
Abstract
Wnt signaling was initially recognized to be vital for tissue development and homeostasis maintenance. Further studies revealed that this pathway is also important for tumorigenesis and progression. Abnormal expression of signaling components through gene mutation or epigenetic regulation is closely associated with tumor progression and poor prognosis in several tissues. Additionally, Wnt signaling also influences the tumor microenvironment and immune response. Some strategies and drugs have been proposed to target this pathway, such as blocking receptors/ligands, targeting intracellular molecules, beta-catenin/TCF4 complex and its downstream target genes, or tumor microenvironment and immune response. Here we discuss the roles of these components in Wnt signaling pathway in tumorigenesis and cancer progression, the underlying mechanisms that is responsible for the activation of Wnt signaling, and a series of drugs targeting the Wnt pathway provide multiple therapeutic values. Although some of these drugs exhibit exciting anti-cancer effect, clinical trials and systematic evaluation should be strictly performed along with multiple-omics technology.
Similar content being viewed by others

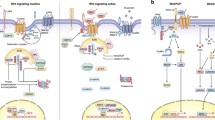

Background
The Wnt signaling cascade is critical for tissue morphogenesis, homeostasis, and regeneration. Wnt signaling activation can go through the canonical or non-canonical pathway depending on the association with the transcription activator beta-catenin. Following the stimulation of the canonical pathway, beta-catenin is translocated from the cytoplasm to the nucleus [1]. By contrast, under the unstimulated condition, the levels of beta-catenin are relatively low in the cytosol due to the degradation complexes consisting of adenomatous polyposis coli (APC), glycogen synthase kinase-3β (GSK-3β), Axin, casein kinase 1(CK1). Degradation of beta-catenin is initiated by phosphorylation via GSK-3β and CK1, followed by β-transducin repeat-containing protein (β-TrCP) E3 ligase mediated ubiquitination [2], and the location and roles of components in the Wnt singalling pathway is summarized in Table 1. The non-canonical pathway includes several cascades, such as Wnt/planar cell polarity signaling, Wnt-cGMP/Ca2+ signaling and Wnt-RAP1 signaling [3]. Studies have shown that the non-canonical pathway plays a role in multiple physiological activities, including maintenance of stem cells in their niche [4].
Wnt signaling also plays significant roles in tumorigenesis and progression. Dysregulated Wnt signaling activity promotes malignant transformation of stem/progenitor cell, leading to increased cell proliferation and abnormal differentiation. Wnt signaling also crosstalks with other signaling pathways (e.g. Hedgehog (Hh), Notch) to synergistically regulate tumor progression [5,6,7]. Abnormal Wnt signaling is associated with poor survival, stress responses, and drug resistance [8, 9]. In addition, canonical Wnt signaling modulates immune cell infiltration in tumors, rendering Wnt signaling a potential immunotherapy target [10]. Here, we review the role of Wnt signaling in cancer development in associated organs, we also review findings on targeting Wnt signaling for cancer therapy.
Aberrant expression of Wnt signaling components during cancer development
Abnormal activation of Wnt signaling cascade is associated with tumorigenesis in several tissues including the esophagus and liver. Canonical Wnt signaling has been shown to promote the self-renewal of cancer stem cells (CSCs) [11, 12]. Esophageal squamous cell carcinoma (ESCC) and adenocarcinoma (EAC) are the two major cancer types of the esophagus. Dysregulated Wnt signaling was found in a significant number of cases of ESCC [13]. For example, WNT2 secreted by tumor fibroblasts promotes tumor cell proliferation and invasion via canonical signaling, and Wnt2-positive ESCC is correlated with lymph-node metastases [14]. In addition, beta-catenin expression was present in a heterogeneous pattern with prominent enrichment in the cell membrane of ESCC samples [15]. Moreover, the transcript and protein levels of beta-catenin and Wnt1 are elevated in ESCC carcinoma cells as compared to the neighboring normal tissues [16]. High levels of Wnt1 and beta-catenin predict lymph node metastasis, advanced pathological stage, and poor prognosis of patients. When combined with Bmi-1, Wnt1 and beta-catenin indicate a relatively worse prognosis [16]. Additionally, hypermethylation of Wnt antagonists/inhibitors including RUNX-3, DKK-3 (Dickkopf-3), and SFRP1 (Secreted Frizzled Related Protein 1) is associated with an elevated chance of ESCC recurrence. Thus, hypermethylation of the promoter of Wnt antagonists/inhibitors can potentially serve as a candidate indicator for ESCC treatment resistance [17].
Wnt signaling influences tumor growth at multiple levels. In squamous cell carcinoma (SCC) cell lines, beta-catenin is a key regulator for the expression of DeltaNp63 [18], a major isoform of p63 which is critical for basal cell proliferation and SCC development [19, 20]. Genes that contribute to regulation of Wnt family members are also associated with cancer progression. For example, Msi2 is a transcriptional regulator that regulates genes involved in development and cell cycle regulation [21, 22]. Overexpression of MSI2 enhances the activities of Wnt/beta-catenin and Hh signaling cascades, leading to increased proliferation of ESCC, epithelial-mesenchymal transition (EMT) and cell migration [23]. The expression levels of Msi2 are correlated with tumor size, differentiation, and lymph-node metastasis in patients with ESCC. Therefore, Msi2 is considered as an independent predictor for overall survival and disease-free survival [23]. In another study, organotypic 3D culture of primary human ESCC was used to show invasive cancer cells exhibiting activation of cyclin D1 and Wnt signaling [24]. Further study using dominant negative Mastermind-like1 to inhibit Notch signaling demonstrates Wnt signaling activation is Notch-independent [24].
In addition, inactivation of Wnt/beta-catenin and PI3K/AKT through depletion of Ras GTPase-activating protein SH3 domain-binding protein 1 (G3BP1) attenuates the proliferative, invasive and migration potential of esophageal carcinoma cells [25]. Wnt signaling can also be modulated by microRNA. For example, Cir-ITCH is a circRNA molecule that targets miR-214, miR-17, and miR-7. Cir-ITCH inhibits cell growth and proliferation of ESCC by enhancing the ITCH expression and inhibiting canonical Wnt signaling [26]. Interestingly, the transcript and protein levels of Dickkopf-1 (DKK1) that attenuate Wnt signaling activities, are up-regulated in esophageal cancerous tissues. Ectopic expression of DKK1 promotes cancer cell proliferation and invasion, although the underlying mechanisms remain to be determined [27]. Additionally, Naked cuticle homolog 2 (NKD2) is commonly methylated in gastric and breast carcinoma. Completely lost or reduced expression of NKD2 through methylation has also been observed in multiple ESCC cell lines and clinical samples. Further study showed that NKD2 attenuates the development of esophageal carcinoma through suppressing Wnt signaling in-vitro and in-vivo, suggesting that NKD2 methylation can potentially be used as a prognostic marker in esophageal carcinoma [28].
Abnormal Wnt signaling is also associated with tumorigenesis of EAC. For example, the mRNA levels of several members of the pathway are frequently altered during the progression of Barrett’s esophagus (BE) towards EAC. Changes in the methylation levels of the APC promoter were found in BE samples and 95% of EAC cases [29]. Methylation of SFRP1, which interacts with Wnts, followed by inhibition of the signaling activation was also detected in BE samples and EAC [29]. In EAC, beta-catenin nuclear-translocation was detected irrespective of APC expression; the upregulation of the WNT2 gene was found along with low-grade dysplasia to EAC [29].
Multiple mechanisms underlie the stimulation of the Wnt/beta-catenin signaling cascade in cancer progression
Activation of the Wnt/beta-catenin signaling cascade is driven by gene mutations
Mutations of CTNNB1 encoding beta-catenin are closely correlated with multiple cancers including hepatocarcinoma, pancreatic cancer, colorectal cancer (CRC), gastroesophageal junction carcinomas and gastric adenocarcinoma. The mutation of p.S45F in beta-catenin is a common mutation, leading to constitutively activated Wnt/beta-catenin signaling. This mutation has been found in a variety of solid tumors as a potential driver mutation, accounting for 3.3–10.4% of the total identified beta-catenin mutations [30]. Moreover, the somatic mutational profiling of 16 genes was analyzed in primary hepatocellular cancer. Somatic mutations occur in TP53 (33%) and CTNNB1 (22%) genes in 55% of the samples as identified through targeted deep sequencing. One protein, CTNNB1(H36P) encoded by the mutated CTNNB1 gene, enables resistance to the degradation of proteins and promotes cell proliferation. Of note is that mutations of TP53 and WNT/beta-catenin signaling cascades co-exist in hepatocellular carcinoma (HCC) [31]. In addition, CTNNB1 mutations are the only actionable genomic lesions in solid pseudopapillary neoplasms relative to other common subtypes of pancreatic tumors [32]. In animal models, mutations in CTNNB1 (exon 3) are predominant events among mutations of multiple genes in colon tumors induced by 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP) and dextrin sulfate sodium (DSS) in humanized CYP1A mice, one type of mice possessing human CYP1A gene to replace mouse Cyp1a gene. Whole exome sequencing revealed mutations on either codon 32 or 34 of exon 3 of the CTNNB1 gene in 39 out of 42 tumors. However, no mutations were detected in either APC or KRAS, suggesting that mutated CTNNB1 is the driver in colon cancer development induced by PhIP/DSS [33]. Besides cancers described above, driver mutations in CTNNB1 gene are also found in other types of cancer including lung cancer [34], endometrioid endometrial carcinoma [35], and mucosal melanomas [36] using whole-exome sequencing analysis and whole-genome sequencing analysis.
Mutation of APC is another important driver for tumor formation. Sixteen recurrently mutated genes were analyzed in 98 advanced CRC patients by next-generation sequencing. Multiple correspondence analysis (MCA) suggested that APC and TP53 mutations are close to the negative outlier group, whereas mutations in other members of WNT signaling are in proximity to the positive outliers. Moreover, patients with tumors harboring TP53mut/APCmut/AMER1wt/TCF7L2wt/FBXW7wt and TP53mut/APCmut/AMER1wt/TCF7L2wt/FBXW7wt/SOX9wt/CTNNB1wt genotypes had shorter progression-free survival. These two signatures are negatively correlated with the overall survival of CRC [37]. In another study, the combined effects of APC and BRAF mutation were tested in mice to determine the mutational landscape of WNT signaling regulators in BRAF mutant cancers. The results show that RNF43 gene is the most mutated WNT signaling regulator (41%), and mutations in the beta-catenin destruction complex are present in 48% of human CRC. 20.8% of CRC cases presented a truncation mutation in APC which is associated with early onset of tumor, advanced stage, and poor prognosis. Double mutations in APC and BRAF are associated with poorer prognosis than an individual mutation. These results suggest that the WNT signaling pathway is commonly mutated in CRC with BRAF mutation. Furthermore, WNT16 and MEN1 may drive aberrant WNT signaling in CRC [38].
Normal human intestinal epithelium-derived organoids have been used to study mutations of Wnt components and other genes associated with tumor. For example, organoids containing mutations of the five genes including SMAD4, APC, TP53, KRAS and/or PIK3CA, can grow independently of niche factors. These organoids also form tumors after implantation under the kidney capsule. Moreover, these organoids give rise to micrometastases post-injection into the mouse spleen, but fail to colonize in the liver [39]. Mouse models possessing numerous combined driver mutations including APC and other genes (KRAS, TRP53, TGFBR2 and FBXW7) have been generated for studying their contribution to the progression of intestinal tumors. Mutation of APC (∆716) causes intestinal adenomas and induces submucosal invasion when combined with TRP53 (R270H) mutation or a TGFBR2 deletion. Addition of the KRAS (G12D) mutation promotes EMT-like morphology and lymph vessel intravasation of the invasive tumors. By contrast, APC (∆716) combined with KRAS (G12D) and FBXW7 mutations induced EMT-like histology but failed to produce submucosal invasion. A combination of KRAS (G12D) with either APC (∆716) plus TRP53 (R270H) or TGFBR2 deletion caused highest incidence of liver metastasis with a genotype of APC (∆716) KRAS(G12D) TGFBR2 (−/−). These findings recapitulate the up-regulated genes observed in human metastatic CRC [40].
The Wnt antagonists SFRP1, SFRP2, DKK2 and Wnt inhibitory factor-1 (WIF-1) are hypermethylated during the transition from colorectal adenoma to carcinoma, whereas mutations in BRAF, APC, and KRAS occur at the transition from normal to adenoma phases, these events may drive the colorectal cancer formation [41]. Among 145 mutations within 31 genes detected in gastroesophageal junction carcinomas and gastric adenocarcinoma, mutations in APC and CTNNB1 are more prevalent among gastric carcinomas, with more than three driver mutations detected in gastric carcinomas. However, TP53 mutations are the most prevalent abnormalities that were detected, especially in gastroesophageal junction cancer [42]. Notably, mutations in either APC or CTNNB1 were also found in colon cancer, leading to increased activity of beta-catenin-Tcf signaling [43].
RNF43 is a negative regulator of Wnt signaling, and mutation of RNF43 is frequently found in CRC and endometrial cancers [38, 44], gastric cancer [45], neoplastic cysts of the pancreas including intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms (MCNs) [46, 47]. Most mutation in RNF43 are truncation mutation with a higher mutation rate in the MSI subtype than in the MSS subtype [45]. Increased Wnt signaling upon the inactivated mutation of RNF43 is key for the transformation phenotype in pancreatic cancer cell lines. Consistently, tumors with an inactivating mutation of RNF43 are more sensitive to Wnt inhibitors, which therefore can be potentially developed as a therapy [48]. TP53 is often mutated in colitis-associated cancer (CAC), while SMAD4, KRAS, and APC have not been commonly mutated. RNF43 is also somatically mutated in 11% of CACs that have been linked with chronic inflammation and long-lasting inflammatory bowel disease (IBD). Many CACs with mutated APC are sporadic colorectal cancers. The expression level of c-Myc along with its target genes is elevated in RNF43-mutated CACs, suggesting that RNF43 is an important CAC driver. Somatic mutations of RNF43 result in genetic variation and are associated with chronic inflammatory process and progression of carcinoma in around 10% of CACs [49]. In another study, mutations of RNF43 were found in 0–17% samples, while other members of Wnt signaling in gastric cancer include CTNNB1 (3.3–9.1%) and APC (3.3–14.9%) [50]. RNF43 is also a tumor suppressor gene in mucinous tumors of the ovary, and RNF43 mutation was found in 21% of mucinous ovarian carcinomas [51]. By contrast, the mutation rates of APC and CTNNB1 are 45.8 to 90.6% and 5 to 7.2% in CRC based on TCGA database, respectively [52, 53].
Mutations of other Wnt family members including ZNRF3 and AXIN. Mutation of ZNRF3, a Wnt signaling suppressor, is often found in adrenocortical carcinoma [54], while AXIN2 mutations induce a predisposition to colorectal cancer [55], its mutations are detected in colorectal cancer, finally leading to the accumulation of beta-catenin in the nuclei [56]. By contrast, AXIN1 mutation is found in HCC, overexpression of wild-type AXIN1 induces apoptosis in HCC and CRC cells with accumulated beta-catenin [57]. General driver mutations of the Wnt cascade in tumorigenesis are listed in Table 2 and Fig.1.

The distribution map of driver mutations on multiple human tissues within Wnt signaling
Wnt/beta-catenin signaling activation is caused by attenuating its inhibitor or via epigenetic modifications
Activation of the Wnt/beta-catenin cascade can also be obtained through suppressing its inhibitor. For example, expression of HOTAIR and secreted Wnt antagonist WIF-1 is inversely correlated in ESCC cell lines and tumor tissues. HOTAIR lowers the expression of WIF-1 by enhancing the methylation of H3K27 (one type of histone of WIF-1) at its promoter site, thereby triggering the activation of Wnt/beta-catenin signaling pathway [58]. Furthermore, the methylation levels of SOX17 (a Wnt signaling suppressor) increase along with the progression of esophageal cancer. SOX17 methylation is also associated with a history of alcohol consumption in esophageal cancer patients [59]. SOX17 methylation leads to the loss of SOX17 expression accompanied by increased expression of beta-catenin and redistribution. Re-expression of SOX17 via exposing 5-aza-2′-deoxycytidine attenuates TCF/beta-catenin-dependent transcription and proliferation of esophageal cancer cell lines. Conversely, SOX17 loss removes the normal inhibition of WNT signaling and enhances esophageal tumorigenesis. MicroRNA 141 can also downregulate the expression of SOX17 and stimulate the WNT signaling cascade [59].
Epigenetic regulation of the WIF-1 is also involved in gastro-intestinal tumorigenesis. Downregulation of WIF-1 caused by CpG island hypermethylation in the promoter region has been found in multiple cancer cell lines and esophageal, gastric, colorectal and pancreatic cancer tissues [60, 61]. Combined treatment of 5-aza-dC with the histone deacetylase inhibitor trichostatin A restores WIF-1 expression, and restoration of WIF-1 inhibits colony formation, cellular proliferation, and anchorage-independent growth of TE-1 ESCC cells or SW48 colon cancer cells [60]. In addition, elevated levels of WIF-1 promoter methylation occurred in EAC tissue samples compared to their matched normal epithelium. Hypermethylation of WIF-1 is more common in BE samples obtained from patients with EAC than in BE samples obtained from patients without EAC, suggesting that WIF-1 silencing caused by promotor hypermethylation underlies the progression of BE towards EAC. Consistently, restoration of WIF-1 in EAC cell lines suppresses the growth of the cells and sensitizes these cells to cisplatin [62].
Epigenetic inactivation of WIF-1 with promoter methylation also existing in ESCC cell lines and tissues. WIF-1 promoter methylation is commonly found in 46% ESCC tissues and 50% cell lines. Removal of promoter methylation with a demethylating agent such as 5-aza-2′-deoxycytidine results in the attenuation of cellular proliferation and migration, along with decreased activity of beta-catenin/T-cell factor-dependent transcription [63]. Consistently, ectopic expression of WIF-1 in nasopharyngeal carcinoma (NPC) and ESCC cells considerably attenuates the colony formation of tumor cells accompanied with significant down-regulation of beta-catenin protein. Therefore, epigenetic inactivation of WIF-1 contributes to the aberrant stimulation of the Wnt cascade in NPC and ESCC. Along this line, WIF-1 methylation may serve as a diagnostic tumor biomarker [64].
Targeting Wnt signaling for therapeutic treatment of cancer
Activation of Wnt signaling is correlated with tumorigenesis in multiple tissues. Therefore, a number of strategies targeting Wnt signaling as therapies have been proposed [8, 65], as summarized in Table 3 and Fig. 2. In Table 3, we described the representative the component of Wnt signal and the molecules are ongoing for drug development.

The Wnt signaling pathway and development of drugs against signaling components
Blocking receptors/ligands of the Wnt signaling cascade
Blockage of Wnt signaling can be realized through ligands and receptors. While Frizzled (Fz)-related proteins (FRPs), Cerberus and WIF act by binding and sequestering Wnts, inhibitors against the receptors like LRP and Frizzled (FZD) also provide useful insights for cancer therapy [96]. For example, DKK1 inhibits Wnt signal transduction by binding to LRP, therefore blocking the interaction of the Wnt ligand to receptors [96,97,98]. LGK974 and IWPs are two potent and selective inhibitors of the Wnt palmitoyltransferase porcupine, resulting in reduced secretion of Wnt ligands. When applied, these two inhibitors decrease the phosphorylation of LRP6 receptors, and thereby inhibit Wnt/beta-catenin signaling [66, 99].
Another approach at attenuating Wnt signaling is through blockade of the receptor FZD [100], since Fzd 7 has been validated to be utilized as a candidate target to inhibit the growth of HCC cells with a small interfering peptides RHPDs [101]. Moreover, Soluble FZD 7 can interact with Wnt3 ligands, thereby competitively inhibiting the binding of a Wnt3 ligand to membrane FZD 7 receptors. This enhances the inhibitory effect on growth of tumor cells caused by doxorubicin via suppression of Wnt/beta-catenin signaling [102]. In addition, OMP-18R5 is a monoclonal antibody that directly binds to the extracellular segment of the FZD receptor members including FZD1, FZD2, FZD5, FZD7 and FZD8, it also reduces the phosphorylation of LRP6 and Wnt3A-induced accumulation of beta-catenin, more importantly, OMP-18R5 exhibits synergistic effect on tumor initiation when combining with chemotherapeutic agents after testing in xenograft model [70]. OMP-54F28, a fused protein that is generated by the fusion of a truncated FZD8 receptor with the IgG1 Fc region, has also been proved to block Wnt signaling and inhibit tumor growth and metastasis, and it also reduce the frequency of CSCs, more importantly, synergistic effect is achieved in breast cancer and pancreatic cancer after combination with other chemotherapeutic agents, such as gemcitabine [71].
Other strategies targeting Wnt ligands or receptors have also been developed. For example, RNAi compounds, monoclonal antibodies and small molecule inhibitors were proposed to target WNT2B [103] and the TNFα-WNT10B signaling loop [104]. Epigenetic disruption or overexpression of Wnt5a and epigenetic modification of its receptor ROR2 are also used to alter WNT5a and expression of ROR2, respectively [105, 106]. Therefore, a repertoire of inhibitors against Wnt receptors or ligands provide many potential choices for treatment of cancers with activated Wnt signaling.
Targeting other intracellular molecules of the Wnt signaling pathway
Multiple intracellular components of Wnt signaling have been targeted for downregulation of the pathway. As a pivotal member of the Wnt signal pathway, reduction of intracellular beta-catenin or inhibition of the translocation of cytoplasmic beta-catenin to the nucleus, can effectively inhibit tumorigenesis and development, representing a pivotal therapeutic target for cancer treatment. Non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin and indomethacin, can down-regulate beta-catenin/TCF activity and their target genes cyclin D1 [107]. An important pathway for beta-catenin degradation has been explored involving Siah, SIP and Ebi, and the expression of Siah is induced by p53 protein, this pathway links genotoxic injury with the destruction of beta-catenin; thereby reducing activity of TCF/LEF transcription factors that contribute to cell cycle arrest [108]. Βeta-catenin and NFκB-p65 proteins bind to the promoter regions of MDR1 in imatinib-sensitive and resistant chronic myleoid leukemia (CML) cells. Nuclear beta-catenin, NFκB-p65 and Akirin-2 levels increase in cells with imatinib resistance. Therefore, targeting Akirin-2, NF-κB and beta-catenin genes may provide an opportunity to treat imatinib resistant CML [109].
Dishevelled (Dvl) is a key scaffold protein that transmits Wnt signals from the cell membrane to the cell. It consists of three conserved functional domains: DIX, PDZ and DEP. Among these domains, the PDZ domain directly binds to the FZD intracellular segment, thereby inhibiting GSK-3β activity, blocking beta-catenin phosphorylation, and causing the accumulation of beta-catenin in the nucleus. Finally, the Wnt signal is transmitted from the cell membrane into the cell [110]. The activity of Dvl is regulated by a series of proteins including and Dapper1 protein. Dapper1 promotes the degradation of Dvl, however, the binding of 14–3-3β to Dapper1 attenuates the ability of Dapper1, and the interaction between 14 and 3-3β and Dapper1 is dependent on protein kinase A (PKA)-mediated phosphorylation of Dapper1 at Ser-237 and Ser-827 [111].
AXIN is a negative regulator of Wnt signal and a scaffold protein involved in Wnt pathway signaling. It has multiple interaction sites with APC, GSK-3β, and CK1 to form beta-catenin destruction complexes, and it can also interact with other components of Wnt signals including Dvl and PP2A [112, 113]. Tankyrase promotes AXIN degradation through the ubiquitin-proteasome pathway and leads to the activation of Wnt pathway. Therefore, inhibition of tankyrase activity can prolong the decay of AXIN and promote the degradation of beta-catenin, leading to the inhibition of the Wnt/beta-catenin signaling pathway [74]. Several small-molecule inhibitor of tankyrase, such as RK-582 [74], XAV939 [75], G007-LK [76], RK-287107 [77], JW55 [78] and IWR [79], have been well developed and demonstrated to possess anti-tumor activity. When the colon cancer cells, SW480 and SW620, were treated with 5-fluorouracil (5-FU)/cisplatin (DDP) alone or in combination with XAV939, XAV939 achieved a synergistic effect on apoptosis with 5-FU/DDP in SW480 cells, suggesting XAV939 can significantly enhance apoptosis induced by 5-FU/DDP. This was accompanied by a changed level of beta-catenin protein, AXIN and CSC markers in colon cancer cells. Hence, AXIN may serve as a potential molecular target for reversing multidrug resistance in colon cancer [80].
Another study revealed that Delta isoform of the CK1 family of serine/threonine kinases (CK1δ), an important mediator of intracellular Wnt signaling, is always amplified, and overexpressed in a subset of breast cancer. Inhibition of CK1δ with knockdown or inhibitors leads to apoptosis, tumor regression and blocking nuclear accumulation of beta-catenin, thus it provides strategy for effective therapy for breast cancer expressing CK1δ [114]. when combined with LiCl or MG-132 treatment, SPINK5 overexpression impedes Wnt signaling by attenuating the phosphorylation of GSK-3β and promoting beta-catenin protein degradation. Consequently, SPINK5 overexpression attenuates the proliferation, migration and invasion of esophageal cancer cells [115].
In another study, significantly higher levels of beta-catenin and lower levels of miR-214 were detected in esophageal carcinoma specimens than paired adjacent tissue. The levels of beta-catenin and miR-214 were inversely correlated in esophageal cancer specimens. Further study revealed that miR-214 regulates the translation of beta-catenin. Overexpression and depletion of miR-214 inhibits and promotes cell growth and invasion in esophageal cancer cell lines, respectively [116]. Furthermore, the level of the sFRP-1 transcript is also regulated by Gli1 in gastric cancer cells. Hence, inhibition of Hh signaling results in the accumulation of Wnt1-mediated beta-catenin in the cytosol [5]. Additionally, other efforts have considered the functional restoration of WIF-1. This might also be developed as a new targeted therapy for the treatment of malignancy, and is correlated with Wnt signal activation.
Targeting the beta-catenin/TCF4 complex and its downstream target genes in cancer
Beta-catenin regulates the expression of downstream target genes via interaction with TCF/LEF family members. Therefore, targeting the formation of beta-catenin-TCF/LEF complex or reduction in the levels of relevant proteins provides potential means to block the transcription of downstream target genes during cancer progression. Traf2- and Nck-interacting kinase (TNIK) is an important regulator of beta-catenin/TCF4 transcription complexes and is also required for the tumor-initiating capability of colorectal cancer stem cells. TNIK-deficient mice showed resistance to colon tumorigenesis induced by azoxymethane, and TNIK (−/−)/APC (min/+) mutant mice form fewer intestinal tumors. The TNIK inhibitor NCB-0846 downregulates the levels of LRP5 and LRP6 and reduces the expression of the Wnt downstream targets AXIN2 and cMYC in HCT116 and DLD-1 colorectal cancer cells [81]. Therefore, small-molecule inhibitors against TNIK can be potential therapeutic reagents for treating colorectal cancer.
The 4-thioureido-benzenesulfonamide derivative LF3 also inhibits canonical Wnt signaling by directly interfering with beta-catenin/TCF-4 interactions. LF3 application reduced cell motility and self-renewal of colon CSCs; it also blocked tumor growth and induced differentiation in mouse xenografts [82]. In addition, Ethacrynic acid exerts anti-tumor effects by binding LEF-1, thereby breaking beta-catenin/LEF-1 transcription complexes and decreasing the expression of cyclin D1 and fibronectin in chronic lymphocytic leukemia (CLL) cells [117]. Significantly, since Ethacrynic acid is currently used as a diuretic in clinics, it can be readily repurposed to treat chronic lymphocytic leukemia and liver cancer [117, 118].
The Wnt downstream target S100A4 is involved in metastasis, promoting cancer cell migration and invasion. High levels of S100A4 predict metastasis and reduced survival in CRC patients. Therefore, S100A4 shRNA or small molecule inhibitors have been tested to treat CRC. In addition, a repurposed anti-helminthic drug niclosamide can inhibit the expression of S100A4 and a prospective phase II clinical trial was carried out for treating CRC patients [119, 120]. Several other reagents that target beta-catenin or its complex are also summarized in Table 3 and Fig. 2.
Targeting Wnt signaling-mediated tumor microenvironment and immune response for cancer therapy
Oncogenic cascade activation in tumor cells affects the local anti-tumor immune reaction. Wnt signaling cross-talks with various immune cells, and thereby exhibits effects on tumor progression. It not only helps the maintenance and renewal of leucocytes, but also promotes immune tolerance, thereby limiting the anti-tumor response [121]. For example, Wnt/beta-catenin signaling activation reduces T-cell recruitment. In contrast, gain-of-function of MYC attenuates the activation and infiltration of T-cells [122]. Wnt/beta-catenin signaling activation leads to the exclusion of T-cell and resistance to immune therapy with anti-PD-L1/anti-CTLA-4 monoclonal antibody in autochthonous mouse melanoma models [123]. In addition, Tumor-infiltrating lymphocytes (TILs) and beta-catenin expression are both up-regulated in hormone receptor negative HER2-enriched and triple negative breast cancer (TNBC) subtypes. CD8(+) T-cells are the main effector cells in anti-tumor immunity. Interestingly, high levels of stromal TILs and CD8+ or FOXP3+ TIL subsets are linked with beta-catenin overexpression in breast carcinoma [124]. Correlations between the Wnt signaling activation and the absence of T-cell infiltration was recently explored in CRC. Studies revealed that tumors with high levels of beta-catenin exhibit decreased CD8+ T-cell infiltration. Therefore, a combination of PD-1-immunotherapy with beta-catenin targeting in CRC will likely be more helpful for therapeutic gains [125].
In addition to beta-catenin, other members such as WNT5a and DKK1 in the canonical Wnt signaling pathway were also explored for cancer immunotherapy. For example, nanoparticle-mediated trapping of WNT5a remodeled the immunosuppressive tumor microenvironment and enhanced the impact of immunotherapy, especially when combined with low-dose doxorubicin [126]. In addition, TGF-β1 silencing suppresses the DKK1 pro-invasive effect, and DKK1 enhances the invasion and migration of HCC via TGF-β1-mediated remodeling of the tumor microenvironment [127].
Progress of drug development in clinical applications for targeting Wnt signaling
Although much progress in tumor suppress has been achieved upon targeting the member of Wnt signaling, no drug targeting this pathway has been approved in clinical applications due to potential side effects. However, some drugs which have been widely used for treating other diseases have been validated to suppress cancer progress, the strategy of analyzing their anti-cancer effect induced by current clinical drugs will save the time and cost. For example, niclosamide is an anthelminthic drug used for treating tapeworms in human, whereas accumulating studies also found that it can also target and inhibit the Wnt signaling in multiple types of cancer, such as ovarian cancer [128], colorectal cancer [129], prostate cancer and breast cancer [130], of note is that niclosamide also target other pathways including nuclear factor-kappaB (NF-kB) and Notch pathways. Besides niclosamide, other drugs comprising nigericin [131], psoralen [132], ethacrynic acid [118] and pyrvinium pamoate [133] are also involved in reducing the growth of cancer cells through inhibiting Wnt signaling. Clinically, nigericin is an antibiotic working by acting as an H+, K+, and Pb2+ ionophore, psoralen, ethacrynic acid and pyrvinium pamoate are used to treat vitiligo, hypertension and oedema, and helminth, respectively.
Another strategy of drug development against Wnt signaling is to screen novel inhibitors. So far, many potential drugs targeting Wnt signaling for cancer therapy have been studied in clinical trials at different stages, and representative drugs in specific phases are summarized in Table 4 after searching in https://clinicaltrials.gov/ct2/results. As shown in Table 4, the drug targeting a specific member of Wnt signaling can be used to treat multiple malignant cancer, and vice versa, patients with specific malignant cancer can also receive treatment programs with multiple drugs targeting different members of Wnt signaling.
Conclusion and perspectives
The Wnt signaling cascade not only controls the development and homeostasis of diverse organs, but also mediates the malignant transformation. Abnormal Wnt activity detected in tumor tissues is associated with tumor initiation, invasion, metastasis, and drug resistance. Further in vitro and in vivo modeling indicates that Wnt signaling can be abnormally activated through genetic and epigenetic modifications. Therefore, several strategies have been proposed for targeting this pathway, such as blocking receptors/ligands, targeting multiple molecules including intracellular molecules, beta-catenin/TCF4 complex and its downstream target genes, or tumor microenvironment and immune response mediated by Wnt signaling. Based on previous attempts, a series of potential drugs have been screened and developed, these drugs comprise small molecules, monoclonal antibody, and other molecules. In addition, some of these drugs also exhibit synergistic effect when combined with chemotherapy. Immunotherapy is a novel therapeutic method, and it also provides strong survival hopes for patients with cancer, especially for those with chemotherapy resistance, thus the combination of Wnt inhibitors with immunotherapy reagents are worth attempting.
Although a series of drugs against the components of Wnt signaling have been obtained, many more drugs targeting this pathway should also be explored with approved drug pool to save time and cost. Moreover, novel pharmacological molecules such as peptide can also be screened from current peptide pool. As described in our previous study, P42, a peptide targeting Sox2 protein, was successfully screened from a peptide library with BiFc method and immunoprecipitation, more importantly, this peptide has inhibitive roles on ESCC progression in vitro and in vivo [134]. Therefore, more novel molecules are worth trying to explore with multiple existing methods.
We have just begun to understand how mutations of the critical Wnt signaling genes (e.g. APC, beta-catenin) cooperate with other tumor-associated genes like p53 and PTEN etc. during initial tumor formation, growth, and metastasis. Similarly, findings of Wnt signaling activation through epigenetic modifications (e.g. methylation or acetylation) are also relatively new. We therefore need more comprehensive models to address the functional relevance of these new findings. In the future, new techniques including CRISPR/Cas9 gene editing, single cell analysis and high-resolution molecule imaging are key to facilitate the establishment of new models, allowing us for deep appreciation of the roles played by Wnt signaling in tumorigenesis. Accordingly, new therapeutic targets and therapies will likely emerge.
Currently, some clinical trials have also been performed with some drugs including niclosamide and LGK974 after searching at website: https://clinicaltrials.gov/ct2/home. However, since Wnt signal also acts in tissue development and homeostasis maintenance, it may also bring some side-effect when use these drugs, thus it is helpful for us to integrate these drugs with nanomaterials to avoid the side-effect on normal tissue development, thereby enhancing their exclusive anti-cancer effects on malignant tissues. Additionally, systematic evaluation on their anti-cancer effects should be strictly performed along with multiple-omics data integration, and the strict clinical studies should be accelerated.
Availability of data and materials
The materials supporting the conclusion of this review is included within the article.
Abbreviations
- APC:
-
Adenomatous polyposis coli
- BE:
-
Barrett’s esophagus
- CAC:
-
Colitis-associated cancer
- CBP:
-
CREB-binding protein
- CK1:
-
Casein kinase 1
- CK2:
-
Casein kinase 2
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myleoid leukemia
- CRC:
-
Colorectal cancer
- CSCs:
-
Cancer stem cells
- Dkk:
-
Dickkopf
- DKK1:
-
Dickkopf-1
- DKK-3:
-
Dickkopf-3
- DSS:
-
Dextrin sulfate sodium
- Dvl:
-
Dishevelled
- EAC:
-
Esophageal adenocarcinoma
- EMT:
-
Epithelial-mesenchymal transition
- ESCC:
-
Esophageal squamous cell carcinoma
- FRPs:
-
Frizzled (Fz)-related proteins
- FZD:
-
Frizzled
- G3BP1:
-
GTPase-activating protein SH3 domain–binding protein 1
- GSK-3β:
-
Glycogen synthase kinase-3
- HCC:
-
Hepatocellular carcinoma
- Hh:
-
Hedgehog
- IBD:
-
Inflammatory bowel disease
- IPMNs:
-
Intraductal papillary mucinous neoplasms
- MCA:
-
Multiple correspondence analysis
- MCNs:
-
Mucinous cystic neoplasms
- MPNSTs:
-
Malignant peripheral nerve sheath tumors
- NKD2:
-
Naked cuticle homolog 2
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- NPC:
-
Nasopharyngeal carcinoma
- PhIP:
-
2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine
- RTKs:
-
Receptor tyrosine kinase
- SCC:
-
Squamous cell carcinoma
- SFRP1:
-
Secreted frizzled related protein 1
- TILs:
-
Tumor-infiltrating lymphocytes
- TNBC:
-
Triple negative breast cancer
- TNIK:
-
Traf2- and Nck-interacting kinase
- β-TrCP:
-
β-transducin repeat-containing protein
- WIF:
-
Wnt inhibitory factor
- WIF-1:
-
Wnt inhibitory factor-1
References
Kiely B, O'Donovan RT, McKenna SL, O'Sullivan GC. Beta-catenin transcriptional activity is inhibited downstream of nuclear localisation and is not influenced by IGF signalling in oesophageal cancer cells. Int J Cancer. 2007;121(9):1903–9.
Kim W, Kim M, Jho EH. Wnt/beta-catenin signalling: from plasma membrane to nucleus. Biochem J. 2013;450:9–21.
Semenov MV, Habas R, Macdonald BT, He X. SnapShot: noncanonical Wnt signaling pathways. Cell. 2007;131:1378.
Sugimura R, He XC, Venkatraman A, Arai F, Box A, Semerad C, et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. 2012;150:351–65.
He J, Sheng T, Stelter AA, Li C, Zhang X, Sinha M, et al. Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J Biol Chem. 2006;281:35598–602.
Clemons NJ, Phillips WA, Lord RV. Signaling pathways in the molecular pathogenesis of adenocarcinomas of the esophagus and gastroesophageal junction. Cancer Biol Ther. 2013;14:782–95.
Moghbeli M, Abbaszadegan MR, Golmakani E, Forghanifard MM. Correlation of Wnt and NOTCH pathways in esophageal squamous cell carcinoma. J Cell Commun Signal. 2016;10:129–35.
Xue G, Romano E, Massi D, Mandala M. Wnt/beta-catenin signaling in melanoma: preclinical rationale and novel therapeutic insights. Cancer Treat Rev. 2016;49:1–12.
Kahn M. Wnt signaling in stem cells and tumor stem cells. Semin Reprod Med. 2015;33:317–25.
Pai SG, Carneiro BA, Mota JM, Costa R, Leite CA, Barroso-Sousa R, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10:101.
Bisson I, Prowse DM. WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009;19:683–97.
Schneider JA, Logan SK. Revisiting the role of Wnt/beta-catenin signaling in prostate cancer. Mol Cell Endocrinol. 2018;462:3–8.
Song Y, Li L, Ou Y, Gao Z, Li E, Li X, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–5.
Fu L, Zhang C, Zhang LY, Dong SS, Lu LH, Chen J, et al. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/beta-catenin signalling pathway. Gut. 2011;60:1635–43.
de Castro J, Gamallo C, Palacios J, Moreno-Bueno G, Rodriguez N, Feliu J, et al. beta-catenin expression pattern in primary oesophageal squamous cell carcinoma. Relationship with clinicopathologic features and clinical outcome. Virchows Arch. 2000;437:599–604.
Lv J, Cao XF, Ji L, Zhu B, Wang DD, Tao L, et al. Association of beta-catenin, Wnt1, Smad4, Hoxa9, and Bmi-1 with the prognosis of esophageal squamous cell carcinoma. Med Oncol. 2012;29:151–60.
Liu JB, Qiang FL, Dong J, Cai J, Zhou SH, Shi MX, et al. Plasma DNA methylation of Wnt antagonists predicts recurrence of esophageal squamous cell carcinoma. World J Gastroenterol. 2011;17:4917–21.
Ruptier C, De Gasperis A, Ansieau S, Granjon A, Taniere P, Lafosse I, et al. TP63 P2 promoter functional analysis identifies beta-catenin as a key regulator of DeltaNp63 expression. Oncogene. 2011;30(46):4656–65.
Yoshida M, Yokota E, Sakuma T, Yamatsuji T, Takigawa N, Ushijima T, et al. Development of an integrated CRISPRi targeting DeltaNp63 for treatment of squamous cell carcinoma. Oncotarget. 2018;9:29220–32.
Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371–86.
Ma X, Tian Y, Song Y, Shi J, Xu J, Xiong K, et al. Msi2 maintains quiescent state of hair follicle stem cells by directly repressing the Hh signaling pathway. J Invest Dermatol. 2017;137(5):1015–24.
Hope KJ, Cellot S, Ting SB, MacRae T, Mayotte N, Iscove NN, et al. An RNAi screen identifies Msi2 and Prox1 as having opposite roles in the regulation of hematopoietic stem cell activity. Cell Stem Cell. 2010;7:101–13.
Li Z, Jin H, Mao G, Wu L, Guo Q. Msi2 plays a carcinogenic role in esophageal squamous cell carcinoma via regulation of the Wnt/beta-catenin and Hedgehog signaling pathways. Exp Cell Res. 2017;361:170–7.
Naganuma S, Whelan KA, Natsuizaka M, Kagawa S, Kinugasa H, Chang S, et al. Notch receptor inhibition reveals the importance of cyclin D1 and Wnt signaling in invasive esophageal squamous cell carcinoma. Am J Cancer Res. 2012;2:459–75.
Zhang LN, Zhao L, Yan XL, Huang YH. Loss of G3BP1 suppresses proliferation, migration, and invasion of esophageal cancer cells via Wnt/beta-catenin and PI3K/AKT signaling pathways. J Cell Physiol. 2019;234:20469–84.
Li F, Zhang L, Li W, Deng J, Zheng J, An M, et al. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/beta-catenin pathway. Oncotarget. 2015;6:6001–13.
Li S, Qin X, Liu B, Sun L, Zhang X, Li Z, et al. Dickkopf-1 is involved in invasive growth of esophageal cancer cells. J Mol Histol. 2011;42:491–8.
Cao B, Yang W, Jin Y, Zhang M, He T, Zhan Q, et al. Silencing NKD2 by promoter region Hypermethylation promotes esophageal Cancer progression by activating Wnt signaling. J Thorac Oncol. 2016;11:1912–26.
Clement G, Braunschweig R, Pasquier N, Bosman FT, Benhattar J. Alterations of the Wnt signaling pathway during the neoplastic progression of Barrett's esophagus. Oncogene. 2006;25:3084–92.
Birkeland AC, Burgin SJ, Yanik M, Scott MV, Bradford CR, McHugh JB, et al. Pathogenetic analysis of Sinonasal Teratocarcinosarcomas reveal actionable beta-catenin overexpression and a beta-catenin mutation. J Neurol Surg B Skull Base. 2017;78:346–52.
Hirotsu Y, Zheng TH, Amemiya K, Mochizuki H, Guleng B, Omata M. Targeted and exome sequencing identified somatic mutations in hepatocellular carcinoma. Hepatol Res. 2016;46:1145–51.
Selenica P, Raj N, Kumar R, Brown DN, Arques O, Reidy D, et al. Solid pseudopapillary neoplasms of the pancreas are dependent on the Wnt pathway. Mol Oncol. 2019;13:1684–92.
Wang H, Zhou H, Liu A, Guo X, Yang CS. Genetic analysis of colon tumors induced by a dietary carcinogen PhIP in CYP1A humanized mice: identification of mutation of beta-catenin/Ctnnb1 as the driver gene for the carcinogenesis. Mol Carcinog. 2015;54:1264–74.
Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet. 2017;49:1693–704.
Liu Y, Patel L, Mills GB, Lu KH, Sood AK, Ding L, et al. Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. J Natl Cancer Inst. 2014;106:dju245.
Newell F, Kong Y, Wilmott JS, Johansson PA, Ferguson PM, Cui C, et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat Commun. 2019;10:3163.
De Nicola F, Goeman F, Pallocca M, Sperati F, Pizzuti L, Melucci E, et al. Deep sequencing and pathway-focused analysis revealed multigene oncodriver signatures predicting survival outcomes in advanced colorectal cancer. Oncogenesis. 2018;7:55.
Fennell LJ, Kane A, Liu C, McKeone D, Fernando W, Su C, et al. APC mutation Marks an aggressive subtype of BRAF mutant colorectal cancers. Cancers (Basel). 2020;12:1171.
Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21:256–62.
Sakai E, Nakayama M, Oshima H, Kouyama Y, Niida A, Fujii S, et al. Combined mutation of Apc, Kras, and Tgfbr2 effectively drives metastasis of intestinal Cancer. Cancer Res. 2018;78:1334–46.
Silva AL, Dawson SN, Arends MJ, Guttula K, Hall N, Cameron EA, et al. Boosting Wnt activity during colorectal cancer progression through selective hypermethylation of Wnt signaling antagonists. BMC Cancer. 2014;14:891.
Li-Chang HH, Kasaian K, Ng Y, Lum A, Kong E, Lim H, et al. Retrospective review using targeted deep sequencing reveals mutational differences between gastroesophageal junction and gastric carcinomas. BMC Cancer. 2015;15:32.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–90.
Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46:1264–6.
Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–82.
Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A. 2011;108:21188–93.
Sakamoto H, Kuboki Y, Hatori T, Yamamoto M, Sugiyama M, Shibata N, et al. Clinicopathological significance of somatic RNF43 mutation and aberrant expression of ring finger protein 43 in intraductal papillary mucinous neoplasms of the pancreas. Mod Pathol. 2015;28:261–7.
Jiang X, Hao HX, Growney JD, Woolfenden S, Bottiglio C, Ng N, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2013;110:12649–54.
Fujita M, Matsubara N, Matsuda I, Maejima K, Oosawa A, Yamano T, et al. Genomic landscape of colitis-associated cancer indicates the impact of chronic inflammation and its stratification by mutations in the Wnt signaling. Oncotarget. 2018;9:969–81.
Lin Y, Wu Z, Guo W, Li J. Gene mutations in gastric cancer: a review of recent next-generation sequencing studies. Tumour Biol. 2015;36:7385–94.
Ryland GL, Hunter SM, Doyle MA, Rowley SM, Christie M, Allan PE, et al. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. J Pathol. 2013;229:469–76.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:l1.
Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet. 2014;46:607–12.
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74(5):1043–50.
Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet. 2000;26:146–7.
Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–50.
Ge XS, Ma HJ, Zheng XH, Ruan HL, Liao XY, Xue WQ, et al. HOTAIR, a prognostic factor in esophageal squamous cell carcinoma, inhibits WIF-1 expression and activates Wnt pathway. Cancer Sci. 2013;104:1675–82.
Jia Y, Yang Y, Zhan Q, Brock MV, Zheng X, Yu Y, et al. Inhibition of SOX17 by microRNA 141 and methylation activates the WNT signaling pathway in esophageal cancer. J Mol Diagn. 2012;14:577–85.
Taniguchi H, Yamamoto H, Hirata T, Miyamoto N, Oki M, Nosho K, et al. Frequent epigenetic inactivation of Wnt inhibitory factor-1 in human gastrointestinal cancers. Oncogene. 2005;24:7946–52.
He B, Reguart N, You L, Mazieres J, Xu Z, Lee AY, et al. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene. 2005;24:3054–8.
Clement G, Guilleret I, He B, Yagui-Beltran A, Lin YC, You L, et al. Epigenetic alteration of the Wnt inhibitory factor-1 promoter occurs early in the carcinogenesis of Barrett's esophagus. Cancer Sci. 2008;99:46–53.
Yang SH, Li SL, Dong ZM, Kan QC. Epigenetic inactivation of Wnt inhibitory factor-1 in human esophageal squamous cell carcinoma. Oncol Res. 2012;20:123–30.
Chan SL, Cui Y, van Hasselt A, Li H, Srivastava G, Jin H, et al. The tumor suppressor Wnt inhibitory factor 1 is frequently methylated in nasopharyngeal and esophageal carcinomas. Lab Investig. 2007;87:644–50.
Nusse R, Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–99.
Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110:20224–9.
Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5:100–7.
Proffitt KD, Madan B, Ke Z, Pendharkar V, Ding L, Lee MA, et al. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013;73:502–7.
Madan B, Ke Z, Harmston N, Ho SY, Frois AO, Alam J, et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene. 2016;35:2197–207.
Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, et al. Wnt pathway inhibition via the targeting of frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109:11717–22.
Le PN, McDermott JD, Jimeno A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54F28. Pharmacol Ther. 2015;146:1–11.
Chen M, Wang J, Lu J, Bond MC, Ren XR, Lyerly HK, et al. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry. 2009;48:10267–74.
Nile AH, de Sousa EMF, Mukund S, Piskol R, Hansen S, Zhou L, et al. A selective peptide inhibitor of frizzled 7 receptors disrupts intestinal stem cells. Nat Chem Biol. 2018;14:582–90.
Shirai F, Mizutani A, Yashiroda Y, Tsumura T, Kano Y, Muramatsu Y, et al. Design and discovery of an orally efficacious Spiroindolinone-based Tankyrase inhibitor for the treatment of Colon Cancer. J Med Chem. 2020;63:4183–204.
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–20.
Lau T, Chan E, Callow M, Waaler J, Boggs J, Blake RA, et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013;73:3132–44.
Mizutani A, Yashiroda Y, Muramatsu Y, Yoshida H, Chikada T, Tsumura T, et al. RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 2018;109:4003–14.
Waaler J, Machon O, Tumova L, Dinh H, Korinek V, Wilson SR, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72:2822–32.
Kulak O, Chen H, Holohan B, Wu X, He H, Borek D, et al. Disruption of Wnt/beta-catenin signaling and Telomeric shortening are inextricable consequences of Tankyrase inhibition in human cells. Mol Cell Biol. 2015;35:2425–35.
Wu X, Luo F, Li J, Zhong X, Liu K. Tankyrase 1 inhibitior XAV939 increases chemosensitivity in colon cancer cell lines via inhibition of the Wnt signaling pathway. Int J Oncol. 2016;48:1333–40.
Masuda M, Uno Y, Ohbayashi N, Ohata H, Mimata A, Kukimoto-Niino M, et al. TNIK inhibition abrogates colorectal cancer stemness. Nat Commun. 2016;7:12586.
Fang L, Zhu Q, Neuenschwander M, Specker E, Wulf-Goldenberg A, Weis WI, et al. A small-molecule antagonist of the beta-catenin/TCF4 interaction blocks the self-renewal of Cancer stem cells and suppresses tumorigenesis. Cancer Res. 2016;76:891–901.
Chen Z, Venkatesan AM, Dehnhardt CM, Dos Santos O, Delos Santos E, Ayral-Kaloustian S, et al. 2,4-Diamino-quinazolines as inhibitors of beta-catenin/Tcf-4 pathway: potential treatment for colorectal cancer. Bioorg Med Chem Lett. 2009;19:4980–3.
Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, et al. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5:91–102.
Tian W, Han X, Yan M, Xu Y, Duggineni S, Lin N, et al. Structure-based discovery of a novel inhibitor targeting the beta-catenin/Tcf4 interaction. Biochemistry. 2012;51:724–31.
Boon EM, Keller JJ, Wormhoudt TA, Giardiello FM, Offerhaus GJ, van der Neut R, et al. Sulindac targets nuclear beta-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br J Cancer. 2004;90:224–9.
Shan J, Shi DL, Wang J, Zheng J. Identification of a specific inhibitor of the dishevelled PDZ domain. Biochemistry. 2005;44:15495–503.
Grandy D, Shan J, Zhang X, Rao S, Akunuru S, Li H, et al. Discovery and characterization of a small molecule inhibitor of the PDZ domain of dishevelled. J Biol Chem. 2009;284:16256–63.
Fiskus W, Sharma S, Saha S, Shah B, Devaraj SG, Sun B, et al. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29:1267–78.
Morrell NT, Leucht P, Zhao L, Kim JB, ten Berge D, Ponnusamy K, et al. Liposomal packaging generates Wnt protein with in vivo biological activity. PLoS One. 2008;3:e2930.
Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol. 2010;6:829–36.
Park CH, Chang JY, Hahm ER, Park S, Kim HK, Yang CH. Quercetin, a potent inhibitor against beta-catenin/Tcf signaling in SW480 colon cancer cells. Biochem Biophys Res Commun. 2005;328:227–34.
Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A. 2004;101:12682–7.
Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, et al. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010;327:459–63.
Jiang G, Xiao X, Zeng Y, Nagabhushanam K, Majeed M, Xiao D. Targeting beta-catenin signaling to induce apoptosis in human breast cancer cells by z-guggulsterone and Gugulipid extract of Ayurvedic medicine plant Commiphora mukul. BMC Complement Altern Med. 2013;13:203.
Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/arrow. Nat Cell Biol. 2001;3:683–6.
Zorn AM. Wnt signalling: antagonistic Dickkopfs. Curr Biol. 2001;11:R592–5.
Bourhis E, Tam C, Franke Y, Bazan JF, Ernst J, Hwang J, et al. Reconstitution of a frizzled8.Wnt3a.LRP6 signaling complex reveals multiple Wnt and Dkk1 binding sites on LRP6. J Biol Chem. 2010;285:9172–9.
Yao H, Ashihara E, Maekawa T. Targeting the Wnt/beta-catenin signaling pathway in human cancers. Expert Opin Ther Targets. 2011;15:873–87.
King TD, Zhang W, Suto MJ, Li Y. Frizzled7 as an emerging target for cancer therapy. Cell Signal. 2012;24:846–51.
Nambotin SB, Lefrancois L, Sainsily X, Berthillon P, Kim M, Wands JR, et al. Pharmacological inhibition of Frizzled-7 displays anti-tumor properties in hepatocellular carcinoma. J Hepatol. 2011;54:288–99.
Wei W, Chua MS, Grepper S, So SK. Soluble Frizzled-7 receptor inhibits Wnt signaling and sensitizes hepatocellular carcinoma cells towards doxorubicin. Mol Cancer. 2011;10:16.
Katoh M. WNT2B: comparative integromics and clinical applications (review). Int J Mol Med. 2005;16:1103–8.
Katoh M. AP1- and NF-kappaB-binding sites conserved among mammalian WNT10B orthologs elucidate the TNFalpha-WNT10B signaling loop implicated in carcinogenesis and adipogenesis. Int J Mol Med. 2007;19:699–703.
Li J, Ying J, Fan Y, Wu L, Ying Y, Chan AT, et al. WNT5A antagonizes WNT/beta-catenin signaling and is frequently silenced by promoter CpG methylation in esophageal squamous cell carcinoma. Cancer Biol Ther. 2010;10:617–24.
Li L, Ying J, Tong X, Zhong L, Su X, Xiang T, et al. Epigenetic identification of receptor tyrosine kinase-like orphan receptor 2 as a functional tumor suppressor inhibiting beta-catenin and AKT signaling but frequently methylated in common carcinomas. Cell Mol Life Sci. 2014;71:2179–92.
Dihlmann S, Siermann A, von Knebel DM. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene. 2001;20:645–53.
Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001;7:915–26.
Karabay AZ, Koc A, Ozkan T, Hekmatshoar Y, Altinok Gunes B, Sunguroglu A, et al. Expression analysis of Akirin-2, NFkappaB-p65 and beta-catenin proteins in imatinib resistance of chronic myeloid leukemia. Hematology. 2018;23:765–70.
Punchihewa C, Ferreira AM, Cassell R, Rodrigues P, Fujii N. Sequence requirement and subtype specificity in the high-affinity interaction between human frizzled and dishevelled proteins. Protein Sci. 2009;18:994–1002.
Chen H, Liu L, Ma B, Ma TM, Hou JJ, Xie GM, et al. Protein kinase A-mediated 14-3-3 association impedes human Dapper1 to promote dishevelled degradation. J Biol Chem. 2011;286:14870–80.
Kikuchi A. Roles of Axin in the Wnt signalling pathway. Cell Signal. 1999;11:777–88.
Luo W, Peterson A, Garcia BA, Coombs G, Kofahl B, Heinrich R, et al. Protein phosphatase 1 regulates assembly and function of the beta-catenin degradation complex. EMBO J. 2007;26:1511–21.
Rosenberg LH, Lafitte M, Quereda V, Grant W, Chen W, Bibian M, et al. Therapeutic targeting of casein kinase 1delta in breast cancer. Sci Transl Med. 2015;7:318ra202.
Wang Q, Lv Q, Bian H, Yang L, Guo KL, Ye SS, et al. A novel tumor suppressor SPINK5 targets Wnt/beta-catenin signaling pathway in esophageal cancer. Cancer Med. 2019;8:2360–71.
Xu Y, Lu S. Regulation of beta-catenin-mediated esophageal cancer growth and invasion by miR-214. Am J Transl Res. 2015;7:2316–25.
Lu D, Liu JX, Endo T, Zhou H, Yao S, Willert K, et al. Ethacrynic acid exhibits selective toxicity to chronic lymphocytic leukemia cells by inhibition of the Wnt/beta-catenin pathway. PLoS One. 2009;4:e8294.
Al-Dali AM, Weiher H, Schmidt-Wolf IGH. Utilizing ethacrynic acid and ciclopirox olamine in liver cancer. Oncol Lett. 2018;16:6854–60.
Dahlmann M, Kobelt D, Walther W, Mudduluru G, Stein U. S100A4 in Cancer metastasis: Wnt signaling-driven interventions for metastasis restriction. Cancers (Basel). 2016;8:59.
Sack U, Walther W, Scudiero D, Selby M, Kobelt D, Lemm M, et al. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. J Natl Cancer Inst. 2011;103:1018–36.
Patel S, Alam A, Pant R, Chattopadhyay S. Wnt signaling and its significance within the tumor microenvironment: novel therapeutic insights. Front Immunol. 2019;10:2872.
Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. 2018;18:139–47.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5.
Ma X, Zhao X, Yan W, Yang J, Zhang H, Hui Y, et al. Tumor-infiltrating lymphocytes are associated with beta-catenin overexpression in breast cancer. Cancer Biomark. 2018;21(3):639–50.
Xue J, Yu X, Xue L, Ge X, Zhao W, Peng W. Intrinsic beta-catenin signaling suppresses CD8(+) T-cell infiltration in colorectal cancer. Biomed Pharmacother. 2019;115:108921.
Liu Q, Zhu H, Tiruthani K, Shen L, Chen F, Gao K, et al. Nanoparticle-mediated trapping of Wnt family member 5A in tumor microenvironments enhances immunotherapy for B-Raf proto-oncogene mutant melanoma. ACS Nano. 2018;12:1250–61.
Fezza M, Moussa M, Aoun R, Haber R, Hilal G. DKK1 promotes hepatocellular carcinoma inflammation, migration and invasion: implication of TGF-beta1. PLoS One. 2019;14:e0223252.
Arend RC, Londono-Joshi AI, Samant RS, Li Y, Conner M, Hidalgo B, et al. Inhibition of Wnt/beta-catenin pathway by niclosamide: a therapeutic target for ovarian cancer. Gynecol Oncol. 2014;134:112–20.
Osada T, Chen M, Yang XY, Spasojevic I, Vandeusen JB, Hsu D, et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011;71:4172–82.
Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/beta-catenin pathway. PLoS One. 2011;6:e29290.
Liu F, Li W, Hua S, Han Y, Xu Z, Wan D, et al. Nigericin exerts anticancer effects on human colorectal Cancer cells by inhibiting Wnt/beta-catenin signaling pathway. Mol Cancer Ther. 2018;17:952–65.
Wang X, Xu C, Hua Y, Cheng K, Zhang Y, Liu J, et al. Psoralen induced cell cycle arrest by modulating Wnt/beta-catenin pathway in breast cancer cells. Sci Rep. 2018;8:14001.
Xu L, Zhang L, Hu C, Liang S, Fei X, Yan N, et al. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int J Oncol. 2016;48:1175–86.
Liu K, Xie F, Zhao T, Zhang R, Gao A, Chen Y, et al. Targeting SOX2 protein with peptide aptamers for therapeutic gains against esophageal squamous cell carcinoma. Mol Ther. 2020;28:901–13.
Acknowledgements
Not applicable.
Funding
This work is supported by the Program from Xiang’an Hospital (No. PM202012220001 to K.L), the China National Natural Science Foundation (No. 81772994, 81302068 to K.L), the China National High Technology Research and Development Program (863-Program, No. 2014AA020541 to K.L), the International Collaborative Project of Fujian Province (No. 2017I0014 to K. L), and the Program for the Top Young Innovative Talents of Fujian Province (No. 2016RCLKC to K. L).
Author information
Authors and Affiliations
Contributions
Zhuo Wang and Tingting Zhao contributed to prepare the figures and literature; Kuancan Liu contributed to write the manuscript. Shihui Zhang, Junkai Wang, Yunyun Chen, Hongzhou Zhao, Yaxin Yang, Songlin Shi and Qiang Chen contributed to revise the manuscript. All authors reviewed and approved this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Z., Zhao, T., Zhang, S. et al. The Wnt signaling pathway in tumorigenesis, pharmacological targets, and drug development for cancer therapy. Biomark Res 9, 68 (2021). https://doi.org/10.1186/s40364-021-00323-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00323-7