
Abstract
Background
By assaying hundreds of thousands of single nucleotide polymorphisms, genome wide association studies (GWAS) allow for a powerful, unbiased review of the entire genome to localize common genetic variants that influence health and disease. Although it is widely recognized that some correction for multiple testing is necessary, in order to control the family-wide Type 1 Error in genetic association studies, it is not clear which method to utilize. One simple approach is to perform a Bonferroni correction using all n single nucleotide polymorphisms ( SNPs) across the genome; however this approach is highly conservative and would "overcorrect" for SNPs that are not truly independent. Many SNPs fall within regions of strong linkage disequilibrium (LD) ("blocks") and should not be considered "independent".
Results
We proposed to approximate the number of "independent" SNPs by counting 1 SNP per LD block, plus all SNPs outside of blocks (interblock SNPs). We examined the effective number of independent SNPs for Genome Wide Association Study (GWAS) panels. In the CEPH Utah (CEU) population, by considering the interdependence of SNPs, we could reduce the total number of effective tests within the Affymetrix and Illumina SNP panels from 500,000 and 317,000 to 67,000 and 82,000 "independent" SNPs, respectively. For the Affymetrix 500 K and Illumina 317 K GWAS SNP panels we recommend using 10-5, 10-7 and 10-8 and for the Phase II HapMap CEPH Utah and Yoruba populations we recommend using 10-6, 10-7 and 10-9 as "suggestive", "significant" and "highly significant" p-value thresholds to properly control the family-wide Type 1 error.
Conclusion
By approximating the effective number of independent SNPs across the genome we are able to 'correct' for a more accurate number of tests and therefore develop 'LD adjusted' Bonferroni corrected p-value thresholds that account for the interdepdendence of SNPs on well-utilized commercially available SNP "chips". These thresholds will serve as guides to researchers trying to decide which regions of the genome should be studied further.
Similar content being viewed by others

Background
Since first proposed in 1996 by Risch and Merikangas [1], it has increasingly been accepted that association studies are powerful to detect modest effects of common alleles involved in complex trait susceptibility. Until recently, genotype-phenotype tests of association have been limited to candidate genes. Recent advances in molecular technologies and the availability of the human genome sequence have revolutionized researchers' ability to catalogue human genetic variation. In addition, the International HapMap project has provided researchers with invaluable information regarding the linkage disequilibrium (LD) structure within the genome [2, 3]. These advances have made genome wide association studies (GWAS) to identify common variants a reality. However many issues regarding the design, analysis and interpretation of results remain to be investigated.
In particular, interpretation of results is not trivial in light of the scale of multiple testing proposed. Testing such a large number of SNPs will require a balance between power and the chance of making false discoveries. There are many methods that have been proposed to address the multiple testing issue. These include false discovery rate (FDR), permutation testing, Bayesian factors (BF) and the Bonferonni correction. The FDR controls the expected proportion of false positives among all rejections, providing a less stringent control of the Type I error [4]. The application of the FDR method specifically in the context of genome wide studies has been proposed [4–6]. Permutation testing, in which the datasets are permuted thousands of times to achieve genomewide significance is another method that has been used in candidate gene studies and now genome wide association studies [7, 8]. Although empirical p-values have a theoretical advantage they may be computationally infeasible with large datasets. Another proposed method is the use of Bayesian Factors (BF) instead of frequentist p-values which need to be interpreted with the power of the study. However, BF also requires an assumption about the effect size, but the major advantage is that it can be compared across studies [9]. A simple method to control the family-wise error rate is the Bonferroni correction, which adjusts the Type 1 error (a) by the total number of tests (a/n). The Bonferroni correction can use the actual number of tests performed (i.e. SNPs genotyped) or a theoretical value based on the total number of tests possible (i.e. all SNPs). One critical, but often overlooked, assumption, of the Bonferroni correction method, is the assumption that all the tests are independent [10]. Biologically, we know that SNPs in close proximity are not independent, and therefore we are "overcorrecting" when we use the traditional Bonferroni method to adjust significance thresholds for multiple testing in GWAS studies [11]. We propose Bonferroni corrected p-value thresholds that account for the interdepdendence of SNPs on commonly used commercially available SNP "chips" (Illumina 317 K and Affymetrix 500 K) and in the HapMap panels. This method is an extension of the Bonferroni correction that accounts for the underlying linkage disequilibrium or dependence in dense SNP panels. These thresholds will be invaluable to researchers as they can be used as a guide to identifying regions of interest or significance in genome wide association studies, which should be studied further.
Methods
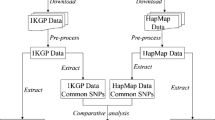
In order to estimate the effective number of "independent" SNPs in 3 autosomal marker panels (HapMap, Illumina 317 K and Affymetrix 500 K) we downloaded genotype data from release 22 of the International HapMap project. We used the non-redundant CEU and YRI data mapped against the "rs strand" of build 36 of the human genome. For the Illumina and Affymetrix marker sets we used a perl script to generate chromosome specific files containing only the subset of specific markers included in the Illumina 317 K or Affymetrix 500 K panels using CEU data. Then for each chromosome of data we used a perl script to generate smaller more manageable files each containing genotype data for approximately 2500 SNPs. We used Haploview version 4.0 to evaluate blocks of linkage disequilibrium (LD) using the 'Solid Spine of LD' algorithm with a minimum D' value of 0.8. The Solid Spine of LD method internal to Haploview defines a block when the first and last markers are in strong LD with all intermediate markers. We also evaluated chromosome 1 for the CEU HapMap data using the "Solid Spine of LD' algorithm and varying the minimum D' value to 0.7 and 0.9 to determine if this value altered the thresholds. In addition, we evaluated chromosome 1 for the CEU HapMap data using the Gabriel and 4-gamete block defining methods. For all analyses we ignored pairwise comparisons of markers >500 kb apart and excluded individuals with >50% missing genotypes. We also excluded markers with a minor allele frequency less than 0.01, a Hardy-Weinberg equilibrium p-value less than 0.001 or a genotype call rate less than 75%. We then summarized across the genome: Total number of SNPs, Total number of Blocks, Total number of SNPs not in a block (inter-block SNPs) and Total number of blocks + interblock SNPs for each panel. Our programs are available upon request so that thresholds can be established per population.
Results and discussion
We established three thresholds that correspond to 1) suggestive association in which we expect 1 false positive association per GWAS 2) significant association in which we expect one false positive association to occur 0.05 times per GWAS and 3) highly significant association in which we expect one false positive association to occur 0.001 times per GWAS. In the CEPH Utah (CEU) population, by considering the interdependence of SNPs, we reduced the total number of effective tests within the Affymetrix and Illumina SNP panels from 500,000 and 317,000 to 67,000 and 82,000 "independent" SNPs, respectively (Tables 1 and 2). This results in p-value thresholds of ≈10-5, 10-7 and 10-8 for both the Affymetrix and Illumina SNP panels (Table 3) compared to ≈10-6, 10-7 and 10-9 if we do not correct for the lack of independence among SNPs. For researchers using these set genome-wide SNP panels this provides valuable thresholds to interpret association results, and to identify SNPs that may be important for replication.
In addition to the established SNP panels, we evaluated the number of "independent" tests within the Phase II HapMap publicly available data for both the CEPH from Utah (CEU) and Yoruba (YRI) populations. Since our proposed thresholds are LD block dependent, they are population specific and the total number of "independent" SNPs may vary across populations and therefore should be considered separately. The publicly available data includes 2.4 million (CEU) and 2.7 million (YRI) SNPs across the genome. We reduced the total number of tests to 164,000 SNPs and 289,000 SNPs for the CEU and YRI, respectively (Tables 4 and 5). This results in p-value thresholds of ≈10-6, 10-7 and 10-9 for both the CEU and YRI populations (Table 3) compared to ≈10-7, 10-8 and 10-10 if we do not correct for the lack of independence among SNPs. The total number of "independent" SNPs for the YRI population is nearly double that for the CEU, however this does not have an impact on the exponent of the p-value. As expected, as the density of SNPs increases, the average number of SNPs within a block also increases. Therefore, it is likely that the additional Affymetrix and Illumina SNP panels (1 million and 650,000 SNPs) will not greatly increase the number of independent SNPs but will increase the number of SNPs within a block. However, using the highly dense HapMap population (Tables 4 and 5) provides us with thresholds that can be used for denser platforms (e.g. 1 million SNPs) or for studies that utilize statistical methods to impute the 2.5 million+ HapMap SNPs.
We also altered the D' value used to define the blocks from 0.7 to 0.9 for Chromosome 1 in the HapMap CEU population to determine if block definition had a large impact on our results. Using a D' value of 0.7 results in 2,039 fewer "independent" SNPs on chromosome 1 which extrapolates to 44,000 fewer "independent" SNPs across the genome. Using a more stringent value of D' = 0.9 results in 2,906 more "independent" SNPs on chromosome 1 which extrapolates to 63,932 more "independent" SNPs across the genome. Although this may increase the range of total SNPs across the genome from 120,000 to 228,000 it does not alter the exponent of the p-value or substantially affect the thresholds (Table 3).
We also defined blocks using two additional block definitions: the Gabriel method and the 4-gamete rule. The Gabriel method creates blocks using stringent criteria of LD with a D' upper bound >0.98 and a lower bound >0.70[12]. This creates smaller blocks with fewer SNPs within a block. The 4-gamete rule of Wang, based on Hudson and Kaplan determines blocks based on presumed recombination[13, 14]. Using pairwise sets of SNPs it determines the frequency of observing all 4 possible 2-SNP haplotypes. If all 4 haplotypes are observed, this method assumes recombination has occurred. Table 6 shows the results of different block definitions for Chromosome 1 for the CEU HapMap samples. The Gabriel method results in a similar number of blocks, but the number of SNPs per block is greatly reduced resulting in more SNPs outside of the block that are still in LD but do not meet the stringent criteria of a "block". The 4-gamete rule results in fewer blocks and more SNPs outside of blocks that represent potential recombination events. To limit the dependence on LD we believe the solid spine of LD is the best method to capture the underlying LD and biological dependence of SNPs, and therefore we base our thresholds on this method.
The method we detail is an extension to the original Bonferroni correction which is widely utilized; however, we have reduced the total number of SNPs to reflect the number of "independent SNPs" since independence is an assumption of the Bonferroni correction. Therefore, our thresholds are based on the original Bonferroni calculation of 1/Total # of SNPs, 0.05/Total # of SNPs and 0.001/Total # of SNPs where the number of SNPs that we use is now a better estimate of the number of independent tests being performed. Therefore, our proposed method allows a Bonferroni correction that has less violation of the assumption of independence.
We have empirically defined thresholds for genome wide association studies to control the family-wise error rate while accounting for the interdependence of SNPs in linkage disequilibrium. The use of actual data provides us an opportunity to unequivocally characterize the underlying linkage disequilibrium structure in these two populations. We considered the use of simulations as has been done for single chromosomes by assigning haplotypes based on frequencies from inferred haplotypes of founders for a set number of replicates [11]. But the reality is that simulation programs have thus far been unable to recreate the complexity of the underlying LD structure of the human genome. While we could use real 500 K genotype data and simulate unassociated traits, we would need to obtain many real 500 K GWAS data sets and then simulate many replicates of unassociated traits in each of them to adequately examine Type I error. Currently, this is a daunting task since the process just for obtaining the data from public databases is quite lengthy and the analysis time required to perform hundreds of GWAS analyses would be prohibitive.
By identifying the "independent" SNPs, we have significantly reduced the total number of SNPs to be used for Bonferroni correction in the set of SNP panels (Affymetrix and Illumina) and in HapMap. These "independent" SNPs provide us with a more accurate number of SNPs to include when adjusting for multiple testing using the Bonferroni correction. In addition, these p-values can assist in determining power for GWAS prior to genotyping so that only studies which can attain suggestive or significant association are pursued. We acknowledge that although we reduce the number of independent SNPS, the corresponding p-value cutoffs are still very low because we are analyzing more than 2 million SNPs without a specific biological hypothesis and stringency is still important. We need to balance identifying a true association while limiting Type 1 error.
We did evaluate the effects of the new thresholds on power using the Genetic Power Calculator to [15] determine the sample sizes we would need using a significance level based on all HapMap SNPs versus only the independent SNPs and blocks, as we recommend here. Table 7 provides different sample sizes using the 'LD adjusted' Bonferroni correction that we suggest here and the unadjusted Bonferroni correction in both CEU and YRI HapMap samples. Using the unadjusted Bonferroni correction would result in a necessary increase in sample size of 358–890 cases depending on the genotype relative risk and population. This increased burden of sample recruitment, collection and genotyping to adjust for "all" SNPs needs to be considered carefully, especially since many of the SNPs will be in strong LD and not contributing increased information.
Conclusion
The emerging trend towards genome wide association studies and large scale SNP genotyping warrants universal thresholds of significance, similar to those established by Lander and Kruglyak for LOD score genetic linkage analyses [16]. The dilemma facing many researchers is which regions to follow-up with dense SNPs or sequencing? To date, the most utilized threshold has been the arbitrary value set by the Wellcome Trust Case Control Consortium of 5 × 10-7 [17]. Interestingly, our Bonferroni LD-adjusted values are similar to these two thresholds (nominal p-value = 3.04 × 10-7 for CEU), but we also provide thresholds for suggestive and highly significant association. We believe the suggestive association threshold should be used to identify SNPs for consideration in follow-up studies, and both the significant and highly significant associations should be considered regions more likely of association. Of course, these thresholds are only guidelines that account for the interdependency of SNPs and investigators should carefully consider any regions with strong candidate genes or biologic plausibility even if they do not meet these thresholds. We also agree with the NHGRI/NCI working group on Replication in Association Studies that all statistically significant regions should be replicated using additional populations with adequate sample size to confirm any GWAS finding [18]. These thresholds should assist in replicating regions of true association.
References
Risch N, Merikangas K: The future of genetic studies of complex human diseases. Science. 1996, 273: 1516-1517. 10.1126/science.273.5281.1516.
A haplotype map of the human genome. Nature. 2005, 437: 1299-1320. 10.1038/nature04226.
Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Shen Y, Sun W, Wang H, Wang Y, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallee C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P, Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe'er I, Price A, Purcell S, Richter DJ, Sabeti P, Saxena R, Schaffner SF, Sham PC, Varilly P, Altshuler D, Stein LD, Krishnan L, Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A, Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR, Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS, Tsunoda T, Mullikin JC, Sherry ST, Feolo M, Skol A, Zhang H, Zeng C, Zhao H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD, Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole IF, Knoppers BM, Foster MW, Clayton EW, Watkin J, Gibbs RA, Belmont JW, Muzny D, Nazareth L, Sodergren E, Weinstock GM, Wheeler DA, Yakub I, Gabriel SB, Onofrio RC, Richter DJ, Ziaugra L, Birren BW, Daly MJ, Altshuler D, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM, Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT, Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M, Wallenburg JC, L'Archeveque P, Bellemare G, Saeki K, Wang H, An D, Fu H, Li Q, Wang Z, Wang R, Holden AL, Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS, Kennedy K, Jamieson R, Stewart J: A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007, 449: 851-861. 10.1038/nature06258.
Benjamini Y, Yekutieli D: Quantitative trait Loci analysis using the false discovery rate. Genetics. 2005, 171: 783-790. 10.1534/genetics.104.036699.
Benjamini Y, Hochberg Y: Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple testing. 1995, 289-300. 57
Storey JD, Tibshirani R: Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003, 100: 9440-9445. 10.1073/pnas.1530509100.
Dudbridge F: A note on permutation tests in multistage association scans. Am J Hum Genet. 2006, 78: 1094-1095. 10.1086/504527.
Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, Barnetson RA, Theodoratou E, Cetnarskyj R, Cartwright N, Semple C, Clark AJ, Reid FJ, Smith LA, Kavoussanakis K, Koessler T, Pharoah PD, Buch S, Schafmayer C, Tepel J, Schreiber S, Volzke H, Schmidt CO, Hampe J, Chang-Claude J, Hoffmeister M, Brenner H, Wilkening S, Canzian F, Capella G, Moreno V, Deary IJ, Starr JM, Tomlinson IP, Kemp Z, Howarth K, Carvajal-Carmona L, Webb E, Broderick P, Vijayakrishnan J, Houlston RS, Rennert G, Ballinger D, Rozek L, Gruber SB, Matsuda K, Kidokoro T, Nakamura Y, Zanke BW, Greenwood CM, Rangrej J, Kustra R, Montpetit A, Hudson TJ, Gallinger S, Campbell H, Dunlop MG: Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008, 40: 631-637. 10.1038/ng.133.
Marchini J, Howie B, Myers S, McVean G, Donnelly P: A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007, 39: 906-913. 10.1038/ng2088.
Sidak Z: Rectangular confidence regions for themeans of multivariate normal distributions. 1967, 626-633.
Nicodemus KK, Liu W, Chase GA, Tsai YY, Fallin MD: Comparison of type I error for multiple test corrections in large single-nucleotide polymorphism studies using principal components versus haplotype blocking algorithms. BMC Genet. 2005, 6 (Suppl 1): S78-10.1186/1471-2156-6-S1-S78.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, Defelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D: The structure of haplotype blocks in the human genome. Science. 2002, 296: 2225-2229. 10.1126/science.1069424.
Hudson RR, Kaplan NL: Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985, 111: 147-164.
Wang N, Akey JM, Zhang K, Chakraborty R, Jin L: Distribution of recombination crossovers and the origin of haplotype blocks: the interplay of population history, recombination, and mutation. Am J Hum Genet. 2002, 71: 1227-1234. 10.1086/344398.
Purcell S, Cherny SS, Sham PC: Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003, 19: 149-150. 10.1093/bioinformatics/19.1.149.
Lander E, Kruglyak L: Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995, 11: 241-247. 10.1038/ng1195-241.
Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. 2007, 661-678. 447
Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, Brooks LD, Cardon LR, Daly M, Donnelly P, Fraumeni JF, Freimer NB, Gerhard DS, Gunter C, Guttmacher AE, Guyer MS, Harris EL, Hoh J, Hoover R, Kong CA, Merikangas KR, Morton CC, Palmer LJ, Phimister EG, Rice JP, Roberts J, Rotimi C, Tucker MA, Vogan KJ, Wacholder S, Wijsman EM, Winn DM, Collins FS: Replicating genotype-phenotype associations. Nature. 2007, 447: 655-660. 10.1038/447655a.
Acknowledgements
This work was supported by the Intramural Program at the National Human Genome Research Institute, National Institutes of Health.
We would like to acknowledge the programming support of NHGRI's Bioinformatics and Scientific Programming Core. Specifically we would like to recognize Suiyuan Zhang.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
PD and EMG participated in the design and analysis of the study, interpreted the data, and participated in writing and revising the manuscript. TNH analyzed the data, and participated in revising the manuscript. JEBW participated in the design and interpretation of the study and participated in revising the manuscript. All authors have read and approved the final manuscript.
Priya Duggal, Elizabeth M Gillanders contributed equally to this work.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Duggal, P., Gillanders, E.M., Holmes, T.N. et al. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics 9, 516 (2008). https://doi.org/10.1186/1471-2164-9-516
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-9-516