtained via somatic embryogenesis and androgenesis, respectively, whereas PE and PA are the progenies of regenerants obtained via somatic embryogenesis and androgenesis, respectively
Androgenesis
Tillers of the donor plants were collected at late microspore stage and kept for 3 weeks in water in darkness at 4 °C. Spikes were surface sterilized, anthers removed from the spikes and plated on solidified medium N6 [68] supplemented with vitamins [69], with 2 mg l−1 2,4-D, 0.5 mg l−1 kinetin and 80 g l−1 maltose. They were cultured in Petri dishes in the dark at 26 °C for 4–6 weeks. Androgenic structures (calli, embryos) were transferred to the regeneration medium 190-2 [70] supplemented with 0.5 mg l−1 NAA and 0.5 mg l−1 kinetin [71]. Cultures were kept under 16/8 h (day/night photoperiod) for 2–4 weeks. Green plantlets were transferred to half-strength MS [72] medium without growth hormones for rooting. Plants were potted, adapted to soil conditions and grown in the greenhouse to maturity under standard conditions.
Somatic embryogenesis
Embryos were excised from immature sterilized caryopses from donor plants (harvested 12–16 days after pollination), plated on MS medium supplemented with 2 mg l −1 2,4-D [73] with the scutellum sidefacing up. The plates were incubated under a 16/8 h (day/night) photoperiod at 26 °C for 3–4 weeks. Embryogenic calli were transferred to regeneration media (the same as for androgenesis) and subcultured every 2 weeks. Rooted plants were transferred to the greenhouse and grown to maturity under standard conditions.
Progeny of regenerants
Four regenerants from androgenesis and four from embryogenesis were self-pollinated. These regenerants were derived from four different donor plants (two regenerants each obtained from one donor plant). Separated seed samples of these eight regenerants were used to derive the first generation progenies of the regenerants (Ps) (Fig. 4).
Plant materials for SSAP and MSTD
Five donor plants (the progeny of DH regenerants), seven regenerants derived via somatic embryogenesis from one of the donor plants and seven progenies obtained from one regenerant was the plant material used in SSAP and MSTD approach. All plants were chosen from those prepared for RP-HPLC analysis.
Genomic DNA extraction
Total genomic DNA was isolated from 100 mg of 7-day-old seedling leaves, using the DNasy Mini Prepkit (Qiagen GmbH, Hilden, Germany). The quantity of DNA was evaluated spectrophotometrically at λ = 260 nm. DNA integrity and purity was verified electrophoretically on 1.2 % agarose gel in 1× TBE, stained with ethidium bromide of final concentration 0.5 μg ml−1. Separation was performed at 160 V for 30 min.
DNA preparation to RP-HPLC
DNA samples (4 μg each) were dried, dissolved in 100 μl of deionised water, denaturated (100 °C for 2 min) and left on ice for 5 min. The mixture was gently stirred after adding 5 μl of 10 mM ZnSO4and 10 μl of 1.0 U ml−1 nuclease P1 in 30 mM NaOAc (pH 5.4) and then incubated at 37 °C for 17 h. After incubation, 10 μl of 0.5 M Tris (pH 8.3) and 10 μl of 10.0 U ml−1 alkaline phosphatase in 2.5 M (NH4)2SO4 were added, and samples were again gently stirred and incubated at 37 °C for 2 h. Samples were centrifuged for 5 min at 12 × 103 rpm.
RP-HPLC c12
RP-HPLC analysis was performed using the Waters 625 LC System (encompassing: Waters 625 Pump, Waters 600 Controller, Waters 717plus Autosampler, Waters Degasser and Waters 996 PDA detector) Synergy Max-RP C12 (250 × 4.6 mm, 4u, Phenomenex) column, combined with Synergy Max-RP C12 pre-column according to an adapted procedure [74, 75]. Separation of nucleosides was conducted in the presence of ‘A’ buffer (0.5 % v/v methanol in 10 mM KH2PO4, pH 3.7) and ‘B’ buffer (10 % v/v methanol in 10 mM KH2PO4, pH 3.7). The pH of the buffers was adjusted with phosphoric acid. The linear gradient used for separation consisted of 100 % ‘A’ buffer to 100 % of ‘B’ buffer for 10 min, next 100 % of ‘B’ buffer for 10–25 min and then, at the end of 25 min program 100 % ‘A’ buffer was pumped for 5 min. Flow rate was 1 ml per min and column temperature was set at 30 °C. UV-detection was used at the wavelength of 280 nm. The external standard consisted of major DNA (0.5–50 µM) and RNA nucleosides (1.5–150 µM) and 5-methyl-2′-deoxycytidine (5mdC) dissolved in deionized water. Peaks corresponding to 2′-deoxycytidine (dC) and 5mdC had retention time equal to 6.5 and 9.3 min, respectively. The contribution of 5mdC was calculated based on Millennium 32 v. 4.0 software (Waters Corporation, Milford, Massachusetts, USA).
Assessment of nucleosides
Quantification of nucleosides was based on the automatically integrated surface areas (μV s−1) of the chromatograph peaks. The amount of cytidine (dC) and 5-methyl-2′-deoxycytidine (5mdC) (also sum of dC and 5mdC) in relation to all nucleosides was assessed using the formula: \({\text{dC}} = {{\text{dC}} \mathord{\left/ {\vphantom {{\text{dC}} {\left( {{\text{dC}} + 5{\text{mdc}} + {\text{dG}} + {\text{dT}} + {\text{dA}}} \right)}}} \right. \kern-0pt} {\left( {{\text{dC}} + 5{\text{mdc}} + {\text{dG}} + {\text{dT}} + {\text{dA}}} \right)}} \times 100\) and \(5{\text{mdc}} = {{5{\text{mdc}}} \mathord{\left/ {\vphantom {{5{\text{mdc}}} {\left( {{\text{dC}} + 5{\text{mdc}} + {\text{dG}} + {\text{dT}} + {\text{dA}}} \right)}}} \right. \kern-0pt} {\left( {{\text{dC}} + 5{\text{mdc}} + {\text{dG}} + {\text{dT}} + {\text{dA}}} \right)}} \times 100.\) The amount of global DNA methylation was calculated as the concentration of 5-methyl-2′-deoxycytidine (5mdC) in relation to the whole amount of cytidine according to the formula: \({{5{\text{mdc}}} \mathord{\left/ {\vphantom {{5{\text{mdc}}} {\left( {5{\text{mdc}} + {\text{dC}}} \right)}}} \right. \kern-0pt} {\left( {5{\text{mdc}} + {\text{dC}}} \right)}} \times 100\). Mean values and standard deviation of the amount of global DNA methylation were evaluated for D, R, P, RA, RE, PA and PE.
MetAFLP approach
The metAFLP procedure followed that described elsewhere [1]. The arrangement of selective primer combinations is given in Table 4.
Sequence-specific amplification polymorphism (SSAP) based on met AFLP platforms
The SSAP approach was based on the metAFLP technique [1]. The DNA samples were digested with the KpnI and MseI endonucleases, following adaptor ligation, pre-selective and selective amplification steps. For the selective amplification step oligonucleotides directed toward BARE-1, NIKITA and SUKKULA sequences and MseI adaptor sequences (Table 4) were used. The selective amplification was followed by electrophoresis on 7 % PAGE and exposure to X-ray film.
Methyl-sensitive transposon display (MSTD) based on metAFLP platforms
The Methyl-sensitive transposon display (MSTD) was based on the metAFLP approach [1]. The markers related to DNA methylation were extracted following the procedure described by Chwedorzewska & Bednarek [76]. Briefly, molecular profiles based on the Acc65I/MseI (A) and KpnI/MseI (K) platforms were juxtaposed and scored in a ‘0–1’ binary matrix with ‘1’ standing for the presence and ‘0’ for the absence of the marker. As the Acc65I/MseI platform is capable of identifying (epi)mutations while KpnI/MseI one only sequence changes, this information was used to extract epimarkers. Markers that were present in the first and missed in the second (or vice versa) metAFLP platform were related to DNA methylation Acc65I/MseI-KpnI/MseI (A–K). Thus, the data from both platforms were used to evaluate the “DNA methylation associated markers” also described as “epimarkers”. Instead of the MseI selective primers applied in metAFLP method, those directed towards BARE-1, NIKITA and SUKKULA elements combined with primers directed to methylation—CpGand CpXpG (Table 4) were used to reflect TEs activity. The other steps were performed as for SSAP.
Statistics
Analysis of variance (ANOVA) was applied for the RP-HPLC results using SAS software version 9.1 [77].
GenAlEx6.501 (Excel add-in software) [78] was used to estimate: the number of bands, the number of unique (individual) bands, a percentage of polymorphic loci (P %) generated by metAFLP platforms. Shannon’s diversity index (I) was applied to characterize marker informativeness. PAST software [79] was used for cluster analysis (Ward’s method) with 1000 bootstrap replicates to estimate the robustness of the branches. Analysis of Molecular Variance—AMOVA (Φ PT index value) in GenAlEx6.501 was applied to SSAP and MSTD data. Reliability of the results was performed using 999 random permutations of the raw data.
Abbreviations
- 2,4-D:
-
2,4-dichlorophenoxyacetic acid
- NAA:
-
1-naphthaleneacetic acid
- 5mdC:
-
5-methyl-2’-deoxycytidine
- MS:
-
Murashige and Skoog basal salt mixture
- MSAP:
-
methylation sensitive amplified polymorphism
- RFLP:
-
restriction fragments length polymorphism
- RP-HPLC:
-
reverse phase-high performance liquid chromatography
- SSAP:
-
sequence-specific amplification polymorphism
- MSTD:
-
methyl-sensitive transposon display
- TE:
-
transposable element
References
Bednarek PT, Orłowska R, Koebner RMD, Zimny J. Quantification of the tissue-culture induced variation in barley (Hordeum vulgare L.). BMC Plant Biol. 2007;7:10.
Agarwal PK, Bhojwani SS. Genetic variability in the progeny of androgenic dihaploid plants and selection of high agronomic performing lines in Brassica juncea. Biol Plantarum. 2004;48:503–8.
Guo W, Gong L, Ding Z, Li Y, Li FX, Zhao SP, et al. Genomic instability in phenotypically normal regenerants of medicinal plant Codonopsis lanceolata Benth. et Hook. f., as revealed by ISSR and RAPD markers. Plant Cell Rep. 2006;25:896–906.
Feng S, Cokus SJ, Zhang X, Chen P-Y, Bostick M, Goll MG, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010;107:8689–94.
Zhang M, Kimatu JN, Xu K, Liu B. DNA cytosine methylation in plant development. J Genet Genom. 2010;37:1–12.
Steimer A, Schöb H, Grossniklaus U. Epigenetic control of plant development: new layers of complexity. Curr Opin Plant Biol. 2004;7:11–9.
Karan R, DeLeon T, Biradar H, Subudhi PK. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE. 2012;7:e40203.
Sianipar NF, Wattimena GA, Thenawijaya Soehartono M, Aswidinnoor H, Toruan-Mathius N, Ginting G. DNA methylation detection of oil palm (Elaeis guineensis Jacq) somatic embryo by randomly amplified DNA finger-printing with methylation–sensitive (RAF-SM) and RP-HPLC. J Biotechnol Res Tropic Reg. 2008;1:1–6.
Akimoto K, Katakami H, Kim H-J, Ogawa E, Sano CM, Wada Y, et al. Epigenetic inheritance in rice plants. Ann Bot-London. 2007;100:205–17.
Causevic A, Delaunay A, Ounnar S, Righezza M, Delmotte F, Brignolas F, Hagège D, Maury S. DNA methylating and demethylating treatments modify phenotype and cell wall differentiation state in sugarbeet cell lines. Plant Physiol Biochem. 2005;43:681–91.
Kaeppler SM, Kaeppler HF, Rhee Y. Epigenetic aspects of somaclonal variation in plants. Plant Mol Biol. 2000;43:179–88.
Koukalova B, Fojtova M, Lim KY, Fulnecek J, Leitch AR, Kovarik A. Dedifferentiation of tobacco cells is associated with ribosomal RNA gene hypomethylation, increased transcription, and chromatin alterations. Plant Physiol. 2005;139:275–86.
Rapp RA, Wendel JF. Epigenetics and plant evolution. New Phytol. 2005;168:81–91.
Sano H. DNA methylation and Lamarckian inheritance. P Jpn Acad B-Phys. 2002;78:293–8.
Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol. 2002;12:1138–44.
Sugimoto K, Gordon SP, Meyerowitz EM. Regeneration in plants and animals: dedifferentiation, transdifferentiation, or just differentiation? Trend Cell Biol. 2011;21:212–8.
Fras A, Maluszynska J. Regeneration of diploid and tetraploid plants of Arabidopsis thaliana via callus. Acta Biol Cracov Bot. 2003;45:145–52.
Vining K, Pomraning KR, Wilhelm LJ, Ma C, Pellegrini M, Di Y, et al. Methylome reorganization during in vitro dedifferentiation and regeneration of Populus trichocarpa. BMC Plant Biol. 2013;13:92.
Leljak-Levanić D, Bauer N, Mihaljević S, Jelaska S. Changes in DNA methylation during somatic embryogenesis in Cucurbita pepo L. Plant Cell Rep. 2004;23:120–7.
Fraga HP, Vieira LN, Caprestano CA, Steinmacher DA, Micke GA, Spudeit DA, et al. 5-Azacytidine combined with 2,4-D improves somatic embryogenesis of Acca sellowiana (O. Berg) Burret by means of changes in global DNA methylation levels. Plant Cell Rep. 2012;31:2165–76.
Tanurdzic M, Vaughn MW, Jiang H, Lee T-J, Slotkin RK, Sosinski B, et al. Epigenomic consequences of immortalized plant cell suspension culture. PLoS Biol. 2008;6:e302.
Smýkal P, Valledor L, Rodríguez R, Griga G. Assessment of genetic and epigenetic stability in long-term in vitro shoot culture of pea (Pisum sativum L.). Plant Cell Rep. 2007;26:1985–98.
Ito H, Gaubert H, Bucher E, Mirouze M, Vaillant I, Paszkowski J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature. 2011;472:115–9.
Jursch T, Miskey C, Izsvák Z, Ivics Z. Regulation of DNA transposition by CpG methylation and chromatin structure in human cells. Mob DNA. 2013;4:15.
Fu C, Li L, Wu W, Li M, Yu X, Yu L. Assessment of genetic and epigenetic variation during long-term Taxus cell culture. Plant Cell Rep. 2012;31:1321–31.
Novero AU, Mabras MB, Esteban HJ. Epigenetic inheritance of spine formation in sago palm (Metroxylon sagu Roettb). Plant Omics. 2012;5:559–66.
Kubis SE, Castilho AM, Vershinin AV, Heslop-Harrison JS. Retroelements, transposons and methylation status in the genome of oil palm (Elaeis guineensis) and the relationship to somaclonal variation. Plant Mol Biol. 2003;52:69–79.
Msogoya T, Grout BW, Roberts A. Reduction in genome size and DNA methylation alters plant and fruit development in tissue culture induced off-type banana (Musa spp.). J Anim Plant Sci. 2011;11:1450–6.
Renau-Morata B, Nebauer SG, Arrillaga I, Segura J. Assessments of somaclonal variation in micropropagated shoots of Cedrus: consequences of axillary bud breaking. Tree Genetics Gen. 2005;1:3–10.
Machczyńska J, Orłowska R, Mańkowski DR, Zimny J, Bednarek PT. DNA methylation changes in triticale due to in vitro culture plant regeneration and consecutive reproduction. Plant Cell Tiss Organ Cult. 2014;119:289–99.
Kaeppler SM, Phillips RL. Tissue culture-induced DNA methylation variation in maize. Proc Natl Acad Sci USA. 1993;90:8773–6.
Ngezahayo F, Xu C, Wang H, Jiang L, Pang J, Liu B. Tissue culture-induced transpositional activity of mPing is correlated with cytosine methylation in rice. BMC Plant Biol. 2009;9:9.
Cheng C, Daigen M, Hirochika H. Epigenetic regulation of the rice retrotransposon Tos17. Mol Genet Genomics. 2006;276:378–90.
Planckaert F, Walbot V. Molecular and genetic characterization of Mu transposable elements in Zea mays: behavior in callus culture and regenerated plants. Genetics. 1989;123:567–78.
Okamoto H, Hirochika H. Efficient insertion mutagenesis of Arabidopsis by tissue culture-induced activation of the tobacco retrotransposon Tto1. Plant J. 2000;23:291–304.
Ishizaki T, Kato A. Introduction of the tobacco retrotransposon Tto1 into diploid potato. Plant Cell Rep. 2005;24:52–8.
Cavrak VV, Lettner N, Jamge S, Kosarewicz A, Bayer LM. Mittelsten Scheid O. How a retrotransposon exploits the plant’s heat stress response for its activation. PLoS Genet. 2014;10:e1004115.
Ikeda K, Nakayashiki H, Takagi M, Tosa Y, Mayama S. Heat shock, copper sulfate and oxidative stress activate the retrotransposons MAGGY resident in the plant pathogenic fungus Magnaporthe grisea. Mol Genet Genomics. 2001;266:318–25.
Peschke VM, Phillips RL, Genggenbach BG. Discovery of transposable element activity among progeny of tissue-culture derived plants. Science. 1987;238:804–7.
Flavell AJ, Dunbar E, Anderson R, Pearce SR, Hartley R, Kumar A. Ty1-copia group retrotransposons are ubiquitous and heterogeneous in higher plants. Nucleic Acids Res. 1992;20:3639–44.
Suoniemi A, Tanskanen J, Schulman AH. Gypsy-like retrotransposons are widespread in the plant kingdom. Plant J. 1998;13:699–705.
Gribbon BM, Pearce SR, Kalendar R, Schulman AH, Paulin L, Jack P, et al. Phylogeny and transpositional activity of Ty1- copia group retrotransposons in cereal genomes. Mol Gen Genet. 1999;261:883–91.
Pearce SR, Harrison G, Heslop-Harrison PJS, Flavell AJ, Kumar A. Characterization and genomic organization of Ty1-copia group retrotransposons in rye (Secale cereale). Genome. 1997;40:617–25.
Campbell BC, LeMare S, Piperidis G, Godwin ID. IRAP, a retrotransposon-based marker system for the detection of somaclonal variation in barley. Mol Breeding. 2011;27:193–206.
Borse N, Chimote VP, Jadhav AS. Stability of micropropagated Musa acuminata cv Grand Naine over clonal generations: a molecular assessment. Sci Hortic Amsterdam. 2011;129:390–5.
Li X, Yu X, Wang N, Feng Q, Dong Z, Liu L, et al. Genetic and epigenetic instabilities induced by tissue culture in wild barley (Hordeum brevisubulatum (Trin.) Link). Plant Cell Tiss Org. 2007;90:153–68.
Machczyńska J, Orłowska R, Zimny J, Bednarek PT. Extended metAFLP approach in studies of the tissue culture induced variation (TCIV) in triticale. Mol Breed. 2014;34:845–54.
Jaligot E, Hooi WY, Debladis E, Richaud F, Beulé T, Collin M, et al. DNA methylation and expression of the EgDEF1 gene and neighboring retrotransposons in mantled somaclonal variants of oil palm. PLoS ONE. 2014;9:e91896.
Jaligot E, Rival A, Beulé T, Dussert S, Verdeil J-L. Somaclonal variation in oil palm (Elaeis guineensis Jacq.): the DNA methylation hypothesis. Plant Cell Rep. 2000;19:684–90.
Fiuk A, Bednarek PT, Rybczyński JJ. Flow cytometry, HPLC-RP, and metAFLP analyses to assess genetic variability in somatic embryo-derived plantlets of Gentiana pannonica Scop. Plant Mol Biol Rep. 2010;28:413–20.
Ma X-F, Gustafson JP. Allopolyploidization-accommodated genomic sequence changes in triticale. Ann Bot. 2008;101:825–32.
Oleszczuk S, Rabiza-Swider J, Zimny J, Lukaszewski AJ. Aneuploidy among androgenic progeny of hexaploid triticale (XTriticosecale Wittmack). Plant Cell Rep. 2011;30:575–86.
Li A, Hu BQ, Xue ZY, Chen L, Wang WX, Song WQ, et al. DNA methylation in genomes of several annual herbaceous and woody perennial plants of varying ploidy as detected by MSAP. Plant Mol Biol Rep. 2011;29:784–93.
Peng J, Korol AB, Fahima T, Röder MS, Ronin YI, Li YC, et al. Molecular genetic maps in wild emmer wheat, Triticum dicoccoides: genome-wide coverage, massive negative interference, and putative quasi-linkage. Genome Res. 2000;10:1509–31.
Bednarek PT, Lewandowska R, Gołas T, Paśnik M. The chromosomal location of rye AFLP bands. Cell Mol Biol Lett. 2003;8:955–61.
Liu ZL, Han FP, Tan M, Shan XH, Dong YZ, Wang XZ, et al. Activation of a rice endogenous retrotransposon Tos17 in tissue culture is accompanied by cytosine demethylation and causes heritable alteration in methylation pattern of flanking genomic regions. Theor Appl Genet. 2004;109:200–9.
Vitte C, Panaud O. LTR retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenet Genome Res. 2005;110:91–107.
Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–8.
Shirasu K, Schulman AH, Lahaye T, Schulze-Lefert P. A contiguous 66-kb barley DNA sequence provides evidence for reversible genome expansion. Genom Res. 2000;10:908–15.
Huang J, Zhang K, Shen Y, Huang Z, Li M, Tang D, et al. Identification of a high frequency transposon induced by tissue culture, nDaiZ, a member of the hAT family in rice. Genomics. 2009;93:274–81.
Smith AM, Hansey CN, Kaeppler SM. TCUP: a novel hAT transposon active in maize tissue culture. Front Plant Sci. 2012;3:6.
Leigh F, Kalendar R, Lea V, Lee DL, Donini P, Schulman AH. Comparison of the utility of barley retrotransposon families for genetic analysis by molecular marker techniques. Mol Genet Genomics. 2003;269:464–74.
Kikuchi K, Terauchi K, Wada M, Hirano H-Y. The plant MITE mPing is mobilized in anther culture. Nature. 2003;421:167–70.
Young WP, Schupp JM, Keim P. DNA methylation and AFLP marker distribution in the soybean genome. Theor Appl Genet. 1999;99:785–92.
Wenzel G. Asexual cell genetics. In: Altman A, editor. Agricultural biotechnology. New York: Marcel Dekker Inc; 1998. p. 89–118.
Stotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–85.
Oleszczuk S, Sowa S, Zimny J. Androgenic response to pre-culture stress in the microspore cultures of barley. Protoplasma. 2006;228:95–100.
Chu CC. The N6 medium and its applications to anther culture of cereal crops. Pekin: Proc Symp Plant Tissue Culture, Science Press; 1978. p. 43–50.
Li H, Qureshi JA, Kartha KK. The influence of different temperature treatments on anther culture response of spring wheat (Triticum aestivum L.). Plant Sci. 1988;57:55–61.
Zhuang JJ, Xu J. Increasing differentiation frequencies in wheat pollen callus. In: Hu H, Vega MR, editors. Cell and tissue culture techniques for cereal crop improvement. Beijing: Science Press; 1983. p. 431–2.
Pauk J, Manninen O, Mattila I, Salo Y, Pulli S. Androgenesis in hexaploid spring wheat F2 populations and their parents using a multiple-step regeneration system. Plant Breeding. 1991;107:18–27.
Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue culture. Physiol Plantarum. 1962;15:473–97.
Sowa S, Oleszczuk S, Zimny J. A simple and efficient method for cryopreservation of embryogenic triticale calli. Acta Physiol Plant. 2005;27:237–43.
Havliš J, Trbušek M. 5-Methylcytosine as a marker for the monitoring of DNA methylation. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;781:373–92.
Johnston JW, Harding K, Bremner DH, Souch G, Green J, Lynch PT, et al. HPLC analysis of plant DNA methylation: a study of critical methodological factors. Plant Physiol Biochem. 2005;43:844–53.
Chwedorzewska KJ, Bednarek PT. Genetic and epigenetic variation in a cosmopolitan grass Poa annua from Antarctic and Polish populations. Pol Polar Res. 2012;33:63–80.
Littell R, Stroup W, Freund R. SAS for linear models. 4th ed. Cary: SAS Publishing; 2002.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics. 2012;28:2537–9.
Schönswetter P, Tribsch A. Vicariance and dispersal in the alpine perennial Bupleurum stellatum L. (Apiaceae). Taxon. 2005;54:725–32.
Authors’ contributions
RO was responsible for the experimental part of the study, participated in writing the ms; JM performed some experiments and drafted the ms; SO, JZ provided plant materials for the analyses, drafted the ms; PTB designed the study, participated in writing the ms. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Dr. D. Mańkowski for running some statistical analyses and to our technician Mr. S. Bany for assistance in the RP-HPLC analysis.
Competing interests
The authors declared that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Orłowska, R., Machczyńska, J., Oleszczuk, S. et al. DNA methylation changes and TE activity induced in tissue cultures of barley (Hordeum vulgare L.). J of Biol Res-Thessaloniki 23, 19 (2016). https://doi.org/10.1186/s40709-016-0056-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40709-016-0056-5