
Abstract
Background
Fatty acids are important traits that affect meat quality and nutritive values in beef cattle. Detection of genetic variants for fatty acid composition can help to elucidate the genetic mechanism underpinning these traits and promote the improvement of fatty acid profiles. In this study, we performed a genome-wide association study (GWAS) on fatty acid composition using high-density single nucleotide polymorphism (SNP) arrays in Chinese Wagyu cattle.
Results
In total, we detected 15 and 8 significant genome-wide SNPs for individual fatty acids and fatty acid groups in Chinese Wagyu cattle, respectively. Also, we identified nine candidate genes based on 100 kb regions around associated SNPs. Four SNPs significantly associated with C14:1 cis-9 were embedded with stearoyl-CoA desaturase (SCD), while three SNPs in total were identified for C22:6 n-3 within Phospholipid scramblase family member 5 (PLSCR5), Cytoplasmic linker associated protein 1 (CLASP1), and Chymosin (CYM). Notably, we found the top candidate SNP within SCD can explain ~ 7.37% of phenotypic variance for C14:1 cis-9. Moreover, we detected several blocks with high LD in the 100 kb region around SCD. In addition, we found three significant SNPs within a 100 kb region showing pleiotropic effects related to multiple FA groups (PUFA, n-6, and PUFA/SFA), which contains BAI1 associated protein 2 like 2 (BAIAP2L2), MAF bZIP transcription factor F (MAFF), and transmembrane protein 184B (TMEM184B).
Conclusions
Our study identified several significant SNPs and candidate genes for individual fatty acids and fatty acid groups in Chinese Wagyu cattle, and these findings will further assist the design of breeding programs for meat quality in cattle.
Similar content being viewed by others

Background
Fatty acids of beef products have received considerable attention for their significance in human health, the improvement of salutary fatty acid (FA) content can offer more economic benefits in the beef market [1,2,3,4,5,6]. Like most economically important traits in beef cattle, FA composition are complex traits influenced by both genetic and environmental factors. Several studies have suggested that FA composition are lowly or moderately heritable traits and can be altered by feeding strategies [7,8,9,10]. However, recent studies from estimates of genetic parameters suggested that investigation of the genetic basis for FA composition can enable us to promote genetic improvement for them [7, 9, 11, 12]. Several studies have also reported genetic parameter analyses and genome-wide association studies of fatty acid profiles from milk in dairy cattle [13, 14]. Application of molecular genetic approaches can provide more opportunities to design genomic selection strategies for meat quality in beef cattle [15, 16].
In the last decade, genome-wide association studies (GWASs) have emerged as a powerful approach for detecting the candidate variants and genes for complex traits in beef cattle [8, 17,18,19]. Many studies have identified candidate markers associated with FA composition in different populations, such as Japanese Black cattle, Hereford, Angus, and Shorthorn [16, 20,21,22,23]. However, most of these studies were carried out using low density SNP arrays. Only a few studies were conducted using the high-density BovineHD (770 K) SNP array in limited breeds [8, 24, 25]. The genetic basis of FA composition may vary among populations, and the use of genomic technologies for improving fatty acid profiles in beef cattle have not been comprehensively addressed [8, 16, 21, 23,24,25,26,27,28,29].
Wagyu is especially well-known for its remarkable marbling score and meat quality [6, 30]. While the marbling is mainly fat tissue containing abundant monounsaturated fatty acids (MUFAs), it also reflects a lower melting point and contributes positively to beef flavor and tenderness [6, 31]. Several studies have been reported to investigate the genetic basis of fatty acids in Wagyu cattle, and a list of associated SNPs and candidate genes have been identified for these traits [6, 16, 32,33,34]. Chinese Wagyu cattle is a hybrid population from Wagyu and Fuzhou cattle, Fuzhou cattle is an indigenous population raised in Liaoning Province, China. Therefore, a GWAS of Chinese Wagyu cattle can contribute valuable knowledge for understanding the genetic basis of fatty acid composition. The objectives of the current study were to 1) identify the associated genomic variants and relative candidate genes for FA composition and 2) elucidate the genetic architecture of FA composition at the whole genome level in Chinese Wagyu cattle.
Materials and methods
Ethics statement
All animals were treated following the guidelines established by the Council of China Animal Welfare. Experimental protocols were approved by the Science Research Department of the Institute of Animal Sciences, Chinese Academy of Agricultural Sciences (CAAS) (Beijing, China).
Animals and phenotypes
The Chinese Wagyu population (464 animals) was established in Dalian, Liaoning Province, China, and all animals were born between 2012 and 2013. After weaning, they were fattened using the same feeding conditions for 20–24 months and slaughtered at an average of 28 months. During slaughtering, we measured meat quality traits in strict accordance with the guidelines proposed by the Institutional Meat Purchase Specifications for fresh beef. Meat samples were selected from the longissimus lumborum muscle, between the 12th and 13th ribs from each animal, after storage for 48 h, and samples were vacuum packed and chilled at − 80 °C. In addition, approximately 10 g of sample were taken for subsequent analyses. Total lipids were extracted from samples according to protocols described previously [35]. Approximately 2 mg of extracted lipid was re-dissolved in 2 mL of n-hexane and 1 mL of KOH (0.4 mol/L) for saponification and methylation. A total of 21 individual fatty acids, including six saturated fatty acids, four monounsaturated FAs, and eleven polyunsaturated fatty acids, were measured using gas chromatography (GC-2014 CAFsc, Shimadzu Scientific Instruments). Each FA was quantified as a weight of percentage of total FAs. In addition, FAs groups were measured as total saturated fatty acid (SFA), total monounsaturated (MUFA), total polyunsaturated (PUFA), total omega 3 (n-3), total omega 6 (n-6), ratio between PUFA and SFA (PUFA/SFA), ratio between n-6 and n-3 (n-6/n-3) and health index (HI). The estimation of various FA groups follows the same process as previously described [8, 11].
Genotyping and quality control
Blood samples were obtained with the regular quarantine inspection of the farms. DNA was extracted from the blood samples using a routine procedure. In total, 464 individuals were genotyped using the Illumina Bovine HD BeadChip (Illumina, Inc., San Diego, CA). SNPs were pre-processed based on the following filters using PLINK v1.07 [36]: Minor allele frequency (> 0.05), proportion of missing genotypes (< 0.05), and the Hardy-Weinberg equilibrium (P > 10E-6). Moreover, individuals with more than 10% missing genotypes were excluded. After quality control, the final data consisted of 364 individuals and 503,579 autosomal SNPs.
Heritability and genetic correlation estimation
Phenotypic and genetic (co)variances of fatty acids were estimated using the pairwise bivariate animal model implemented in the ASReml v3.0 package [37]. The model is
where y1 and y2 are vectors of phenotypic values of trait 1 and 2, respectively; X1 and X2 are incidence matrices for fixed effects; b1 and b2 are vectors of the fixed effects; Z1 and Z2 are incidence matrices relating the phenotypic observations to vectors of the polygenic (a1 + a2) effects for two traits; and e1 and e2 are random residuals for two traits. Variances of the random effects are defined as \( V(a)=\mathbf{G}{\sigma}_a^2 \) for the polygenes and \( V(e)=\mathbf{I}{\sigma}_e^2 \) for the residuals, where G is the additive genetic relationship matrix, I is the identity matrix, \( {\sigma}_a^2 \) is the additive genetic variance, and \( {\sigma}_e^2 \) is the residual variance. The G matrix was constructed using the SNP genotypes based on the proportion of total loci shared by two individuals [38], which was defined as G = MM′/ ∑ 2pi(1 − pi), where M is an n × m matrix of the number of animals (n) and number of marker loci (m), and it specifies the marker genotype coefficient at each locus, pi is the frequency of allele A of SNP, and (1 − pi) is the frequency of allele B. In MM′ the number of alleles shared by relatives was reported on the off-diagonals and an individual’s relationship with itself was reported on the diagonals. Farm and carcass grade were considered as fixed effects in the model. In addition, the duration of fattening and back-fat thickness were considered as covariates in the model. Genomic heritability of each trait was estimated using \( {h}^2={\sigma}_a^2/\left({\sigma}_a^2+{\sigma}_e^2\right) \). The phenotypic and genetic correlation coefficients were calculated using \( {r}_P={\mathit{\operatorname{cov}}}_{P_{XY}}/\sqrt{\sigma_{P_X}^2\times {\sigma}_{P_Y}^2} \) and\( {r}_G={\mathit{\operatorname{cov}}}_{G_{XY}}/\sqrt{\sigma_{G_X}^2\times {\sigma}_{G_Y}^2} \), where rP and rG are phenotypic and genetic correlation coefficients, respectively. \( {\sigma}_{P_X}^2 \) and \( {\sigma}_{P_Y}^2 \) are the phenotypic variance of trait X and trait Y. \( {\sigma}_{P_X}^2 \) and \( {\sigma}_{P_Y}^2 \) are the additive genetic variance of trait X and trait Y. \( {\mathit{\operatorname{cov}}}_{P_{XY}} \) and \( {\mathit{\operatorname{cov}}}_{G_{XY}} \) are the phenotypic and genetic covariance.
Genome-wide association analysis using FarmCPU
Fatty acid composition were adjusted for fixed effects and covariates using linear mixed models. The Fixed and random model Circulating Probability Unification (FarmCPU) model was used to test the single-SNP association. This algorithm takes into account the confounding problem between covariates using both the Fixed Effect Model (FEM) and Random Effect Model (REM) [39, 40]. The first three principal components were calculated using GAPIT, which were considered as the covariates [41]. The quantile-quantile (Q-Q) plot was generated to assess population stratification [42]. The linkage disequilibrium (r2) was estimated using PLINK v1.07 software. Linkage disequilibrium between SNPs around the target regions were estimated and visualized using Haploview v4.3 software [43]. Region plots were generated using the asplot function in the R package “gap” [44]. Positional candidate genes were investigated for 100 kb windows around SNPs using UCSC Genome Browser, which was based on the Bos taurus genome assembly UMD 3.1. The proportion of phenotypic variance explained by each SNP was calculated as follows:
where pi and qi represent the frequencies of two alleles for the ith SNP, βi is the effect of the ith SNP, and \( {\sigma}_p^2 \) denotes the phenotypic variance.
Results
Descriptive statistics and heritability estimations of fatty acid compositions
Descriptive statistics and heritability estimate results were presented in Table 1 for 21 individual FAs and eight FA groups. We observed that the most abundant individual saturated FAs were C18:1 cis-9 (46.87%), and C16:0 (27.46%), while relatively high proportions of the total FAs were found for C18:0 (11.96%), C16:1 cis-9 (4.22%), and C18:2 n-6 (3.84%) among the 21 individual FAs. In contrast, saturated FAs (C20:0, C22:0, C24:0), monounsaturated FAs (C14:1 cis-9, C20:1 cis-11), and polyunsaturated FAs (C18:2 c-9 t-11, C18:2 c-12 t-10, C18:3 n-6, C18:3 n-3, C20:2 n-6, C20:3 n-3, C20:4 n-6, C20:5 n-3, C22:5 n-3, C22:6 n-3) occupy relatively low proportions. For fatty acid groups, Saturated fatty acids (SFA), monounsaturated fatty acids (MUFA), and polyunsaturated fatty acids (PUFA) account for 42.5%, 52.51%, and 4.96% of total FAs, respectively. In general, the relative proportions of FAs and FA groups were consistent with our previous findings in Chinese Simmental cattle [8].
The heritability varied remarkably among 21 individual fatty acids. Among them, C14:1 cis-9 showed the highest heritability (0.43), while six fatty acids, including C14:0, C18:0, C22:0, C18:1 cis-9, C20:4 n-6, and C22:6 n-3, displayed moderate heritability (0.2~0.4). The estimated heritability for 14 out of 21 other FAs were less than 0.2. For eight FAs groups, high heritability were estimated for SFA, health index (HI), and MUFA (0.51, 0.43 and 0.40), while the estimated heritability for PUFA, total omega 3, and total omega 6 were 0.13, 0.07, and 0.14, respectively.
Phenotypic and genetic correlations between fatty acids
We estimated the phenotypic and genetic correlations among 21 individual FAs, which were shown in Additional file 1: Table S1. For six individual SFAs, high positive phenotypic and genetic correlations were observed between each pairwise comparison of C14:0, C16:0, and C18:0. Strong positive genetic correlations for C20:0 vs. C22:0 (0.994) and C20:0 vs. C24:0 (0.990) were observed, while strong negative genetic correlations between C22:0 and C24:0 was −0.99. Also, we observed a weak correlation between C20:0 vs. C14:0 (−0.098) and C20:0 vs. C16:0 (0.013) (Table S1). For individual MUFA, methyl myristoleate (C14:1 cis-9) showed a high positive genetic correlation with C16:1 cis-9 (0.866), C18:1 cis-9 (0.789), and C20:1 cis-11 (0.864). Also, C18:1 cis-9 has a highly positive genetic correlation with C16:1 cis-9 (0.839). For individual PUFA, several highly positive and negative genetic correlations were estimated among these individual groups. For instance, high positive correlations were observed among C22:6 n-3 vs. C22:5 n-3 (0.992), C22:6 n-3 vs. C24:0 n-6 (0.99), C18:3 n-3 vs. C20:3 n-3 (0.99), and C20:2 n-6 vs. C14:1 cis-9 (0.991). In contrast, negative correlations were observed among C18:3 n-3 vs. C20:5 n-3 (−0.976), C20:2 n-6 vs. C20:5 n-3 (−0.966), and C20:4 n-6 vs. C20:5 n-3 (−0.978).
GWAS results and candidate regions
We performed GWAS for 21 individual FAs and 8 FA groups using FarmCPU, and only results for traits with genomic heritability ≥0.10 were reported in the current study. As the Bonferroni correction was highly conservative for the GWAS using the high-density SNP array, we considered P < 1.36E-06 (0.1/73,531) as the suggestive significant level which was proposed by Zhu et al. [8] and Duggal et al. [45]. This strategy evaluates the approximate number of “independent” SNPs by counting one SNP per linkage disequilibrium (LD) block, plus all SNPs outside of the LD blocks (inter-block SNPs). The summary of the results from the GWAS using FarmCPU methods were shown in Table 2. In total, we identified 15 and 8 candidate SNPs for individual FAs and FA groups, respectively.
Seven SNPs associated with C22:6 n-3 were detected that located at six chromosomes. Among them, three significant SNPs (P-value = 3.38E-10, 9.19E-08, and 1.31E-07) overlapped with phospholipid scramblase family member 5 (PLSCR5), CLIP-associating protein 1 (CLASP1), and Chymosin (CYM), respectively, while no genes were found for other four SNPs (Fig. 1i). Notably, we observed that four candidate SNPs at BTA26 within SCD for C14:1 cis-9 had significant P values (P = 1.02E-07) (Fig. 1c). Among them, three SNPs were imbedded within the intron of SCD, while one SNP located at its exon region. Our results revealed that the highly significant SNPs contribute ~ 7.37% of phenotypic variance for C14:1 cis-9, and some nearby SNPs display high LD around the SCD gene. In addition, we observed one block with 6 kb at this region (Fig. 2). Thus, these identified SNPs are possible candidate markers for further application of maker assisted selection. In addition, one SNP at 42.65 Mb in BTA8 (P = 4.89E-07) for C20:1 cis-11 overlapped with SWI/SNF Related, Matrix Associated, Actin Dependent Regulator of Chromatin, Subfamily A, Member 2 (SMARCA2) (Fig. 1g). We also found one SNP (at BTA6:63.66 Mb) associated with C22:0 (Fig. 1a) and two SNPs (at BTA6:64.22 Mb and BTA6:64.28 Mb) were associated with C18:1 cis-9, respectively (Fig. 1e). However, no putative candidate gene was identified near this SNP. Q-Q plots for five FAs were presented in Fig. 1b, d, f, h, and j.

a Manhattan plot of association results for C22:0, where the Y-axis was defined as -log10(P) and the genomic position was represented along the X-axis. The green line indicated P = 1.36E-06. b Quantile-quantile plot of 503,579 SNPs in the genome-wide association study for C22:0. c Manhattan plot showing P-values of association for C14:1 cis-9. d Quantile-quantile plot for C14:1 cis-9. e Manhattan plot of association results for C18:1 cis-9. f Quantile-quantile plot for C18:1 cis-9. g Manhattan plot showing P-values of association for C20:1 cis-11. h Quantile-quantile plot of for C20:1 cis-11. i Manhattan plot showing P-values of association for C22:6 n-3. j Quantile-quantile plot for C22:6 n-3

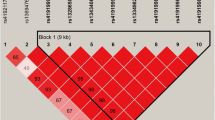
a Regional plots of candidate region at 21.05–21.21 Mb on BTA26 for C14:1 cis-9, the top SNP was highlighted by blue solid circles. Different levels of linkage disequilibrium (LD) between the most significant SNP and surrounding SNPs were indicated in different colors. b LD block for the region located at 21.05 Mb - 21.25 Mb on BTA26
We also detected eight associated SNPs for five FA groups (Table 2 and Fig. 3). Of these SNPs, we observed three, two, one, one, and one candidate SNPs for PUFA/SFA, MUFA, PUFA, SFA, and n-6, respectively. QQ plots for five FA groups were presented in Fig. 3b, d, f, h, and j. Interestingly, we found one SNP (BovineHD0500031844) associated with pleiotropic effects for multiple FA groups (PUFA, n-6, and PUFA/SFA). Additionally, three candidate genes were identified at 100 kb widows around this SNP. Among them, one gene, BAIAP2L2, was located in the upstream region, while two genes, MAFF and TMEM184B, were located at the downstream region. Moreover, we found several large blocks around the significant SNPs, ranging from 110.3 to 110.5 Mb (Fig. 4d).

a Manhattan plot of association results for SFA, where the Y-axis was defined as -log10(P) and the genomic position was represented along the X-axis. The green line indicated P = 1.36E-06. b Quantile-quantile plot of 503,579 SNPs in the genome-wide association study for SFA. c Manhattan plot showing P-values of association for PUFA. d Quantile-quantile plot for PUFA. e Manhattan plot showing P-values of association for MUFA. f Quantile-quantile plot for MUFA. g Manhattan plot showing P-values of association for n-6. h Quantile-quantile plot for n-6. i Manhattan plot showing P-values of association for PUFA/SFA. j Quantile-quantile plot for PUFA/SFA

a Regional plot of candidate region at 110.34–110.53 Mb on BTA5 for PUFA. b Regional plot of candidate region at 110.34–110.53 Mb on BTA5 for n-6 group. c Regional plot of candidate region at 110.34–110.53 Mb on BTA5 for PUFA/SFA. d LD block for the region located at 110.34–110.53 Mb on BTA5
Discussion
Fatty acids have generally been recognized as essential contributors to the tenderness and flavor of meat [46]. To our knowledge, this study is the first attempt to investigate the molecular mechanisms underpinning FAs using a high-density SNP array in Chinese Wagyu cattle. Our analyses showed that the estimated heritabilities varied among FAs, which is in agreement with previous publications [47,48,49]. This difference could be explained by the genetic architecture of the studied traits, and the effects of candidate SNPs may vary among diverse populations [25, 47]. In the present study, C14:1 cis-9 has the highest heritability (0.43) among MUFAs. Inoue et al. [50] and Ekine et al. [11] estimated that the heritability for C14:1 cis-9 were 0.86 and 0.51, which were also the highest among the MUFAs. These reports suggested that the amount of C14:1 cis-9 is likely to be influenced by genetic factors more than other MUFAs.
The heritability for C18:1 cis-9 was 0.25 in our analysis, whereas a relatively high heritability for C18:1 cis-9 (0.42 to 0.78) were reported in Japanese Black cattle [12, 32, 50]. Indeed, previous studies have reported that the SNPs within the SCD gene showing significant associations with C18:1 cis-13 of Canadian commercial steers as well as in Spanish breeds [51]. The heritability for eight FA groups were higher than those reported by previous studies [9, 10]. However, our results are similar to previous reports for the FA groups in Japanese Black cattle [12, 32, 50]. These results may suggest that the genetic structure of FAs in our population was more similar to Japanese Black cattle. The estimates of heritability for the FAs and FA groups are different across studies. This is particularly evident when the studied breeds are different, which may indicate differences in the genetic architecture of FAs in different populations. However, other factors, such as sample sizes and the statistical models, may also contribute to the difference of heritability estimates across studies [11]. We also observed high SE for some of FA except C16:0, C20:0, C18:2 n-6, C20:2 n-6 and C20:5 n-3, this is probably be explained by the small sample size. Also, population genetic structure and environmental conditions can affect the estimation of heritability.
For SFAs, strong positive genetic and phenotypic correlations between C22:0 and C20:0 were observed, which may indicate their similar origins of de novo synthesis from carbohydrates, amino acids, and volatile FA precursors [52]. The high positive phenotypic correlations between C22:0 and C20:0 also suggested that environmental conditions had similar effects on these FAs. Kim and Ntambi [53] proposed that C16:0 and stearic C18:0 fatty acids were the preferred substrates for SCD and can be converted to C16:1 cis-9 and C18:1 cis-9, respectively. C14:1 cis-9 is a major type of MUFA. We observed C14:1 cis-9 had a high positive genetic correlation with C16:1 cis-9, C18:1 cis-9, and C20:1 cis-9 (0.866, 0.789, 0.864), suggesting that the synthesis of MUFAs may be regulated by the same genes. C18:1 cis-9, as a type of FA proposed to be beneficial to human health, it had strong negative genetic correlations with the two most harmful saturated FA, C14:0 (−0.828) and C16:0 (−0.984). Genetic improvement of C18:1 cis-9 can possibly lead to a much healthier FA profile [5]. C16:1 cis-9, C18:1 cis-9, and C20:1 cis-9 also had strongly negative genetic correlations with C14:0, C16:0, C18:0, and C20:0. These results suggest that the synthesis of MUFAs may lead to a reduction of the concentration of SFAs.
In total, we identified 23 SNPs associated with nine candidate genes for FA composition in Chinese Wagyu cattle. Among them, we observed four significant SNPs for C14:1 cis-9 located at 21.14 Mb on BTA26, these newly identified SNPs were embedded with Stearoyl-CoA desaturase (SCD). Also, several LD blocks were observed within this gene (Fig. 2). SCD is the key enzyme involved in the endogenous synthesis of conjugated linoleic acid (CLA) and the conversion of saturated fatty acids into mono-unsaturated fatty acids (MUFA) in mammalian adipocytes [33]. Many SNPs near SCD have been reported, which are associated with C14:1 cis-9, C16:1 cis-9, C18:1 cis-9, and C18:2 n-6 in different cattle populations [6, 20, 23, 54]. For instance, two associated SNPs for C14:1 cis-9 were detected at 17.39 Mb and 17.58 Mb on BTA26 in Gifu cattle [6]. Three associated SNPs at 18.99~21.26 Mb on BTA26 for C14:1 cis-9, C16:1 cis-9, C18:1 cis-9, and C18:2 n-6 were detected in Angus and Hereford-Angus crossbred populations in Canada [23]. We suspected that different candidate SNPs for FA composition may be due to different genetic bases across populations. However, no significant SNPs around SCD was found that associated with C18:1 cis-9 in Chinese Wagyu cattle, which was consistent with previous findings in other populations [16, 20, 21, 51]. In addition, the direct effect of polymorphism within SCD on FA composition of milk has been extensively reported [6, 8, 18, 21,22,23,24,25]. Changes in the enzymatic activity as a result of SCD polymorphism and regulation have been recognized to cause diet-independent variations of CLA content in milk [55].
We also identified three genes, namely, CLIP-associating protein 1 (CLASP1), chymosin (CYM), and phospholipid scramblase family member 5 (PLSCR5) for C22:6 n-3. Among them, CLASP1 located within a QTL region that was related to Warner-Bratzler shear force in Nelore beef cattle [56]. A previous study reported that the CYM is related to immune response and milk fat percentage [57]. In addition, we observed one SNP at 123 Mb in PLSCR, which was associated with C22:6 n-3. PLSCR has been previously reported to be related to reproductive traits in pigs [58, 59]. In our study, highly positive phenotypic and genetic correlations were observed between each pairwise comparison of PUFA, PUFA/SFA, and n-6. The phenotypic correlation coefficients between PUFA vs. PUFA/SFA, PUFA vs. n-6, and PUFA/SFA vs. n-6 were 0.825, 0.99, and 0.817, while the genetic correlation coefficients were 0.751, 0.948, and 0.632, respectively. These results suggest that FA groups with high positive correlations may be regulated by the same genes. Indeed, we identified three genes, including BAIAP2L2, MAFF, and TMEM184B, for multiple FA groups (PUFA, n-6, and PUFA/SFA), which may imply their pleiotropic effects for these traits in beef cattle. Among them, we found that MAFF has a high expression in fat tissues as reported by Fagerberg et al. [60]. Another gene, named BAIAP2L2, has been previously reported as related to the marbling score in Korean cattle, and the differentially expressed pattern of this gene is correlated with the expression of several miRNAs [61].
Previous studies suggested that Fatty acid synthase (FASN) and Elongation of very long chain fatty acids protein 5 (ELOVL5) are associated with fatty acid compositions including C14:0, C14:1 cis-9, C16:0, C16:1 cis-9, C18:0, and C18:1 cis-9 in cattle [8]. However, these two genes were not identified in the present study, thus we suspected that heterogeneous genetic architecture of fatty acids exists among different populations. We accurately detected one SNP at 120 Mb on BTA1 for C20:4 n-6, this SNP was embedded with ELOVL7 (suggested P-value = 1.05E-05). In addition, several studies have found candidate SNPs within ELOVL7 associated with FA composition in porcine muscle and abdominal fat tissues [62, 63]. In mammals, seven enzymes have been identified in the ELOVL family (ELOVL1–7). The ELOVL enzyme has a distinct distribution in different tissues, and different enzymes exhibit different preferences for the FA substrate. The ELOVL5 and ELOVL6 genes are involved in the production/synthesis of palmitic (C16:0), palmitoleic (C16:1 cis-9), stearic (C18:0), and oleic (C18:1 cis-9) fatty acids. Therefore, the role of ELOVL5 and ELOVL6 in the synthesis of FAs is of great importance for beef cattle breeding programs [24, 64, 65]. The investigation into the molecular mechanism of the ELOVL gene family can provide valuable insight into improving the composition of beneficial FA in cattle and expanding our knowledge of transcriptional regulation mechanisms in domestic animals [66].
Conclusions
We identified several candidate SNPs and genes for individual FAs and FA groups in Chinese Wagyu cattle. Our findings highlight four novel SNPs in SCD with large effects for C14:1 cis-9 and one SNP with pleiotropic effects related to FA groups.
Abbreviations
- BTA:
-
Bos taurus autosome
- FA:
-
Fatty acids
- GWAS:
-
Genome-wide association study
- HI:
-
Health index
- MUFA:
-
Total monounsaturated fatty acids
- n-3:
-
Total omega-3 fatty acids
- n-6:
-
Total omega-6 fatty acids
- n-6/n-3:
-
Ratio between n-6 and n-3
- PUFA:
-
Total polyunsaturated fatty acid
- QTL:
-
Quantitative trait loci
- SE:
-
Standard error
- SFA:
-
Total saturated fatty acids
- SNP:
-
Single nucleotide polymorphism
References
Vahmani P, Mapiye C, Prieto N, Rolland DC, McAllister TA, Aalhus JL, et al. The scope for manipulating the polyunsaturated fatty acid content of beef: a review. J Anim Sci Biotechnol. 2015;6(1):29. https://doi.org/10.1186/s40104-015-0026-z.
Wood JD, Enser M, Fisher AV, Nute GR, Sheard PR, Richardson RI, et al. Fat deposition, fatty acid composition and meat quality: a review. Meat Sci. 2008;78(4):343–58. https://doi.org/10.1016/j.meatsci.2007.07.019.
Woodside JV, Kromhout D. Fatty acids and CHD. Proc Nutr Soc. 2005;64(04):554–64. https://doi.org/10.1079/PNS2005465.
Schwingshackl L, Hoffmann G. Monounsaturated fatty acids and risk of cardiovascular disease: synopsis of the evidence available from systematic reviews and meta-analyses. Nutrients. 2012;4(12):1989–2007. https://doi.org/10.3390/nu4121989.
Jimenez-Colmenero F, Carballo J, Cofrades S. Healthier meat and meat products: their role as functional foods. Meat Sci. 2001;59(1):5–13.
Ishii A, Yamaji K, Uemoto Y, Sasago N, Kobayashi E, Kobayashi N, et al. Genome-wide association study for fatty acid composition in Japanese black cattle. Anim Sci J. 2013:675–82. https://doi.org/10.1111/asj.12063.
De Smet S, Raes K, Demeyer D. Meat fatty acid composition as affected by fatness and genetic factors: a review. Anim Res. 2004;53(2):81–98. https://doi.org/10.1051/animres:2004003.
Zhu B, Niu H, Zhang W, Wang Z, Liang Y, Guan L, et al. Genome wide association study and genomic prediction for fatty acid composition in Chinese Simmental beef cattle using high density SNP array. BMC Genomics. 2017;18(1):464. https://doi.org/10.1186/s12864-017-3847-7.
Malau-Aduli AEO, Edriss MA, Siebert BD, Bottema CDK, Deland MPB, Pitchford WS. Estimates of genetic parameters for triacylglycerol fatty acids in beef cattle at weaning and slaughter. J Anim Physiol Anim Nutr. 2000;83(4–5):169–80.
Pitchford WS, Deland MP, Siebert BD, Malau-Aduli AE, Bottema CD. Genetic variation in fatness and fatty acid composition of crossbred cattle. J Anim Sci. 2002;80(11):2825–32.
Ekine-Dzivenu C, Chen L, Vinsky M, Aldai N, Dugan MER, McAllister TA, et al. Estimates of genetic parameters for fatty acids in brisket adipose tissue of Canadian commercial crossbred beef steers. Meat Sci. 2014;96(4):1517–26. https://doi.org/10.1016/j.meatsci.2013.10.011.
Nogi T, Honda T, Mukai F, Okagaki T, Oyama K. Heritabilities and genetic correlations of fatty acid compositions in longissimus muscle lipid with carcass traits in Japanese black cattle. J Anim Sci. 2011;89(3):615–21. https://doi.org/10.2527/jas.2009-2300.
Li X, Buitenhuis AJ, Lund MS, Li C, Sun D, Zhang Q, et al. Joint genome-wide association study for milk fatty acid traits in Chinese and Danish Holstein populations. J Dairy Sci. 2015;98(11):8152–63. https://doi.org/10.3168/jds.2015-9383.
Hein L, Sørensen LP, Kargo M, Buitenhuis AJ. Genetic analysis of predicted fatty acid profiles of milk from Danish Holstein and Danish Jersey cattle populations. J Dairy Sci. 2018;101(3):2148–57. https://doi.org/10.3168/jds.2017-13225.
Goddard ME, Hayes BJ. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet. 2009;10(6):381–91. https://doi.org/10.1038/nrg2575.
Uemoto Y, Abe T, Tameoka N, Hasebe H, Inoue K, Nakajima H, et al. Whole-genome association study for fatty acid composition of oleic acid in Japanese black cattle. Anim Genet. 2011;42(2):141–8. https://doi.org/10.1111/j.1365-2052.2010.02088.x.
Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, et al. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 2014;10(3):e1004198. https://doi.org/10.1371/journal.pgen.1004198.
Li C, Sun D, Zhang S, Wang S, Wu X. Genome wide association study identifies 20 novel promising genes associated with milk fatty acid traits in Chinese Holstein; 2014.
Liu A, Wang Y, Sahana G, Zhang Q, Liu L, Lund MS, et al. Genome-wide association studies for female fertility traits in chinese and nordic holsteins. Sci Rep. 2017;7(1). https://doi.org/10.1038/s41598-017-09170-9.
Kelly MJ, Tume RK, Fortes M, Thompson JM. Whole-genome association study of fatty acid composition in a diverse range of beef cattle breeds. J Anim Sci. 2014;92(5):1895–901. https://doi.org/10.2527/jas.2013-6901.
Saatchi M, Garrick DJ, Tait RG, Mayes MS, Drewnoski M, Schoonmaker J, et al. Genome-wide association and prediction of direct genomic breeding values for composition of fatty acids in Angus beef cattlea. BMC Genomics. 2013;14(1):730. https://doi.org/10.1186/1471-2164-14-730.
Ahlberg CM, Schiermiester LN, Howard TJ, Calkins CR, Spangler ML. Genome wide association study of cholesterol and poly- and monounsaturated fatty acids, protein, and mineral content of beef from crossbred cattle. Meat Sci. 2014;98(4):804–14. https://doi.org/10.1016/j.meatsci.2014.07.030.
Chen L, Ekine-Dzivenu C, Vinsky M, Basarab J, Aalhus J, Dugan MER, et al. Genome-wide association and genomic prediction of breeding values for fatty acid composition in subcutaneous adipose and longissimus lumborum muscle of beef cattle. BMC Genet. 2015;16(1). https://doi.org/10.1186/s12863-015-0290-0.
Lemos MVA, Chiaia HLJ, Berton MP, Feitosa FLB, Aboujaoud C, Camargo GMF, et al. Genome-wide association between single nucleotide polymorphisms with beef fatty acid profile in Nellore cattle using the single step procedure. BMC Genomics. 2016;17(1). https://doi.org/10.1186/s12864-016-2511-y.
Cesar AS, Regitano LC, Mourao GB, Tullio RR, Lanna DP, Nassu RT, et al. Genome-wide association study for intramuscular fat deposition and composition in Nellore cattle. BMC Genet. 2014;15:39. https://doi.org/10.1186/1471-2156-15-39.
Xie YR, Busboom JR, Gaskins CT, Johnson KA, Reeves JJ, Wright RW, et al. Effects of breed and sire on carcass characteristics and fatty acid profiles of crossbred wagyu and angus steers. Meat Sci. 1996;43(2):167–77.
Chiaia HLJ, Peripoli E, Silva RMDO, Aboujaoude C, Feitosa FLB, Lemos MVAD, et al. Genomic prediction for beef fatty acid profile in Nellore cattle. Meat Sci. 2017;128:60–7. https://doi.org/10.1016/j.meatsci.2017.02.007.
Yang A, Larsen TW, Powell VH, Tume RK. A comparison of fat composition of Japanese and long-term grain-fed Australian steers. Meat Sci. 1999;51(1):1–9.
Lee KT, Chung WH, Lee SY, Choi JW, Kim J, Lim D, et al. Whole-genome resequencing of Hanwoo (Korean cattle) and insight into regions of homozygosity. BMC Genomics. 2013;14:519. https://doi.org/10.1186/1471-2164-14-519.
Matsuhashi T, Maruyama S, Uemoto Y, Kobayashi N, Mannen H, Abe T, et al. Effects of bovine fatty acid synthase, stearoyl-coenzyme a desaturase, sterol regulatory element-binding protein 1, and growth hormone gene polymorphisms on fatty acid composition and carcass traits in Japanese Black cattle1. J Anim Sci. 2011;89(1):12.
Smith SB, Lunt DK, Chung KY, Choi CB, Tume RK, Zembayashi M. Adiposity, fatty acid composition, and delta-9 desaturase activity during growth in beef cattle. Anim Sci J. 2006;77(5):478–86. https://doi.org/10.1111/j.1740-0929.2006.00375.x.
Yokota S, Sugita H, Ardiyanti A, Shoji N, Nakajima H, Hosono M, et al. Contributions of FASN and SCD gene polymorphisms on fatty acid composition in muscle from Japanese black cattle. Anim Genet. 2012;43(6):790–2. https://doi.org/10.1111/j.1365-2052.2012.02331.x.
Taniguchi M, Utsugi T, Oyama K, Mannen H, Kobayashi M, Tanabe Y, et al. Genotype of stearoyl-CoA desaturase is associated with fatty acid composition in Japanese black cattle. Mamm Genome. 2004;15(2):142–8. https://doi.org/10.1007/s00335-003-2286-8.
Sasago N, Abe T, Sakuma H, Kojima T, Uemoto Y. Genome-wide association study for carcass traits, fatty acid composition, chemical composition, sugar, and the effects of related candidate genes in Japanese black cattle. Anim Sci J. 2017;88(1):33–44. https://doi.org/10.1111/asj.12595.
Jordi F, Lees M, Stanley GHS. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226(1):497–509.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Gilmour AG, Cullis B, Thompson R. ASREML user guide release 3.0 in Hemel Hempstead, HP1 1ES. UK: VSN International Ltd.; 2009.
VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91(11):4414–23. https://doi.org/10.3168/jds.2007-0980.
Liu X, Huang M, Fan B, Buckler ES, Zhang Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016;12(2):e1005767. https://doi.org/10.1371/journal.pgen.1005767.
Zhang X, Chu Q, Guo G, Dong G, Li X, Zhang Q, et al. Genome-wide association studies identified multiple genetic loci for body size at four growth stages in Chinese Holstein cattle. PLoS One. 2017;12(4):e175971. https://doi.org/10.1371/journal.pone.0175971.
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, et al. GAPIT: genome association and prediction integrated tool. Bioinformatics. 2012;28(18):2397–9. https://doi.org/10.1093/bioinformatics/bts444.
Arora S, Singh N, Kaur S, Bains NS, Uauy C, Poland J, et al. Genome-wide association study of grain architecture in wild wheat aegilops tauschii. Front Plant Sci. 2017;8. https://doi.org/10.3389/fpls.2017.00886.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. https://doi.org/10.1093/bioinformatics/bth457.
Zhao JH. Gap: Genetic analysis package. J Stat Softw. 2007;23(8):1–18.
Duggal P, Gillanders EM, Holmes TN, Bailey-Wilson JE. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics. 2008;9:516. https://doi.org/10.1186/1471-2164-9-516.
Melton SL, Amiri M, Davis GW, Backus WR. Flavor and chemical characteristics of ground beef from grass-, forage-grain-and grain-finished steers. J Anim Sci. 1982;55:77–87.
Inoue K, Shoji N, Honda T, Oyama K. Genetic relationships between meat quality traits and fatty acid composition in Japanese black cattle. Anim Sci J. 2017;88(1):11–8. https://doi.org/10.1111/asj.12613.
Aboujaoude C, Pereira ASC, Feitosa FLB, Antunes De Lemos MV, Chiaia HLJ, Piatto Berton M, et al. Genetic parameters for fatty acids in intramuscular fat from feedlot-finished Nelore carcasses. Anim Prod Sci. 2018;58(2):234. https://doi.org/10.1071/AN16107.
Sakuma H, Saito K, Kohira K, Ohhashi F, Shoji N, Uemoto Y. Estimates of genetic parameters for chemical traits of meat quality in Japanese black cattle. Anim Sci J. 2017;88(2):203–12. https://doi.org/10.1111/asj.12622.
Inoue K, Kobayashi M, Shoji N, Kato K. Genetic parameters for fatty acid composition and feed efficiency traits in Japanese black cattle. Animal. 2011;5(07):987–94. https://doi.org/10.1017/S1751731111000012.
Li C, Aldai N, Vinsky M, Dugan MER, McAllister TA. Association analyses of single nucleotide polymorphisms in bovine stearoyl-CoA desaturase and fatty acid synthase genes with fatty acid composition in commercial cross-bred beef steers. Anim Genet. 2012;43(1):93–7. https://doi.org/10.1111/j.1365-2052.2011.02217.x.
Mapiye C, Aldai N, Turner TD, Aalhus JL, Rolland DC, Kramer JKG, et al. The labile lipid fraction of meat: from perceived disease and waste to health and opportunity. Meat Sci. 2012;92(3):210–20. https://doi.org/10.1016/j.meatsci.2012.03.016.
Kim Y, Ntambi JM. Regulation of Stearoyl-CoA desaturase genes: role in cellular metabolism and preadipocyte differentiation. Biochem Biophys Res Commun. 1999;266(1):1–4. https://doi.org/10.1006/bbrc.1999.1704.
Berton MP, Fonseca LFS, Gimenez DFJ, Utembergue BL, Cesar ASM, Coutinho LL, et al. Gene expression profile of intramuscular muscle in Nellore cattle with extreme values of fatty acid. BMC Genomics. 2016;17(1). https://doi.org/10.1186/s12864-016-3232-y.
Rincon G, Islas-Trejo A, Castillo AR, Bauman DE, German BJ, Medrano JF. Polymorphisms in genes in the SREBP1 signalling pathway and SCD are associated with milk fatty acid composition in Holstein cattle. J Dairy Res. 2012;79(01):66–75. https://doi.org/10.1017/S002202991100080X.
Tizioto PC, Decker JE, Taylor JF, Schnabel RD, Mudadu MA, Silva FL, et al. Genome scan for meat quality traits in Nelore beef cattle. Physiol Genomics. 2013;45(21):1012–20. https://doi.org/10.1152/physiolgenomics.00066.2013.
Makina SO, Muchadeyi FC, van Marle-Koster E, Taylor JF, Makgahlela ML, Maiwashe A. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet Sel Evol. 2015;47:92. https://doi.org/10.1186/s12711-015-0173-x.
Onteru SK, Fan B, Du Z, Garrick DJ, Stalder KJ, Rothschild MF. A whole-genome association study for pig reproductive traits. Anim Genet. 2012;43(1):18–26. https://doi.org/10.1111/j.1365-2052.2011.02213.x.
Lucot KL. Genomic predictions for age at puberty and reproductive longevity in sows using bayesian methods, University of Nebraska; 2014.
Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406. https://doi.org/10.1074/mcp.M113.035600.
Seong J, Yoon H, Kong HS. Identification of microRNA and target gene associated with marbling score in Korean cattle (Hanwoo). Genes Genomics. 2016;38(6):529–38. https://doi.org/10.1007/s13258-016-0401-y.
Zhang W, Zhang J, Cui L, Ma J, Chen C, Ai H, et al. Genetic architecture of fatty acid composition in the longissimus dorsi muscle revealed by genome-wide association studies on diverse pig populations. Genet Sel Evol. 2016;48(5). https://doi.org/10.1186/s12711-016-0184-2.
Yang B, Zhang W, Zhang Z, Fan Y, Xie X, Ai H, et al. Genome-wide association analyses for fatty acid composition in porcine muscle and abdominal fat tissues. PLoS One. 2013;8(6):e65554. https://doi.org/10.1371/journal.pone.0065554.
Green CD, Ozguden-Akkoc CG, Wang Y, Jump DB, Olson LK. Role of fatty acid elongases in determination of de novo synthesized monounsaturated fatty acid species. J Lipid Res. 2010;51(7):1871–7. https://doi.org/10.1194/jlr.M004747.
Tamura K, Makino A, Hullin-Matsuda F, Kobayashi T, Furihata M, Chung S, et al. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer Res. 2009;69(20):8133–40. https://doi.org/10.1158/0008-5472.CAN-09-0775.
Chen S, He H, Liu X. Tissue expression profiles and transcriptional regulation of elongase of very long chain fatty acid 6 in bovine mammary epithelial cells. PLoS One. 2017;12(4):e175777. https://doi.org/10.1371/journal.pone.0175777.
Acknowledgments
The authors would like to thank all staffs at the cattle experimental unit in Beijing for animal care and sample collection. We acknowledge TopEdit LLC for the linguistic editing and proofreading during the preparation of this manuscript.
Funding
This study was supported by the National Natural Science Foundations of China (31372294 and 31702084), Chinese Academy of Agricultural Sciences of Technology Innovation Project (CAAS-XTCX2016010, CAAS-ZDXT2018006, ASTIP-IAS-TS-9, ASTIP-IAS-TS-16 and ASTIP-IAS03) for the design of the study and data collection. The project was also partly supported by Beijing City Board of Education Foundation (PXM2016_014207_000012) for the data analysis and interpretation of the study.
Availability of data and materials
The data and computing programs used in this manuscript are available from the corresponding author on request.
Author information
Authors and Affiliations
Contributions
JL and LX conceived and designed the study. ZW performed statistical analyses. ZW and LX wrote the paper. BZ, HG, and LX participated in data analyses. XG, WZ, SZ, YC and HN carried out quantification of fatty acids. LZ and BZ participated in the design of the study and contributed to acquisition of data. ZW performed SNP and gene annotation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Animal experiments were approved by the Science Research Department of the Institute of Animal Sciences, Chinese Academy of Agricultural Sciences (CAAS) (Beijing, China). There was no use of human participants, data or tissues.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional files
Additional file 1:
Table S1. Estimates of phenotypic correlations (upper diagonals) and genetic correlation (lower diagonals) between 21 phenotypes in Chinese Wagyu beef cattle. (XLSX 199 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, Z., Zhu, B., Niu, H. et al. Genome wide association study identifies SNPs associated with fatty acid composition in Chinese Wagyu cattle. J Animal Sci Biotechnol 10, 27 (2019). https://doi.org/10.1186/s40104-019-0322-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-019-0322-0