
Abstract
Background
Dioscorea opposita Thunb, as a dual-purpose edible plant with excellent nutritional and medical value, accumulates abundant bioactive compounds, including polysaccharides. Polysaccharides from D. opposita have been widely recognized and studied due to various biological activities, such as anti-inflammatory and anti-cancer, which are influenced by their complex structures. In recent years, numerous studies have investigated the structures and activities of D. opposita polysaccharides, and the relationships between them. However, the detailed structures remain different because the structures are susceptible to various species, producing areas, growth conditions (weather, soil, temperature, moisture, etc.), and extraction and separation methods. In this study, the structures of purified polysaccharides from D. opposita were determined and the biological activities were preliminarily investigated including anti-inflammatory and anti-tumor activities.
Results
The crude polysaccharides DOP1 and DOP2 were isolated and purified via DEAE-52 and Sephadex G-100 to obtain two purified polysaccharides fractions (DOP1-1 and DOP2-1), with the molecular weights of 15.63 kDa and 32.32 kDa, respectively. The structures of DOP1-1 and DOP2-1 were analyzed by HPLC, FT-IR, methylation, and 1D (1H, 13C) and 2D (HSQC, COSY, TOCSY, and ROESY) NMR spectra. The results indicated the presence of → 2)-β-Manp-(1 → 4)-α-Glcp-(1 → 2)-β-Manp-(1 → , → 1)-β-Manp-(2 → 1)-α-GalpA-(4 →, and → 2)-β-Manp-(1 → 2)-β-Manp-(1 → in DOP1-1, and the ratio was 2:11:5. The bone structure of DOP2-1 was predicted as → 1)-β-d-Galp-(4 → , along with two main branching structures composed of arabinose of different degrees of polymerization. Preliminary anti-inflammatory and anti-tumor activities were investigated. DOP1-1 and DOP2-1 demonstrated inhibitory effects on macrophages cells (RAW264.7). DOP2-1 exhibited anti-tumor effects against hepatocellular carcinoma (SNU-739 and HepG2).
Conclusions
The structures and bioactivities of DOP1-1 and DOP2-1 were analyzed to provide a basis for the establishment of structure/function relationships. The polysaccharides derived from Dioscorea opposita could be potentially employed in functional food, health care, and pharmaceutical industries.
Graphical Abstract

Similar content being viewed by others


Introduction
Dioscorea opposita Thunb. (Chinese yam, family: Dioscoreaceae) accumulates abundant bioactive compounds, making it a dual-purpose edible plant with excellent nutritional and medical value [1]. D. opposita Thunb. contains polysaccharides, flavonoids, allantoin, phenolic compounds, and many other bioactive substances [2,3,4,5]. Among them, polysaccharides of D. opposita have attracted much attention due to their high bioactivity and good biocompatibility [6].
Polysaccharides of D. opposita are a kind of carbohydrate polymer, composed of more than ten monosaccharides connected by glycosidic linkages, with anti-inflammatory, anti-cancer, antioxidant, immunoregulation, cardioprotective, and other effects [7,8,9]. The bioactivities of polysaccharide are influenced by the complex structure, including molecular weights, monosaccharide compositions, glycosidic linkages, triple helix conformations, substituents, and degree of branching [10,11,12]. For example, acidic polysaccharide obtained from tea showed markedly increased effect, while neutral polysaccharides obtained from tea exhibited markedly inhibitory effects at 400 μg/mL on the proliferation of HeLa cells [13].
However, the structures and bioactivities of D. opposita polysaccharides show significant difference under different species and growth conditions, which greatly increases the difficulty of structure analysis [1]. In recent years, the bioactivities of purified polysaccharides from Chinese yam with different structures have been investigated. The water-soluble polysaccharide (YP-1) purified from D. opposite roots by Zhao et al. showed immunomodulating effects, main chain of which is (1 → 3)-α-glucopyranose, with the side chain of -β-galactopyranose-[(1 → 2)-α-mannopyranose]3-(1 → 2)-α-mannopyranose-(1 → 6)- [14]. Yang et al. stated that the purified Chinese yam polysaccharide containing β-1,3-glucose, α-1,6-galactose, and α-1-galactose presented antioxidant activity [15]. Zhu et al. revealed that the purified polysaccharides from Chinese yams (CYS-1 and CYS-2) were mainly galactose, which possessed antioxidant and anti-cancer effects. The backbone of CYS-1 had two linkages, β-(1 → 2) and β-(1 → 4), and overall, there were two α and four β glycosidic linkages. CYS-2 had two α and three β glycosidic linkages [1].
Researches on polysaccharides substantially thrived in 2021. Zhou et al. identified the structure of purified polysaccharide from Chinese yam as having α-d-Gluc-(1 → 4) glycosidic bond, and the C2 hydroxyl group was replaced by ethoxyl group [16]. However, Huo and Tian commented on this study and alerted readers to consider residue ethanol solvent signals in NMR at 16.89 ppm and 57.49 ppm [17]. Meanwhile, Tang et al. also characterized the structure of polysaccharides of Chinese yam as a low methyl-esterified pectin containing homogalacturonan (approximately 59.1%) and highly branched rhamnogalacturonan (approximately 38.1%) with side chains embracing arabinogalactan and rabinan-/arabino-galactan. Additionally, the homogalacturonan backbone consists of → 4)-α-GalpA-(1 → and → 4)-α-GalpA-6-O-methyl-(1 → with molar ratio of 2:1, and repeating unit of arabinogalactan side chain composed of α-Araf-(1 → and → 3,6)-β-Galp-(1 → [18].
Although structural analysis of D. opposita polysaccharides (DOP) are available in literature, the detailed structures remain different because the structure is susceptible to various species, producing areas, growth conditions (weather, soil, temperature, and moisture etc.), and extraction and separation methods. In the present study, five polysaccharides’ fractions (DOP1-1, DOP1-2, DOP1-3, DOP2-1, and DOP2-2) were isolated and purified from crude polysaccharides of D. opposita. The structures of purified polysaccharides were determined via monosaccharide analysis (high-performance liquid chromatography, HPLC), Fourier transform infrared (FT-IR) spectroscopy, methylation analysis, and extensive nuclear magnetic resonance (NMR) studies (1H, 13C, heteronuclear single quantum correlation [HSQC], correlation spectroscopy [COSY], total correlation spectroscopy [TOCSY], and rotating-frame nuclear overhauser effect spectroscopy [ROESY]). Additionally, biological activities including anti-inflammatory and anti-cancer activities were investigated.
Materials and methods
Materials and animals
Dried slices of D. opposita were purchased from Bao He Tang (Jiaozuo) Pharmaceutical Co., Ltd. in November 2019 (Lot No.: BHT20191019154427). DEAE-Cellulose A52 and Sephadex G-100 were procured from Whatman Co. (Maidstone, Kent, UK) and Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China), respectively. The standard samples and dialysis membranes were purchased from Beijing Solarbio Science & Technology Co., Ltd. All other reagents were purchased from Sigma-Aldrich Co., Ltd. and Tianjin Kemiou Chemical Reagent Co. Ltd., China. All chemicals used were of analytical grade.
Male and female Sprague–Dawley (SD) rats (140–180 g) and the standard diet were purchased from the Center of Laboratory Animals of Henan Province (Zhengzhou, China). All animals were housed in a room maintained at 22 ± 3 °C under natural light/dark conditions, with free access to food and water. All experimental animal procedures were approved by the Henan University Institutional Animal Care and Use Committee (No. 410975211100011935).
Extraction and purification of polysaccharides
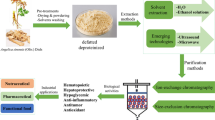
Crude polysaccharides from D. opposita were extracted as previously described [6, 19], and a representative flowchart is shown in Fig. 1. The dried slices of D. opposita were ground and sifted through a 40-mesh sieve. The dried D. opposita powder (1.0 kg) was extracted twice for 3 h at 80 °C in a water bath with 8.0 L of anhydrous ethanol and then filtrered. The precipitation was extracted twice for 3 h at 80 °C in a water bath with 8.0 L of distilled water. The aqueous extract was concentrated to 1/3 of the volume of the primary extracted solution; then anhydrous ethanol was added until the concentration of ethanol (Ce) reached approximately 20% v/v. The solution was precipitate for 24 h to obtain residue III (discarded). Anhydrous ethanol (1/3 volume of supernatant I) was added to the supernatant I until Ce was approximately 40% v/v, and the crude polysaccharides (S1) were centrifuged after 24 h. The crude polysaccharides (DOP1) were obtained after deproteinizing using Sevag reagent (1-butanol: chloroform = 1:4 v/v), dialyzing, and lyophilizing [20]. The same method was used to obtain S2 (Ce = 60% v/v), which was further purified to obtain DOP2. The samples were preserved in vacuum desiccators over P2O5 until further investigation.

Flowchart of extraction and purification of polysaccharides from Dioscorea opposita Thunb
The polysaccharides DOP1 and DOP2 were purified using column chromatography (Fig. 1). Briefly, DOP1 and DOP2 (20.0 mg/mL) were filtered through a 0.45 μm membrane and eluted with 0.05 M, 0.1 M and 0.2 M NaCl on a DEAE-Cellulose 52 chromatography column (2.5 cm × 50 cm) at a flow rate of 1.00 mL/min, respectively. The contents of polysaccharides in eluent were determined by phenol–sulfuric acid colorimetric method [21], and the high contents were eluted with distilled water on a Sephadex G-100 gel permeation chromatography column (1.6 cm × 100 cm) at a flow rate of 0.5 mL/min. The eluates were collected, concentrated, and lyophilized to obtain purified polysaccharides DOP1-1, DOP1-2, DOP1-3, DOP2-1, and DOP2-2.
Structural characterization of purified polysaccharides
Chemical composition analysis
Total carbohydrate content was determined by the phenol–sulfuric acid assay [21]. Uronic acid content was measured via the meta-hydroxydiphenyl assay [22]. Sulfated groups content was determined using the methods described by Dodgson and Price [23].
Determination of molecular weight
For purified DOP samples, the weight-average molecular weights (Mw) were determined by high-performance size exclusion chromatography attached to a multiangle laser light scattering and refractive index detector (HPSEC-MALLS-RID, Wyatt Technology Co., USA) with an OHpak SB-802.5 HQ column (8.0 mm × 300 mm, Shodex Co., Japan). The mobile phase was 0.1 M NaNO3 at a flow rate of 0.5 mL/min, with 50.0 μL of sample solution (1.8 mg/mL) injected. The chromatogram was analyzed using ARTRAV software (Wyatt Technology Co., USA) [6].
Determination of monosaccharides composition
Monosaccharides of purified DOP samples were hydrolyzed with sulfuric acid and analyzed using the 1-phenyl-3-methyl-5-pyrazolone (PMP) pre-column HPLC method [24, 25]. The purified DOP sample (40 mg) was added to 4 mL sulfuric acid (2 mol/L), hydrolyzed at 100 °C for 8 h, and then 4 mol/L NaOH was added to neutralize pH value (pH 7). The hydrolysate was then derivatized by PMP and analyzed using HPLC (Agilent, 1260 infinity, USA) with a Spursil C18 column (5 µm, 4.6 × 250 mm). A mixture of a 0.05 M phosphate buffer solution (pH 6.7) and acetonitrile (84:16, v/v) was used as the mobile phase at a flow rate of 1 mL/min. The temperature of the column was maintained at 25 °C and absorbance was measured using a UV–visible detector (VWD) at 245 nm.
Fourier transform infrared spectroscopy (FT-IR)
Purified DOP samples were mixed with potassium bromide, milled, pressed into a pellet (7 mm diameter), and analyzed using an FT-IR spectrometer (Vertex 70, Bruker, Germany) in a frequency range of 400–4000 cm−1 [26].
SEM
A scanning electron microscope (SEM, JSM-7001, Japan Electron Optics Laboratory Co Ltd., Tokyo, Japan) was used to characterize the morphology of samples according to previous studies [27]. The freeze-dried sample powders were distributed on a copper with double-side adhesive, sprayed with gold powder, and examined in a vacuum specimen chamber.
Methylation analysis
To determine the glycosidic linkages in purified DOP samples, the polysaccharides samples were methylated as previously described [28, 29]. Briefly, 15 mg of a dried sample was dissolved into 1 mL distilled water, and then 0.2 mL NaOH mixed in 0.2 mL methanol was added. The mixture was diluted with 6.0 mL dimethyl sulfoxide (DMSO), followed by sonication in an ultrasonic bath for 10 min. The solution was stirred at room temperature (20 °C) for 1 h, and 1.0 mL iodomethane was added slowly. The reaction mixture was placed in darkness for 2 h, followed by the addition of distilled water (2.0 mL). The methylated polysaccharide was extracted with chloroform (2.0 mL) three times, dried, and collected. The methylated polysaccharide was analyzed using FT-IR to confirm the disappearance of the hydroxyl stretching vibration absorptive band in the range of 3400–3600 cm−1. The methylated polysaccharide was dissolved in 2 M trifluoroacetic acid (TFA) at 105 °C for 6 h. The solvent was removed by adding methanol and 2.0 mL NaBH4 (25 mg/mL) for 3 h at room temperature (20 °C). The solution was neutralized with 25% acetic acid, and the remains were dried and acetylated with acetic anhydride (3.0 mL) at 100 °C for 1 h. The residue was extracted with chloroform, and the organic phase was washed with distilled water (1:1 v/v) three times. The resulting alditol acetates were injected into a gas chromatography–mass spectrometer (GC–MS, Agilent, 1260, USA) with Agilent 122-2932 DB 225 (0.25 mm × 30 m × 0.25 μm).
Nuclear magnetic resonance (NMR) spectroscopy
Purified DOP samples (30 mg) were dissolved in D2O, centrifuged, filtered, and analyzed on a Bruker AVANCE-III-600 spectrometer (Bruker, Germany). The 1H NMR and 13C NMR, HSQC, COSY, TOCSY, and ROESY were recorded using Bruker software [30].
Anti-inflammatory activity of purified polysaccharides
Cell culture
Murine macrophage RAW264.7 cells were obtained from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology (Chinese Academy of Sciences, China) and cultivated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified atmosphere of 5% CO2.
Cell viability
In brief, cells were seeded in a 96-well plate (1 × 104 cells/mL) cultured for 24 h to obtain a confluent monolayer and then treated with 10 μL culture medium (blank group) or 10 μL purified DOP samples (100 μg/mL or 200 μg/mL, extract groups) with 10 μL LPS (lipopolysaccharide, 1.0 μg/mL) for 48 h. Cells treated with LPS (1.0 μg/mL, cultivated for 48 h) were used as the control group. Then, cells were incubated with MTT (10 μL, 5 mg/mL in phosphate-buffered saline [PBS]) for 4 h at 37 °C in a humidified atmosphere of 5% CO2; subsequently, cells were lysed in dimethyl sulfoxide (DMSO) with gentle vibration for 10 min. The formation of formazan was measured at 590 nm using a microplate reader (CLARIOstar, BMG LABTECH, Germany), and the percentage of cells (cell viability) showing cytotoxicity was measured relative to the control group [31]. Each viability inhibition experiment was replicated at least three times.
Cell morphology
In brief, cells were inoculated into six-well plates (1 × 105 cells/well) and treated with purified DOP samples (100 μg/mL and 200 μg/mL, respectively) and LPS for 48 h. Then, the cell morphology was observed and photographed using an inverted fluorescence microscope (Leica, Germany).
Measurement of nitric oxide (NO) production
RAW264.7 cells were seeded in a 96-well plate (1 × 105 cells/well) and incubated with culture medium (blank group) or purified DOP samples at different concentrations (100 μg/mL and 200 μg/mL, respectively), along with LPS (1.0 μg/mL) for 48 h. Cells treated with LPS (1.0 μg/mL) for 48 h were used as the control group. CheKine™ nitric oxide (NO) assay kit (Abbkine, Wuhan, China) was employed, and absorbance was measured at 590 nm. Nitrite concentrations in the supernatants were determined by comparing with a sodium nitrite (NaNO2) standard curve [32].
Phagocytosis activity
Macrophage cells (5 × 105 cells/well) seeded in 96-well plates were pre-incubated in complete DMEM, followed by stimulation with culture medium or purified DOP samples (100 μg/mL and 200 μg/mL, respectively) and LPS for 48 h. Cells treated with LPS (1.0 μg/mL) were used as a positive control. After each treatment, neutral red (0.075%, w/v) was added to each well, and cells were incubated for another 1 h. Then, cells were washed three times with PBS and resuspended in a mixture of ethanol, glacial acetic acid, and water (1:1:1, v/v/v). After incubation for 1 h, the absorbance of the culture medium was measured at 540 nm using a microplate reader (CLARIOstar, BMG LABTECH, Germany) [32].
λ-Carrageenan (Carr)-induced rat paw edema
In vivo anti-inflammatory activity was evaluated using a Carr-induced hind paw edema model [33]. After a 2-week adaptation period, SD rats (190 ± 20 g) were assigned to seven groups (n = 8), including a λ-Carr-induced group (Carr-induced), and Carr + DOP1, and Carr + DOP2 with three dose groups (50 mg/kg, 200 mg/kg, and 400 mg/kg) respectively. The Carr-induced group received 1% Carr (0.1 mL), with normal saline intragastrically administered. DOP1 and DOP2, at doses of 50 mg/kg, 200 mg/kg, and 400 mg/kg, were administered orally, 30 min before the intraplantar injection of 1% Carr (0.1 mL) into the right hind paw of the rat. Paw volume was measured immediately after the Carr injection at 1, 2, 3, 4, 5, and 6 h intervals. The difference in volume (ΔmL) between hind paws was considered paw edema.
Anti-tumor activity against hepatocellular carcinoma
Cell culture
Human hepatocyte cell line HL-7702, human hepatocellular carcinoma cell line SNU-739, and HepG-2 cells were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology (Chinese Academy of Sciences, China). HL-7702 and SNU-739 cells were cultured in RPMI-1640 (Roswell Park Memorial Institute) medium, and HepG-2 cells were cultured in DMEM. All cultures were supplemented with 10% heat-inactivated FBS, NaHCO3 (2 g/L), 100 U/mL penicillin, and 10 μg/mL streptomycin. Cultures were incubated at 37 °C under a humidified atmosphere with 5% CO2 [34, 35].
Cell viability
Cell viability was detected by colorimetric MTT assay [34]. In brief, the cells suspension (5 × 104 cells/mL) was seeded into a 96-well plate and incubated at 37 °C in a 5% CO2 atmosphere for 24 h. Then, the cells were incubated with various concentrations of purified DOP samples (50 μg/mL, 100 μg/mL, 200 μg/mL, and 400 μg/mL) for 24 h or 48 h. Following drug exposure, 10 μL of CCK-8 solution was added and incubated at 37 °C for 4 h. Absorbance was measured at 570 nm using a microplate reader (CLARIOstar, BMG LABTECH, Germany). Six wells were analyzed for each concentration, and the optical density of control (untreated) cells was considered 100% viability. Cell viability was calculated using the following equation:
where Asample is the optical density value of samples with cells, Ablank1 is the optical density value of samples with medium, Acontrol is the optical density value of control with cells, and Ablank2 is the optical density value of control with medium [36].
Statistical analysis
Data values are presented as the mean ± standard deviation, and statistical analysis was performed by employing one-way analysis of variance (ANOVA) plus post hoc Duncan’s test. Statistical significance was defined at P < 0.05.
Results and discussion
Isolation and purification
After hot-water extraction, sequential alcohol precipitation, deproteinization and dialysis, the crude polysaccharides from D. opposita (DOP1 and DOP2) were obtained with yields of about 1.71% and 0.38%, respectively. The DOP1 and DOP2 were isolated by DEAE-52 cellulose column and eluted using 0.05 M, 0.1 M, and 0.2 M NaCl, respectively, to obtain DOP1-1, DOP1-2, DOP1-3, DOP2-1, and DOP2-2 (Fig. 2a, b). The elution peaks of DOP1-1 and DOP2-1 obtained by Sephadex G-100 were high and sharp (Fig. 2c, d), and the peaks were collected for further analysis. The 0.1 M NaCl was employed to purify DOP2, but the elution peak was relatively low and the yield was approximately zero.

Chromatographic isolation of polysaccharides from Dioscorea opposita Thunb. on the DEAE-52 column (a, b); chromatographic isolation of DOP1-1 (c) and DOP2-1 (d) on Sephadex G-100 column
Based on yield, NMR and bioactivity results, the structures of DOP1-1 and DOP2-1 were further characterized. The structures of DOP1-2, DOP1-3, and DOP2-2 require further investigation (data not shown). The carbohydrate content of DOP1-1 was 50.32%, including 13.96% uronic acid and 0.20% sulfated groups. The carbohydrate content of DOP2-1 was 82.00%, including 0.78% uronic acid and 0.12% sulfated groups (Table 1).
Molecular weight and monosaccharides determination
As shown in Table 1, the molecular weights (Mw) of DOP1-1 and DOP2-1 were 15.63 kDa and 32.32 kDa, respectively, the number-average molecular weight (Mn) of DOP1-1 and DOP2-1 were 9.45 kDa and 18.45 kDa, respectively, and the polydispersity (Mw/Mn) values of both samples were 1.653 and 1.751, respectively. According to Xu et al., an Mw/Mn value close to 1 indicated that Mw lies within a small range owing to the narrow distribution. Therefore, results suggested that the molecular weights of DOP1-1 and DOP2-1 were distributed in a relatively wide range [24].
Standard samples containing ten monosaccharides (mannose, ribose, rhamnose, glucuronic acid, galacturonic acid, glucose, galactose, xylose, arabinose, and fucose) were used to analyze the monosaccharide compositions of DOP1-1 and DOP2-1 by PMP pre-column derivatization. Accordingly, DOP1-1 was composed of mannose, ribose, rhamnose, galacturonic acid, glucose, galactose, arabinose, and fucose, and the main molar ratio was Man:GalA:Glc:Fuc:Gal = 155.71:66.44:12.25:10.05:6.32, respectively. DOP2-1 demonstrated similar types of monosaccharides, except fucose, and the molar ratio of DOP2-1 was Gal:Man:Glc:Ara:Rha = 100.65:64.20:49.61:32.42:5.74, respectively (Table 1; Fig. 3a).

HPLC chromatograms of monosaccharides, DOP1-1, and DOP2-1 PMP derivative mixtures (a); FT-IR spectra of DOP1-1 and DOP2-1 (b); SEM images of DOP1-1 (c) and DOP2-1 (d)
FT-IR
FT-IR spectrum is commonly used to identify the organic functional groups in the polysaccharide structure (Fig. 3b). The strong hydroxyl stretching vibration peak at 3391 cm−1 indicates the presence of hydroxyl groups. The absorption at 2980 cm−1 illustrated the C–H stretching peak [37]. The absorptive peaks at 1634 cm−1, the stretching vibration of a free carboxylic carbonyl group, suggested that DOP1-1 and DOP2-1 are acidic polysaccharides [38]. The signals at 1417 cm−1 were attributed to the deformation mode of CH2 [30]. In the range of 1200–1000 cm−1, peaks at 1150 cm−1, 1089 cm−1, and 1049 cm−1 illustrated the presence of C–OH side groups and COC– glycosidic bond, indicating the presence of pyranose [27, 39, 40]. The weak bands near 882 cm−1 and 803 cm−1 demonstrated that glucosyl residues could be present in both β- and α-configurations in the DOP1-1 and DOP2-1. The peak at 821 cm−1 for DOP2-1 suggested the presence of a furan ring [41].
SEM
Previous studies have shown that surface topography, structure, and properties of polysaccharides may be affected by the conditions of extraction, purification, and preparation [42]. As shown in Fig. 3c, d, the microstructures of the samples were analyzed by SEM, and the results showed that the polysaccharides had similar branched structures with slight differences. They presented loose and dendritic-like structures which suggested that the polysaccharides coagulate more easily and flocculate together to form higher molecular weight polymers.
Methylation analysis
Methylation analysis provides further structural information of polysaccharides by GC–MS after hydrolysis, reduction, and acetylation [38]. As shown in Table 2, DOP1-1 was measured to be 1,2,5-Ac3-3,4,6-Me3-D-Man, 2,3,6-Me3-Gal, and 2,3,6-Me3-Glc with molar ratios of 63.2:26.7:5.3; DOP2-1 derivatives were identified as 2,3,6-Me3-Gal, 1,4,5-Ac3-2,3,6-Me3-D-Man, 2,3,6-Me3-Glc, 1,3,5,6-Ac4-2,4-Me2-D-Man, 1,4,5-Ac3-2,3-Me3-D-Ara, and 3,4-Me2-Rha, with molar ratios of 32.3:27.1:17.8:7.4:5.3:1.7. The results suggested that DOP1-1 contained three linkages: → 2) Manp (1 → , → 4) Galp (1 → , and → 4) Glcp (1 → . DOP2-1 contained six linkages: → 4) Galp (1 → , → 4) Manp (1 → , → 4) Glcp (1 → , → 3,6) Manp (1 → , → 5) Araf (1 → , and → 2) Rhap (1 → . The subsequent NMR analysis corroborated the conclusions drawn from the methylation analysis.
NMR analysis
The monosaccharide composition, α- or β-anomeric configuration, linkage pattern, and sugar unit sequence were further determined by NMR spectroscopy. The 1H, 13C, HSQC, COSY, TOCSY, and ROESY NMR spectra of DOP1-1 and DOP2-1 at 600 MHz were investigated (Figs. 4, 5).

1H spectrum (a); 13C spectrum (b); HSQC spectrum (c); COSY spectrum (d); ROESY spectrum (e); predicted structure of DOP1-1 (f)

1H spectrum (a); 13C spectrum (b); HSQC spectrum (c); COSY spectrum (d); ROESY spectrum (e); TOCSY spectrum (f); the predicted structures of two branching chains of DOP2-1 (g)
NMR spectroscopy analysis of DOP1-1
Three anomeric proton and anomeric carbon signals were observed in the 1H and 13C NMR and HSQC spectra (Fig. 4a–c), which were δ 5.13/98.61, 4.61/96.17, and 5.11/99.01, respectively, and named residues A, B, and C. Moreover, the carboxyl signal of 175.46 indicated the presence of uronic acid in DOP1-1. The chemical shifts of other protons were successfully obtained from the COSY spectrum (Fig. 4d). The corresponding chemical shifts of carbon atoms were obtained by HSQC spectrum (Fig. 4c) and are shown in Table 3. Compared with literature data, the result showed that residue A was → 4)-α-d-Glcp-(1 → [43,44,45,46], the residue B was 1,2-β-Manp [47, 48], and the residue C was → 4)-α-d-GalpA-(1 → [49,50,51].
The sequence of glycosidic linkages between DOP1-1 residues was determined by ROESY spectrum (Fig. 4e). The results indicated the presence of → 2)-β-Manp-(1 → 4)-α-Glcp-(1 → 2)-β-Manp-(1 → , → 1)-β-Manp-(2 → 1)-α-GalpA-(4 → and → 2)-β-Manp-(1 → 2)-β-Manp-(1 → in DOP1-1. Based on the monosaccharide composition of DOP1-1, the ratio of three linkages was estimated to be 2:11:5. Hence, the structures of DOP1-1 were predicted as shown in Fig. 4f, considering the comprehensive results of monosaccharide compositions, methylation analysis and NMR analysis.
NMR spectroscopy analysis of DOP2-1
As shown in Fig. 5, the structure of DOP2-1 was further analyzed by NMR spectroscopy (1H, 13C, HSQC, COSY, ROESY, and TOCSY), and the corresponding hydrocarbon signals are shown in Table 3. The 13C and 1H NMR spectra of DOP2-1 showed five anomeric carbon and five anomeric proton signals at δ 5.29/109.30, 5.12/107.43, 5.11/98.92, 4.69/104.36, and 4.55/103.22, named A, B, C, D, and E. The signal at approximately 1.29 of C-6 methyl proton of rhamnose was observed in the 1H spectrum [43]. Residue A was identified as T-α-Araf by comparison with the literature data [51, 52]. The residue B was (1 → 5)-α-Araf according to the downfield shifts of C-5 (δ 68.09) [53]. The residues C, D, and E were identified as → 4)-α-d-Glcp-(1 → , 1,4-β-d-Galp, and → 4)-Manp-(1 → by comparison of proton and carbon resonances with the literature data, respectively [43, 54].
The inter-residual and intra-residual connections of DOP2-1 were determined by ROESY and TOCSY spectra (Fig. 5e, f). Results indicated the presence of → 1)-α-Arbf-(5 → T-α-Arbf and → 1)-Glcp-(4 → 1)-α-Arbf-(5 → . Therefore, based on the NMR information, monosaccharide compositions as well as methylation analysis, the bone structure of DOP2-1 was speculated to be → 1)-β-d-Galp-(4 → , with two main branching structures proposed in Fig. 5g.
The results show that DOP1-1 obtains → 1)-β-Manp-(2 → , → 4)-α-Glcp-(1 → and → 1)-α-GalpA-(4 → , and the backbone of DOP2-1 consists of → 1)-β-d-Galp-(4 → , which is consistent with the finding of Zhu et al. that the backbone of the CY polysaccharides contains β-(1 → 2) and β-(1 → 4) linkages [1]. According to Tang et al. [18], the CY polysaccharides are a low methyl-esterified pectin with galacturonan and rhamnogalacturonan, which are also partially congruous with our finding. Although we were not able to detect few linkages such as → 3,6)-β-Galp-(1 → in the literature, → 1)-α-Arbf-(5 → was found to supplement the results of Tang et al. The structure of CY polysaccharides proposed by Zhou, Huang, and Chen comprises α-d-Gluc-(1 → 4) glycosidic linkages which present in our predicted structures [16]. It is worth noting that the structures of purified polysaccharides DOP1-1 and DOP2-1 predicted in this study were different from previous researches [1, 14, 16, 18], which may be caused by the planting area, weather, the year and months of harvest, and the extraction, separation, and purification method of Dioscorea opposita.
Anti-inflammatory activity
Effects on proliferation
To identify the bioactivities of purified polysaccharides from D. opposita, the anti-inflammatory effects of DOP1-1 and DOP2-1 against macrophage RAW264.7 cells were investigated. The cell proliferation of cells in the control group which was incubated without LPS or DOP was considered as 100%. Following treatment with DOP1-1 or DOP2-1, the cell proliferation rate of LPS-stimulated RAW264.7 cells decreased correlating positively with the concentration employed (Fig. 6b). Thus, DOP1-1 and DOP2-1 were preliminarily considered to possess anti-inflammatory effects.

Effects of the DOP1-1 and DOP2-1 on the general morphology of the LPS-induced RAW264.7 cells (a); RAW264.7 cells were pretreated with LPS, and cell proliferation rate was measured by MTT assay (b); effects of the DOP1-1 and DOP2-1 on NO levels of RAW264.7 cells induced by LPS (c); effects of DOP1-1 and DOP2-1 on phagocytosis index of RAW264.7 cells induced by LPS (d); effect of DOP2-1 on the viability of HL-7702 cells (e); SNU-739 cells (f); HepG2 cells (g). Data are represented as mean ± standard deviation (SD, n = 6). *P < 0.05, **P < 0.01
Morphology
As shown in Fig. 6a, following LPS treatment for 48 h, cells were swollen and differentiated, presenting numerous pseudopods. Meanwhile, the cytoplasm and nuclei became blurred, with white transparent vesicles observed in the cytoplasm. However, the degree of cell differentiation (pseudopods, blurred cytoplasm and nuclei, and white transparent vesicles) decreased significantly at concentrations of 100 μg/mL or 200 μg/mL for DOP1-1 and DOP2-1. Compared with 100 μg/mL, the degree of cell differentiation decreased more obviously at the concentrations of 200 μg/mL, indicating that the decrease correlated with increased concentration.
NO production
Inhibition of NO production is an important way to treat various inflammatory diseases [55]. CheKineTM nitric oxide assay kit was used to measure NO production following reduction of nitrate to nitrite by Griess method. NO production of unstimulated RAW264.7 cells was undetected, and LPS stimulation significantly increased the NO concentration (approximately 17.5 ± 1.3 μM). The ability of DOP1-1 and DOP2-1 to decrease the NO concentration in LPS-activated RAW264.7 macrophages was evaluated (Fig. 6c). Upon LPS stimulation, following treatment with DOP1-1 or DOP2-1 at a concentration of 100 μg/mL, the NO concentrations were significantly decreased to approximately 16.7 ± 2.7 μM or 15.5 ± 0.7 μM, respectively; on treating with DOP1-1 or DOP2-1 at 200 μg/mL, the NO concentrations dropped extremely to approximately 11.8 ± 1.2 μM and 11.6 ± 0.6 μM, respectively. That is to say, RAW264.7 cells treated with DOP1-1 (100 μg/mL or 200 μg/mL) could significantly inhibit NO production mediated by LPS-stimulated RAW264.7 cells (4.43% and 32.84%, respectively). Similarly, DOP2-1 (100 μg/mL or 200 μg/mL) can also significantly inhibit NO production (10.94% and 33.32%, respectively).
Macrophage phagocytosis
Under stimulation, macrophage phagocytosis is activated to eliminate pathogens and cell debris [32]. As shown in Fig. 6d, following treatment with DOP1-1 or DOP2-1, macrophage phagocytosis (LPS-stimulated RAW264.7) was significantly decreased in a dose-dependent manner. The results suggested that LPS-stimulated macrophages were significantly inhibited, and with increasing concentration, the phagocytic index decreased more sharply. Compared with DOP1-1, cells treated with DOP2-1 inhibited macrophage phagocytosis (P < 0.05). The results of the phagocytosis assay were consistent with NO production, indicating the anti-inflammatory effects of DOP1-1 and DOP2-1.
λ-Carrageenan (Carr)-induced rat paw edema
Carr, a polysaccharide, produces clinical symptoms such as edema immediately after injection, inducing inflammatory responses. The inflammatory response is measured by an increase in paw size. DOP1-1 and DOP2-1 were further purified from crude polysaccharides DOP1 and DOP2 with much lower yields. Therefore, the anti-inflammatory effects of DOP1 and DOP2 were evaluated by injected Carr into the right hind paw of rats to induce paw edema.
As shown in Table 4, compared with the Carr-induced group, symptoms were aggravated after administration of DOP1 at oral dosage of 50 mg/kg or 200 mg/kg, which may be due to the complexity of the crude polysaccharides, such as glycoprotein. However, the symptom was significantly reduced and edema subsided when the oral dose of DOP1 reached 400 mg/kg. Oral administration of 50 mg/kg DOP2 showed no significant difference, and a reduction in edema regression was observed with an oral dosage of 200 mg/kg DOP2. The results indicated that a dosage of 50 mg/kg was too low to be effective, and 400 mg/kg of DOP2 was considered to be an ideal dosage. Ultimately, DOP2 showed a superior effect than DOP1 in terms of inhibiting inflammatory edema. These results suggest that DOP1 and DOP2 both present anti-inflammatory effects and could be developed as a novel health-care food.
Anti-tumor effects against hepatocellular carcinoma
To identify the anti-tumor activity of purified polysaccharides, the inhibitory effects of DOP1-1, DOP1-2, DOP1-3, DOP2-1, and DOP2-2 against hepatocellular carcinoma (HepG2 and SNU-739), colorectal carcinoma (HCT-116), and cervical carcinoma (HeLa) were investigated. Only DOP2-1 exhibited inhibitory effects against hepatocellular carcinoma cells.
A comparative study on the cytotoxicity of human normal hepatic cells (HL-7702), human hepatocellular carcinoma cell line SNU-739, and HepG-2 cells treated with various DOP2-1 concentrations (50 μg/mL, 100 μg/mL, 200 μg/mL, and 400 μg/mL) for 24 h or 48 h was performed (Fig. 6e–g). After incubation for 24 h, SNU-739 cell viability decreased from 89.77% with 50 μg/mL to 63.76% with 400 μg/mL DOP2-1. After incubation for 48 h, the SNU-739 cell viability decreased from 88.15% (50 μg/mL) to 40.79% (400 μg/mL) in a dose-dependent manner. For HepG2 cells, cell viability decreased from 83.32% (50 μg/mL) to 43.12% (400 μg/mL) after 24 h incubation, and from 78.35% (50 μg/mL) to 22.44% (400 μg/mL) after incubation for 48 h, in a dose-dependent manner. Overall, compared with the anti-proliferative effect at 48 h, the inhibition was lower in the group at 24 h, suggesting that the inhibitory effect of fractions increases with time [56]. The anti-proliferative effects of DOP2-1 on HepG2 were stronger than those on SNU-739 cells.
DOP2-1 did not affect HL-7702 cells in the range of 0–100 μg/mL and instead promoted cell proliferation. At 200 μg/mL, the effect of DOP2-1 was not significant. At 400 μg/mL, DOP2-1 had an inhibitory effect on cells, presenting viabilities of 82.27% (24 h) and 64.61% (48 h). Therefore, 200 μg/mL was preliminarily considered a safe dose. Cell viabilities of SNU-739 and HepG2 were 75.69% and 56.23%, respectively, after 24 of incubation, and 64.29% and 47.64%, respectively, after 48 h of incubation. Hence, at a 200 μg/mL dose, DOP2-1 is safe and can inhibit tumor cell proliferation to a certain extent, which could be developed as a safe anti-tumor health product.
Conclusions
In the present study, water-soluble polysaccharides DOP1-1 and DOP2-1 were isolated and purified from D. opposita, and their structures were assessed by HPSEC–MALLS–RID, HPLC, FT-IR, methylation analysis, and NMR. According to the results, DOP1-1 (15.63 kDa) existed as → 2)-β-Manp-(1 → 4)-α-Glcp-(1 → 2)-β-Manp-(1 → , → 1)-β-Manp-(2 → 1)-α-GalpA-(4 → and 1 → 2)-β-Manp-(1 → 2)-β-Manp-(1 → , with a ratio of 2:11:5. The bone structure of DOP2-1 (32.32 kDa) was hypothesized to be → 1)-β-d-Galp-(4 → , and contained two main branches composed of arabinose with different degrees of polymerization.
Additionally, preliminary anti-inflammatory and anti-tumor activities were investigated. After treatment with DOP1-1 and DOP2-1 (100 μg/mL or 200 μg/mL), macrophage phagocytosis (LPS-stimulated RAW264.7) was significantly decreased in a dose-dependent manner. More importantly, DOP2-1 showed a stronger inhibition on macrophage phagocytosis than DOP1-1. Meanwhile, DOP2-1 can inhibit the proliferation of tumor cells (SNU-739 and HepG2) to a certain extent at the dose of 200 μg/mL, which can be used as a safe anti-tumor health product. Therefore, the structures and bioactivity results of polysaccharides could help elucidate the use of D. opposita in pharmacological fields and functional health-care foods, while also providing novel insights into the future development of Chinese yam polysaccharides.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Carr-:
-
λ-Carrageenan-induced
- COSY:
-
Correlation spectroscopy
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- DOP:
-
Polysaccharides of Dioscorea opposita Thunb.
- FBS:
-
Fetal bovine serum
- FT-IR:
-
Fourier transform infrared spectroscopy
- HPLC:
-
High-performance liquid chromatography
- HPSEC-MALLS-RID:
-
High-performance size exclusion chromatography attached to multiangle laser light scattering and refractive index detector
- HSQC:
-
Heteronuclear single quantum correlation
- LPS:
-
Lipopolysaccharide
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- M w :
-
Weight-average molecular weight
- Mn:
-
Number-average molecular weight
- NMR:
-
Nuclear magnetic resonance
- PBS:
-
Phosphate-buffered saline
- PMP:
-
1-Phenyl-3-methyl-5-pyrazolone
- ROESY:
-
Rotating-frame nuclear Overhauser effect spectroscopy
- SD rat:
-
Sprague–Dawley rat
- TOCSY:
-
Total correlation spectroscopy
References
Zhu P, Yang L, Zhang C, Tian Y, Zhang F, Li X. Structural and functional analyses of three purified polysaccharides isolated from Chinese Huaishan-yams. Int J Biol Macromol. 2018;120:693–701. https://doi.org/10.1016/j.ijbiomac.2018.08.143.
Alves RML, Grossmann MVE, Silva RSSF. Gelling properties of extruded yam (Dioscorea alata) starch. Food Chem. 1999;67(2):123–7. https://doi.org/10.1016/S0308-8146(99)00064-3.
Brunnschweiler J, Luethi D, Handschin S, Farah Z, Escher F, Petit BC. Isolation, physicochemical characterization and application of yam (Dioscorea spp.) starch as thickening and gelling agent. Strach. 2005;57:107–17. https://doi.org/10.1002/star.200400327.
Hou WC, Lee MH, Chen HJ, Liang WL, Han CH, Liu YW, Lin YH. Antioxidant activities of dioscorin, the storage protein of yam (Dioscorea batatas Decne) tuber. J Agr Food Chem. 2001;49(10):4956–60. https://doi.org/10.1021/jf010606m.
Zava DT, Dollbaum CM, Blen M. Estrogen and progestin bioactivity of foods, herbs, and spices. P Soc Exp Biol Med. 1998;217(3):369–78. https://doi.org/10.3181/00379727-217-44247.
Ma F, Zhang Y, Wen Y, Yao Y, Zhu J, Liu X, Bell AE, Tikkanen-Kaukanen C. Emulsification properties of polysaccharides from Dioscorea opposita Thunb. Food Chem. 2017;221:919–25. https://doi.org/10.1016/j.foodchem.2016.11.073.
Hao L, Zhao X. Immune activities of the water-soluble yam (Dioscorea opposite Thunb.) polysaccharides as affected by thermal, acidic and enzymatic treatments. CyTA J Food. 2016;14(2):1–5. https://doi.org/10.1080/19476337.2015.1095803.
Huang R, Xie J, Yu Y, Shen M. Recent progress in the research of yam mucilage polysaccharides: isolation, structure and bioactivities. Int J Biol Macromol. 2020;155:1262–9. https://doi.org/10.1016/j.ijbiomac.2019.11.095.
Ju Y, Xue Y, Huang JL, Zhai QZ, Wang XH. Antioxidant Chinese yam polysaccharides and its pro-proliferative effect on endometrial epithelial cells. Int J Biol Macromol. 2014;66:81–5. https://doi.org/10.1016/j.ijbiomac.2014.01.070.
Ji X, Peng Q, Yuan Y, Shen J, Xie X, Wang M. Isolation, structures and bioactivities of the polysaccharides from jujube fruit (Ziziphus jujuba Mill.): a review. Food Chem. 2017;227:349–57. https://doi.org/10.1016/j.foodchem.2017.01.074.
Jin M, Zhao K, Huang Q, Xu C, Shang P. Isolation, structure and bioactivities of the polysaccharides from Angelica sinensis (Oliv.) diels: a review. Carbohydr Polym. 2012;89(3):713–22. https://doi.org/10.1016/j.carbpol.2012.04.049.
Tu W, Zhu J, Bi S, Chen D, Song L, Wang L, Zi J, Yu R. Isolation, characterization and bioactivities of a new polysaccharide from Annona squamosa and its sulfated derivative. Carbohydr Polym. 2016;152:287–96. https://doi.org/10.1016/j.carbpol.2016.07.012.
Gao Y, Zhou Y, Zhang Q, Zhang K, Peng P, Chen L, Xiao B. Hydrothermal extraction, structural characterization, and inhibition HeLa cells proliferation of functional polysaccharides from Chinese tea Zhongcha 108. J Func Foods. 2017;39:1–8. https://doi.org/10.1016/j.jff.2017.09.057.
Zhao G, Kan J, Li Z, Chen Z. Structural features and immunological activity of a polysaccharide from Dioscorea opposita Thunb roots. Carbohydr Polym. 2005;61(2):125–31. https://doi.org/10.1016/j.carbpol.2005.04.020.
Yang W, Wang Y, Li X, Yu P. Purification and structural characterization of Chinese yam polysaccharide and its activities. Carbohydr Polym. 2015;117(5):1021–7. https://doi.org/10.1016/j.carbpol.2014.09.082.
Zhou S, Huang G, Chen G. Extraction, structural analysis, derivatization and antioxidant activity of polysaccharide from Chinese yam. Food Chem. 2021;361:130089. https://doi.org/10.1016/j.foodchem.2021.130089.
Huo Z, Tian J. Comment on recent article “Extraction, structural analysis, derivatization and antioxidant activity of polysaccharide from Chinese yam”, published in food chemistry (2021): The need to consider residue ethanol solvent signals in the NMR spectra interpretation of polysaccharides. Food Chem. 2022;368:130867. https://doi.org/10.1016/j.foodchem.2021.130867.
Tang W, Liu D, Wang J, Huang X, Yin J, Geng F, Nie P. Isolation and structure characterization of a low methyl-esterified pectin from the tuber of Dioscorea opposita Thunb. Food Chem. 2021;359:129899. https://doi.org/10.1016/j.foodchem.2021.129899.
Ma F, Zhang Y, Liu N, Zhang J, Tan G, Kannan B, Liu X, Bell AE. Rheological properties of polysaccharides from Dioscorea opposita Thunb. Food Chem. 2017;221:919–25. https://doi.org/10.1016/j.foodchem.2017.01.072.
Staub AM. Removeal of protein—Sevag method. Method Carbohydr Chem. 1965;5:5–6.
Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Colorimetric method for determination of sugars and related substances. Anal Chem. 1956;28(3):350–6. https://doi.org/10.1021/ac60111a017.
Blumenkrantz N, Asboe-Hansen G. New method for quantitative determination of uranic acids. Anal Biochem. 1973;54:484–9. https://doi.org/10.1016/0003-2697(73)90377-1.
Dodgson KS, Price RG. A note on the determination of the ester sulphate content of sulphated polysaccharides. Biochem J. 1962;84(1):106–10. https://doi.org/10.1042/bj0840106.
Xu Z, Li X, Feng S, Liu J, Zhou L, Yuan M, Ding C. Characteristics and bioactivities of different molecular weight polysaccharides from camellia seed cake. Int J Biol Macromol. 2016;91:1025–32. https://doi.org/10.1016/j.ijbiomac.2016.06.067.
Wang H, Zhao J, Li D, Song S, Song L, Fu Y, Zhang L. Structural investigation of a uronic acid-containing polysaccharide from abalone by graded acid hydrolysis followed by PMP-HPLC-MSn and NMR analysis. Carbohydr Res. 2015;402(12):95–101. https://doi.org/10.1016/j.carres.2014.10.010.
Hua D, Zhang D, Huang B, Yi P, Yan C. Structural characterization and DPPH· radical scavenging activity of a polysaccharide from Guava fruits. Carbohydr Polym. 2014;103:143–7. https://doi.org/10.1016/j.carbpol.2013.12.009.
Yu XH, Liu Y, Wu XL, Liu LZ, Fu W, Song DD. Isolation, purification, characterization and immunostimulatory activity of polysaccharides derived from American ginseng. Carbohydr Polym. 2017;156:9–18. https://doi.org/10.1016/j.carbpol.2016.08.092.
Wang H, Li N, Zhu C, Shi S, Jin H, Wang S. Anti-complementary activity of two homogeneous polysaccharides from Eclipta prostrata. Biochem Bioph Res Commun. 2017;493:887–93. https://doi.org/10.1016/j.bbrc.2017.09.126.
Zhang C, Chen X, Ding K. Structural characterization of a galactan from Dioscorea opposita Thunb. and its bioactivity on selected Bacteroides strains from human gut microbiota. Carbohydr Polym. 2019;218:299–306. https://doi.org/10.1016/j.carbpol.2019.04.084.
Chen Y, Wang T, Zhang X, Zhang F, Linhardt RJ. Structural and immunological studies on the polysaccharide from spores of a medicinal entomogenous fungus Paecilomyces cicadae. Carbohydr Polym. 2021;254:117462. https://doi.org/10.1016/j.carbpol.2020.117462.
Chiu C, Deng J, Chang H, Chen Y, Lee M, Hou W, Lee C, Huang S, Huang G. Antioxidant and anti-inflammatory properties of Taiwanese yam (Dioscorea japonica Thunb. Var pseudojapanica (Hayata) Yamam.) and its reference compounds. Food Chem. 2013;141:1087–96. https://doi.org/10.1016/j.foodchem.2013.04.031.
Liu J, Wu C, Li X, Yan Q, Reaney MJT, Jiang Z. Xylose rich heteroglycan from flaxseed gum mediates the immunostimulatory effects on macrophages via TLR2 activation. Carbohydr Polym. 2019;213(5):9–69. https://doi.org/10.1016/j.carbpol.2019.02.080.
Silva FRF, Dore CMPG, Marques CT, Nascimento MS, Benevides NMB, Rocha HAO, Chavante SF, Leite EL. Anticoagulant activity, paw edema and pleurisy induced carrageenan: action of major types of commercial carrageenans. Carbohydr Polym. 2010;79:26–33. https://doi.org/10.1016/j.carbpol.2009.07.010.
Li Y, Lin D, Jiao B, Xu C, Qin J, Ye G, Su G. Purification, antioxidant and hepatoprotective activities of polysaccharides from Cissus pteroclada Hayata. Int J Biol Macromol. 2015;77:307–13. https://doi.org/10.1016/j.ijbiomac.2015.03.039.
Perumal A, AlSalhi MS, Kanakarajan S, Devanesan S, Selvaraj R, Tamizhazhagan T. Phytochemical evaluation and anticancer activity of rambutan (Nephelium lappaceum) fruit endocarp extracts against human hepatocellular carcinoma (HepG-2) cells. Saudi J Biol Sci. 2021;28:1816–25. https://doi.org/10.1016/j.sjbs.2020.12.027.
Ren D, Wang N, Guo J, Yuan L, Yang X. Chemical characterization of Pleurotus eryngii polysaccharide and its tumor-inhibitory effects against human hepatoblastoma HepG-2 cells. Carbohydr Polym. 2016;138:123–33. https://doi.org/10.1016/j.carbpol.2015.11.051.
Jeddou KB, Chaari F, Maktouf S, Nouri-Ellouz O, Helbert CB, Ghorbel RE. Structural, functional, and antioxidant properties of water-soluble polysaccharides from potatoes peels. Food Chem. 2016;205:97–105. https://doi.org/10.1016/j.foodchem.2016.02.108.
Lu Y, Xu L, Cong Y, Song G, Han J, Wang G, Zhang P, Chen K. Structural characteristics and anticancer/antioxidant activities of a novel polysaccharide from Trichoderma kanganensis. Carbohydr Polym. 2019;205:63–71. https://doi.org/10.1016/j.carbpol.2018.09.068.
Anvari M, Tabarsa M, Cao R, You S, Joyner HS, Behnam S, Rezaei M. Compositional characterization and rheological properties of an anionic gum from Alyssum homolocarpum seeds. Food Hydrocolloid. 2016;52:766–73. https://doi.org/10.1016/j.foodhyd.2015.07.030.
Yang LQ, Zhang LM. Chemical structural and chain conformational characterization of some bioactive polysaccharides isolated from natural sources. Carbohydr Polym. 2009;76:349–61. https://doi.org/10.1016/j.carbpol.2008.12.015.
Li F, Wei Y, Liang L, Huang L, Yu G, Li Q. A novel low-molecular-mass pumpkin polysaccharide: Structural characterization, antioxidant activity, and hypoglycemic potential. Carbohydr Polym. 2021;251:117090. https://doi.org/10.1016/j.carbpol.2020.117090.
Nep EI, Conway BR. Characterization of grewia gum, a potential pharmaceutical excipient. J Excip Food Chem. 2010;1(1):30–40.
Gao X, Qi J, Ho C, Li B, Mu J, Zhang Y, Hu H, Mo W, Chen Z, Xie Y. Structural characterization and immunomodulatory activity of a water-soluble polysaccharide from Ganoderma leucocontextum fruiting bodies. Carbohydr Polym. 2020;249:116874. https://doi.org/10.1016/j.carbpol.2020.116874.
Ma J, Liu H, Han C, Zeng S, Xu X, Lu D, He H. Extraction, characterization and antioxidant activity of polysaccharide from Pouteria campechiana seed. Carbohydr Polym. 2020;229:115409. https://doi.org/10.1016/j.carbpol.2019.115409.
Malinowska E, Klimaszewska M, Strączek T, Schneider K, Kapusta C, Podsadni P, Łapienis G, Dawidowski M, Kleps J, Górska S, Pisklak DM, Turło J. Selenized polysaccharides—biosynthesis and structural analysis. Carbohydr Polym. 2018;198:407–17. https://doi.org/10.1016/j.carbpol.2018.06.057.
Teng C, Qin P, Shi Z, Zhang W, Yang X, Yao Y, Ren G. Structural characterization and antioxidant activity of alkali-extracted polysaccharides from quinoa. Food Hydrocoll. 2020;113:106392. https://doi.org/10.1016/j.foodhyd.2020.106392.
Ližičárová I, Matulová M, Capek P, Machová E. Human pathogen Candida dubliniensis: a cell wall mannan with a high content of β-1,2-linked mannose residues. Carbohydr Polym. 2007;70(1):89–100. https://doi.org/10.1016/j.carbpol.2007.03.007.
Suzuki A, Takata Y, Oshie A, Tezuka A, Shibata N, Kobayashi H, Okawa Y, Suzuki S. Detection of β-l,2-mannosyltransferase in Candida albicans cells. FEBS Lett. 1995;373(3):275–9. https://doi.org/10.1016/0014-5793(95)01061-I.
Ji XL, Cheng YQ, Tian JY, Zhang SQ, Jing YS, Shi MM. Structural characterization of polysaccharide from jujube (Ziziphus jujuba Mill.) fruit. Chem Biol Technol Agric. 2021;8(1):54. https://doi.org/10.1186/s40538-021-00255-2.
Lin X, Ji X, Wang M, Yin S, Peng Q. An alkali-extracted polysaccharide from Zizyphus jujuba cv. Muzao: structural characterizations and antioxidant activities. Int J Biol Macromol. 2019;136:607–15. https://doi.org/10.1016/j.ijbiomac.2019.06.117.
Shakhmatov EG, Belyy VA, Makarova EN. Structure of acid-extractable polysaccharides of tree greenery of Picea abies. Carbohydr Polym. 2018;199:320–30. https://doi.org/10.1016/j.carbpol.2018.07.027.
Kang J, Cui SW, Phillips GO, Chen J, Guo Q, Wang Q. New studies on gum ghatti (Anogeissus latifolia) Part III: structure characterization of a globular polysaccharide fraction by 1D, 2D NMR spectroscopy and methylation analysis. Food Hydrocoll. 2011;25:1999–2007. https://doi.org/10.1016/j.foodhyd.2010.11.020.
Ji XL, Guo JH, Ding DQ, Gao J, Hao LR, Guo XD. Structural characterization and antioxidant activity of a novel high-molecular-weight polysaccharide from Ziziphus Jujuba cv. Muzao Food Measure. 2022;16:2191–200. https://doi.org/10.1007/s11694-022-01288-3.
Cardoso SM, Ferreira JA, Mafra I, Silva AMS, Coimbra MA. Structural ripening-related changes of the Arabian-rich pectic polysaccharides from olive pulp cell walls. J Agr Food Chem. 2007;55(17):7124–30. https://doi.org/10.1021/jf070769w.
Korhonen R, Lahti A, Kankaanranta H, Moilanen E. Nitric oxide production and signaling in inflammation. Curr Drug Targets-Inflamm Allergy. 2005;4(4):471–9. https://doi.org/10.2174/1568010054526359.
Li S, Gao A, Dong S, Chen Y, Sun S, Lei Z, Zhang Z. Purification, antitumor and immunomodulatory activity of polysaccharides from soybean residue fermented with Morchella esculenta. Int J Biol Macromol. 2017;96:26–34. https://doi.org/10.1016/j.ijbiomac.2016.12.007.
Acknowledgements
We are grateful to the platform of Key Laboratory of Natural Medicine and Immuno-Engineering of Henan Province, Henan International Joint Laboratory of Medicinal Plants Utilization, State Key Laboratory of Crop Stress Adaptation and Improvement, School of Life Sciences, and College of Chemistry and Molecular Sciences, Henan University.
Funding
We are grateful for financial support from the National Natural Science Foundation of China (No. 31701553), Key Scientific and Technological Project of Henan Province (No. 212102311043 and 202102110144), Foundation of Educational Department of Henan Province (No. 20A550002), and Postgraduate Education Innovation Program of Henan University (SYLYC2022119).
Author information
Authors and Affiliations
Contributions
FM, RW, YZ, and QL designed the experiment. RW, YZ, JB, HF, WM, and WL prepared the samples and performed the experiments. FM, QL, and RW performed the experiments, analyzed data, and wrote the paper. FM, QL, XL, and RW reviewed and checked all the details. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, F., Wang, R., Zhang, Y. et al. Polysaccharides from Dioscorea opposita Thunb.: isolation, structural characterization, and anti-inflammatory and anti-tumor effects against hepatocellular carcinoma. Chem. Biol. Technol. Agric. 10, 43 (2023). https://doi.org/10.1186/s40538-023-00421-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-023-00421-8