
Abstract
Non-Hodgkin lymphomas (NHLs) are heterogeneous and are among the most common hematological malignancies worldwide. Despite the advances in the treatment of patients with NHLs, relapse or resistance to treatment is anticipated in several patients. Therefore, novel therapeutic approaches are needed. Recently, natural killer (NK) cell-based immunotherapy alone or in combination with monoclonal antibodies, chimeric antigen receptors, or bispecific killer engagers have been applied in many investigations for NHL treatment. The functional defects of NK cells and the ability of cancerous cells to escape NK cell-mediated cytotoxicity within the tumor microenvironment of NHLs, as well as the beneficial results from previous studies in the context of NK cell-based immunotherapy in NHLs, direct our attention to this therapeutic strategy. This review aims to summarize clinical studies focusing on the applications of NK cells in the immunotherapy of patients with NHL.
Similar content being viewed by others

Introduction
B-cell non-Hodgkin lymphoma (NHL) collectively represents the most common type of hematologic malignancy [1]. While advances in chemotherapy, monoclonal antibodies, and stem cell transplantation have improved survival rates, many NHL patients remain resistant to therapy or experience relapse. This highlights the necessity for finding novel curative therapeutic options for these patients [2,3,4].
Recently, novel therapeutic approaches, such as chimeric antigen receptor (CAR)-T cell therapy, have been utilized in several clinical trials for patients with relapsed/refractory (R/R) B-cell NHL, resulting in promising clinical responses [5,6,7]. However, this therapeutic approach is expensive and associated with unique and severe side effects such as cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and graft-versus-host disease (GVHD) in allogenic settings [8, 9]. Other immune cells, such as natural killer (NK) cells, which exhibit significant cytotoxic activity against cancer cells and possess a safer immune profile, can be used as alternative approaches for immunotherapy of patients with R/R NHL. Moreover, it is also feasible to equip NK cells with a CAR structure [10, 11].
This study will delve into the B-cell NHL tumor microenvironment (TME) and the interaction between NK cells and malignant cells. Additionally, we provide a comprehensive review of clinical trials focused on the utilization of NK cells in patients with R/R NHL. Finally, new approaches recently used to increase NK cells effectiveness for B-cell NHL immunotherapy are summarized.
NK cells: biology, receptors, and functions
NK cells, a specialized subset of innate lymphoid cells (ILCs), can distinguish between self-cells and non-self-cells through the recognition of self-major histocompatibility complex (MHC) I molecules [12, 13]. They constitute approximately 10–15% of the lymphocyte population in peripheral blood, and characterized as large granular lymphocytes with kidney-shaped nuclei, a high cytoplasm-to-nucleus ratio, and large azurophilic granules in cytoplasm [14, 15]. NK cell development and maturation primarily occur in the bone marrow, where common lymphoid progenitors (CLPs) differentiate into NK precursors (NKPs), immature NK cells, and finally mature NK cells [12]. Notably, recombinant interleukin (rIL)-15 plays a crucial role in NK cell development from hematopoietic stem cells [16]. In humans, CD122 expression on NKPs is crucial for NK cell lineage commitment, and CD56 expression is a final step in the differentiation of NKPs into NK cells [17]. NK cells are typically identified by the expression of CD56 and CD16, and the absence of CD3 (T cell marker) [18].
Human NK cells can be classified into two main subsets: the CD56bright subset, which is characterized by immaturity, limited cytolytic activity, but high cytokine production; and the CD56dim subset, which is mature, exhibits higher cytolytic activity, but lower cytokine production [19].
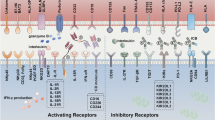
NK cells are equipped with various germline-encoded activating and inhibiting receptors [20]. The function of NK cells is delicately regulated by the balance of the activating and inhibitory signals that are transmitted through their receptors [21]. Table 1 provides an overview of NK cell receptors.
NK cells express the HLA-specific activating receptors such as KIRs/CD158 (2DS1–2DS5 and 3DS1), NKG2C, and NKG2E. NKG2C and NKG2E are expressed as heterodimers with CD94. Upon interaction with HLA-E, they transmit activating signals through the DNAX-activation protein (DAP)-12 adaptor molecule [22,23,24]. The natural cytotoxicity receptors (NCRs) including NKp46, NKp44, and NKp30 are the major non-HLA specific activating NK receptors, which evoke an immune response upon detection of cognate viral and cellular ligands [20, 25]. NKG2D is another non-HLA-specific activating NK cell receptor [26]. The UL16-binding protein (ULBP) and MHC class I chain-related proteins A and B (MICA/B), which are increased in the tumor, stressed, and infected cells, are representative of NKG2D ligands [27]. Additionally, some other molecules, such as 2B4, NTB-A, CD59, NKp80, and DNAX accessory molecule-1 (DNAM-1), are essentially coreceptors; in fact, they can intensify the NK cell triggering induced by NCRs or NKG2D (See Table 1) [28,29,30,31,32]. NK cells are also equipped with the CD16a (FcγRIIIa, a low-affinity Fc), which plays a crucial role in their antibody-dependent cell-mediated cytotoxicity (ADCC) effector function. It is worth noting that CD16 is the only receptor that can activate NK cells without the need for further activation from other receptors [33].
Besides activating receptors, NK cells express inhibitory receptors that modulate the strength of activating receptors and contribute to regulating immune responses and tolerance [34]. The CD94/NKG2A (CD94/CD159a) heterodimer and members of the KIR/CD158 family are two distinct classes of HLA-specific inhibitory receptors [35, 36]. The LIR-1/ILT2/CD85 is an inhibitory receptor with broad specificity for both classical and non-classical MHC molecules [37,38,39]. Program-cell death receptor 1 (PD-1), Sialic acid recognizing Immunoglobulin-like Lectins (Siglecs)/p75/AIRM1/CD328, leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1)/p40/CD305, and IRp60/CD300a known as another non-HLA-specific inhibitory receptor that hamper NK cell-mediated antitumor immunity through the recognition of different ligands on the surface of cancerous cells (See Table 1) [40, 41].
NK cells have diverse functions, including their natural antitumor and antiviral activities, as well as their regulatory roles in modulating immune responses and promoting tissue growth. They are abundant in the TME, where they kill cancer cells in a variety of ways [21]. The antitumor functions of NK cells include missing self-mechanisms, direct cytotoxicity, and activation of adaptive immune responses (Fig. 1) [42,43,44,45,46,47].

Natural killer (NK) cell function within tumor microenvironment (TME). A) Missing-self recognition against tumor cells lacking MHC class I ligands for inhibitory NK receptors. B) Direct cytotoxicity against tumor cells mediated by releasing cytotoxic granules containing perforin/granzymes, IFN-γ and TNF-α production, antibody-dependent cell-mediated cytotoxicity (ADCC) via CD16, and induction of apoptosis pathway through death receptor ligands like TRAIL/FasL. C) Triggering the adaptive antitumor immunity by recruiting dendritic cells via chemokines and then amplifying CD4+ and CD8+ T cells antitumor immune response
TME in NHLs: composition and functions
The development of B-cell lymphoma involves an intricate interplay between tumor cells and the surrounding TME (Fig. 2). The microenvironment in B-cell lymphoma is fascinating because it has crucial functions in regulating the survival and growth of tumor cells, promoting immune evasion, and contributing to the development of resistance to treatment [48,49,50,51,52]. It is worth mentioning that different cells within the TME can display pro-tumorigenic or anti-tumorigenic functions, as shown in Table 2.

Schematic representation of tumor microenvironment (TME) constituents in non-Hodgkin lymphomas (NHLs). The TME comprises cellular and noncellular components. The cellular microenvironment consists of immune and nonimmune cells that can play pro- or antitumorigenic roles within the NHL milieu (see Table 2 for more details about the pro-tumorigenic and anti-tumorigenic cells in the TME). CTLs, effector CD4.+ T cells (TH1, TH2, TH17, TFH, and Treg cells), follicular dendritic cells (FDCs), natural killer (NK) cells, tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), and myeloid-derived suppressor cells (MDSCs) constitute the immune microenvironment. On the other hand, mesenchymal stromal cells (MSCs) and cancer-associated fibroblasts (CAFs) are involved in nonimmune microenvironments. The noncellular components include the extracellular matrix (ECM) as well as various cytokines, chemokines, and molecules produced by cancerous and noncancerous cells and executed by these cells to induce their stimulatory or inhibitory effect on bystander cells (see Sect. "TME in NHLs: Composition and Functions" for more details about the interaction between cells inside the TME)
Intratumoral T lymphocytes constitute 50% of the total cells within the TME and are categorized based on the expression of CD4 or CD8. Typically, CD4+ T lymphocytes support other immune cells, while CD8+ cytotoxic T lymphocytes (CTLs) are known to trigger target cell killing by releasing perforin and granzyme B, expression of death ligands and IFN-γ and TNF-α production [53, 54]. CTLs are activated by antigen presentation through MHC-I molecules and the interaction of costimulatory molecules (B7-1 and B7-2) with CD28 [55, 56]. Conversely, inhibitory signals from molecules like CTLA-4, PD-L1/PD-L2, and LAG-3 regulate the activation of CTLs [57, 58]. Clinical studies have shown that higher numbers of intratumoral CD8+ T cells are linked to longer overall survival (OS) and disease-specific survival, regardless of other prognostic factors [59]. CD4+ T cells play a crucial role in regulating the immune response by boosting Ab production, attracting granulocytes to areas of inflammation, and supporting an efficient immune response by generating cytokines and chemokines [53]. These cells were categorized into effector CD4+ T cells, follicular helper T (TFH) cells, and regulatory T cells (Tregs). Effector CD4+ T cells polarized to TH1, TH2, and TH 17 cells based on the patterns of various cytokines (See Table 2) [60, 61]. TH1 cells aid the activation of macrophages, NK cells, and CTL, while TH2 cells facilitate humoral immune responses by promoting B-cell growth and antibody production [62,63,64]. In B-cell NHL, the expression of both TH1 and TH2 cytokines at high mRNA levels has been reported [65]. Traditionally, the TH1 immune response is considered more effective at promoting antitumor immunity, while the TH2 immune response may support tumor growth by promoting angiogenesis and inhibiting the TH1-mediated immune response [66]. Higher levels of IL-4, indicative of a TH2 response, are associated with longer survival, while increased levels of IL-12, a cytokine involved in TH1 immunity, are linked to poorer prognosis in certain NHL types, suggesting that malignant B cells can modulate the effects of TH1 and TH2 cells in different lymphoma types [52, 67]. Besides, defects in TH17 cells have been observed in B cell NHL [52, 68, 69]. TFH cells play specific roles in B-cell clonal selection, maturation, and differentiation into memory cells or plasma cells within germinal centers [70]. TFH cells also facilitate B-cell activation, prevent malignant B cells from undergoing spontaneous apoptosis, and stimulate the proliferation of lymphoma cells [52, 71]. Tregs have a critical role in cancer by restricting immune activation and specific immune responses [72]. In lymphoma biopsy samples, Treg cells are abundant and have been shown to suppress antitumor immunity by inhibiting other intratumoral CD4+ and CD8+ T-cell populations. TGF-β produced by lymphoma cells can stimulate the expression of FoxP3, a specific marker for Tregs, which can result in the conversion of CD4 + /CD25- T cells into Tregs [73].
In addition to T cells, the immune TME in NHLs also contains tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), and myeloid-derived suppressor cells (MDSCs). TAMs are classified to anti-tumorigenic (M1 macrophages) and pro- tumorigenic (M2 macrophages) [74]. M1 macrophages support the growth and differentiation of TH1 cells and NK cells, trigger CTLs cytotoxicity, and mediate ADCC [75,76,77]. Conversely, M2 macrophages hinder antitumor immunity and promote TH2 and Treg function, as well as CTL suppression [76, 78]. In B-cell lymphoma, TAMs play an important role in tumor progression, drug resistance, and recurrence via multiple mechanisms [78]. Similarly, in the early stages of tumor formation, TANs primarily exhibit the N1 phenotype in the presence of INF-β resulting in CTL activation and recruitment and triggering ADCC. However, as the tumor progresses, TGF-β triggers the transition of TANs to the N2 phenotype. Neutrophils with an N2-like phenotype accompanied by tumor proliferation, blood vessel formation, extracellular matrix (ECM) degradation, and hinder T-cell activation. In addition to their functions, the N1 phenotype and N2 phenotype differ from each other in terms of cell surface markers (See Table 2) [50, 78,79,80,81]. MDSCs are a diverse group of cells that include monocytic MDSCs (M-MDSCs) and granulocytic MDSCs (PMN-MDSCs). MDSCs suppress CD4 + T cells, CD8 + T cells, and NK cells through direct cell contact and the production and activation of inhibitory molecules. Furthermore, MDSCs regulate the expansion and activation of Tregs, support tumor angiogenesis and metastasis, and can transform into TAMs at the tumor site. A high prevalence of the M-MDSC subpopulation has been linked to disease progression and decreased OS in B-cell NHL patients [50, 52, 73, 82].
TME also contains stromal cells such as mesenchymal stromal cells (MSCs), which reduce cell death and support tumor growth by secretion of immune molecules. Within the lymphoma TME, MSCs recruit monocytes, macrophages, and neutrophils to the tumor site. Moreover, MSCs can differentiate into cancer-associated fibroblast (CAF)-like cells and secrete diverse chemokines that contribute to the homing and adhesion of lymphoma B cells [52, 83, 84]. CAFs, as another stromal cell, influence a variety of biological processes that advance cancer, including angiogenesis as well as the production and release of growth factors, cytokines, and exosomes. CAFs actively stimulate tumor cell growth, invasion, and inflammation, and contribute to resistance to treatment [50, 52].
Exosomes are an important part of the TME. In lymphomas, exosomes can decrease NK cell-mediated cytotoxicity, trigger immune cell death, and increase treatment resistance through the delivery of various molecules such as interleukins, PGE2, TGF-β, and microRNAs. Additionally, tumor-derived exosomes expedite the activation and growth of MDSCs [50, 52, 85, 86]. Apart from exosomes, chemokines and cytokines present in the TME can also support tumor growth and development. Many studies have shown that the serum level of soluble IL-2Rα, which is produced by CD4+ CD25+ T cells, is greater in B-cell NHL patients and is associated with a poorer prognosis [73, 87]. The secretion of TGF-β malignant B cells leads to the suppression of TH1 and TH17 cell growth and hinders the proliferation of T cells [73, 88]. Serum IL-10 levels have been demonstrated to be increased in B-cell NHL patients and to be negatively correlated with prognosis [73]. In addition, chemokines released by MSCs in lymphoma, including CXCL13, CCL19/CCL21, and CXCL12, facilitate B-cell adhesion and homing [52].
NK cell defects in NHL
The NK cell defects in the TME of NHL include quantitative deficiency, distribution abnormalities, functional deficiency, the presence of an immunosuppressive TME, and tumor cell escape from NK cell surveillance. Figure 3 summarizes NK cell defects in the TME of NHLs.

Possible mechanisms of natural killer (NK) cell defects in non-Hodgkin lymphomas (NHLs). The NK cell defects within the TME of NHLs include functional deficiency, the presence of an immunosuppressive TME, and tumor cell escape from NK cell surveillance (as well as quantitative deficiency and distribution abnormalities that are not shown in Fig. 2). See Sect. "NK cell defects in NHL" for more information). A) NK cell functional defects include decreased expression of activating receptors, overexpression of suppressor receptors, decreased ADCC mechanisms, decreased IFN-γ production, and impaired degranulation capacity. B) Immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and M2 macrophages hinder NK cell function through the production of immunosuppressive factors or the expression of inhibitory receptors. C) NHL cells evade NK cell-mediated cytotoxicity via resistance to the perforin/granzyme-mediated apoptosis pathway, resistance to death receptor-mediated apoptosis pathways, and inhibition of NK cell activation
Quantitative deficiency
The absolute NK cell count (A-NKC) decreases in (Diffuse large B cell lymphoma) DLBCL patients. According to Plonquet et al. investigation, one-third of DLBCL patients who present with 2 or 3 adverse prognostic factors of aaIPI have low NKC at diagnosis. Furthermore, they established that NKC was associated with a poorer response to treatment and shorter event-free survival (EFS) [89]. Similarly, flow cytometry analysis of CD3−CD56+ and/or CD16+ cells in the peripheral blood of DLBCL and (follicular lymphoma) FL patients demonstrated that both DLBCL and FL patients had low NKC, which was correlated with a reduction in progression-free survival (PFS) and OS [90]. Furthermore, a deficit in NKC was detected in other NHLs, such as primary central nervous system lymphoma (PCNSL). Lin et al. conducted a study on 161 patients with PCNSL and found that individuals who responded to treatment had a higher median of circulating NKC and NK cell proportion compared to those who did not respond. Their study revealed that PCNSL patients who have a higher baseline NKC display longer OS than those with a low NKC [91]. Consequently, NKC in NHL patients represents a prognostic biomarker for the assessment of clinical outcomes.
Functional deficiency
Cancer cells have been observed to employ a wide range of mechanisms to escape from the innate immune pressure exerted by NK cells, including abnormalities in NK cytotoxicity function [92]. In the context of NHLs, NK cells have impaired degranulation capacity. According to previous studies, NK cells exhibit defects in the production and exocytosis of cytotoxic granules containing perforin and granzyme [93, 94]. In a protein quantification study involving 12 patients with NHL, it was discovered that while the gene expression levels of perforin and granzyme B were higher in NHL patients compared to the control group, the intracellular levels of perforin in NK cells were lower in NHL patients [93]. Furthermore, a separate study conducted by Baychelier et al. found that patients who developed NHL after undergoing lung transplantation exhibited an accumulation of NK cells with low expression of perforin and impaired degranulation against NHL target cells [94]. Other aspects of NK cell hypofunction include decreased cytokines production, e.g. IFN-γ and TNF-α, the overexpression of suppressor receptors such as T-cell immunoreceptors with immunoglobulin and ITIM domains (TIGIT), and the decreased expression of activating receptors such as TIM-3 [95]. It should be noted that TIGIT has been associated with NK cell exhaustion [96]. In addition, decreased CD16 expression in NK cells and impaired ADCC activity were observed in newly diagnosed and refractory NHL patients [95, 97].
NK cells in refractory NHL exhibit downregulated expression of activating receptors, including NKp30, NKp46, and NKG2D. Further investigation revealed that de novo NHL development was correlated with increased NKG2A and CD62L expression but reduced inhibitory KIR and CD57 receptor expression [94]. Essa et al. demonstrated that DLBCL patients with advanced stages of the disease have significantly lower NKp44 levels than patients with earlier stages of DLBCL. This decrease in NKp44 may be attributed to the high level of IL6 and TGF-β in the advanced stages of the disease, which in turn downregulate NK activating receptors [98]. The expression of CD16 and NKG2D activating receptors on the surface of CD56dim cells was also reported to be decreased after rituximab treatment [99] (Fig. 3A).
Immunosuppressive TME
Successful interaction between NK cells and dendritic cells (DCs) and the production of chemokines are required to induce effective antitumor immunity by NK cells (Fig. 1). This process is negatively affected by TME, especially cellular and soluble components of the TME, which are associated with the escape of cancer cells due to the lack of effective immune responses [100]. Several immune suppressive cells, like MDSC, TAMs, and Tregs negatively interfere with NK cell activation and function. In a phase 2 clinical trial conducted by Bachanova et al., the frequencies of MDSCs and Tregs were investigated about adoptive NK cell therapy response in patients with NHL. Results from the trial indicated that patients who exhibited higher frequencies of MDSCs and Tregs, along with the adoptive NK cells, had a poorer response to therapy [95]. Similarly, Sato et al. demonstrated that the accumulation of MDSCs leads to NK cell depletion in NHL patients [101]. Increased numbers of MDSCs were reported in DLBCL, MZL, MCL, high‐grade B‐cell lymphoma (HGBL), PCNSL, and FL. Interestingly, MDSCs are markedly increased in high-grade NHLs and may be a potential prognostic marker [102]. The inhibitory effect of MDSCs on NKG2D expression and IFN-γ production in NK cells was confirmed in both in vivo and in vitro experiments [103]. The downregulation of other types of NK cell activating receptors, such as NKp30 and NKp46, was also detected [104, 105]. Further analysis in a murine lymphoma model revealed that MDSCs, which can secrete IL-10, reduced the frequency of NK cells [101]. Additionally, the coculture of MDSCs with NK cells has been shown to negatively affect the degranulation capacity of these cells through the TIGIT/CD155 pathway [105].
Tregs are another immunosuppressive cell type that limits adoptive NK cell therapy in NHL patients. Increased numbers of Tregs expressing high levels of Foxp3 following high-dose chemotherapy and IL2 administration before adaptive NK cell infusion interfere with NK cell expansion [106]. Treg infiltration in the TME could be justified via Indoleamine-2,3-dioxygenase (IDO). IDO is an immunosuppressive enzyme that catalyzes the conversion of tryptophan to kynurenine [107]. NHL patients who overexpress IDO simultaneously exhibit increased levels of FoxP3, a Treg marker [108]. In addition, IDO not only inhibits NK cell proliferation but also decreases activating receptors [107]. In a study conducted by Ninomiya et al., it was found that 32% of DLBCL patients exhibit overexpression of IDO, which is associated with unfavorable clinical outcomes [109]. Additionally, Yoshikawa et al. reported elevated levels of tryptophan-derived kynurenine in DLBCL patients [110].
Crosstalk between M2 macrophages and NK cells is another barrier to NK cell function in the TME. M2 macrophages limit NK cells' function by triggering the expression of inhibitory receptors immunoglobulin-like transcript 2 (ILT2/ CD85j), an NK cell inhibitory receptor [111]. In NHL, a high density of M2 macrophages in DLBCL of the central nervous system (CNS-DLBCL) has been detected and accounted for poor clinical outcomes [15].
Among other immunosuppressive factors, TGF-β has been investigated in NHL, and previous studies revealed TGF-β signaling Dysregulation in mantle cell lymphoma (MCL), FL, and DLBCL. The TGF-β signaling cascade is dysregulated through various mechanisms, such as altered receptor expression, disrupted SMAD signaling, and disturbances in epigenetic and genetic processes [112]. TGF-β inhibits IFN-γ expression, affects the metabolic pathway of NK cells, and reduces NKG2D and NKp30 expression, which are essential for tumor cell recognition and elimination, as well as for the effective interaction between natural NK cells and DCs [113, 114]. Interestingly, MDSCs and M2 macrophages participate in NK cell exhaustion by producing TGF-β1 (Fig. 3B) [103, 115].
Evasion mechanism
Resistance to apoptosis
Tumor cells in NHLs may escape from NK cell-mediated cytotoxicity through resistance to perforin/granzyme-mediated apoptosis. For this purpose, tumor cells may exhibit elevated intrinsic levels of proteinase inhibitor 9 (PI9), which functions to restrict the proteolytic action of granzyme B and secure their survival [116]. In this line, Bladergroen et al. verified that P19 was overexpressed in different types of T/B-NHL, such as extranodal T-cell NHL, enteropathy type T-cell NHL, NK/T-cell nasal-type lymphoma, and DLBCL [116]. Furthermore, cancer cells may escape apoptosis by inactivating apoptotic pathways activated by death receptors. The death ligands FasL/CD95L and TRAIL, which are members of the TNF family, are expressed in NK cells. These ligands interact with their respective receptors, Fas/CD95 and TRAIL-R, present on the surface of target cells. Upon interaction, the death domain (DD) is activated, initiating the apoptotic signaling cascade and ultimately leading to apoptosis [117]. According to previous studies, loss of Fas/CD95 expression was found in some FL and diffuse B/T-cell lymphomas [118], mucosa-associated lymphoid tissue lymphomas (MALTLs) [119], and cutaneous B-cell lymphomas (CBCLs) [120], which are associated with poor prognosis. In addition, mutations in Fas/CD95 have been reported in GC-derived B-cell lymphomas, such as primary nodal DLBCL, MALT-type lymphomas, FL, and anaplastic large cell lymphoma (ALCL) [121, 122]. A somatic mutation in TRAIL-R, which is correlated with the loss of chromosome 8p21-22, has also been detected in NHLs (Fig. 3C) [123].
Inhibition of NK cell activation
CD58 or lymphocyte-function antigen 3 (LFA-3) is known as an NK cell activator that interacts with CD2 on the NK cell surface [124,125,126]. Based on this speculation, mutations or deletions in CD58 also prevent NK cell function, which has been reported in DLBCL and FL [125, 126]. HLA-G is an inhibitory molecule in both membrane-bound and soluble isoforms that suppresses NK cells through interaction with its ILT2 [127]. The serum level of soluble HLA-G increased in NLHs such as DLBCL, FL, and peripheral T-cell lymphoma, which may disrupt NK cell function and be involved in lymphoma development [128]. Notably, HLA-G expression in lymphoma is a double-edged sword with protective and destructive effects [129]. To explore the evasion mechanisms, strategies that disturb NK cell receptors are also considered. The investigation by Satwani et al. revealed that, incubation of NHL cells with romidepsin enhanced NK cell cytotoxicity. Subsequently, they reported that romidepsin increases the surface expression of the NKG2D ligands MIC A/B on lymphoma cells. Based on the results of this study, impairment of NK cell function may be related to decreased expression of activating receptor ligands such as MIC A/B [130]. The immune checkpoints PD-1 and PD-L1 also restrict NK cell function, and PD-1/PD-L1 axis blockade unleashes NK cell cytotoxicity [127]. Research conducted by Laurent et al. revealed that DLBCL cells exhibit notably elevated levels of PD-1 and PD-L1/2 compared to FL cells. Notably, some DLBCL tumor cells coexpress both PD-1 and PD-L1/2. Interestingly, there are more PD-L1/2-positive lymphoma cells in the activated B-cell (ABC) subtype of DLBCL (ABC-DLBCL) than in the GC subtype (GC-DLBCL) [129]. Similarly, Kiyasu et al. reported that PL-1 is frequently expressed in tumor cells in DLBCL and is associated with poor prognosis [131].
NK cell immunotherapy in NHLs
In NHL, the A-NKC of the autograft directly influences clinical outcomes of following HSCT [132]. In a randomized, double-blind phase III clinical trial, patients with NHL who received an autograft with an A-NKC ≥ 0.5 × 109 cells/kg demonstrated 5-year OS and 5-year PFS rates of 87% and 71%, respectively. In contrast, patients infused with an autograft A-NKC < 0.5 × 109 cells/kg experienced 5-year OS rates of 55% and 5-year PFS rates of 32% [133]. With a 10.6-year median follow-up in the final update, the 13-year OS rates demonstrated a significant difference between groups, with a rate of 46% for the cohort infused with autograft A-NKC ≥ 0.09 × 109 cells/kg compared to 36% for the group infused with A-NKC < 0.09 × 109 cells/kg (P-value < 0.02) [134]. Faster and robust recovery of NK cells following HSCT is another factor that can affect clinical outcomes [135]. Porrata et al. reported that NHL patients with an A-NKC ≥ 80 cells/µL on day 15 after autologous HSCT had longer OS and PFS than patients with lower counts (not reached vs 5 months, p0.001; not reached vs 3 months, p0.0001, respectively) [136]. These findings suggest that the early post-HSCT recovery of NK cells may play a crucial antitumor role in the potential graft-versus-tumor (GVT) effect, given that NK cells are the only immune effector cells that reach normal numbers and function post-HSCT [137].
Several early studies have employed the administration of low-dose subcutaneous rlL-2 to promote the recovery and cytotoxic activity of NK cells as an effective approach to eradicate residual disease and prevent relapse following autologous HSCT in NHL patients [138, 139]. In a clinical trial involving patients with R/R high-grade NHL, researchers demonstrated that the administration of a low dose of rlL2 early after autologous HSCT for a duration of one year is well-tolerated and leads to the in vivo expansion of CD16+/CD56+ NK cells. Significantly, compared to their baseline quantity and function before starting treatment, the expanded CD56bright NK cell subsets exhibited enhanced activity against K562 cells (an NK-sensitive cell line) and CD16-mediated redirected killing activity against P815 target cells (an NK-resistant cell line). All ten patients who participated in the trial remained free from relapse for a period ranging from 5 to 34 months (median 16 months) after initiating rIL2 therapy. Notably, two patients who still had residual disease following HSCT experienced complete disease disappearance after rIL2 treatment [138]. Building upon the encouraging outcomes of these early studies, clinical trials have explored the adoptive transfer of ex vivo activated autologous NK cells or lymphokine-activated killer cells as a therapeutic approach for patients with lymphoma [140, 141]. The adoptive transfer of autologous NK cells was found to be feasible and safe, although only a limited antitumor effect was observed [142]. This limitation primarily stemmed from the matching of inhibitory receptors on autologous NK cells with self-MHC class I present on tumor cells, leading to "self" recognition signals that dampen NK cell activation and subsequent antitumor effects [142]. Furthermore, the adoptive transfer of autologous NK cells is costly and frequently requires multiple apheresis procedures, and the dose of injected NK cells is limited to approximately 107/kg [143]. To overcome these limitations, researchers have recently used allogeneic NK cells for lymphoma immunotherapy. In the phase 1 clinical trial conducted by Green Cross LabCell Corporation, the safety and possible efficacy of allogeneic NK cells were assessed in patients with malignant lymphoma or advanced solid tumors. In this study, allogeneic NK cells (namely, MG 4101) were obtained from random healthy unrelated donors and expanded in culture bags supplemented with IL-2, irradiated autologous feeder cells, and OKT3. Multiple doses of MG4101 were administered in the dose range of 1 × 106 cells/kg to 3 × 107 cells/kg without any signs of GVHD or serious toxicity. Among the 17 evaluable patients, only 8 exhibited stable disease (SD), while the disease progressed in the remaining patients. The median PFS for patients with SD was 4 months, ranging from 2 to 18 months [144]. The results of this study indicated that the use of alloreactive NK cells alone was not sufficient to eliminate the disease mass completely. As a result, researchers explored the combination of NK cells with other strategies to enhance their therapeutic effectiveness in subsequent studies [135].
NK cells combined with mAbs
Over the past two decades, the therapeutic effects of at least 570 monoclonal antibodies (mAbs) have been investigated in clinical trials. Among them, 79 therapeutic mAbs, including 30 mAbs for the treatment of hematological malignancies, have received approval from the United States food and drug administration (FDA) and are currently commercially available [145]. When mAbs bind to their targets, they can kill cancer cells through a variety of mechanisms, including programmed cell death (PCD), complement-dependent cytotoxicity (CDC), and ADCC [146]. Among these mechanisms, ADCC is an effective immune mechanism that is triggered when therapeutic mAbs are employed to eliminate cancer cells [147]. During the ADCC process, the FC region of the antibody is ligated to its corresponding FC receptor (FcR) on the plasma membrane of immune effector cells, while the Fab portion of the antibody attaches to target antigens on the surface of the cancer cell [148]. Human NK cells serve as crucial effector cells in the context of ADCC by expressing CD16A, which is a low-affinity receptor for IgG1 and IgG3 antibodies [149]. Given the likelihood that the efficacy of ADCC-mediated tumor cell elimination relies on the ratio of effector to target cells, the number and function of NK cells have been investigated as potential biomarkers to predict the response to anti-CD20 immunotherapy in NHL patients [90, 150]. Klanova et al. reported that low peripheral blood NKC in FL and DLBCL patients receiving anti-CD20 mAbs (rituximab or obinutuzumab) plus chemotherapy were linked to shorter PFS in both FL and DLBCL patients and diminished OS specifically in FL patients [90] Hence, the number of NK cells in individuals with lymphoma is important for determining their prognosis [151].
The administration of adoptive NK cells to enhance the ADCC capabilities of mAbs is a growing area of intervention that has been explored in recent years [152]. There are several ongoing and completed clinical trials exploring the safety and effectiveness of combining mAbs with infusions of autologous or allogeneic NK cells in patients with NHL (Tables 3 and 4). In a recent phase I study, Tanaka et al. investigated the infusion of ex vivo-expanded autologous NK cells in combination with rituximab-containing chemotherapy in patients with relapsed CD20+ malignant lymphoma [153]. Expanded autologous NK cells with high expression of NKp30, NKp44 and CD16 were intravenously infused (up to 10 × 106 cells/kg) into lymphoma patients one day after rituximab-combined salvage chemotherapy. The combination was safe and feasible, and among the nine lymphoma patients, seven achieved complete response (CR), with a median duration of 44 months (range: 6–56 months). However, it is difficult to determine the precise contribution of autologous NK cells to the clinical response, given that chemotherapy was administered to eight of nine patients after NK cell infusion [153]. In another study on chemotherapy-refractory NHL patients, allogeneic NK cell therapy (dose of 0.5–3.27 × 107 cells/kg) in combination with IL-2 and rituximab was found to be safe and effective in 4 of 15 evaluable patients, with 2 patients achieving CR lasting 3 and 9 months and 2 patients obtaining partial response (PR) [95]. Moreover, in a recent phase I study employing ex vivo-expanded allogeneic NK cells (namely, MG4101) plus rituximab after lymphodepleting chemotherapy for R/R NHL patients, Yoon et al. demonstrated that the treatment was well tolerated and led to a PR in 4 patients and a CR in 1 patient, yielding an overall response rate (ORR) of 55.6% [154]. Notably, one patient achieved a lasting CR that extended beyond 806 days [154].
In addition to autologous or allogeneic peripheral blood (PB)-derived NK cells, an increasing number of clinical trials have scrutinized the safety and efficacy of other NK cell sources, including cord blood (CB) [6, 155, 157], induced pluripotent stem cells (iPSCs) [158], and immortalized NK cell lines [162], for NHL immunotherapy. For example, in a phase 1/2 clinical trial, the safety and clinical activity of AB-101 (an allogeneic, nongenetically modified, CB-NK cell product) has been evaluated as a monotherapy and combined with rituximab for the treatment of R/R NHL patients [155]. The results from this study indicated that the concurrent administration of both agents was safe, resulting in an ORR of 67% in 6 patients (CR observed in 3 patients and PR in 1 patient), in contrast to an ORR of 27% in cohorts receiving AB-101 alone [155]. Another study by Katayoun Rezvani’s group assessed the efficacy of ex vivo-expanded CB-NKs in combination with rituximab and high-dose chemotherapy in NHL patients who were candidates for autologous HSCT [157]. Patients received rituximab and high-dose chemotherapy from days 13 through 7, lenalidomide from days 7 through 2, and CB-NK cells (108/kg) on day 5 before to autologous HSCT. CB-NK cells were detectable in vivo for two weeks, regardless of their HLA mismatch status. Importantly, no adverse events attributable to the CB-NK cells were observed. At a median follow-up of 47 months, the rates of relapse free survival (RFS) and OS were 53% and 74%, respectively [157].
NK-92 is an immortalized IL-2-dependent CD16− NK cell line that was isolated and successfully established by Klingman et al. in 1992 from a patient suffering from lymphoma. NK-92 cells exhibit potent cytotoxicity against several cancer cells, a phenomenon primarily ascribed to the overexpression of numerous activating receptors, concurrent downregulation of almost all inhibitory receptors, and heightened expression of perforin and granzyme. Furthermore, NK-92 cells can continuously proliferate with a doubling time of 2–4 days, are easily obtainable, and have a homogeneous phenotype [163, 164]. However, due to their cancerous nature, NK-92 cells must be mitotically inactivated prior to infusion into patients to inhibit undesired clonal proliferation, which restricts their persistence and expansion in vivo, and allogeneic administration demands very high doses of NK-92 cells [165]. In 2008, Arai et al. demonstrated for the first time the feasibility and safety of administering NK-92 cells (up to 3 × 109) to cancer patients [166]. Recently, a phase I dose-escalation study using NK-92 cells (1 × 109 cells/m2, 3 × 109 cells/m2 and 5 × 109 cells/m2) for refractory hematological malignancies that relapsed after autologous HSCT was conducted by Williams et al. [162]. A total of 12 patients were enrolled in this trial, including 2 patients with HL and 5 patients with NHL. The infusions of irradiated NK-92 cells were well-tolerated even at high doses and resulted in CR in one HL patient and a minor response (defined as 10–30% regression of target tumor lesions without the occurrence of new lesions or progression of nontarget lesions) in 2 NHL patients. Notably, in this study, no NK-92 cells were detected more than 15 min after infusion [162]. As mentioned earlier, NK-92 cells are highly dependent on exogenous IL-2 for survival and lack the CD16 receptor, thus impeding their capacity to mediate ADCC [167]. To address this, NK-92 cells have been modified to internally express IL-2 and the high-affinity CD16 receptor [168, 169]. Currently, this product, designated high-affinity NK (haNK), is being investigated in several clinical trials for solid tumors [170, 171]. Furthermore, preclinical data indicated that the combination of haNK cells withmAbs, such as daratumumab for multiple myeloma (MM) and rituximab for NHL, may have a synergistic effect. However, further clinical investigation is required to validate these approaches for NHL [163].
NK cells derived from iPSCs (iNKs) are another promising avenue for NK cell therapy and have the potential to address challenges commonly encountered with other sources of NK cells (Fig. 4) [172]. To generate iNK cells, somatic cells are first differentiated into iPSCs and then into CD34+ hematopoietic stem and progenitor cells (HSPCs). Subsequently, NK cells differentiate from HSPCs using cytokines (IL-3, IL-7, IL-15, SCF, and FLT3L) or stromal-based feeder cell lines and are then cocultured with feeder cells for further expansion [173, 174]. Currently, iPSC-based NK cell platforms have been evaluated in several clinical trials as monotherapies or in combination with mAbs for the treatment of hematological malignancies or solid tumors [158, 175,176,177]. As an example, FT516 is an iPSC-derived NK cell product modified to express high-affinity, cleavage-resistant Fc receptor (CD16A), with a preliminary report of 18 patients with R/R B-cell lymphoma in combination with rituximab demonstrating safety, with no evidence of GVHD, ICANS or CRS. Patients received two cycles of treatment consisting of a conditioning regimen (fludarabine and cyclophosphamide, each for 3 days), a single dose of rituximab and three weekly cycles of FT516 (four patients received 90 million cells/dose, seven patients received 300 million cells/dose, and seven patients received 900 million cells/dose) accompanied by IL-2 (6 MIU after each dose of FT516). Of the 18 patients, 10 patients were naive to treatment with autologous CD19-targeted CAR-T cells, and eight patients were previously treated with autologous CD19-targeted CAR-T-cell therapy. A total of 8/10 naive patients achieved an ORR (including 5 patients who achieved CR), and 3/8 patients previously treated with CD19-targeted CAR-T-cell therapy achieved an OR and CR [158, 177].

Overview of the advantages and limitations of different sources of NK cells
NK cells combined with bispecific antibodies
Bispecific killer engagers (BiKEs) were created with the intention of having one "arm" that binds to CD16 on NK cells and the other "arm" that targets a specific antigen on tumor cells [178]. The engager serves as a replacement for traditional antibody-Fc interactions in facilitating the immunological synapse between tumor cells and NK cells, thereby promoting NK activation and the killing of tumor cells [179]. Therefore, the use of BiKEs could enhance the function of NK cells by creating a stronger interaction when binding to anti-CD16 compared to the interaction between CD16 and the natural Fc portion of antibodies [180]. Moreover, BiKEs are nonimmunogenic and have rapid clearance properties, making them easy to engineer to target known tumor antigens. In addition to these advantages, BiKEs may offer advantages over mAbs due to their smaller size, which allows for better distribution in the body. This approach is especially beneficial for treating solid tumors [181,182,183]. Currently, several clinical trials are being conducted to evaluate the effectiveness of BiKEs in combination with NK cell therapy as a treatment for patients with lymphoma. Some of these trials focused on AFM13 [159, 184]. AFM13 is a tetravalent, bispecific innate cell engager that targets CD16A/CD30 and activates innate immune cells such as NK cells and macrophages [185]. AFM13 acts as a mediator by binding to CD16A on NK cells and to CD30 on lymphoma cells, which aids in the recruitment and activation of NK cells in proximity to tumor cells [186]. AFM13 was initially tested as a single therapy in a phase 1 clinical study for patients with R/R lymphoma [187]. The study showed that AFM13 treatment was safe and well-tolerated and led to positive tumor responses in several patients [187]. CB-NK cells precomplexed with AFM13 were recently tested within an ongoing phase I/II clinical trial for patients with refractory CD30-positive lymphomas. Forty-two patients (37 patients with HL and 5 patients with NHL with a median of seven prior lines of therapy) received fludarabine/cyclophosphamide followed by CB-NK cells precomplexed with AFM13 and three weekly IV infusions of AFM13. The results of this study showed that AFM13 in combination with NK cells was safe for patients with no instances of CRS, ICANS, or GVHD and resulted in an ORR of 92.8% and a CR rate of 66.7%. All four patients who had previously failed CD30 CAR-T-cell therapy achieved a CR [159].
NK cells combined with CAR structure
The CAR construct plays a crucial role in activating cells that have been transduced with CAR. The CARs employed in CAR-NK cells are often analogous to those utilized in CAR-T cells. A CAR consists of four essential components: an extracellular binding domain, a hinge region, a transmembrane domain, and one or more intracellular signaling domains (Fig. 5). Single-chain antibody variable fragments (scFvs) originate from a tumor-specific antibody and have the ability to bind to a particular antigen displayed on the surface of cancer cells. Moreover, the intracellular signaling domains are obtained from the cytoplasmic domains of ITAMs found in TCRs or other stimulating receptors [188]. The extracellular binding domain of CAR-modified effector cells enhances specificity by targeting tumor-associated antigens (TAAs). The hinge region serves as a connection between the extracellular binding domain and the transmembrane domain. The intracellular signaling domains in different generations of CARs possess different compositions, which affects the potency of the activation signal transmitted and consequently influences the cytotoxic capability against tumor cells (Fig. 5) [189]. The first generation of CARs consisted of only the CD3-ζ activation signaling domain. Subsequent generations of CARs incorporated one or two supplementary costimulatory molecules, including CD28, ICOS, 4-1BB, CD27, OX40, and CD40. CD28 and 4-1BB are the predominant molecules utilized among this group of molecules [190, 191]. Researchers have utilized other molecules as activation signaling domains for NK cells, in addition to the commonly used CARs that are applicable for both CAR-T cells and CAR-NK cells. CD244 (2B4), a member of the signaling lymphocyte activation molecule (SLAM) family, can also serve as a costimulatory molecule. The overexpression of 2B4 in NK cells leads to an enhanced ability to amplify signals and increased innate cytotoxicity against tumor cells [192]. DAP-12 is present on NK cells and plays a role in transmitting signals through the NK-activating receptors NKG2C and NKp44. Additionally, DAP-10 is involved in signal transmission through NKG2D [193, 194]. Hence, DAP-12 and DAP-10 can transmit intracellular signals in CAR-NK cells. In addition, NK cells modified with DAP-12-based CARs exhibited superior performance compared to that of NK cells modified with CD3-ζ-based CARs [193]. Recent research has indicated that NKG2D ligands are overexpressed in several hematological malignancies. Hence, the NKG2D-DAP-10-CD3-ζ CAR, which specifically targets NKG2D ligands, holds significant promise for the treatment of blood malignancies [195].

An overview of (A) CAR-NK cell therapy workflow in NHLs, (B) CAR structure and generation and (C, D) various methods of delivering CAR into NK cells
Transduction of the CAR gene into NK cells encompasses viral transduction, namely, retrovirus-based and lentivirus-based approaches, as well as transfection techniques such as electroporation, lipofection, and their combination with transposon systems (Fig. 5) [11]. CAR constructs are commonly integrated into a retrovirus or lentivirus-based expression vector. These vectors are then used to transduce primary NK cells or NK cell lines, with NK-92 being the most frequently used. The transduction of retroviral vectors shows a high level of effectiveness (ranging from 43 to 93%) in primary NK cells. However, the occurrence of insertional mutagenesis and its negative consequences significantly limit the use of this method in clinical applications [196]. However, lentivirus-based transduction is considered to be a safer method. Although its transduction efficiency in peripheral blood mononuclear cell (PBMC)-derived NK cells ranges from 8 to 16%, there is still an opportunity for improvement [197]. RNA transfection methods are economical strategies that have greater efficacy in transferring genes. However, the production of CAR constructs using this method is temporary, lasting for approximately 3–5 days. Although the short therapeutic time frame is a limitation, the temporary nature of CAR therapy may lower the occurrence of CAR-associated adverse effects, such as on-target off-tumor effects [195, 197, 198]. The integration of DNA into cells using transposon systems, such as PiggyBac (PB) and sleeping beauty (SB), in combination with transfection methods has emerged as an appealing strategy for generating cells that express transgenes in a safer and more stable manner [199, 200]. The SB transposon vector has proven to be a cost-effective and efficient means of gene transfer. However, its suitability for use with CAR-NK cells has not yet been evaluated [201].
CAR-NK cells are safer than CAR-T cells. The enhanced safety of CAR-NK cells can be attributed to two primary factors. CRS and neurotoxicity are frequently adverse effects of CAR-T-cell therapy [202]. The cytokine storm triggered by CAR-T cells, specifically TNF-α, is primarily facilitated by proinflammatory cytokines such as IL-1 and IL-6 [203]. CAR-NK cells secrete a variety of cytokines, such as IFN-γ and GM-CSF, which differ from the cytokines produced by CAR-T cells. Second, CAR-T cells can cause life-threatening GVHD due to HLA limitations. On the other hand, NK cells, which are considered important cells that initiate the GVT response early on, can potentially prevent GVHD by eliminating recipient antigen-presenting cells and CTLs [204]. Furthermore, CAR-NK cells may exhibit superior effectiveness in targeting and destroying cancerous cells compared to CAR-T cells. CAR-NK cells possess the ability to identify and execute their cytotoxic functions via both their designed and innate killing capabilities. By utilizing CARs, effector cells can enhance their ability to selectively target and eliminate a specific antigen with greater efficiency. Unlike CAR-T cells, CAR-NK cells retain the inherent ability of NK cells to destroy target tumor cells even when the expression of specific tumor antigens is reduced [205]. Moreover, the production of CAR-NK cells is more convenient than that of CAR-T cells. Due to the absence of the risk of GVHD, NK cells can be obtained from either a donor who is a match or a donor who has an HLA mismatch, hence expanding the pool of potential donors and enhancing the overall quality of the end products [206].
Recently, CAR-NK cell therapy has been assessed in various clinical trials for the treatment of lymphoma (Tables 5 and 6). CB-CAR-NK cells are presently employed in a clinical trial at MD Anderson Cancer Center, specifically targeting CD19 cells, and yielding highly favorable outcomes. 37 patients with R/R CD19-positive malignancies were enrolled in this trial and treated with CB-CAR-NK cells in two phases: a dose-escalation phase and an expansion phase. In the dose-escalation phase (n = 11), patients received a conditioning regimen (fludarabine and cyclophosphamide, each for 3 consecutive days) followed by the infusion of CB-CAR-NK cells (three patients received 10 × 104 cells/kg, four patients received 10 × 105 cells/kg, and four patients received 10 × 106 cells/kg). In the expansion phase (n = 26), patients were first treated with 10 × 106 cells/kg CB-CAR-NK. Then, the trial was amended to include a second expansion cohort in which patients received a single flat dose of 8 × 108 cells/kg CB-CAR-NK. A retroviral vector including an anti-CD19-CD28-CD3-ζ CAR, an IL-15 gene, and a suicide switch was utilized for transduction. None of the patients developed neurotoxicity or GVHD, and only one patient developed mild CRS (grade I). The ORR (including PR and CR) on days 30 and 100 for the 37 patients was 48.6%. The 1-year OS and PFS were 68% and 32%, respectively. Compared with non-responders, patients who achieved OR had higher levels and longer persistence of CB-CAR-NK cells [6, 207].
Goodridge et al. created a CAR-NK product called FT596. This product was derived from iPSCs. The iPSCs were modified to consistently produce anti-CD19 CAR, a high affinity and non-cleavable CD16 Fc receptor, and a combination of a membrane-bound IL-15 and an IL-15Rα fusion protein. In a Raji xenograft mouse model, the combination of FT596 with rituximab resulted in a substantial increase in the elimination of Raji tumor cells. In addition, when a mouse model that had been engrafted with human CD34 cells, FT596 showed enhanced longevity and safety compared to primary CAR19 T cells [211]. This platform has been translated into a multicenter, phase I clinical trial as monotherapy or in combination with rituximab to treat patients with R/R B-cell lymphoma [210]. A total of 20 patients underwent two treatment regimens, including 10 in regimen A (FT596 alone) and 10 in regimen B (FT596 cells combined with rituximab). Among the 17 evaluable patients, clinical response was observed in 9 patients (5 from regimen A and 4 from regimen B), 7 of whom achieved CR. Notably, no dose-limiting toxicity, ICANS, or GVHD of any grade was observed. Interestingly, 2/4 of patients treated with CAR-T-cell therapy at doses ≥ 9 × 107 cells/kg achieved CR [210]. An extended follow-up period will provide insight into the durability and efficacy of this platform. More recently, similar peripheral blood-derived anti-CD19 CAR-NK cells (named NKX019, a cryopreserved product utilizing OX40/CD3-ζ signaling domains and expressing a membrane-bound form of IL-15 for activation) were investigated in a phase I trial as a monotherapy for 19 patients with R/R B-cell malignancies. Patients received a daily lymphodepletion regimen of fludarabine and cyclophosphamide for 3 days. Next, they received three infusions of NKX019 at 3 dose levels, with doses ranging from 300 million to 1.5 billion cells per infusion. During the follow-up period, no dose-limiting toxicity, neurotoxicity, CRS or GVHD was reported. Among the 14 patients with NHL, 8 achieved CR; however, 3 patients with indolent lymphoma subsequently experienced relapse after a remission period of greater than 6 months [208, 212].
What’s Next? CIML NK cells
NK cells following exposure to happens, viral infection or a combination of cytokines achieve memory properties. NK cells preactivated with IL-12/15/18 have been described as cytokine-induced memory-like (CIML) NK cells [213]. CIML NK cells present distinctive characteristics, such as high proliferative capacity, sensitivity to low doses of IL-2, increased IFN-γ production, resistance to TGF-β, elevated glycolysis, and oxidative phosphorylation, which distinguishes them from conventional NK cells (cNK cells) [214,215,216,217]. In addition, the long-term life span and adaptive immune features of CIML NK cells have drawn attention to the use of these cells in cancer immunotherapy. Recent findings from preclinical and clinical trials have shown that CIML NK-based immunotherapy has produced promising results and also offers a safe approach to preventing GVHD, CRS, and neurotoxicity [218]. In the context of hematological malignancies, CIML NK cell-based immunotherapy has aided in the discovery of novel treatments for various cancers, particularly myeloid disorders. Similarly, adoptively transferred CIML NK cells trigger CR in 44% of R/R acute myeloid leukemia (AML) patients [219].
Another clinical trial by Shapiro et al. revealed that CIML NK cell infusion into an immune-compatible microenvironment in posttransplant relapsed AML, MDS, and MPN patients resulted in satisfactory expansion and persistence [220]. Similarly, CIML NK cells injected into pediatric/young adults with post-HCT-relapsed AML patients significantly expand and persist in a compatible milieu. Furthermore, this clinical trial established that AML patients were treated with donor lymphocyte infusions (DLIs), and CIML NK cells showed promising outcomes [221].
Unlike for myeloid disease, the therapeutic approach involving CIML NK cells in lymphoid malignancies has received less attention. One of these few studies was performed on a rat model of T-ALL, namely, Roser leukemia (RL). In this in vivo experiment, RL was treated with cNK cells, and NK cells were stimulated with IL12/15/18 (CIML NK cells). Based on these results, RL is resistant to cNK cells but not to CIML NK cells. Therefore, CIML NK cells could be introduced as a possibility for immunotherapeutic clinical trials in T-ALL patients [222].
The role of CIML NK cells in lymphoma was studied by Ni et al. in mice injected with RMA-S lymphoma cells. Tumor-bearing mice were treated with IL-12/15/18–preactivated NK cells and IL-15–pretreated NK cells. The results highlighted that compared with IL-15–pretreated NK cells, IL-12/15/18–preactivated NK cells display greater frequency, persistence, proliferation, and functional killing activity at the tumor site [223]. In another study, Gang et al. further investigated CIML NK cell incorporation in lymphoma. They preactivated NK cells with IL-12/15/18 and then developed them to express the anti-CD19 CAR structure. The 19-CAR-CIML NK cells exhibited improved in vitro cytotoxicity against Raji cells and CD19+ primary lymphoma cells, as illustrated by elevated IFN-γ production and degranulation capacity. In addition, 19-CAR-CIML NK cells exhibited satisfactory durability, expansion, and effector function in a human lymphoma xenograft mouse model [224].
Finally, the challenges in NK cell-based immunotherapy in NHLs, the highly appreciated features of CIML NK cells, and promising results from current preclinical studies have prompted us to develop new therapeutic options based on CIML NK cells. Hopefully, we will witness a fundamental revolution in the management of patients with NHL.
NK cell expansion
NK cells offer significant potential for immunotherapy in NHL treatment, but there are still obstacles to overcome to harness their full therapeutic benefits. There are still efforts to obtain a considerable number of NK cells for therapeutic purposes and to ensure that the obtained NK cells are fully functional and capable of effectively targeting and killing abnormal cells. This requires careful selection and expansion of NK cells, which can be technically challenging in the laboratory [225, 226]. Most PB-derived NK cell expansion protocols can be categorized into feeder-cell or feeder-free systems [227].
Feeder cells
The production of a significant amount of NK cells from a small initial quantity relies on feeder cells. These feeder cells, whether naturally or through additional modifications, present ligands for NK cell receptors. When combined with cytokines, this interaction drives a substantial expansion of NK cells outside the body, enabling the generation of a large number of NK cells for therapeutic purposes [228]. Various types of cells, such as EBV-transformed lymphoblastoids and genetically engineered HEK293 or K562 cell lines, are utilized as feeder cells. Among these, genetically modified K562 cells are the most commonly employed [227]. For example, when a mixed lymphocyte population is infected with Epstein‒Barr virus (EBV) in vitro, it results in an immortalized cell line that exhibits characteristics similar to those of proliferating B cells. With the expression of different ligands (4-1BBL), CD155, CD48, and CD58) that have specific receptors (4-1BB, DNAM-1, 2B4, and CD2, respectively) on activated NK cells, EBV-bearing lymphoblastoid cell lines (LCLs) play essential roles in NK cell expansion and stimulation [228]. Using this method, an average of 1,000–2,000-fold expansion of NK cells was reported to be observed over a period of 14 days [229]. The addition of IL-21 and IL-2 reportedly improved the expansion efficacy [227, 230, 231]. In another method, irradiated feeder cells were employed to amplify NK cells in laboratory settings. The K562 leukemia cell line has been altered to display particular ligands linked to antigen-presenting cells (CD64, CD86, and truncated CD19, CD137L, 4-1BB ligand, and membrane-bound IL-21). The irradiated K562-mbIL21-41BBL cells seemed to be very effective at rapidly increasing the number of NK cells in RPMI media (containing 10% FBS). These modified cells expanded NK cells 47,967-fold in 21 days [232]. Nevertheless, using feeder cells can pose challenges due to licensing intricacies, difficulties in sourcing, and the requirement for their elimination from the culture. Challenges such as incomplete irradiation of feeder cells (which might lead to teratoma) and separating and thoroughly eliminating cancer cells from the culture environment to avoid injection into patients are additional difficulties [233].
Feeder-free expansion methods
Expanding NK cells without the need for feeder cells has benefits compared to traditional methods, especially in terms of lower contamination risks and improved regulatory compliance. Additionally, other benefits, such as reducing costs through a more straightforward process and even decreasing cytotoxicity, have been reported [227]. Feeder-free NK cell expansion systems rely on cytokines and stimulating supplements or antibodies. Ex vivo cultured NK cells treated with IL-15 and nicotinamide exhibited stable CD62L expression, which was linked to increased FOXO1 levels. Nicotinamide enhanced NK cell metabolism, cytotoxicity, and cytokine production, leading to improved outcomes in adoptive transfer experiments. Recently, Cichocki et al. performed a phase 1 clinical trial in patients with relapsed or refractory NHL using rituximab in association with NK cells expanded with IL-15 and nicotinamide. The final result showed a 74% response rate in 19 patients [156]. Gluk et al. conducted two phase I studies to assess the combination therapy of rituximab and IL-2 (4.5–14 million international units) in relapsed or refractory B-cell NHL to boost ADCC through NK cell activation. The results showed that adding IL-2 to rituximab treatment is safe and effective, particularly with thrice-weekly IL-2 dosing, leading to increased NK cell counts and associated with treatment response [234]. In conclusion, obtaining a sufficient number of functional NK cells for therapeutic purposes remains a challenge. Despite the advancements made in feeder-free NK cell expansion, further investigations are needed to optimize this process and ensure its utility in clinical applications.
Conclusion
In this comprehensive review, we first provided an overview of NK cells, including their function, characteristics, development, and maturation. We then delved into the complex tumor microenvironment and the interplay between various presented cells that can either support or hinder the antitumor activity of NK cells in NHL.
Building on these findings, we explored various strategies to enhance the therapeutic potential of NK cells because based on the findings reported in the literature, the function and number of NK cells are defective in NHL patients. Therefore, it would be beneficial to bolster the innate immune response by injecting and activating NK cells. Also, combinations of NK cells with multiplex immunotherapy strategies such as mAbs, BiKEs, and CARs could be effective and have been investigated in numerous clinical trials. The mAbs and BiKEs augmented NK cell-killing activity mediated by ADCC. However, BiKEs simultaneously bind to the tumor antigen and the NK cell surface Fc receptor, potentially creating a bridge between NK cells and tumor cells and allowing them to act more effectively than mAbs. The use of NK cells engineered with a CAR structure is another type of NK cell-based immunotherapy for NHLs. CAR-NK cells, when equipped with cytokine receptors or cytokine genes, have demonstrated enhanced proliferation and prolonged survival in the patient's bloodstream. They can target TAAs with particular specificity and result in improved treatment responses. CIML NK cells with adaptive immune characteristics and long lifespans are also appropriate for this application, but they have not been well assessed in NHLs.
By reviewing the available clinical trial data, we concluded that NK cell-based approaches are generally well-tolerated, with no major safety concerns observed specifically GVHD. Overall, the available clinical trial data provide an encouraging foundation for the continued investigation and development of NK cell-based immunotherapies for the management of NHLs. The safety profile demonstrated in these studies, coupled with the potential for improved clinical outcomes, warrants further exploration of NK cell-based approaches, either as standalone therapies or in combination with other modalities, to improve the treatment landscape for patients with this complex hematological malignancy.
Availability of data and materials
Not applicable.
Abbreviations
- HL:
-
Hodgkin lymphoma
- NHL:
-
Non-Hodgkin lymphoma
- mAb:
-
Monoclonal antibody
- SCT:
-
Stem cell transplantation
- CAR:
-
Chimeric antigen receptor
- R/R:
-
Relapsed/refractory
- CRS:
-
Cytokine release syndrome
- ICANS:
-
Immune effector cell-associated neurotoxicity syndrome
- GVHD:
-
Graft-versus-host disease
- NK cell:
-
Natural killer cell
- TME:
-
Tumor microenvironment
- DLBCL:
-
Diffuse large B-cell lymphoma
- MCL:
-
Mantle cell lymphoma
- BL:
-
Burkitt lymphoma
- FL:
-
Follicular lymphoma
- MZL:
-
Marginal zone lymphoma
- CLL/SLL:
-
Chronic lymphocytic leukemia/small-cell lymphocytic lymphoma
- HSCT:
-
Hematopoietic stem cell transplantation
- CTL:
-
Cytotoxic T-lymphocyte
- BiTE:
-
Bispecific T cell engager
- ILC:
-
Innate lymphoid cell
- HSC:
-
Hematopoietic stem cell
- MHC:
-
Major histocompatibility complex
- LGL:
-
Large granular lymphocyte
- TCR:
-
T cell receptor
- CLP:
-
Common lymphoid progenitor
- NKP:
-
NK precursor
- rIL:
-
Recombinant interleukin
- NKG2:
-
Natural killer group 2
- CCR:
-
Chemokine (C–C motif) receptor
- KIR:
-
Killer-cell immunoglobulin-like receptor
- DAP:
-
DNAX-activation protein
- NCR:
-
Natural cytotoxicity receptor
- ULBP:
-
UL16-binding protein
- MICA/B:
-
MHC class I chain-related protein A and B
- IDO:
-
Indoleamine-pyrrole 2,3-dioxygenase
- PGE2:
-
Prostaglandin E2
- DNAM-1:
-
DNAX accessory molecule-1
- GPI:
-
Glycosylphosphatidylinositol
- FcγRIII:
-
IgG Fc region receptor III
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- PD-1:
-
Program-cell death receptor 1
- PD-L1/2:
-
PD-1/2 Ligand
- Siglecs:
-
Sialic acid recognizing immunoglobulin-like lectins
- LAIR-1:
-
Leukocyte-associated immunoglobulin-like receptor-1
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- FasL:
-
Fas ligand
- TRADD:
-
TNF receptor-associated death domain
- TH:
-
Helper T cell
- Treg:
-
Regulatory T cell
- DC:
-
Dendritic cell
- TAM:
-
Tumor-associated macrophage
- TAN:
-
Tumor-associated neutrophil
- MDSC:
-
Myeloid-derived suppressor cell
- TFH:
-
Follicular helper T cell
- MSC:
-
Mesenchymal stromal cell
- CAF:
-
Cancer-associated fibroblast
- ECM:
-
Extracellular matrix
- IFN:
-
Interferon
- TNF:
-
Tumor necrosis factor
- CTLA-4:
-
Cytotoxic T lymphocyte-associated antigen 4
- LAG-3:
-
Lymphocyte activation gene 3
- TGF:
-
Tumor growth factor
- CXCR:
-
Chemokine (C-X-C motif) receptor
- TFR:
-
Follicular regulatory T cell
- FoxP3:
-
Forkhead box P3
- TIM-3:
-
T-cell immunoglobulin and mucin domain-containing 3
- FDC:
-
Follicular dendritic cell
- GC:
-
Germinal center
- Ag-Ab complex:
-
Antigen-Antibody complex
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- CXCL:
-
Chemokine (C-X-C motif) ligand
- APRIL:
-
A proliferation-inducing ligand
- ICAM-1:
-
Intercellular adhesion molecule 1
- VEGF:
-
Vascular endothelial growth factor
- M-MDSC:
-
Monocytic MDSC
- PMN-MDSC:
-
Granulocytic MDSC
- Arg1:
-
Arginase1
- iNOS:
-
Inducible nitric oxide synthase
- ROS:
-
Reactive oxygen species
- A-NKC:
-
Absolute NK cell count
- EFS:
-
Event-free survival
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
- PCNSL:
-
Primary central nervous system lymphoma
- TIGIT:
-
T-cell immunoreceptors with Ig and ITIM domain
- HGBL:
-
High‐grade B‐cell lymphoma
- ILT2:
-
Inhibitory receptors immunoglobulin-like transcript 2
- CNS-DLBCL:
-
DLBCL of the central nervous system
- DD:
-
Death domain
- MALTL:
-
Mucosa-associated lymphoid tissue lymphoma
- CBCL:
-
Cutaneous B-cell lymphoma
- ALCL:
-
Anaplastic large cell lymphoma
- LFA-3:
-
Lymphocyte-function antigen 3
- HLA:
-
Human leukocyte antigen
- ABC-DLBCL:
-
Activated B-cell-DLBCL
- GC-DLBCL:
-
Germinal center-DLBCL
- GVT:
-
Graft-versus-tumor
- SD:
-
Stable disease
- FDA:
-
Food and drug administration
- CR:
-
Complete response
- PR:
-
Partial response
- ORR:
-
Overall response rate
- PB:
-
Peripheral blood
- CB:
-
Cord blood
- iPSC:
-
Induced pluripotent stem cell
- RFS:
-
Relapse free survival
- haNk:
-
High affinity NK cell
- MM:
-
Multiple myeloma
- iNK:
-
NK cells derived from iPSCs
- HSPC:
-
Hematopoietic stem and progenitor cell
- BiKE:
-
Bispecific killer engager
- scFv:
-
Single-chain antibody variable fragment
- TAA:
-
Tumor-associated antigen
- SLAM:
-
Signaling lymphocyte activation molecule
- PBMC:
-
Peripheral blood mononuclear cell
- cNK cell:
-
Conventional NK cell
- CIML NK cell:
-
Cytokine-induced memory-like NK cells
- AML:
-
Acute myeloid leukemia
- DLI:
-
Donor lymphocyte infusion
- RL:
-
Roser leukemia
- ALL:
-
Acute lymphocytic leukemia
- LCL:
-
Lymphoblastoid cell line
References
Barreno-Rocha SG, Guzmán-Silahua S, Cardona-Muñoz EG, Zavala-Cerna MG, Muñoz Gaytan DE, Riebeling-Navarro C, et al. Frequency of autoantibodies on non-Hodgkin lymphoma. Healthcare. 2023;11(15):2210.
Ayyappan S, Maddocks K. Novel and emerging therapies for B cell lymphoma. J Hematol Oncol. 2019;12(1):82.
St-Pierre F, Gordon LI. CAR T-cell therapy for relapsed/refractory non-Hodgkin’s lymphoma: a comprehensive review. Clin Adv Hematol Oncol. 2022;20(5):309–18.
Kumar S, Sharma A, Pramanik R, Pathak N, Gogia A, Kumar A, et al. Long-term outcomes and safety trends of autologous stem-cell transplantation in non-Hodgkin lymphoma: a report from a tertiary care center in India. JCO Glob Oncol. 2022;8:e2100383.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–42.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377(26):2531–44.
Yamshon S, Gribbin C, Chen Z, Demetres M, Pasciolla M, Alhomoud M, et al. Efficacy and toxicity of CD19 chimeric antigen receptor T cell therapy for lymphoma in solid organ transplant recipients: a systematic review and meta-analysis. Transplant Cell Ther. 2024;30(1):73.e1-.e12.
Khanmohammadi S, Rezaei N. CAR-NK cells: a promising cellular immunotherapy in lymphoma. Expert Opin Biol Ther. 2023;23(1):37–47.
Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975.
Zhang B, Yang M, Zhang W, Liu N, Wang D, Jing L, et al. Chimeric antigen receptor-based natural killer cell immunotherapy in cancer: from bench to bedside. Cell Death Dis. 2024;15(1):50.
Tsao TM, Tsai MJ, Hwang JS, Cheng WF, Wu CF, Chou CK, et al. Health effects of a forest environment on natural killer cells in humans: an observational pilot study. Oncotarget. 2018;9(23):16501–11.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–10.
Jonges LE, Albertsson P, van Vlierberghe RL, Ensink NG, Johansson BR, van de Velde CJ, et al. The phenotypic heterogeneity of human natural killer cells: presence of at least 48 different subsets in the peripheral blood. Scand J Immunol. 2001;53(2):103–10.
Gharehbaghian A, Haque KM, Truman C, Newman J, Bradley BA. Quantitation of natural killer cell precursors in man. J Immunol Methods. 2002;260(1–2):69–77.
Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity. 2018;49(5):971-86.e5.
Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. 2009;126(4):458–65.
Crome SQ, Lang PA, Lang KS, Ohashi PS. Natural killer cells regulate diverse T cell responses. Trends Immunol. 2013;34(7):342–9.
Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. 2019;16(5):430–41.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(2):85–100.
Parham P, Norman PJ, Abi-Rached L, Guethlein LA. Human-specific evolution of killer cell immunoglobulin-like receptor recognition of major histocompatibility complex class I molecules. Philos Trans R Soc Lond B Biol Sci. 2012;367(1590):800–11.
Siemaszko J, Marzec-Przyszlak A, Bogunia-Kubik K. Activating NKG2C Receptor: Functional Characteristics and Current Strategies in Clinical Applications. Arch Immunol Ther Exp (Warsz). 2023;71(1):9.
Miyashita R, Tsuchiya N, Hikami K, Kuroki K, Fukazawa T, Bijl M, et al. Molecular genetic analyses of human NKG2C (KLRC2) gene deletion. Int Immunol. 2004;16(1):163–8.
Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013;34(4):182–91.
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285(5428):727–9.
Mistry AR, O’Callaghan CA. Regulation of ligands for the activating receptor NKG2D. Immunology. 2007;121(4):439–47.
Sivori S, Parolini S, Falco M, Marcenaro E, Biassoni R, Bottino C, et al. 2B4 functions as a co-receptor in human NK cell activation. Eur J Immunol. 2000;30(3):787–93.
Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity. 1996;4(6):573–81.
Marcenaro E, Augugliaro R, Falco M, Castriconi R, Parolini S, Sivori S, et al. CD59 is physically and functionally associated with natural cytotoxicity receptors and activates human NK cell-mediated cytotoxicity. Eur J Immunol. 2003;33(12):3367–76.
Bottino C, Falco M, Parolini S, Marcenaro E, Augugliaro R, Sivori S, et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J exp med. 2001;194(3):235–46.
Vitale M, Falco M, Castriconi R, Parolini S, Zambello R, Semenzato G, et al. Identification of NKp80, a novel triggering molecule expressed by human NK cells. Eur J Immunol. 2001;31(1):233–42.
Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214:73–91.
Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. 2008;224:70–84.
Parham P, Guethlein LA. Genetics of Natural Killer Cells in Human Health, Disease, and Survival. Annu Rev Immunol. 2018;36:519–48.
Dizaji Asl K, Velaei K, Rafat A, Tayefi Nasrabadi H, Movassaghpour AA, Mahdavi M, et al. The role of KIR positive NK cells in diseases and its importance in clinical intervention. Int Immunopharmacol. 2021;92:107361.
Vitale M, Castriconi R, Parolini S, Pende D, Hsu ML, Moretta L, et al. The leukocyte Ig-like receptor (LIR)-1 for the cytomegalovirus UL18 protein displays a broad specificity for different HLA class I alleles: analysis of LIR-1 + NK cell clones. Int Immunol. 1999;11(1):29–35.
Colonna M, Navarro F, Bellón T, Llano M, García P, Samaridis J, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J exp med. 1997;186(11):1809–18.
Ponte M, Cantoni C, Biassoni R, Tradori-Cappai A, Bentivoglio G, Vitale C, et al. Inhibitory receptors sensing HLA-G1 molecules in pregnancy: decidua-associated natural killer cells express LIR-1 and CD94/NKG2A and acquire p49, an HLA-G1-specific receptor. Proc Natl Acad Sci USA. 1999;96(10):5674–9.
Beldi-Ferchiou A, Lambert M, Dogniaux S, Vély F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget. 2016;7(45):72961–77.
Sivori S, Della Chiesa M, Carlomagno S, Quatrini L, Munari E, Vacca P, et al. Inhibitory Receptors and Checkpoints in Human NK Cells, Implications for the Immunotherapy of Cancer. Front immunol. 2020;11:2156.
Paul S, Lal G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front immunol. 2017;8:1124.
Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128(1):7–15.
Wang R, Jaw JJ, Stutzman NC, Zou Z, Sun PD. Natural killer cell-produced IFN-γ and TNF-α induce target cell cytolysis through up-regulation of ICAM-1. J Leukoc Biol. 2012;91(2):299–309.
Dixon KJ, Wu J, Walcheck B. Engineering anti-tumor monoclonal antibodies and Fc receptors to enhance ADCC by human NK Cells. Cancers. 2021;13(2):312.
Sordo-Bahamonde C, Lorenzo-Herrero S, Payer ÁR, Gonzalez S, López-Soto A. Mechanisms of apoptosis resistance to NK cell-mediated cytotoxicity in cancer. Int J Mol Sci. 2020;21(10):3726.
Wang F, Lau JKC, Yu J. The role of natural killer cell in gastrointestinal cancer: killer or helper. Oncogene. 2021;40(4):717–30.
Ingravallo G, Tamma R, Opinto G, Annese T, Gaudio F, Specchia G, et al. The effect of the tumor microenvironment on lymphoid neoplasms derived from b cells. Diagnostics. 2022;12(3):573.
Fowler NH, Cheah CY, Gascoyne RD, Gribben J, Neelapu SS, Ghia P, et al. Role of the tumor microenvironment in mature B-cell lymphoid malignancies. Haematologica. 2016;101(5):531.
Liu Y, Zhou X, Wang X. Targeting the tumor microenvironment in B-cell lymphoma: challenges and opportunities. J Hematol Oncol. 2021;14(1):1–17.
Menter T, Tzankov A, Dirnhofer S. The tumor microenvironment of lymphomas: Insights into the potential role and modes of actions of checkpoint inhibitors. Hematol Oncol. 2021;39(1):3–10.
Khurana A, Ansell SM. Role of microenvironment in non-Hodgkin lymphoma: understanding the composition and biology. Cancer J. 2020;26(3):206–16.
Ansell SM, Vonderheide RH. Cellular composition of the tumor microenvironment. Am Soc Clin Oncol Educ Book. 2013;33(1):e91–7.
Hay ZLZ, Slansky JE. Granzymes: the molecular executors of immune-mediated cytotoxicity. Int J Mol Sci. 2022;23(3):1833.
Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer. 2021;124(2):359–67.
Ito H, Seishima M. Regulation of the induction and function of cytotoxic T lymphocytes by natural killer T cell. J Biomed Biotechnol. 2010;2010:641757.
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68.
Ye B, Li X, Dong Y, Wang Y, Tian L, Lin S, et al. Increasing LAG-3 expression suppresses T-cell function in chronic hepatitis B: a balance between immunity strength and liver injury extent. Medicine. 2017;96(1):e5275.
Wahlin BE, Sander B, Christensson B, Kimby E. CD8+ T-cell content in diagnostic lymph nodes measured by flow cytometry is a predictor of survival in follicular lymphoma. Clin Cancer Res. 2007;13(2 Pt 1):388–97.
Sallusto F, Lanzavecchia A. Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity. Eur J Immunol. 2009;39(8):2076–82.
Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. 2018;87:1–15.
Walsh KP, Mills KH. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 2013;34(11):521–30.
Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–89.
Ahyi AN, Chang HC, Dent AL, Nutt SL, Kaplan MH. IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J Immunol. 2009;183(3):1598–606.
Jones EA, Pringle JH, Angel CA, Rees RC. Th1/Th2 cytokine expression and its relationship with tumor growth in B cell non-Hodgkin’s lymphoma (NHL). Leuk Lymphoma. 2002;43(6):1313–21.
Andreu-Sanz D, Kobold S. Role and potential of different T helper cell subsets in adoptive cell therapy. Cancers. 2023;15(6):1650.
Yang Z-Z, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest. 2012;122(4):1271–82.
Yang Z-Z, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Malignant B cells skew the balance of regulatory T cells and TH17 cells in B-cell non-Hodgkin’s lymphoma. Cancer res. 2009;69(13):5522–30.
Lu T, Yu S, Liu Y, Yin C, Ye J, Liu Z, et al. Aberrant Circulating Th17 Cells in Patients with B-Cell Non-Hodgkin’s Lymphoma. PLoS One. 2016;11(1):e0148044.
Mintz MA, Cyster JG. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol Rev. 2020;296(1):48–61.
Amé-Thomas P, Le Priol J, Yssel H, Caron G, Pangault C, Jean R, et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia. 2012;26(5):1053–63.
Yang J, Bae H. Drug conjugates for targeting regulatory T cells in the tumor microenvironment: guided missiles for cancer treatment. Exp Mol Med. 2023;55(9):1996–2004.
Yang Z-Z, Liang A-B, Ansell SM. T-cell-mediated antitumor immunity in B-cell non-Hodgkin lymphoma: activation, suppression and exhaustion. Leuk Lymphoma. 2015;56(9):2498–504.
Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front immunol. 2020;11:1731.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96.
van Dalen FJ, van Stevendaal M, Fennemann FL, Verdoes M, Ilina O. Molecular Repolarisation of Tumour-associated macrophages. Molecules. 2018;24(1):9.
Ng WL, Ansell SM, Mondello P. Insights into the tumor microenvironment of B cell lymphoma. J Exp Clin Cancer Res. 2022;41(1):362.
Zhang W, Liu M, Li W, Song Y. Immune cells in the B-cell lymphoma microenvironment: From basic research to clinical applications. Chin Med J. 2024;13:776–90.
Masucci MT, Minopoli M, Carriero MV. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol. 2019;9:1146.
Palano MT, Gallazzi M, Cucchiara M, De Lerma Barbaro A, Gallo D, Bassani B, et al. Neutrophil and Natural Killer Cell Interactions in Cancers: dangerous liaisons instructing immunosuppression and angiogenesis. Vaccines. 2021;9(12):1488.
Garley M, Jabłońska E. Heterogeneity among neutrophils. Arch Immunol Ther Exp. 2018;66(1):21–30.
Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68–77.
Ghaneialvar H, Soltani L, Rahmani HR, Lotfi AS, Soleimani M. Characterization and classification of mesenchymal stem cells in several species using surface markers for cell therapy purposes. Indian J Clin Biochem. 2018;33(1):46–52.
Lee MJ, Park SY, Ko JH, Lee HJ, Ryu JS, Park JW, et al. Mesenchymal stromal cells promote B-cell lymphoma in lacrimal glands by inducing immunosuppressive microenvironment. Oncotarget. 2017;8(39):66281.
Xu B, Wang T. Intimate cross-talk between cancer cells and the tumor microenvironment of B-cell lymphomas: the key role of exosomes. Tumor Biology. 2017;39(6):1010428317706227.
Tian X, Shen H, Li Z, Wang T, Wang S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J Hematol Oncol. 2019;12:1–18.
Yang Z-Z, Grote DM, Ziesmer SC, Manske MK, Witzig TE, Novak AJ, et al. Soluble IL-2Rα facilitates IL-2–mediated immune responses and predicts reduced survival in follicular B-cell non-Hodgkin lymphoma. Blood. 2011;118(10):2809–20.
Yang Z-Z, Grote DM, Ziesmer SC, Xiu B, Yates NR, Secreto FJ, et al. Soluble and membrane-bound TGF-β-mediated regulation of intratumoral T cell differentiation and function in B-cell non-Hodgkin lymphoma. PLoS ONE. 2013;8(3):e59456.
Plonquet A, Haioun C, Jais JP, Debard AL, Salles G, Bene MC, et al. Peripheral blood natural killer cell count is associated with clinical outcome in patients with aaIPI 2–3 diffuse large B-cell lymphoma. Ann Oncol. 2007;18(7):1209–15.
Klanova M, Oestergaard MZ, Trněný M, Hiddemann W, Marcus R, Sehn LH, et al. Prognostic impact of natural killer cell count in follicular lymphoma and diffuse large B-cell lymphoma patients treated with immunochemotherapy. Clin Cancer Res. 2019;25(15):4634–43.
Lin Z, Ma J, Ma Y, Li Q, Kang H, Zhang M, et al. Prognostic impact of peripheral natural killer cells in primary central nervous system lymphoma. Front immunol. 2023;14:1191033.
Baier C, Fino A, Sanchez C, Farnault L, Rihet P, Kahn-Perlès B, et al. Natural killer cells modulation in hematological malignancies. Front immunol. 2013;4:459.
Souza BMB, De Vito FB, Calado ML, Silva MV, Oliveira LR, Rodrigues-Júnior V, et al. Evaluation of the cytotoxic response mediated by perforin and granzyme B in patients with non-Hodgkin lymphoma. Leuk Lymphoma. 2018;59(1):214–20.
Baychelier F, Achour A, Nguyen S, Raphael M, Toubert A, Besson C, et al. Natural killer cell deficiency in patients with non-Hodgkin lymphoma after lung transplantation. J Heart Lung Transplant. 2015;34(4):604–12.
Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother. 2018;67(3):483–94.
Yu L, Liu X, Wang X, Yan F, Wang P, Jiang Y, et al. TIGIT(+) TIM-3(+) NK cells are correlated with NK cell exhaustion and disease progression in patients with hepatitis B virus-related hepatocellular carcinoma. Oncoimmunology. 2021;10(1):1942673.
Danielou-Lazareth A, Henry G, Geromin D, Khaznadar Z, Briere J, Tamouza R, et al. At diagnosis, diffuse large B-cell lymphoma patients show impaired rituximab-mediated NK-cell cytotoxicity. Eur J Immunol. 2013;43(5):1383–8.
Essa ES, Tawfeek GA, El Hassanin SA, Emara KGM. Modulation the expression of natural killer cell activating receptor (NKp44) in the peripheral blood of diffuse large B-cell lymphoma patients and the correlation with clinic pathological features. Clin Immunol. 2018;188:38–44.
Cox MC, Battella S, La Scaleia R, Pelliccia S, Di Napoli A, Porzia A, et al. Tumor-associated and immunochemotherapy-dependent long-term alterations of the peripheral blood NK cell compartment in DLBCL patients. Oncoimmunology. 2015;4(3):e990773.
Seliger B, Koehl U. Underlying mechanisms of evasion from NK cells as rationale for improvement of NK cell-based immunotherapies. Front immunol. 2022;13:910595.
Sato Y, Shimizu K, Shinga J, Hidaka M, Kawano F, Kakimi K, et al. Characterization of the myeloid-derived suppressor cell subset regulated by NK cells in malignant lymphoma. Oncoimmunology. 2015;4(3):e995541.
Wang Y, Wang J, Zhu F, Wang H, Yi L, Huang K, et al. Elevated circulating myeloid-derived suppressor cells associated with poor prognosis in B-cell non-Hodgkin’s lymphoma patients. Immun Inflamm Di. 2022;10(5):e616.
Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182(1):240–9.
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799–807.
Yue J, Li J, Ma J, Zhai Y, Shen L, Zhang W, et al. Myeloid-derived suppressor cells inhibit natural killer cells in myelodysplastic syndromes through the TIGIT/CD155 pathway. Hematology. 2023;28(1):2166333.
Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother. 2010;59(11):1739–44.
Jia H, Yang H, Xiong H, Luo KQ. NK cell exhaustion in the tumor microenvironment. Front immunol. 2023;14:1303605.
Liu X-Q, Lu K, Feng L-L, Ding M, Gao J-M, Ge X-L, et al. Up-regulated expression of indoleamine 2, 3-dioxygenase 1 in non-Hodgkin lymphoma correlates with increased regulatory T-cell infiltration. Leuk Lymphoma. 2014;55(2):405–14.
Ninomiya S, Hara T, Tsurumi H, Hoshi M, Kanemura N, Goto N, et al. Indoleamine 2,3-dioxygenase in tumor tissue indicates prognosis in patients with diffuse large B-cell lymphoma treated with R-CHOP. Ann Hematol. 2011;90(4):409–16.
Yoshikawa T, Hara T, Tsurumi H, Goto N, Hoshi M, Kitagawa J, et al. Serum concentration of L-kynurenine predicts the clinical outcome of patients with diffuse large B-cell lymphoma treated with R-CHOP. Eur j of haematol. 2010;84(4):304–9.
Nuñez SY, Ziblat A, Secchiari F, Torres NI, Sierra JM, Raffo Iraolagoitia XL, et al. Human M2 Macrophages Limit NK Cell Effector Functions through Secretion of TGF-β and Engagement of CD85j. J Immunol. 2018;200(3):1008–15.
Timmins MA, Ringshausen I. Transforming Growth Factor-Beta Orchestrates Tumour and Bystander Cells in B-Cell Non-Hodgkin Lymphoma. Cancers. 2022;14(7):1772.
Regis S, Dondero A, Caliendo F, Bottino C, Castriconi R. NK Cell Function Regulation by TGF-β-Induced Epigenetic Mechanisms. Front immunol. 2020;11:311.
Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A. 2003;100(7):4120–5.
Krneta T, Gillgrass A, Poznanski S, Chew M, Lee AJ, Kolb M, et al. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J Leukoc Biol. 2017;101(1):285–95.
Bladergroen BA, Meijer CJ, ten Berge RL, Hack CE, Muris JJ, Dukers DF, et al. Expression of the granzyme B inhibitor, protease inhibitor 9, by tumor cells in patients with non-Hodgkin and Hodgkin lymphoma: a novel protective mechanism for tumor cells to circumvent the immune system? Blood. 2002;99(1):232–7.
Ramírez-Labrada A, Pesini C, Santiago L, Hidalgo S, Calvo-Pérez A, Oñate C, et al. All About (NK Cell-Mediated) Death in Two Acts and an Unexpected Encore: Initiation, Execution and Activation of Adaptive Immunity. Front immunol. 2022;13:896228.
Kondo E, Yoshino T, Yamadori I, Matsuo Y, Kawasaki N, Minowada J, et al. Expression of Bcl-2 protein and Fas antigen in non-Hodgkin’s lymphomas. Am J Pathol. 1994;145(2):330–7.
Chatzitolios A, Venizelos I, Tripsiannis G, Anastassopoulos G, Papadopoulos N. Prognostic significance of CD95, P53, and BCL2 expression in extranodal non-Hodgkin’s lymphoma. Ann Hematol. 2010;89:889–96.
Zoi-Toli O, Meijer CJ, Oudejans JJ, de Vries E, van Beek P, Willemze R. Expression of Fas and Fas ligand in cutaneous B-cell lymphomas. J Pathol. 1999;189(4):533–8.
Grønbæk K, Straten PT, Ralfkiaer E, Ahrenkiel V, Andersen MK, Hansen NE, et al. Somatic Fas mutations in non-Hodgkin’s lymphoma: association with extranodal disease and autoimmunity. Blood. 1998;92(9):3018–24.
Scholl V, Stefanoff CG, Hassan R, Spector N, Renault IZ. Mutations within the 5′ region of FAS/CD95 gene in nodal diffuse large B-cell lymphoma. Leuk Lymphoma. 2007;48(5):957–63.
Lee SH, Shin MS, Kim HS, Lee HK, Park WS, Kim SY, et al. Somatic mutations of TRAIL-receptor 1 and TRAIL-receptor 2 genes in non-Hodgkin’s lymphoma. Oncogene. 2001;20(3):399–403.
Zhang Y, Liu Q, Yang S, Liao Q. CD58 Immunobiology at a Glance. Front immunol. 2021;12:705260.
Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, et al. Combined genetic inactivation of β2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell. 2011;20(6):728–40.
Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell rep. 2014;6(1):130–40.
Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L. The Immune Checkpoint PD-1 in Natural Killer Cells: Expression, Function and Targeting in Tumour Immunotherapy. Cancers. 2020;12(11):3285.
Park Y, Kim SJ, Lee SJ, Kim BS. Serum level of soluble human leukocyte antigen-G molecules in non-Hodgkin lymphoma: does it have a prognostic value? Leuk Lymphoma. 2008;49(8):1623–6.
de Charette M, Houot R. Hide or defend, the two strategies of lymphoma immune evasion: potential implications for immunotherapy. Haematologica. 2018;103(8):1256–68.
Satwani P, Bavishi S, Saha A, Zhao F, Ayello J, van de Ven C, et al. Upregulation of NKG2D ligands in acute lymphoblastic leukemia and non-Hodgkin lymphoma cells by romidepsin and enhanced in vitro and in vivo natural killer cell cytotoxicity. Cytotherapy. 2014;16(10):1431–40.
Kiyasu J, Miyoshi H, Hirata A, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood. 2015;126(19):2193–01.
Porrata LF. Natural Killer cells are key host immune effector cells affecting survival in autologous peripheral blood hematopoietic stem cell transplantation. Cells. 2022;11(21):3469.
Porrata LF, Burgstaler EA, Winters JL, Jacob EK, Gastineau DA, Suman VJ, et al. Immunologic autograft engineering and survival in non-hodgkin lymphoma. Biol Blood Marrow Transplant. 2016;22(6):1017–23.
Kansagra A, Inwards DJ, Ansell SM, Micallef IN, Johnston PB, Hogan WJ, et al. Infusion of autograft natural killer cell/CD14(+)HLA-DR(DIM) cell ratio predicts survival in lymphoma post autologous stem cell transplantation. Bone Marrow Transplant. 2018;53(2):146–54.
Chu Y, Lamb M, Cairo MS, Lee DA. The future of natural killer cell immunotherapy for B Cell Non-Hodgkin Lymphoma (B Cell NHL). Curr Treat Options Oncol. 2022;23(3):381–403.
Porrata LF, Inwards DJ, Ansell SM, Micallef IN, Johnston PB, Gastineau DA, et al. Early lymphocyte recovery predicts superior survival after autologous stem cell transplantation in non-Hodgkin lymphoma: a prospective study. Biol Blood Marrow Transplant. 2008;14(7):807–16.
Porrata LF. Clinical evidence of autologous graft versus tumor effect. Am J Immunology. 2009;5(1):1–7.
Raspadori D, Lauria F, Ventura MA, Tazzari PL, Ferrini S, Miggiano MC, et al. Low doses of rIL2 after autologous bone marrow transplantation induce a “prolonged” immunostimulation of NK compartment in high-grade non-Hodgkin’s lymphomas. Ann Hematol. 1995;71(4):175–9.
Miller JS, Tessmer-Tuck J, Pierson BA, Weisdorf D, McGlave P, Blazar BR, et al. Low dose subcutaneous interleukin-2 after autologous transplantation generates sustained in vivo natural killer cell activity. Biol Blood Marrow Transplant. 1997;3(1):34–44.
Margolin KA, Aronson FR, Sznol M, Atkins MB, Ciobanu N, Fisher RI, et al. Phase II trial of high-dose interleukin-2 and lymphokine-activated killer cells in Hodgkin’s disease and non-Hodgkin’s lymphoma. J Immunother. 1991;10(3):214–20.
Lister J, Rybka WB, Donnenberg AD, deMagalhaes-Silverman M, Pincus SM, Bloom EJ, et al. Autologous peripheral blood stem cell transplantation and adoptive immunotherapy with activated natural killer cells in the immediate posttransplant period. Clinical Cancer res. 1995;1(6):607–14.
Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front immunol. 2019;10:1205.
Koepsell SA, Miller JS, McKenna DH Jr. Natural killer cells: a review of manufacturing and clinical utility. Transfusion. 2013;53(2):404–10.
Yang Y, Lim O, Kim TM, Ahn YO, Choi H, Chung H, et al. Phase I Study of Random Healthy Donor-Derived Allogeneic Natural Killer Cell Therapy in Patients with Malignant Lymphoma or Advanced Solid Tumors. Cancer Immunol Res. 2016;4(3):215–24.
Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1.
Tang L, Huang Z, Mei H, Hu Y. Immunotherapy in hematologic malignancies: achievements, challenges and future prospects. Signal Transduct Target Ther. 2023;8(1):306.
Deng X, Terunuma H, Nieda M, Xiao W, Nicol A. Synergistic cytotoxicity of ex vivo expanded natural killer cells in combination with monoclonal antibody drugs against cancer cells. Int Immunopharmacol. 2012;14(4):593–605.
Pinto S, Pahl J, Schottelius A, Carter PJ, Koch J. Reimagining antibody-dependent cellular cytotoxicity in cancer: the potential of natural killer cell engagers. Trends Immunol. 2022;43(11):932–46.
Walcheck B, Wu J. iNK-CD64/16A cells: a promising approach for ADCC? Expert Opin Biol Ther. 2019;19(12):1229–32.
Huo Z, Chen F, Zhao J, Liu P, Chao Z, Liu K, et al. Prognostic impact of absolute peripheral blood NK cell count after four cycles of R-CHOP-like regimen treatment in patients with diffuse large B cell lymphoma. Clin Exp Med. 2023;23(8):4665–72.
Tanaka J. Recent advances in cellular therapy for malignant lymphoma. Cytotherapy. 2021;23(8):662–71.
Capuano C, Pighi C, Battella S, De Federicis D, Galandrini R, Palmieri G. Harnessing CD16-Mediated NK cell functions to enhance therapeutic efficacy of tumor-targeting mAbs. Cancers. 2021;13(10):2500.
Tanaka J, Tanaka N, Wang YH, Mitsuhashi K, Ryuzaki M, Iizuka Y, et al. Phase I study of cellular therapy using ex vivo expanded natural killer cells from autologous peripheral blood mononuclear cells combined with rituximab-containing chemotherapy for relapsed CD20-positive malignant lymphoma patients. Haematologica. 2020;105(4):e190–3.
Yoon DH, Koh Y, Jung M, Kwak JE, Shin EC, Hwang YK, et al. Phase I study: safety and efficacy of an ex vivo-expanded allogeneic natural killer cell (MG4101) with rituximab for relapsed/refractory B Cell Non-Hodgkin Lymphoma. Transplant Cell Ther. 2023;29(4):253.e1-e9.
Khanal R, Mehta A, Maly JJ, Holmes H, Saultz JN, Hamdan A, et al. AB-101, an Allogeneic, Non-Genetically Modified, Natural Killer (NK) Cell Therapy, Evaluated as Monotherapy or in Combination with Rituximab in R/R Non-Hodgkin Lymphoma. J Clin Oncol. 2023;41:7529.
Cichocki F, Zhang B, Wu C-Y, Chiu E, Day A, O’Connor RS, et al. Nicotinamide enhances natural killer cell function and yields remissions in patients with non-Hodgkin lymphoma. Sci Transl Med. 2023;15(705):eade3341.
Nieto Y, Banerjee P, Kaur I, Kim KH, Fang D, Thall PF, et al. Ex Vivo Expanded Cord Blood Natural Killer Cells Combined with Rituximab and High-Dose Chemotherapy and Autologous Stem Cell Transplantation for B Cell Non-Hodgkin Lymphoma. Transplant Cell Ther. 2024;30(2):203.e1-e9.
Patel K, Bachanova V, Goodman AM, Pagel JM, Griffis K, Anderson M, et al. Phase I study of FT516, an off-the-shelf iPSC-derived NK cell therapy, in combination with rituximab in patients with relapsed/refractory B-cell lymphoma. Blood. 2021;138:3873.
Nieto Y, Banerjee P, Kaur I, Griffin L, Barnett M, Ganesh C, et al. Innate Cell Engager (ICE®) AFM13 Combined with Preactivated and Expanded (P+ E) Cord Blood (CB)-Derived Natural Killer (NK) Cells for Patients with Refractory CD30-Positive Lymphomas: Final Results. Blood. 2023;142(Supplement 1):774.
Mamo T, Williams SM, Kinney S, Tessier KM, DeFor TE, Cooley S, et al. Infusion reactions in natural killer cell immunotherapy: a retrospective review. Cytotherapy. 2021;23(7):627–34.
Lamers-Kok N, Panella D, Georgoudaki AM, Liu H, Özkazanc D, Kučerová L. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J Hematol Oncol. 2022;15(1):164.
Williams BA, Law AD, Routy B, denHollander N, Gupta V, Wang XH, et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget. 2017;8(51):89256–68.
Lin C, Horwitz ME, Rein LAM. Leveraging natural killer cell innate immunity against hematologic malignancies: from stem cell transplant to adoptive transfer and beyond. Int J Mol Sci. 2022;24(1):204.
Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy - advantages of the NK-92 cell line over blood NK Cells. Front immunol. 2016;7:91.
Ramos-Mejia V, Arellano-Galindo J, Mejía-Arangure JM, Cruz-Munoz ME. A NK cell odyssey: from bench to therapeutics against hematological malignancies. Front immunol. 2022;13:803995.
Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10(6):625–32.
Fabian KP, Hodge JW. The emerging role of off-the-shelf engineered natural killer cells in targeted cancer immunotherapy. Mol Ther Oncolytics. 2021;23:266–76.
Boissel L, Klingemann H, Campbell K, Nichols K, Toneguzzo F, Marcus P, et al. An ‘off the shelf’,GMP-grade, IL-2-independent NK cell line expressing the high-affinity Fc-receptor to augment antibody therapeutics. Cancer res. 2016;76(14_Supplement):2302-.
Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM 2nd, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. 2016;7(52):86359–73.
Shankar K, Capitini CM, Saha K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res Ther. 2020;11(1):234.
Kistler M, Nangia C, To C, Sender L, Lee J, Jones F, et al. Abstract P5–04–02: Safety and efficacy from first-in-human immunotherapy combining NK and T cell activation with off-the-shelf high-affinity CD16 NK cell line (haNK) in patients with 2nd-line or greater metastatic triple-negative breast cancer (TNBC). Cancer res. 2020;80(4_Supplement):P5-04-2-P5--2.
Chen X, Jiang L, Liu X. Natural killer cells: the next wave in cancer immunotherapy. Front immunol. 2022;13:954804.
Berrien-Elliott MM, Jacobs MT, Fehniger TA. Allogeneic natural killer cell therapy. Blood. 2023;141(8):856–68.
Lin X, Sun Y, Dong X, Liu Z, Sugimura R, Xie G. IPSC-derived CAR-NK cells for cancer immunotherapy. Biomed Pharmacother. 2023;165:115123.
Fate TA. Encouraging Interim, Phase 1 Data for iPSC-Derived NK Cell, Programs in Relapsed/Refractory, Acute Myeloid, Leukemia. https://ir.fatetherapeutics.com/news-releases/news-release-details/fate-therapeutics-announces-encouraging-interim-phase-1-data
Gutierrez M, Patel M, Liu F, Szabo P, Valamehr B, Chu Y-W, et al. 726 Phase I results of FT516, an off-the-shelf, iPSC-derived NK cell therapy expressing a high-affinity, non-cleavable CD16 (hnCD16) combined with avelumab in patients with advanced solid tumors. BMJ Specialist Journals. 2022.
Fate therapeutics highlights positive durability of response data from FT516 Phase 1 study for B-cell lymphoma and announces FDA regen erative medicine advanced therapy designation granted to FT516 for relapsed/refractory DLBCL. Available from: https://ir.fatetherapeutics.com/news-releases/news-release-details/fate-therapeutics-highlights-positive-durability-response-data.
Allen C, Zeidan AM, Bewersdorf JP. BiTEs, DARTS, BiKEs and TriKEs-are antibody based therapies changing the future treatment of AML? Life. 2021;11(6):465.
Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. Methods Mol Biol. 2016;1441:333–46.
Moore GL, Bautista C, Pong E, Nguyen DH, Jacinto J, Eivazi A, et al. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs. 2011;3(6):546–57.
Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126–36.
Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–33.
Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–87.
Moskowitz A, Harstrick A, Emig M, Overesch A, Pinto S, Ravenstijn P, et al. AFM13 in Combination with Allogeneic Natural Killer Cells (AB-101) in Relapsed or Refractory Hodgkin Lymphoma and CD30+ Peripheral T-Cell Lymphoma: A Phase 2 Study (LuminICE). Blood. 2023;142:4855.
Bartlett NL, Chen RW, Domingo-Domenech E, Forero-Torres A, Garcia-Sanz R, Armand P, et al. A phase 1b study investigating the combination of the tetravalent bispecific NK cell engager AFM13 and pembrolizumab in patients with relapsed/refractory Hodgkin lymphoma after brentuximab vedotin failure: updated safety and efficacy data. Blood. 2018;132:1620.
Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K, et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs. 2014;6(3):728–39.
Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015;125(26):4024–31.
Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3(4):356–67.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27.
Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. 2017;5:22.
Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology. 2019;8(5):e1049.
Buller CW, Mathew PA, Mathew SO. Roles of NK Cell Receptors 2B4 (CD244), CS1 (CD319), and LLT1 (CLEC2D) in Cancer. Cancers. 2020;12(7):1755.
Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol (Baltimore, Md : 1950). 2015;194(7):3201–12.
Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat Immunol. 2003;4(6):557–64.
Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther. 2019;27(6):1114–25.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83.
Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, et al. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma. 2012;53(5):958–65.
Schmidt P, Raftery MJ, Pecher G. Engineering NK Cells for CAR Therapy-Recent Advances in Gene Transfer Methodology. Front immunol. 2020;11:611163.
Hudecek M, Ivics Z. Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr Opin Genet Dev. 2018;52:100–8.
Kim A, Pyykko I. Size matters: versatile use of PiggyBac transposons as a genetic manipulation tool. Mol Cell Biochem. 2011;354(1–2):301–9.
Hu Y, Tian ZG, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. 2018;39(2):167–76.
Hay KA. Cytokine release syndrome and neurotoxicity after CD19 chimeric antigen receptor-modified (CAR-) T cell therapy. Br J Haematol. 2018;183(3):364–74.
Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147.
Zhang L, Chu J, Yu J, Wei W. Cellular and molecular mechanisms in graft-versus-host disease. J Leukoc Biol. 2016;99(2):279–87.
Ingegnere T, Mariotti FR, Pelosi A, Quintarelli C, De Angelis B, Tumino N, et al. Human CAR NK Cells: a new non-viral method allowing high efficient transfection and strong tumor cell killing. Front immunol. 2019;10:957.
Geller MA, Miller JS. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy. 2011;3(12):1445–59.
Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J, et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19(+) B cell tumors: a phase 1/2 trial. Nat Med. 2024;30(3):772–84.
Dickinson M, Hamad N, Bryant C, Kothari N, Ojeras P, Vohra A, et al. First in human data of NKX019, an allogeneic CAR NK for the treatment of relapsed/refractory (R/R) B-cell malignancies. Hematol Oncol. 2023;41(S2):526–7.
Ramachandran I, Rothman S, Clausi M, Mcfadden K, Salantes B, Jih G, et al. Multiple Doses of Cnty-101, an iPSC-Derived Allogeneic CD19 Targeting CAR-NK Product, are safe and result in tumor microenvironment changes associated with response: a case study. Blood. 2023;142:1654.
Bachanova V, Ghobadi A, Patel K, Park JH, Flinn IW, Shah P, et al. Safety and efficacy of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood. 2021;138:823.
Goodridge JP, Mahmood S, Zhu H, Gaidarova S, Blum R, Bjordahl R, et al. FT596: translation of first-of-kind multi-antigen targeted off-the-shelf CAR-NK cell with engineered persistence for the treatment of B cell malignancies. Blood. 2019;134:301.
Dickinson M, Hamad N, Bryant CE, Borthakur G, Hosing C, Shook D, Tan J, Rajangam K, Liu H, Kennedy GA, McSweeney PA. Phase 1 Study of NKX019, a CD19 Chimeric Antigen Receptor Natural Killer (CAR NK) Cell Therapy, in Subjects with B-cell Malignancies. Blood. 2021;138:3868.
Terrén I, Orrantia A, Astarloa-Pando G, Amarilla-Irusta A, Zenarruzabeitia O, Borrego F. Cytokine-Induced Memory-Like NK Cells: From the Basics to Clinical Applications. Front immunol. 2022;13:884648.
Ghofrani J, Lucar O, Dugan H, Reeves RK, Jost S. Semaphorin 7A modulates cytokine-induced memory-like responses by human natural killer cells. Eur J Immunol. 2019;49(8):1153–66.
Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, et al. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. 2006;24(5):575–90.
Terrén I, Orrantia A, Mosteiro A, Vitallé J, Zenarruzabeitia O, Borrego F. Metabolic changes of Interleukin-12/15/18-stimulated human NK cells. Sci Rep. 2021;11(1):6472.
Leong JW, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA, et al. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transplant. 2014;20(4):463–73.
Tarannum M, Romee R. Cytokine-induced memory-like natural killer cells for cancer immunotherapy. Stem Cell Res Ther. 2021;12(1):592.
Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123.
Shapiro RM, Birch GC, Hu G, Vergara Cadavid J, Nikiforow S, Baginska J, et al. Expansion, persistence, and efficacy of donor memory-like NK cells infused for posttransplant relapse. J Clin Invest. 2022;132(11):e154334.
Bednarski JJ, Zimmerman C, Berrien-Elliott MM, Foltz JA, Becker-Hapak M, Neal CC, et al. Donor memory-like NK cells persist and induce remissions in pediatric patients with relapsed AML after transplant. Blood. 2022;139(11):1670–83.
Boieri M, Ulvmoen A, Sudworth A, Lendrem C, Collin M, Dickinson AM, et al. IL-12, IL-15, and IL-18 pre-activated NK cells target resistant T cell acute lymphoblastic leukemia and delay leukemia development in vivo. Oncoimmunology. 2017;6(3):e1274478.
Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J exp med. 2012;209(13):2351–65.
Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136(20):2308–18.
Lee DA. Cellular therapy: Adoptive immunotherapy with expanded natural killer cells. Immunol Rev. 2019;290(1):85–99.
Lapteva N, Szmania SM, van Rhee F, Rooney CM. Clinical grade purification and expansion of natural killer cells. Crit Rev Oncog. 2014;19(1–2):121–32.
Fang F, Wang W, Chen M, Tian Z, Xiao W. Technical advances in NK cell-based cellular immunotherapy. Cancer Biol Med. 2019;16(4):647–54.
Gurney M, Kundu S, Pandey S, O’Dwyer M. Feeder Cells at the Interface of Natural Killer Cell Activation. Expansion and Gene Editing Front immunol. 2022;13:802906.
Granzin M, Soltenborn S, Müller S, Kollet J, Berg M, Cerwenka A, et al. Fully automated expansion and activation of clinical-grade natural killer cells for adoptive immunotherapy. Cytotherapy. 2015;17(5):621–32.
Granzin M, Stojanovic A, Miller M, Childs R, Huppert V, Cerwenka A. Highly efficient IL-21 and feeder cell-driven ex vivo expansion of human NK cells with therapeutic activity in a xenograft mouse model of melanoma. Oncoimmunology. 2016;5(9):e1219007.
Bae DS, Lee JK. Development of NK cell expansion methods using feeder cells from human myelogenous leukemia cell line. Blood research. 2014;49(3):154–61.
Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE. 2012;7(1):e30264.
Johnson CDL, Zale NE, Frary ED, Lomakin JA. Feeder-Cell-Free and Serum-Free Expansion of Natural Killer Cells Using Cloudz Microspheres, G-Rex6M, and Human Platelet Lysate. Front immunol. 2022;13:803380.
Gluck WL, Hurst D, Yuen A, Levine AM, Dayton MA, Gockerman JP, et al. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clinical Cancer res. 2004;10(7):2253–64.
Acknowledgements
The authors would like to express their gratitude to Shahid Beheshti University of Medical Sciences (Tehran, Iran) for supporting this study.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MB: literature and clinical trial searches, conceptualization, writing – original draft, design of figures, tables preparations and editing. SY: writing – original draft, tables preparations and editing. SK: writing – original draft and editing. FAS, MS, ASJ and FD: writing – original draft. SMS: writing – original draft and editing. AI and SP: Editing of the final manuscript. AG, ER and AH: Supervision and validation. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bakhtiyaridovvombaygi, M., Yazdanparast, S., Kheyrandish, S. et al. Harnessing natural killer cells for refractory/relapsed non-Hodgkin lymphoma: biological roles, clinical trials, and future prospective. Biomark Res 12, 66 (2024). https://doi.org/10.1186/s40364-024-00610-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-024-00610-z