
Abstract
Background
Single-variant associations with age-related macular degeneration (AMD), one of the most prevalent causes of irreversible vision loss worldwide, have been studied extensively. However, because of a lack of refinement of these associations, there remains considerable ambiguity regarding what constitutes genetic risk and/or protection for this disease, and how genetic combinations affect this risk. In this study, we consider the two most common and strongly AMD-associated loci, the CFH-CFHR5 region on chromosome 1q32 (Chr1 locus) and ARMS2/HTRA1 gene on chromosome 10q26 (Chr10 locus).
Results
By refining associations within the CFH-CFHR5 locus, we show that all genetic protection against the development of AMD in this region is described by the combination of the amino acid-altering variant CFH I62V (rs800292) and genetic deletion of CFHR3/1. Haplotypes based on CFH I62V, a CFHR3/1 deletion tagging SNP and the risk variant CFH Y402H are associated with either risk, protection or neutrality for AMD and capture more than 99% of control- and case-associated chromosomes. We find that genetic combinations of CFH-CFHR5 haplotypes (diplotypes) strongly influence AMD susceptibility and that individuals with risk/protective diplotypes are substantially protected against the development of disease. Finally, we demonstrate that AMD risk in the ARMS2/HTRA1 locus is also mitigated by combinations of CFH-CFHR5 haplotypes, with Chr10 risk variants essentially neutralized by protective CFH-CFHR5 haplotypes.
Conclusions
Our study highlights the importance of considering protective CFH-CFHR5 haplotypes when assessing genetic susceptibility for AMD. It establishes a framework that describes the full spectrum of AMD susceptibility using an optimal set of single-nucleotide polymorphisms with known functional consequences. It also indicates that protective or preventive complement-directed therapies targeting AMD driven by CFH-CFHR5 risk haplotypes may also be effective when AMD is driven by ARMS2/HTRA1 risk variants.
Similar content being viewed by others


Background
Age-related macular degeneration (AMD) is the leading cause of irreversible vision loss in the USA [1, 2] and affects close to 200 million individuals worldwide [3]. Prevalence among individuals over 45 years of age ranges from approximately 7.5% in Asians and Africans to 12.3% among individuals with European ancestry [3]. AMD is characterized by a gradual loss of visual acuity [4, 5], a decrease in contrast sensitivity [6,7,8,9] and delays in dark adaptation [10,11,12], which are associated with progressive photoreceptor loss [13, 14] and impaired retinal pigment epithelium (RPE) metabolism in the macula, the region of the primate eye responsible for high-acuity vision. Patients in the early and intermediate stages of AMD typically present with pigmentary abnormalities, drusen formation and/or pigment epithelium detachments in the fundus. A fraction of patients [3] ultimately progress to the late form of the disease, which is characterized by the gradual atrophy of regions of the retina (geographic atrophy, GA) and/or the abnormal growth of choroidal and/or retinal vessels (neovascular AMD) [15]. Therapeutic options are currently limited to patients with neovascular AMD, although these therapies suffer from inconsistent clinical outcomes [16,17,18,19,20,21].
Genetic associations with AMD have been extensively studied and are well documented. The most common genetic contributors to AMD are variants associated with a cluster of genes near complement factor H (CFH) – complement factor H-related (CFHR) 5 on chromosome 1q32 (Chr1 locus) [22,23,24,25,26,27], and with age-related maculopathy susceptibility 2 (ARMS2) and high-temperature requirement factor A1 (HTRA1), two tightly-linked genes located on chromosome 10q26 (Chr10 locus) [28, 29]. Genome-wide association studies (GWAS) have identified 32 additional loci associated with AMD, which include C3, C2/CFB and CFI, genes involved in the regulation of the complement system, and genes involved in lipid metabolism and extracellular matrix remodeling. These associations are independent from risk variants on Chr1 and Chr10 [30,31,32,33], and only account for a small number of patients with AMD [34,35,36]. Variants associated with CFH-CFHR5 and ARMS2/HTRA1 account for approximately 70% of the variability in AMD explained by additive genetic effects. While they may modulate disease, the other associated loci have a marginal effect when assessing AMD susceptibility [34, 35].
All genetic risk at the Chr10 locus is attributable to the variant rs10490924 (ARMS2) or to single-nucleotide polymorphisms (SNPs) in strong linkage disequilibrium (LD) with it [28, 29, 37]. In contrast, multiple SNPs characterize AMD genetic associations within the CFH-CFHR5 locus [22, 27]. The association between the CFH Y402H variant (rs1061170) and increased disease susceptibility was the first to be reported in this region [22, 24, 26]. These early studies found that all common risk haplotypes within the CFH-CFHR5 region are related to a single risk haplotype with a C allele at CFH Y402H [22,23,24] and that certain haplotypes were associated with a lower risk for AMD [22]. While investigated in multiple studies [22, 25, 27, 30, 38], genetic protection against AMD remains poorly defined, often overlooked [39], and no consensus on causative variants currently exists. This is partly caused by the use of CFH-CFHR5 risk variants and haplotypes as references when assessing genetic associations with AMD [34, 40, 41] within this locus, which results in the absence of clear distinction between lack of risk and genetic protection. The common missense CFH I62 (rs800292) polymorphism is the only amino-acid altering variant that confers protection against AMD [22]. Another form of genetic protection independent of CFH Y402H is associated with a common haplotype containing the deletion of CFHR3 and CFHR1 (CFHR3/1 deletion) [25, 38, 42,43,44]. Haplotypes containing the CFH Y402H and CFH I62V polymorphisms and the deletion of CFHR3/1 account for more than 90% of the genetic variability within the CFH-CFHR5 locus [45]. Some of these haplotypes confer an increased risk for AMD, and two of them confer protection against the development of disease. The remaining common haplotypes are present with similar frequencies in cases and controls; they are therefore associated with a lack of risk and referred to as neutral [42, 45,46,47,48,49]. The noncoding variant rs1410996 (or any perfect proxy), which is more strongly associated with AMD than CFH Y402H, is also associated with protection against AMD [27, 30, 34, 36]. However, since this variant is shared by protective haplotypes containing the minor allele at CFH I62V or the deletion of CFHR3/1, the protection associated with it is not entirely independent from that associated with rs800292 or the CFHR3/1 deletion [33, 42]. To date, true causality between genetic protection and rs1410996, CFH I62V or the deletion of CFHR3/1 remains to be established [40, 50]. A GWAS performed by the International AMD Genetic Consortium (IAMDGC) defined 8 credible sets of variants within the extended CFH-CFHR5 region independently associated with AMD [34]. Out of the 8 index SNPs describing the credible sets, one is a proxy for rs1410996 (rs10922109, IAMDGC Locus 1.1, r2 = 0.9919, Dʹ = 0.9959), one is a proxy for rs1061170 (rs570618, IAMDGC Locus 1.2, r2 = 0.9914, Dʹ = 1) and four (rs121913059, IAMDGC Locus 1.3; rs148553336, IAMDGC Locus 1.4, rs35292876, IAMDGC Locus 1.7 and rs191281603, IAMDGC Locus 1.8) are rare (frequency < 1%). Out of the two remaining common index SNPs, one is associated with risk for AMD (rs187328863, IAMDGC Locus 1.5) while the other (rs61818925, IAMDGC Locus 1.6), is associated with reduced risk. A haplotype analysis of the extended CFH-CFHR5 region was recently performed using 7 out of the 8 locus SNPs defined in this GWAS and a SNP tagging the deletion of CFHR3/1 [41]. The study identified an association between haplotypes based on a proxy for rs1410996 and the index SNP for locus 1.6 and circulating levels of complement factor-related 4 protein. This study and others [51,52,53,54] highlight the need to adequately account for genetic protection at Chr1 to fully elucidate the genetic etiology and pathophysiology of AMD.
While many studies have assessed the effect of heterozygosity and homozygosity for CFH-CFHR5 risk variants on AMD susceptibility [22], very few have considered haplotype combinations (diplotypes) at this locus. In particular, the effect of combinations of CFH-CFHR5 protective and risk haplotypes on disease susceptibility remains to be determined. Many investigations have established that the effect of CFH-CFHR5 and ARMS2/HTRA1 risk variants on AMD susceptibility were independent and additive [28,29,30,31, 55, 56]. Individuals homozygous for risk variants at both Chr1 and Chr10 are approximately 32 times more likely to develop AMD as compared to subject with no risk alleles at either locus [56]. So far, no study has considered whether the presence of protective CFH-CHFR5 haplotypes decrease disease incidence in individuals with risk genotypes at ARMS2/HTRA1.
In this study, we first identify the smallest set of variants accounting for common genetic risk and protection against AMD. Analyses of haplotypes and diplotypes based on these SNPs are then performed to assess how protective haplotypes affect AMD susceptibility. We finally characterize the combined effect of Chr1 diplotypes and Chr10 risk variants on AMD risk.
Results
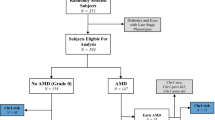
Cohort
The case/control cohort included 4787 individuals (Utah: 3306; Iowa: 1481) with a median age of \(77.4\) (IQR 12.7; see Table 1). Approximately one-third of the cohort (1587 individuals) consisted of controls. The remaining two-thirds (3200 individuals) presented with AMD in at least one eye. A majority of cases (61.9%) presented with late AMD in at least one eye, with subjects with early or intermediate AMD accounting for 11.7% and 10% of all cases, respectively. The minor allele frequency (MAF) of the variants considered in this study is summarized in Table 2. Frequencies among controls were consistent with frequencies among individuals with European ancestry (EUR) from the 1000 Genomes Project (denoted 1000 G), and among the 17,832 controls used in the IAMGC GWAS [34]. MAF among cases were also consistent with the 16,144 cases from the IAMGC GWAS. The frequency among cases and controls of CFH-CFHR5 haplotypes based on common IAMDGC index variants (Locus #1.1, #1.2, #1.5, #1.6 with the addition of Locus #1.7) and the deletion of CFHR3/1 [41] and their associated effect sizes were similar to those of the IAMDGC cohort [41] (see Table 3).
Protection at the CFH-CFHR5 locus is explained entirely by the combination of CFH I62V and CFHR3/1 deletion
Genetic protection within the CFH-CFHR5 extended region is generally accounted for by the noncoding variant rs1410996 (or any proxy) [27, 30, 34, 36], which is the SNP most strongly associated with AMD protection at this locus (OR 0.41 [0.37; 0.45], p = 1.04e−66 in our cohort); see Fig. 1a. The CFH I62V polymorphism (rs800292, OR: 0.52 [0.46; 0.58], p = 1.83e−29), the SNP tagging the deletion of CFHR3/1 (rs12144939, OR: 0.45 [0.39; 0.51], p = 1.43e−34) and the index SNP for the IAMDGC Locus 1.6 (rs61818925, OR: 0.65 [0.59; 0.72], p = 1.68e−17) are also associated with protection, but with comparatively larger odd ratios. Out of these four SNPs, we sought to identify the set of variants that accounts for all common genetic protection against AMD. To do so, we compared regression models including additive combinations of rs800292, rs12144939, rs1410996 and rs61818925 while controlling for age and sex using log-likelihood tests (see Fig. 1b). We found that the model including the two variants rs800292 and rs12144939 only was a significantly better fit than the model including rs1410996 only (p = 0.0063). When conditioning on CFH Y402H, the model including rs800292 and rs12144939 only was superior to the one including rs1410996 only (p = 0.013), which was consistent with a previous observation [50]. Adding rs61818925 to the model including rs800292, rs12144939 and CFH Y402H did not significantly increase the log-likelihood (p = 0.93).

Common protection at the CFH-CFHR5 locus is entirely described by the combination of CFH I62V and the deletion of CFHR3/1. a Manhattan plot showing the four common variants associated with protection against AMD (CFH I62V, rs1410996, the CFHR3/1-tagging rs12144939 and IAMDGC Locus 1.6) and the risk-conferring CFH Y402H. b Heatmap of the log-likelihood of additive regression models conditioning variables on the vertical axis to those of the horizontal axis. The combination of CFH I62V, the CFHR3/1 deletion and CFH Y402H yields the best model (boxed and highlighted). c Haplotype analysis using the four common variants associated with protection against AMD and CFH Y402H. Frequency for rs12144939 among Caucasians from the 1000 Genomes Project was based on rs6677604. *Score statistic based on the \({\rm X}^{2}\) test statistic with 1 degree of freedom. **Bonferroni correction for multiple testing of 9 haplotypes = 0.0056 (0.05/9)
To elucidate why CFH Y402H, CFH I62V and the deletion of CFHR3/1 are a superior set of variants to describe common risk and protection against AMD, we performed a haplotype analysis of the CFH-CFHR5 locus using rs800892, rs1061170, rs140996, rs12144939 and rs61818925 (see Fig. 1c). We identified three common protective haplotypes (H2, H3 and H8) with a frequency > 1% in our cohort. All of these haplotypes carry a protective allele (A) at rs800292 (H2, H8) or have the CFHR3/1 deletion (T allele at rs12144939, H3). Nearly all (98.4%) rs1410996 chromosomes with the protective A allele contain either the protective allele at CFH I62V or the deletion of CFHR3/1. This variant (and any SNP in LD with it) is therefore a proxy for the combination of CFH I62V and the deletion of CFHR3/1. One haplotype (H5), with frequency of 1.9% among our controls and 1% among cases, does not carry the protective allele at rs1410996 despite having a protective allele at CFH I62V (see Additional file 1: Table S2). In addition, one haplotype (H7) with frequency > 1% among individuals with European ancestry of the 1000 Genomes Project (but rare among our cases and controls) was not associated with protection against AMD despite carrying the minor allele at rs1410996 (p = 0.10). The protective allele at rs61818925 (T) is only part of one protective haplotype (H2), which contains the A allele at CFH I62V. The T allele at rs61818925 is also part of a haplotype with a C allele at CFH Y402H (H6) that is twice as frequent among our cases than in our controls, and of haplotypes with no significant association with AMD (H4 and H9). The presence of this allele on risk, neutral and protective haplotypes, in addition to the fact that it provides no additional information to our regression models, led us to exclude rs61818925 from further analyses.
Haplotypes based on CFH I62V, CFHR3/1 deletion and CFH Y402H differentiate disease susceptibility at the CFH-CFHR5 locus
The risk allele (T) for the IAMDGC Locus 1.5 (rs187328863 OR: 2.12 [1.63; 2.74], p = 1.29e−8), which was independently associated with increased risk for AMD in a previous GWAS [34], exists exclusively on a low-frequency haplotype containing a C (risk) allele at rs1061170 (see Table 3). While this variant may modulate risk, its frequency and effect size suggest that this risk is generally accounted for by haplotypes with a C allele at rs1061170.
The combination of rs800292, rs1061170 and rs12144939 yields four common haplotypes, which capture 99% of control- and 99.5% of case-associated chromosomes in our cohort (see Table 4 and Additional file 1: Table S3). Compared to a haplotype analysis of the extended CFH-CFHR5 region using 7 out of the 8 IAMDGC Locus SNPs (including four rare variants) and a SNP tagging the deletion of CFHR3/1 [41], we can estimate that haplotypes based on these 4 variants comprise at least 96.7% of control- and 92.8% of case-associated chromosomes of the IAMDGC cohort. A common haplotype (H3) has a frequency similar (approximately 20%) among cases and controls (\({\chi }^{2}=-1.31,\) p = 0.19) and is therefore neutral against AMD. Because this haplotype describes the absence of genetic risk or protection for developing AMD, it was used as a reference to describe the full spectrum of AMD susceptibility in place of the most common haplotype. The most common haplotype (H1) is associated with an increased risk for AMD when compared to H3 and carries a C allele at rs1061170 (OR = 1.61 [1.42; 1.83], p = 1.2e−13). Two common protective haplotypes carry either a protective allele at CFH I62V (referred to as Prot-I62, OR = 0.61 [0.52; 0.71], p = 1.9e−10) or the deletion of CFHR3/1 (referred to as Prot-Del, OR = 0.53 [0.45; 0.62], p = 3.9e−14). A fifth haplotype (H5) with frequency of 1.1% in the 1000 Genomes Project carries both a protective allele at CFH I62V and the CFHR3/1 deletion. While rare in our cohort, this haplotype is twice as frequent in our controls than in our cases (See Additional file 1: Table S3).
Genetic risk at the CFH-CFHR5 locus is determined by combinations of protective and risk haplotypes on Chr1
Ten CFH-CFHR5 haplotype combinations (diplotypes) with frequencies higher than 1% were present among our cases and controls (see Fig. 2 and Additional file 1: Table S4). The frequency of combinations of neutral haplotypes (Neutral/Neutral diplotype) did not differ significantly between cases and controls (frequency of 4%, \({\chi }^{2}=0.06,\) p = 0.80). This diplotype is therefore neutral and was used as a reference to differentiate disease susceptibility in our cohort. Overall, we found that combinations of CFH-CFHR5 haplotypes strongly influence AMD susceptibility. Risk/Risk (OR: 2.56 [1.8; 3.6]) and Risk/Neutral (OR: 1.4 [1.0; 2.0]) diplotypes confer an increased risk for AMD. Notably, individuals with combinations of risk and protective haplotypes are generally protected against AMD, with odds ratios ranging from 0.71 (CI [0.5; 1.0]) for Risk/Prot-Del diplotypes to 0.79 (CI [0.5; 1.1]) for Risk/Prot-I62 diplotypes. The strongest form of genetic protection is found among individuals homozygous for the Prot-Del haplotype (OR: 0.34 [0.2; 0.6]).

Association between haplotypes combinations (diplotypes) based on the protection conferring CFHR3/1 deletion and CFH I62V and the risk variant CFH Y402H. The common neutral diplotype Neutral/Neutral, which describes the absence of genetic risk or protection, was used as the reference diplotype to describe the full spectrum of AMD susceptibility at the CFH-CFHR5 locus. *Bonferroni correction for multiple testing of 10 diplotypes = 0.005 (0.05/10)
Risk and protective CFH-CFHR5 haplotypes strongly influence risk at the ARMS2/HTRA1 locus
Since protective CFH-CFHR5 haplotypes strongly influence AMD susceptibility on Chr1, we sought to determine if they had any effect on risk associated with the second most common and strongly AMD-associated locus, the ARMS2/HTRA1 gene. Risk at this locus is tagged by the variant rs10490924 (OR 2.33 [2.10; 2.59], p = 1.59e−56 in our cohort). Consistent with previous reports [28,29,30,31, 55, 56], there is no evidence of epistasis between CFH-CFHR5 and ARMS2/HTRA1, and the contribution of risk and protective CFH-CFHR5 haplotypes to Chr10 risk is additive in nature (see Fig. 3a and Additional file 1: Fig. S1). In the absence of risk or protective haplotypes on Chr1 (Neutral/Neutral diplotype), odds ratios in our case control cohort range from 0.64 (95% CI [0.41; 1.01], p = 0.049) among individuals with no ARMS2/HTRA1 risk alleles to 1.34 (95% CI [0.78; 2.45], p = 0.076) when one ARMS2/HTRA1 risk allele is present and 2.89 (95% CI [1.03; 14.04], p = 0.076) among subjects with two ARMS2/HTRA1 risk alleles.

Association between CFH-CFHR5 and ARMS2/HTRA1 diplotype combinations and AMD susceptibility. The effect size was calculated using Firth’s bias-reduced logistic regression [57] while adjusting for age and gender, and were used to define the colormap in all sub-figures. Bonferroni correction for multiple testing of 18 diplotype combinations = 0.0028 (0.05/18). a Odds ratios and frequency among cases and controls for CFH-CFHR5 and ARMS2/HTRA1 diplotype combinations. b Effect of CFH-CFHR5 diplotypes on AMD susceptibility among individuals with zero (left circle), one (middle circle) or two (right circle) ARMS2/HTRA1 risk alleles and associated odds ratios (provided in circles). AMD susceptibility moves towards protection with protective haplotypes on Chr1. c Effect of ARMS2/HTRA1 risk alleles on AMD susceptibility among individuals with Prot/Prot (left circle), Neutral/Neutral (middle circle) or Risk/Risk (right circle) CFH-CFHR5 diplotypes and associated odds ratios (in circles)
Overall, AMD susceptibility among individuals with 0, 1 or 2 ARMS2/HTRA1 risk alleles moves towards protection when protective haplotypes are present on Chr1 (see Fig. 3b). When compared to individuals with two CFH-CFHR5 Neutral/Neutral diplotypes, the presence of two protective haplotypes (Prot-I62 or Prot-Del, combined in Fig. 3) reduces odds ratios 2.3-fold (OR reduced from 2.89 to 1.24; two ARMS2/HTRA1 risk alleles) or 3.3-fold (OR reduced from 1.34 to 0.41; one ARMS2/HTRA1 risk allele). When considering the effect size associated with combinations of risk, neutral and protective haplotypes on Chr1 and risk alleles on Chr10, we find that, as expected for additive genetic contributions, ARMS2/HTRA1 risk alleles are counteracted by protective CFH-CFHR5 haplotypes in an approximately one-to-one manner (see Fig. 3b and Additional file 1: Fig. S2). At the other end of the AMD susceptibility spectrum, the presence of two CFH-CFHR5 risk haplotypes increases odds ratios associated with the Neutral/Neutral diplotype 2.8-fold (OR increased from 0.64 to 1.78; no ARMS2/HTRA1 risk alleles) to 4.7-fold (OR increased from 2.89 to 13.57; two ARMS2/HTRA1 risk alleles). In comparison, AMD susceptibility only moves towards risk with ARMS2/HTRA1 risk alleles, regardless of diplotypes on Chr1 (see Fig. 3c). This is because the spectrum of susceptibility associated with this locus only ranges from lack of risk (no risk alleles) to risk (one or two risk alleles).
Discussion
Genetic protection within the 1q32 CFH-CFHR5 extended region is generally accounted for by the noncoding variant rs1410996 (IAMDGC Locus 1.1). This variant tags two independent protective haplotypes that include the CFH I62 allele or genetic deletion of CFHR3/1, but rarely both [40, 42, 50]. It is likely that rs1410996 was identified as the most likely causal variant by the IAMDGC GWAS (for signal 1.1) [34] and as an independent AMD-associated variant by others [25, 30] precisely because of these combined haplotype effects. To demonstrate this, we can apply the same methodology as the one used by the IAMDGC GWAS authors to illustrate a counterexample of credible set variants able to depict the most likely causal variants in the presence of haplotype effects (Supplementary Figure S4 of the original publication [34]). Our haplotype analysis (Additional file 1: Table S2) indicates that one protective haplotype, with a frequency of 1.9% among controls and 1% among cases, carries the protective allele at CFH I62V but not at rs1410996. In addition, one haplotype with frequency > 1% among individuals with European ancestry of the 1000 Genomes Project (but rare among our cases and controls) is not associated with protection against AMD despite carrying the minor allele at rs1410996 (p = 0.10). This suggests that it is not the A allele of rs1410996 that carries protection but rather its coinciding with the protective allele of CFH I62V and the CFHR3/1 deletion. Our analysis therefore indicates that causality for protection at the CFH-CFHR5 locus is more likely to originate from CFH I62 or the deletion of CFHR3/1 than from rs1410996. This idea is further supported by the fact that unlike rs1410996 or variants in LD with it, both of these variants are protein altering. The minimal number of CFH-CFHR5 SNPs used to define risk, neutrality and protection associated with Chr1 could effectively be reduced to rs1061170 and rs1410996 without losing many chromosomes. However, these two variants are not sufficient to identify the origin of genetic protection, which is essential to elucidate the pathophysiology of AMD and identify viable therapeutic targets.
While being associated with a lower risk for AMD [34], rs61818925 (IAMDGC Locus 1.6) shows no added protection against AMD and does not explain any risk or protection that could not be attributed to CFH 402H, CFH I62 or the CFHR3/1 deletion. It is likely that the significance of the association between rs61818925 and AMD results from its partial LD with CFH I62V (r2 = 0.29, Dʹ = 0.78). Our results are consistent with a published haplotype analysis performed using the IAMDGC cohort [41]. In this study, IAMDGC Locus 1.6 showed no added protection in H4 (vs H5), H7 (vs H3) or H8 (vs H1) (see Table 3). The only other haplotype containing the minor allele at rs61818925 is H2. This haplotype contains the minor allele at rs1410996 without the CFHR3/1 deletion and therefore predominantly contains the protective allele at rs800292. In agreement with a previous analyses [41], we found that risk associated with rs187328863 (IAMDGC Locus 1.5) is accounted for by haplotypes with a C allele at rs1061170 (see Table 3). It is unclear if this intronic variant, which is located within the KCNT2 gene, has any functional consequences.
Narrowing the number of variants necessary to define genetic susceptibility at CFH-CFHR5 allows for the analysis of diplotypes and the assessment of AMD susceptibility in attainable sample sizes. Considering CFH-CFHR5 diplotypes is a robust and accurate way of assessing AMD susceptibility at this locus. For instance, whereas Risk/Neutral and Risk/Prot diplotypes are both associated with a C/T genotype when considering rs1061170 only, our study shows that these two combinations are in fact associated with very distinct susceptibilities for AMD (risk and protection, respectively). The concept of risk, neutrality and protection, which has been used in other studies [42, 45,46,47,48,49], provides an intuitive framework to understand how variants affect AMD outcomes. Using the risk haplotype H1 as a reference [34, 40, 41] obscures the fact that some haplotypes are present with similar frequencies among cases and controls, and are therefore not associated with AMD. Conversely, the use of neutral haplotypes as a reference simplifies the identification of risk haplotypes, which are more common in cases, and protective haplotypes, which are more common in controls. Unlike most genetic disease-associated loci where risk and protection are binary, lack of risk at CFH-CFHR5 does not imply protection and vice-versa. This is especially important in light of our finding that Chr1 protective haplotypes lower risk originating from the presence of one or two risk alleles at the ARMS2/HTRA1 locus, the other major driver of AMD.
Our study demonstrates that CFH I62V, CFH Y402H and a CFHR3/1 deletion tagging-SNP form the smallest set of variants necessary to fully differentiate the most common AMD susceptibility associated with the CFH-CFHR5 extended region. It also suggests that associations of common variants within this locus [22,23,24, 26,27,28,29,30, 34, 36, 37] can be traced back to the protein function altering changes at position 62 and 402 affecting the CFH protein and its splice variant, factor H-like protein 1 (FHL-1), and to the loss of the FHR-1 and FHR-3 proteins. The CFH/FHL-1 H402 allotype has been shown to alter the binding specificity of the CFH protein at the interface between the retinal pigment epithelium and Bruch’s membrane [58,59,60,61], which is the relevant location of AMD pathology, and for glycosaminoglycans [62, 63]. The CFH and FHL-1 I62 allotype is associated with increased complement co-factor activity, which may result in reduced complement activation and protection against AMD [46, 53]. Several studies have shown that FHR-1 and FHR-3 proteins compete with CFH and FHL-1 for binding to C3b and other ligands [42, 64]. Their loss is associated with an enhanced regulation by CFH/FHL-1 that leads to protection against AMD. A recent study reported that lower circulating levels of complement factor-related 4 (FHR-4) protein were associated with a lower risk for AMD [41]. Previous work showed that the protective rs1410996 allele had the strongest association with reduced FHR-4 levels within the CFH-CFHR5 region and that this association was independent from the loss of the FHR-1 and FHR-3 [65]. Our results indicate that the association between lower FHR-4 levels and reduced AMD risk is likely driven by CFH I62V or SNPs in LD with it, although more work is necessary to confirm this. These studies and others [41, 51] highlight the importance of considering the effect of CFH I62V, CFH Y402H and CFHR3/1 deletion in genotype/phenotype association studies.
The mechanisms associated with AMD driven by ARMS2/HTRA1 risk variants have yet to be elucidated. The AMD-associated region within this locus has recently been narrowed to a block of SNPs overlapping ARMS2 exon 1 and intron 1 [37]. Due to conflicting reports, it is not yet clear if the Arms2 protein is present in human tissue and cells [66,67,68,69]. The HtrA1 protein functions as both a secreted serine protease and an extracellular chaperone [70], and cleaves a variety of extracellular matrix (ECM) proteins, proteoglycans and growth factors [71, 72]. Evidence suggests that the biological and disease initiation events associated with AMD driven by risk at Chr10 are distinct from Chr1-directed AMD [51, 73]. However, the observed mitigating effect of protective CFH-CFHR5 on ARMS2/HTRA1 risk indicates that therapeutic interventions targeting the complement system may potentially modulate risk on Chr10. We did not have the power to investigate the effect of protective CFH-CFHR5 haplotypes on risk associated with loci other than ARMS2/HTRA1, but by showing that protection in the CFH-CFHR5 region alleviates risk in the two loci responsible for the majority of AMD, our study does indicate that targeting the complement system may have beneficial effects even when AMD is driven by other loci.
Conclusions
Our study demonstrates that all associations between common CFH-CFHR5 variants and AMD reported to date can be explained by the variants that alter CFH protein function (CFH I62V and CFH Y402H) and by the genetic deletion of CFHR3/1. It also shows that genetic susceptibility to AMD associated with the CFH-CFHR5 and ARMS2/HTRA1 loci is mitigated by protective CFH-CFHR5 haplotypes. These protective haplotypes counteract CFH-CFHR5 risk haplotypes significantly, so much so that individuals with risk/protective haplotype combinations are generally protected against the development of AMD. They also essentially neutralize the effect of ARMS2/HTRA1 risk polymorphisms, which indicates that protective complement-directed therapies designed to prevent AMD driven by CFH-CFHR5 risk haplotypes may also be effective when AMD is driven by ARMS2/HTRA1 risk variants.
Methods and materials
Cohort
Subjects were recruited between 2009 and 2019 at the Steele Center for Translational Medicine, John A. Moran Eye Center, University of Utah, USA, and between 1999 and 2009 at the University of Iowa, Iowa City, Iowa, USA, as part of a case/control study of the genetic etiology of AMD. The study adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Review Boards of the University of Utah and University of Iowa. All participants provided informed written research consent at the two locations. All subjects were Caucasian, unrelated and older than 55. Venous blood and/or saliva and demographic data including age, gender, ethnicity, smoking history were collected at the time of recruitment.
Grading
For each subject, both eyes were graded by the same two independent experienced observers based on fundus photographs and/or spectral domain optical coherence tomography volume scans collected at the time of recruitment. Grading was based on the international classification of mutually exclusive stages of age-related maculopathy introduced by the Rotterdam Group [74] and is detailed in Additional file 1: Table S1. Patients with no clinically observable signs of AMD were classified as controls (grade 0). Patients were classified as cases by the presence of drusen less than 63 µm in diameter, soft distinct drusen (≥ 63 µm in diameter) with or without pigmentary changes, isolated pigmentary changes without drusen (≥ 63 µm in diameter), soft indistinct drusen (≥ 125 µm in diameter) with or without pigmentary changes, geographic atrophy and/or neovascular AMD.
SNP selection
We selected SNPs within the CFH-CFHR5 and ARMS2/HTRA1 regions on the basis of previous genetic association analyses [22,23,24, 26,27,28,29,30, 37] including genome wide association studies [34, 36]. We considered SNPs with minor allele frequency > 1% and set a minimum threshold value of 0.8 for the r2 linkage disequilibrium parameter. Therefore, the SNPs selected accounted for all common variants associated with AMD within the CFH-CFHR5 and ARMS2/HTRA1 regions. Three SNPs, including rs10490924, were genotyped within the ARMS2/HTRA1 region in all samples. Thirty-seven SNPs were genotyped in the extended CFH-CFHR5 region; they included CFH I62V (rs800292), CFH Y402H (rs1061170), rs1410996 and the CFHR3/1 deletion tagging SNP rs12144939. Copy number variant assays were performed to validate the use of rs12144939 as a tagging SNP for the CFHR3/1 deletion. Common (frequency > 1%) index variants identified by Fritsche [34] that were not in LD (r2 > 0.8) with these SNPs were also genotyped. These included rs187328863 (IAMGC Locus 1.5) and rs61818925 (IAMDGC Locus 1.6).
Genotyping
Genomic DNA was isolated from peripheral blood leukocytes with QIAamp DNA Blood Maxi kits (Qiagen, Valencia, CA). Genotyping was performed by TaqMan assays (Applied Biosystems, Foster City, California) using 10 ng of template DNA in a 5µL reaction. When available, pre-designed assays were used. When pre-designed assays were unavailable, custom assays were designed using the manufacturer’s design software. The thermal cycling conditions in the 384-well thermocycler (PTC-225, MJ Research) consisted of an initial hold at 95 °C for 10 min, followed by 40 cycles of a 15-s 95 °C denaturation step and a 1-min 60 °C annealing and extension step. Plates were read in the 7900HT Fast Real-Time PCR System (Applied Biosystems).
Quality control
Data cleaning and quality control checks were performed using PLINK (v1.9) [75]. Heterogeneity between subjects recruited in Utah and Iowa was assessed using Cochran’s Q-statistic [76] and the I2 metric [77] while adjusting for age and sex. We found no evidence of heterogeneity when considering SNPs and haplotypes in the CFH-CFHR5 region (\({I}^{2}=0\%, p > 0.47\)). Low heterogeneity was detected when considering the rs10490924 (\({I}^{2}=42\%\)) with a non-significant Q-statistic (\(p=0.19\)). The Utah and Iowa cohorts were therefore combined without resorting to meta-analyses approaches. Linkage disequilibrium analyses were performed using the R package LDlinkR [78] and populations of Caucasian descent from the 1000 Genomes Project phase 3 [79].
Allele frequencies
Minor allele frequencies were generated using PLINK (v1.9) [75]. Allele frequencies among Caucasians were obtained from the 1000 Genomes Project phase 3 [79] (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) and the R [80] package LDlinkR [78]. Estimates were obtained by combining Utah residents in the USA (CEU), residents from Toscani in Italy (TSI), Finnish individuals in Finland (FIN), British individuals in England and Scotland (GBR) and subjects from the Iberian population in Spain (IBS). When available, frequencies among cases and controls of the International AMD Genetic Consortium (IAMDGC) were collected from publicly available sources [34, 41]. In the 1000 Genomes Project phase 3 and IAMDGC study, the frequency of the CFHR3/1 deletion was determined based on rs6677604 (minor allele A).
Haplotype phasing and analyses
Haplotype analyses were performed in R [80] using the package haplo.stats [81]. The package uses an EM algorithm to analyze indirectly measured haplotypes and assumes that all subjects are unrelated. The haplo.glm function was used to perform generalized regressions of AMD status on haplotype effects. The function uses the posterior probabilities of pairs of haplotypes per subjects as weights to update regression coefficients. Phasing was generated for our cohort and for Caucasians from the 1000 Genomes Project phase 3 [79] (genotype data for the SNPs of interest were downloaded from https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) using the same R package.
Association analyses
Association analyses were performed using PLINK (v1.9) [75] and R [80]. Associations between AMD single variants, haplotypes and diplotypes were assessed using the \({\chi }^{2}\) test for association and logistic regressions under additive models including sex and age as covariates. Epistasis between genes was assessed by including a multiplicative interaction term in the logistic regression models. Odds ratios and confidence intervals for CFH-CFHR5 and ARMS2/HTRA1 diplotype combinations were estimated by applying Firth’s bias-reduced logistic regression [57] using the R package brglm2 [82]. Multiple testing was accounted for by adjusting the significance level using Bonferroni corrections. Manhattan plots were generated using the R package CMplot [83]. When considering multiple variants, the most parsimonious best-fit regression was determined on the basis of likelihood ratio test statistics.
Comparison with IAMDGC GWAS
Frequencies and effect sizes of variants and haplotypes of interest were compared to those of the GWAS published in 2016 by the International Age-related Macular Degeneration Genomics Consortium (IAMDGC) [34]. The study used 16,144 cases and 17,832 controls of European descent. The publicly available summary statistics include genotyped SNPs, p-values and directions of associations. Frequencies for proxies of CFH Y402H, rs1410996, rs12144939 and rs10490924 were collected from previous investigations [34, 41]. Frequencies for CFH I62V could not be inferred from publicly available resources. Since effect sizes for rs12144939 and CFH I62V proxies were not available, we used a published and validated method [84, 85] to infer beta coefficients and standard errors (SE) from p-values for these two SNPs. Briefly, the method uses the fact that the sample size was the same for each variant to convert p-values to z-scores. Under the assumption that SE of the beta coefficient from a logistic regression is proportional to \(1-\sqrt{\mathrm{MAF}(1-\mathrm{MAF})}\), where MAF is the minor allele frequency, then \(\mathrm{SE}\times \sqrt{\mathrm{MAF}(1-\mathrm{MAF})}\) should be constant for all variants. We took the average of this term for the 34 genome-wide significant variants for which beta coefficients and SE were provided by the consortium (Table 1 of the published manuscript [34]). The SE for the remaining variants was then estimated by dividing the term by \(\sqrt{\mathrm{MAF}(1-\mathrm{MAF})}\). The accuracy of this approach was validated using the 34 genome-wide significant variants [84].
Availability of data and materials
All relevant data and summary statistics are included in the manuscript or its supplementary materials. Access to genotype information is not provided to protect participants from identification. Additional data available upon request.
References
Friedman DS, O’Colmain BJ, Muñoz B, Tomany SC, McCarty C, de Jong PTVM, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–72. https://doi.org/10.1001/archopht.122.4.564.
Klein R, Klein BEK. The prevalence of age-related eye diseases and visual impairment in aging: current estimates. Investig Ophthalmol Vis Sci. 2013;54:ORSF5–13. https://doi.org/10.1167/iovs.13-12789.
Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng C-Y, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2:e106–16. https://doi.org/10.1016/S2214-109X(13)70145-1.
Vinding T. Visual impairment of age-related macular degeneration. An epidemiological study of 1000 aged individuals. Acta Ophthalmol (Copenh). 1990;68:162–7.
Klein R, Wang Q, Klein BE, Moss SE, Meuer SM. The relationship of age-related maculopathy, cataract, and glaucoma to visual acuity. Investig Ophthalmol Vis Sci. 1995;36:182–91.
Midena E, Segato T, Blarzino MC, Degli AC. Macular drusen and the sensitivity of the central visual field. Doc Ophthalmol. 1994;88:179–85. https://doi.org/10.1007/BF01204616.
Stangos N, Voutas S, Topouzis F, Karampatakis V. Contrast sensitivity evaluation in eyes predisposed to age-related macular degeneration and presenting normal visual acuity. Ophthalmologica. 1995;209:194–8. https://doi.org/10.1159/000310612.
Sunness JS, Massof RW, Johnson MA, Bressler NM, Bressler SB, Fine SL. Diminished foveal sensitivity may predict the development of advanced age-related macular degeneration. Ophthalmology. 1989;96:375–81.
Kleiner RC, Enger C, Alexander MF, Fine SL. Contrast sensitivity in age-related macular degeneration. Arch Ophthalmol. 1988;106:55–7. https://doi.org/10.1001/archopht.1988.01060130061028.
Owsley C, Jackson GR, White M, Feist R, Edwards D. Delays in rod-mediated dark adaptation in early age-related maculopathy. Ophthalmology. 2001;108:1196–202. https://doi.org/10.1016/S0161-6420(01)00580-2.
Owsley C, McGwin G, Jackson GR, Heimburger DC, Piyathilake CJ, Klein R, et al. Effect of short-term, high-dose retinol on dark adaptation in aging and early age-related maculopathy. Investig Ophthalmol Vis Sci. 2006;47:1310–8. https://doi.org/10.1167/iovs.05-1292.
Owsley C, McGwin G, Clark ME, Jackson GR, Callahan MA, Kline LB, et al. Delayed rod-mediated dark adaptation is a functional biomarker for incident early age-related macular degeneration. Ophthalmology. 2016;123:344–51. https://doi.org/10.1016/j.ophtha.2015.09.041.
Curcio CA, Medeiros NE, Millican CL. Photoreceptor loss in age-related macular degeneration. Investig Ophthalmol Vis Sci. 1996;37:1236–49.
Curcio CA. Photoreceptor topography in ageing and age-related maculopathy. Eye (London). 2001;15(Pt 3):376–83. https://doi.org/10.1038/eye.2001.140.
Bird AC, Bressler NM, Bressler SB, Chisholm IH, Coscas G, Davis MD, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. Surv Ophthalmol. 1995;39:367–74. https://doi.org/10.1016/S0039-6257(05)80092-X.
Comparison of Age-related Macular Degeneration Treatments Trials (CATT) Research Group, Martin DF, Maguire MG, Fine SL, Ying G, Jaffe GJ, et al. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012;119:1388–98. https://doi.org/10.1016/j.ophtha.2012.03.053.
Kloeckener-Gruissem B, Barthelmes D, Labs S, Schindler C, Kurz-Levin M, Michels S, et al. Genetic association with response to intravitreal ranibizumab in patients with neovascular AMD. Investig Ophthalmol Vis Sci. 2011;52:4694–702. https://doi.org/10.1167/iovs.10-6080.
Brown DM, Michels M, Kaiser PK, Heier JS, Sy JP, Ianchulev T, et al. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study. Ophthalmology. 2009;116:57-65.e5. https://doi.org/10.1016/j.ophtha.2008.10.018.
Byun YJ, Lee SJ, Koh HJ. Predictors of response after intravitreal bevacizumab injection for neovascular age-related macular degeneration. Jpn J Ophthalmol. 2010;54:571–7. https://doi.org/10.1007/s10384-010-0866-1.
Rosenfeld PJ, Shapiro H, Tuomi L, Webster M, Elledge J, Blodi B, et al. Characteristics of patients losing vision after 2 years of monthly dosing in the phase III ranibizumab clinical trials. Ophthalmology. 2011;118:523–30. https://doi.org/10.1016/j.ophtha.2010.07.011.
Comparison of Age-related Macular Degeneration Treatments Trials (CATT) Research Group, Maguire MG, Martin DF, Ying G-S, Jaffe GJ, Daniel E, et al. Five-year outcomes with anti-vascular endothelial growth factor treatment of neovascular age-related macular degeneration: the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2016;123:1751–61. https://doi.org/10.1016/j.ophtha.2016.03.045.
Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–32. https://doi.org/10.1073/pnas.0501536102.
Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. https://doi.org/10.1126/science.1109557.
Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. https://doi.org/10.1126/science.1110189.
Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–7. https://doi.org/10.1038/ng1890.
Zareparsi S, Branham KEH, Li M, Shah S, Klein RJ, Ott J, et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet. 2005;77:149–53. https://doi.org/10.1086/431426.
Li M, Atmaca-Sonmez P, Othman M, Branham KEH, Khanna R, Wade MS, et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet. 2006;38:1049–54. https://doi.org/10.1038/ng1871.
Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, Meitinger T, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14:3227–36. https://doi.org/10.1093/hmg/ddi353.
Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005;77:389–407. https://doi.org/10.1086/444437.
Maller J, George S, Purcell S, Fagerness J, Altshuler D, Daly MJ, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055–9. https://doi.org/10.1038/ng1873.
Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39:1200–1. https://doi.org/10.1038/ng2131.
Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100–4. https://doi.org/10.1038/ejhg.2008.140.
Grassmann F, Fritsche LG, Keilhauer CN, Heid IM, Weber BHF. Modelling the genetic risk in age-related macular degeneration. PLoS ONE. 2012;7: e37979. https://doi.org/10.1371/journal.pone.0037979.
Fritsche LG, Igl W, Bailey JNC, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48:134–43. https://doi.org/10.1038/ng.3448.
Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genom Hum Genet. 2014;15:151–71. https://doi.org/10.1146/annurev-genom-090413-025610.
Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, et al. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013;45:433–9. https://doi.org/10.1038/ng.2578.
Grassmann F, Heid IM, Weber BHF, International AMD Genomics Consortium (IAMDGC). Recombinant haplotypes narrow the ARMS2/HTRA1 association signal for age-related macular degeneration. Genetics. 2017;205:919–24. https://doi.org/10.1534/genetics.116.195966.
Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med. 2006;38:592–604.
Harper AR, Nayee S, Topol EJ. Protective alleles and modifier variants in human health and disease. Nat Rev Genet. 2015;16:689–701. https://doi.org/10.1038/nrg4017.
Raychaudhuri S, Ripke S, Li M, Neale BM, Fagerness J, Reynolds R, et al. Associations of CFHR1-CFHR3 deletion and a CFH SNP to age-related macular degeneration are not independent. Nat Genet. 2010;42:553–5. https://doi.org/10.1038/ng0710-553 (author reply 555).
Cipriani V, Lorés-Motta L, He F, Fathalla D, Tilakaratna V, McHarg S, et al. Increased circulating levels of factor H-related protein 4 are strongly associated with age-related macular degeneration. Nat Commun. 2020;11:778. https://doi.org/10.1038/s41467-020-14499-3.
Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet. 2010;19:4694–704. https://doi.org/10.1093/hmg/ddq399.
Schmid-Kubista KE, Tosakulwong N, Wu Y, Ryu E, Hecker LA, Baratz KH, et al. Contribution of copy number variation in the regulation of complement activation locus to development of age-related macular degeneration. Investig Ophthalmol Vis Sci. 2009;50:5070–9. https://doi.org/10.1167/iovs.09-3975.
Spencer KL, Hauser MA, Olson LM, Schmidt S, Scott WK, Gallins P, et al. Deletion of CFHR3 and CFHR1 genes in age-related macular degeneration. Hum Mol Genet. 2008;17:971–7. https://doi.org/10.1093/hmg/ddm369.
de Jorge EG, Lera AL, Bayarri-Olmos R, Yebenes H, Lopez-Trascasa M, de Córdoba SR. Common and rare genetic variants of complement components in human disease. Mol Immunol. 2018;102:42–57. https://doi.org/10.1016/j.molimm.2018.06.011.
Martínez-Barricarte R, Recalde S, Fernández-Robredo P, Millán I, Olavarrieta L, Viñuela A, et al. Relevance of complement factor H-related 1 (CFHR1) genotypes in age-related macular degeneration. Investig Ophthalmol Vis Sci. 2012;53:1087–94. https://doi.org/10.1167/iovs.11-8709.
Spencer KL, Hauser MA, Olson LM, Schnetz-Boutaud N, Scott WK, Schmidt S, et al. Haplotypes spanning the complement factor H gene are protective against age-related macular degeneration. Investig Ophthalmol Vis Sci. 2007;48:4277–83. https://doi.org/10.1167/iovs.06-1427.
Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007;204:1249–56. https://doi.org/10.1084/jem.20070301.
de Córdoba SR, de Jorge EG. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clin Exp Immunol. 2008;151:1–13. https://doi.org/10.1111/j.1365-2249.2007.03552.x.
Hughes AE, Orr N, Cordell HJ, Goodship T. Reply to “Associations of CFHR1–CFHR3 deletion and a CFH SNP to age-related macular degeneration are not independent.” Nat Genet. 2010;42:555–6. https://doi.org/10.1038/ng0710-555.
Zouache MA, Bennion A, Hageman JL, Pappas C, Richards BT, Hageman GS. Macular retinal thickness differs markedly in age-related macular degeneration driven by risk polymorphisms on chromosomes 1 and 10. Sci Rep. 2020;10:21093. https://doi.org/10.1038/s41598-020-78059-x.
Keenan TDL, Toso M, Pappas C, Nichols L, Bishop PN, Hageman GS. Assessment of proteins associated with complement activation and inflammation in maculae of human donors homozygous risk at chromosome 1 CFH-to-F13B. Investig Ophthalmol Vis Sci. 2015;56:4870–9. https://doi.org/10.1167/iovs.15-17009.
Tortajada A, Montes T, Martínez-Barricarte R, Morgan BP, Harris CL, de Córdoba SR. The disease-protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum Mol Genet. 2009;18:3452–61. https://doi.org/10.1093/hmg/ddp289.
Ansari M, McKeigue PM, Skerka C, Hayward C, Rudan I, Vitart V, et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum Mol Genet. 2013;22:4857–69. https://doi.org/10.1093/hmg/ddt336.
Francis PJ, Zhang H, Dewan A, Hoh J, Klein ML. Joint effects of polymorphisms in the HTRA1, LOC387715/ARMS2, and CFH genes on AMD in a Caucasian population. Mol Vis. 2008;14:1395–400.
Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314:992–3. https://doi.org/10.1126/science.1133811.
Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993;80:27–38. https://doi.org/10.1093/biomet/80.1.27.
Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, et al. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J Biol Chem. 2010;285:30192–202. https://doi.org/10.1074/jbc.M110.103986.
Skerka C, Lauer N, Weinberger AAWA, Keilhauer CN, Sühnel J, Smith R, et al. Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol Immunol. 2007;44:3398–406. https://doi.org/10.1016/j.molimm.2007.02.012.
Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol. 2007;178:3831–6. https://doi.org/10.4049/jimmunol.178.6.3831.
Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci USA. 2006;103:17456–61. https://doi.org/10.1073/pnas.0606234103.
Clark SJ, Higman VA, Mulloy B, Perkins SJ, Lea SM, Sim RB, et al. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem. 2006;281:24713–20. https://doi.org/10.1074/jbc.M605083200.
Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, et al. Structural basis for complement factor H linked age-related macular degeneration. J Exp Med. 2007;204:2277–83. https://doi.org/10.1084/jem.20071069.
Alic L, Papac-Milicevic N, Czamara D, Rudnick RB, Ozsvar-Kozma M, Hartmann A, et al. A genome-wide association study identifies key modulators of complement factor H binding to malondialdehyde-epitopes. Proc Natl Acad Sci USA. 2020;117:9942–51. https://doi.org/10.1073/pnas.1913970117.
Cipriani V, de Motta LL, He F, Fathalla D, McHarg S, Bayatti N, et al. Factor H-Related Protein 4 (FHR-4) drives complement dysregulation in age-related macular degeneration. Investig Ophthalmol Vis Sci. 2019;60(9):3870–3870.
Fritsche LG, Loenhardt T, Janssen A, Fisher SA, Rivera A, Keilhauer CN, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40:892–6. https://doi.org/10.1038/ng.170.
Kanda A, Chen W, Othman M, Branham KEH, Brooks M, Khanna R, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci USA. 2007;104:16227–32. https://doi.org/10.1073/pnas.0703933104.
Kortvely E, Hauck SM, Duetsch G, Gloeckner CJ, Kremmer E, Alge-Priglinger CS, et al. ARMS2 is a constituent of the extracellular matrix providing a link between familial and sporadic age-related macular degenerations. Investig Ophthalmol Vis Sci. 2010;51:79–88. https://doi.org/10.1167/iovs.09-3850.
Wang G, Spencer KL, Court BL, Olson LM, Scott WK, Haines JL, et al. Localization of age-related macular degeneration-associated ARMS2 in cytosol, not mitochondria. Investig Ophthalmol Vis Sci. 2009;50:3084–90. https://doi.org/10.1167/iovs.08-3240.
De Luca A, De Falco M, Severino A, Campioni M, Santini D, Baldi F, et al. Distribution of the serine protease HtrA1 in normal human tissues. J Histochem Cytochem. 2003;51:1279–84. https://doi.org/10.1177/002215540305101004.
Hansen G, Hilgenfeld R. Architecture and regulation of HtrA-family proteins involved in protein quality control and stress response. Cell Mol Life Sci. 2013;70:761–75. https://doi.org/10.1007/s00018-012-1076-4.
Zurawa-Janicka D, Wenta T, Jarzab M, Skorko-Glonek J, Glaza P, Gieldon A, et al. Structural insights into the activation mechanisms of human HtrA serine proteases. Arch Biochem Biophys. 2017;621:6–23. https://doi.org/10.1016/j.abb.2017.04.004.
Williams BL, Seager NA, Gardiner JD, Pappas CM, Cronin MC, di San Filippo CA, et al. Chromosome 10q26-driven age-related macular degeneration is associated with reduced levels of HTRA1 in human retinal pigment epithelium. Proc Natl Acad Sci USA. 2021;118:e2103617118. https://doi.org/10.1073/pnas.2103617118.
van Leeuwen R, Klaver CCW, Vingerling JR, Hofman A, de Jong PTVM. The risk and natural course of age-related maculopathy: follow-up at 6 1/2 years in the Rotterdam study. Arch Ophthalmol. 2003;121:519–26. https://doi.org/10.1001/archopht.121.4.519.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. https://doi.org/10.1086/519795.
Cochran WG. The combination of estimates from different experiments. Biometrics. 1954;10:101. https://doi.org/10.2307/3001666.
Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–58. https://doi.org/10.1002/sim.1186.
Myers TA, Chanock SJ, Machiela MJ. Ldlinkr: an R package for rapidly calculating linkage disequilibrium statistics in diverse populations. Front Genet. 2020;11:157. https://doi.org/10.3389/fgene.2020.00157.
1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. https://doi.org/10.1038/nature15393.
Team RC. R: a language and environment for statistical computing.
Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–34. https://doi.org/10.1086/338688.
Kosmidis I, Pagui ECK, Sartori N. Mean and median bias reduction in generalized linear models. Stat Comput. 2019;1:17. https://doi.org/10.1007/s11222-019-09860-6.
Yin L, Zhang H, Tang Z, Xu J, Yin D, Zhang Z, et al. rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. BioRxiv. 2020. https://doi.org/10.1101/2020.08.20.258491.
Burgess S, Davey SG. Mendelian randomization implicates high-density lipoprotein cholesterol-associated mechanisms in etiology of age-related macular degeneration. Ophthalmology. 2017;124:1165–74. https://doi.org/10.1016/j.ophtha.2017.03.042.
Han X, Gharahkhani P, Mitchell P, Liew G, Hewitt AW, MacGregor S. Genome-wide meta-analysis identifies novel loci associated with age-related macular degeneration. J Hum Genet. 2020;65:657–65. https://doi.org/10.1038/s10038-020-0750-x.
Acknowledgements
The authors would like to thank the laboratory of Dr. Mark Leppert (Department of Human Genetics, University of Utah), including Jeff Stevens, Nori Matsunami, Lisa Baird, Tami Leppert and Brith Otterud, for research assistance and advice.
Funding
This work was supported in part by the National Eye Institute of the National Institutes of Health under award numbers R24EY017404 (GSH). Additional support from donations to the Steele Center for Translational Medicine and an Unrestricted Grant from Research to Prevent Blindness, New York, NY, to the Department of Ophthalmology and Visual Sciences, University of Utah (GSH and MAZ).
Author information
Authors and Affiliations
Contributions
CP and MAZ contributed equally to the article as first authors. CP, MAZ and GSH supervised the study. CP and MAZ designed the protocol for the study. MAZ, CDF, BLW and BTR contributed analysis tools. MAZ wrote the manuscript. CP, MAZ, BLW, BTR and GSH reviewed the manuscript. MAZ and CP performed all statistical analyses. CP and SM processed and genotyped all samples from the cohort and performed quality controls. JLH managed recruitment of cases and controls and collected all relevant demographic information. GSH and JLH determined the AMD status of all cases and controls. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Review Boards of the University of Utah and University of Iowa, where all research was performed. All participants provided informed written research consent at the time of recruitment.
Consent for publication
All participants provided informed written research consent at the time of recruitment, which covered publication of said research.
Competing interests
GSH is a shareholder, consultant and co-founder of Voyant Biotherapeutics, LLC. GSH, CP, BLW and BTR are inventors on patents and patent applications owned by the University of Utah. JLH is the spouse of GSH. None of the other authors have any relevant proprietary interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary tables and figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pappas, C.M., Zouache, M.A., Matthews, S. et al. Protective chromosome 1q32 haplotypes mitigate risk for age-related macular degeneration associated with the CFH-CFHR5 and ARMS2/HTRA1 loci. Hum Genomics 15, 60 (2021). https://doi.org/10.1186/s40246-021-00359-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-021-00359-8