
Abstract
3-Ketosteroid 9α-hydroxylase (KSH, consisting of KshA and KshB), a key enzyme in steroid metabolism, can catalyze the transformation of 4-androstene-3,17-dione (AD) to 9α-hydroxy-4-androstene-3,17-dione (9OHAD) with NADH as coenzyme. In this work, KSH from Mycobacterium neoaurum JC-12 was successfully cloned and overexpressed in Bacillus subtilis 168. The expression and purification of KSH was analyzed by SDS-PAGE and KSH activity assay. Preliminary characterization of KSH was performed using purified KshA and KshB. The results showed that KSH was very unstable, and its activity was inhibited by most metal ions, especially Zn2+. The whole-cells of recombinant B. subtilis, co-expression of KSH and glucose 1-dehydrogenase (GDH), were used as biocatalyst to convert AD to 9OHAD. The biocatalyst, in which the intracellular NADH was regenerated, efficiently catalyzed the bioconversion of AD to 9OHAD with a conversion rate of 90.4 % and productivity of 0.45 g (L h)−1, respectively. This work proposed a strategy for efficiently producing 9OHAD by using B. subtilis as a promising whole-cell biocatalyst host and co-expressing KSH and GDH to construct a NADH regeneration system.
Similar content being viewed by others
Background
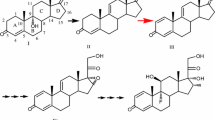
9α-Hydroxylated steroids are important precursors in the synthesis of steroidal hormone pharmaceuticals, which have been attracted increasing attention (Donova and Egorova 2012; Donova 2007; Fernandes et al. 2003). Highly specific reactions are required to produce functionalized compounds with therapeutic use and commercial value. Due to the high region- and stereo-selectivity of the reactions, together with the mild conditions required, the high yield biological production process, which are more environmentally friendly than their chemical synthesis counterparts, has been developed (Fernandes et al. 2003). Microbial fermentation has been wildly used to accumulate some important steroids intermediates, such as 4-androstene-3,17-dione (AD), androst-1,4-diene-3,17-dione (ADD) and 9α-hydroxy-4-AD (9OHAD) (Zhang et al. 2013; Shao et al. 2015a; Yuan et al. 2015). The 3-ketosteroid 9α-hydroxylase (KSH) and 3-ketosteroid-∆1-dehydrogenase (KSDD) are key enzymes in the process of microbial steroids degradation. KSH catalyzes the 9α-hydroxylation reaction of AD/ADD to 9OHAD/9α-hydroxy-4-ADD (9OHADD), whereas KSDD catalyzes the reaction of ∆1-dehydrogenation of AD/9OHAD to ADD/9OHADD. In this process, however, 9OHADD could subsequently form 3-hydroxy-9,10-secoandrost-1,3,5(10)-triene-9,17-dione (3HSA) by B-ring cleavage, spontaneously (Martin 1977; Kieslich 1985) (Fig. 1). Therefore, KSH combined with KSDD lead to the opening of the B-ring of steroid degradation.

Bioconversion of AD to 9ODAD and their degradation pathway. AD 4-androstene-3,17-dione, ADD androst-1,4-diene-3,17-dione, 9OHAD 9α-hydroxy-4-AD, 9OHADD 9α-hydroxy-4-ADD, KSH 3-ketosteroid 9α-hydroxylase, KSDD 3-ketosteroid-∆1-dehydrogenase, 3HSA 3-hydroxy-9,10-secoandrost-1,3,5(10)-triene-9,17-dione
The activity of KSH has been found in various bacterial genera, such as Mycobacterium (Wovcha et al. 1978; Brzostek et al. 2005; Van der Geize et al. 2007), Nocardia (Strijewski 1982), Rhodococcus (Van der Geize et al. 2002; Petrusma et al. 2009; Datcheva et al. 1989) and Arthrobacter (Dutta et al. 1992). Heterologous expression of ksh and characterization of KSH have been reported, and the conserved sequences analysis demonstrated that KSH is a Rieske monooxygenase. It belongs to class IA monooxygenase, including a terminal oxygenase (KshA) and a ferredoxin reductase (KshB) (Petrusma et al. 2009; Capyk et al. 2009; Arnell et al. 2007). It has been certified that KshA and KshB are essential for KSH activity by gene deletion studies of kshA and kshB (Andor et al. 2006). There were some reports about microbial fermentation from phytosterols to 9OHAD. However, due to low enzyme activities of steroids degradation pathway, they took long fermentation durations (about 120–144 h). For example, it has been reported that the mutant Mycobacterium sp. 2–4 M can be used to produce 9OHAD as a major product from sitosterol, with a 50 % molar yield (Donova et al. 2005). By using the resting Rhodococcus sp. cells to transform AD to 9OHAD, the substrate conversion ratio reached to about 85 % (Angelova et al. 1996). Generally, it is difficult to accumulate 9OHAD using fermentation method despite bacteria that can degrade steroids, because 9OHAD could be ∆1-dehydrogenated to 9OHADD and then spontaneously form 3HSA in these strains. Since the present of KSDD isoenzymes prevented the accumulation of intermediates (Van der Geize et al. 2000), deletion of all ksdd genes and overexpression of kshA resulted in accumulation of 9OHAD in Mycobacterium neoaurum (about 6.78–7.33 g L−1). However, the fermentation duration was more than 150 h (Yao et al. 2014). Thus, the strains which could accumulate 9OHAD, might be lack of KSDD or deficiency in KSDD (Seidel and Horhold 1992). Hence, double-stage fermentation method was developed to produce 9OHAD. The first step was the side-chain cleavage of sterols to form AD by one strain, and then the second step was 9α-hydroxylation of AD accomplished by another strain (Seidel and Horhold 1992).
Our laboratory has been devoted to using microorganisms to produce steroids intermediates with non-pollution and non-toxic biological technology (Shao et al. 2015a, 2016a). For example, we have co-expressed human 17β-hydroxysteroid dehydrogenase type 3 (17β-HSD3) and Saccharomyces cerevisiae glucose 6-phosphate dehydrogenase (G6PDH) to construct the NADPH regeneration system for efficient testosterone (TS) production form AD (Shao et al. 2016b). The M. neoaurum JC-12 (CCTCC No. M208135), capable of producing AD and ADD from phytosterol or cholesterol by fermentation method, was isolated with phytosterol as the sole carbon source from soil (Zhang et al. 2013). Genes of steroids degradation pathway from M. neoaurum JC-12 had been heterologous over-expressed to construct bioconversion system for steroids intermediates production. For example, cholesterol oxidase gene (choM) had been over-expressed in Bacillus subtilis for bioconversion of cholesterol to 4-cholesten-3-one (Shao et al. 2015b). 3-ketosteroid-∆1-dehydrogenase (ksdd) gene had been over-expressed in B. subtilis for bioconversion of AD to ADD (Zhang et al. 2013). The previous work indicated that B. subtilis might be a preferred host for bioconversion of steroids intermediates as compared with M. neoaurum strains. Hence, in this study, we cloned kshA and kshB gene from M. neoaurum JC-12 and first heterologously co-expressed them in B. subtilis 168. For efficiently bioconversion of AD to 9OHAD, glucose 1-dehydrogenase (GDH, EC 1.1.1.47, encoded by gdh gene) was co-expressed with KSH to construct a NADH regeneration system (Additional file 1: Fig. S1). The intracellular NADH concentration and the whole-cell bioconversion capability of recombinant B. subtilis were detected. This work provided a new reference for 9OHAD production.
Methods
Bacterial strains, plasmids and culture conditions
Mycobacterium neoaurum JC-12 (AD and ADD producing strain) was stored in our laboratory. B. subtilis 168 was purchased from Bacillus Genetic Stock Center (BGSC). The E. Coli and B. Subtilis shuttle vector pMA5 (HpaII, ColE1, repB, AmpR, KmR) was preserved in our lab (Zhang et al. 2013b). B. subtilis strains were cultivated at 37 °C and 160 rpm in LB medium (Luria–Bertani broth) with 0.5 % (w/v, weight/volume) glucose. Kanamycin (50 mg L−1) was added to the growth medium for selecting the recombinants. M. neorarum JC-12 was grown at 30 °C and 160 rpm in liquid medium containing 0.5 % (w/v) glucose, 0.5 % (w/v) tryptone, 0.3 % (w/v) beef extract, 1.5 % (w/v) glycerol and 1.5 % (w/v) NaCl. 2 % (w/v) agar was added during cultivation on solid medium.
Gene cloning and sequencing
Restriction enzymes and T4 DNA ligase were purchased from TaKaRa Co. (Dalian, China). Extraction and purification of plasmids were carried out by Mini Plasmid Rapid Isolation Kit (Sangon Biotech Co., Ltd., Shanghai, China). Isolation of DNA restriction fragment from agarose gels was done using the Mini DNA Rapid Purification Kit (Sangon Biotech Co., Ltd., Shanghai, China). Nucleotide sequence of kshA and kshB were analyzed by Sangon Biotech Co., Ltd. Shanghai, China. Protein and nucleotide sequences alignment were performed using the function of the BLAST server at NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Construction of recombinant B. subtilis 168/pMA5-ksh and B. subtilis 168/pMA5-ksh–gdh
Primers used in this work are listed in Table 2. The construction steps of these plasmids are shown in Additional file 1: Fig. S2. The kshA was cloned from chromosomal DNA of M. neoaurum JC-12 with the forward primer KshA-F and reverse primer KshA-R. Primers were originally designed with NdeI and MluI restriction sites to clone kshA into the plasmid pMA5. Gene cloning of kshB was performed with forward primer KshB-F and reverse primer KshB-R1. The PCR product was ligated onto pMA5 after digested by BamHI. The identification of pMA5-kshB was performed by digestion of MluI. Then, we got HpaII-kshB from pMA5-kshB with the forward primer HpaII-F1 and reverse primer KshB-R2 (containing His-Tag). The construction of pMA5-ksh was performed by ligation of HpaII-kshB, digested by EcoRI and SmaI, onto pMA5-kshA. For co-expression of ksh and gdh to construct a NADH regeneration system, plasmid of pMA5-gdh (previously constructed by us, homologous over-expression of GDH from B. subtilis 168) was used as the template and primer pair HpaII-F2/Gdh-R was used to amplify HpaII-gdh. The construction of B. subtilis/pMA5-ksh–gdh was performed by ligation of HpaII-gdh onto pMA5-ksh, which were digested by KpnI and HindIII. The primer sequences were listed in Table 1. Transformation of B. subtilis cells were carried out according to the procedure described by Anagnostopoulos and Spizizen (1961). The recombinant B. subtilis with pMA5-ksh was selected by resistance to kanamycin and confirmed by DNA sequencing.
Co-expression of KshA and KshB in B. subtilis 168 and protein purification
The recombinant plasmid pMA5-ksh was introduced into B. subtilis 168. Transformants were obtained after growing overnight on selective LB medium supplement with 50 mg L−1 of kanamycin. The recombinant cells were grown in LB medium (50 mL) for 24 h at 37 °C. Cell pellets (8000 rpm; 10 min; 4 °C) were washed with 100 mL of 50 mM Tris–HCl buffer (pH 7.0). Cell pellets were then resuspended in Tris–HCl buffer added with 5 mg lysozyme for 30 min and then sonicated for 10 min at 4 °C. Cell extracts were centrifuged for 30 min at 10,000 rpm in an SIGAMA 3K-15 centrifuge to remove cell debris. The purification of KSH-His was carried out by procedure described previously (Zhang et al. 2014). The final samples were verified via SDS-PAGE (12 % acrylamide).
Enzyme activity assay
The KSH activity was detected according to the procedure described previously (Petrusma et al. 2009). For KSH enzyme activity assay, the reaction mixture consists of 50 mmol L−1 Tris–HCl buffer (pH 7.0), 105 µmol L−1 NADH, 250 µmol L−1 AD dissolved in 2 % methanol, and 20–25 µg co-expressed KSH (KshA and KshB). For KshB enzyme activity assay, the reaction mixture consists of 50 mmol L−1 Tris–HCl buffer (pH 7.0), 0.1 mmol L−1 2,6-dichlorophenolindophenol, 0.25 mmol L−1 NADH, and 1–2 µg KshB was added to the assay. Assays were performed at room temperature. GDH activity was detected as the procedure described before (Hilt et al. 1991).
Determination of NADH and NAD+ concentrations
The intracellular NADH and NAD+ concentrations of recombinant B. subtilis strains were determined according to the manufacturers’ instructions of Amplite Fluorimetric NAD/NADH Ratio Assay Kit (15263) (Sunnyvale, USA). Sample preparation was followed by the procedure described previously (Bao et al. 2015).
Bioconversion of AD by recombinant B. subtilis 168
The bioconversion of AD was performed in shake flasks with the recombinant B. subtilis 168. The growth condition of B. subtilis strains was as previously described. After growing until late exponential phase (OD600 = 4–6), cells were collected by an SIGMA 3 K-15 centrifuge. Cell pellets were washed with 200 mL of 50 mmol L−1 Tris–HCl buffer (pH 7.0) for twice and then resuspended in 20 mL Tris–HCl buffer. AD and 0.2 % Tween-80 were then added into the bioconversion system. When using the NAD regeneration system as whole-cell biocatalyst, 1.5 % glucose was added as the substrate of GDH. Steroids extracted from the bioconversion solution (1 mL) by ethyl acetate were used for high-performance liquid chromatography (HPLC). For HPLC analysis, samples were diluted 5 times with ethyl acetate and filtered. Steroids were analyzed by HPLC with a reversed phase Diamonsil C18 at 35 °C using methanol–water (80: 20, v/v) solvent as mobile phase with a flow rate of 1 mL min−1, and subsequent detected via determination of UV absorption at 254 nm.
Results and discussion
kshA and kshB clone and sequence analysis
The amplification and sequences alignment of kshA and kshB genes were conducted as described in Materials and methods. The kshA gene of M. neoaurum JC-12 encodes a protein of 395 amino acids. Protein sequence analysis showed that typical conserved sequences of class IA terminal oxygenase (Van der Geize et al. 2002; Petrusma et al. 2009), the Rieske Fe2S2 binding domain (CXHX16CX2H, residues 65–87) and the non-heme Fe2+ motif (DX3DX2HX4H, residues 172–186) were found in this KshA protein. The kshB gene encodes 351 amino acids of KshB protein, which contains typical class IA monooxygenase reductase domains, a flavin-binding domain (RXYSL, residues 65–69), an NAD-binding domain (GSGITP, residues 129–134), and a [Fe2S2Cys4] domain (CX4CX2CX29C, residues 288–336). The sequences of kshA (GenBank: KR611532.1) and kshB (GenBank: KR611533.1) genes were then submitted to the GenBank database.
Co-expression, purification and characterization of KshA and KshB
The over-expressed KSH from M. neoaurum JC-12 in E. coli with the vector pET28a had been executed in our previous study, the recombinant proteins mainly existed in the form of inclusion bodies and no KSH activity was detected. Thus, the recombinant plasmids of pMA5-kshA, pMA5-kshB and pMA5-ksh (i.e., pMA5-kshA–kshB), which allowed the gene kshA and kshB to be expressed under the control of HpaII promoter, were constructed. After transformation of the recombinant plasmids into B. subtilis 168, they were then selected by using kanamycin as the selectable marker.
In this study, the possible expression of KSH by recombinant B. subtilis was investigated by SDS-PAGE (Fig. 2), and the analysis of proteins showed that KshA and KshB were successfully expressed. The enzyme activity of KshB, the reductase component of KSH, accepts electrons from NAD(P)H and transfers it to oxygenase component KshA, had been successfully detected in B. subtilis 168/pMA5-kshB with B. subtilis 168 as control (data not shown). The previous study strongly suggested that the cooperation of KshA and KshB are critically important for maintaining KSH activity (Petrusma et al. 2009). Hence, KSH activity could be only detected when KshA and KshB were co-expressed (Table 2). The KSH activity of B. subtilis 168/pMA5-ksh was 0.57 U (mg total protein)−1. However, KSH activity of M. neoaurum JC-12 were only about 0.02 U (mg total protein)−1, suggesting co-expression of KshA and KshB in engineered B. subtilis successfully improved KSH activity.

SDS-PAGE analysis of cell-free extract and purified KshA and KshB. Lanes: M protein marker (Takara Biotechnology Co., Ltd., Dalian, China); 1 cell-free extract of B. subtilis 168; 2 cell-free extract of B. subtilis 168/pMA5-kshA; 3 cell-free extract of B. subtilis 168/pMA5-kshB; 4 cell-free extract of B. subtilis 168/pMA5-ksh; 5 purified KshA (45.1 kDa); 6 purified KshB (37.8 kDa); 7 copurified KshA and KshB
KshA and KshB were purified with the C-terminal His-tag and showed KSH activity of 2.41 U mg−1. The maximum KSH activity of co-purified KshA and KshB was observed at 33 °C and pH 7.0. Measurements showed that a rather narrow pH range was needed for KSH activity. However, the enzyme was not stable when stored at −20 and 0 °C, and the KSH activity was reduced by 28 and 67 % after 24 h, respectively. No metal ions were found enhanced the KSH activity conspicuously. On the contrary, however, most metal ions were inhibitors of KSH, such as Fe3+, Co2+, Zn2+, Cu2+, Zn2+ and Ni2+, in which Zn2+ could inhibit KSH activity completely.
Construction of NADH regeneration system for biocatalysis of AD to 9OHAD
GDH from B. subtilis 168 and KSH from M. neoaurum JC-12 were co-expressed to construct the NADH regeneration system (B. subtilis 168/pMA5-ksh–gdh). SDS-PAGE analysis proved GDH and KSH were successfully co-expressed in B. subtilis (Fig. 3). Compared to B. subtilis 168, of which GDH activity was 0.03 U (mg total protein)−1, the KSH and GDH activities of B. subtilis 168/pMA5-ksh–gdh were 0.53 and 0.35 U (mg total protein)−1, respectively, indicating the functional over-expression of GDH.

SDS-PAGE analysis of KSH and GDH co-expressed in B. subtilis. Lanes: M protein marker (Takara Biotechnology Co., Ltd., Dalian, China); 1 cell-free extract of B. subtilis 168; 2 cell-free extract of B. subtilis 168/pMA5-ksh–gdh
The significant role of cofactors in the biocatalysts was proved by comparing the intracellular concentrations of NAD+ and NADH in the recombinant B. subtilis strains (Fig. 4). The results showed that the intracellular NADH pool in recombinant B. subtilis 168/pMA5-ksh–gdh was improved (17 %) as compared with B. subtilis 168/pMA5-ksh by over-expression of GDH, suggesting the NADH regeneration system was successfully constructed.

The intracellular NADH and NAD+ concentrations of recombinant strains B. subtilis 168/pMA5-ksh (a) and B. subtilis 168/pMA5-ksh–gdh (b) (all assays were performed by three independent biological repeats, and the standard deviations of the biological replicates were represented by error bars)
Steroid transformation analysis of the recombinant B. subtilis 168/pMA5-ksh and B. subtilis 168/pMA5-ksh–gdh
The biosynthesized 9OHAD in M. neoaurum strains could be subsequently transformed to 9OHADD, which then undergoes a nonenzymatic ring cleavage to form 3HSA. However, the recombinant B. subtilis 168 could catalyze the transformation of 9OHAD from AD in one step without any degradation of 9OHAD. Moreover, B. subtilis has been wildly used and regarded as a safe strain in industries (Schallmey et al. 2004).
While using whole-cells as biocatalyst, 1 g L−1 AD was added as substrate to validate the bioconversion efficiency of different strains. The results showed that B. subtilis 168/pMA5-ksh and B. subtilis 168/pMA5-ksh–gdh successfully catalyzed the transformation of AD to 9OHAD (Fig. 5). However, as controls, no 9OHAD was detected during AD bioconversion by whole-cells of M. neoaurum JC-12, B. subtilis 168, B. subtilis 168/pMA5-kshA and B. subtilis 168/pMA5-kshB. Although both B. subtilis 168/pMA5-ksh and B. subtilis 168/pMA5-ksh–gdh showed significant improved KSH activity, NAD+ was only continuously regenerated by GDH in B. subtilis 168/pMA5-ksh–gdh to keep a persistently 9OHAD productivity. The maximum conversion rate (g g−1) of B. subtilis 168/pMA5-ksh was 70.1 % at 16 h, while B. subtilis 168/pMA5-ksh–gdh showed a conversion rate (g g−1) of 96.3 % at 2 h, indicating cofactor regeneration increased the conversion rate of the biocatalyst. After the batch conversion of B. subtilis 168/pMA5-ksh–gdh, the pH of the reaction mixture was decreased from 7.0 to about 6.3. Thus, the pH variation had little effect on this system.

Conversion rate of AD to 9ODAD by recombinant strains B. subtilis 168/pMA5-ksh (X1) and B. subtilis 168/pMA5-ksh–gdh (X2) (all assays were performed by three independent biological experiments, and the standard deviations of the biological replicates were represented by error bars)
From above experiments, B. subtilis 168/pMA5-ksh–gdh was expected a good candidate for transforming of AD to 9OHAD. However, due to the low solubility of the steroid substrates and products in aqueous conversion system, the repeated batch strategy of bioconversion from AD to 9OHAD was done in this work. While using 1 g L−1 AD as substrate for repeated batch bioconversion, the whole-cells of B. subtilis 168/pMA5-ksh–gdh could continuously transform total 8 g L−1 AD to about 7.23 g L−1 9OHAD within 16 h with a conversion rate (g g−1) of 90.4 % and productivity of 0.45 g (L h)−1 (Fig. 6). However, the enzyme activity of whole-cell biocatalyst decreased greatly (left about 38.9 % KSH activity and 30.6 % GDH activity) after 16 h. Thus the conversion duration was controlled within 16 h. In summary, the successful expression of M. neoaurum KSH in B. subtilis and the construction of NADH regeneration system made it possible to achieve one-step efficient transformation of AD to 9OHAD.

Repeated batch conversion of AD to 9ODAD by recombinant strain B. subtilis 168/pMA5-ksh–gdh. Y1: 9ODAD production of batch conversion, Y2: total 9ODAD production of repeated conversion (all assays were performed by three independent biological experiments, and the standard deviations of the biological replicates were represented by error bars)
Conclusions
9OHAD, an important precursor in the synthesis of steroid pharmaceuticals, can be produced by microorganisms using fermentation method. However, due to the long fermentation duration and low substrate conversion rate, the productivity of biosynthesis of 9OHAD cannot meet the need of industrial production. This work cloned and over-expressed M. neoaurum KSH and B. subtilis GDH to construct a NADH regeneration system (B. subtilis pMA5-ksh–gdh), which could efficiently transform AD to 9OHAD. By using the NADH regeneration system as a biocatalyst integrated the repeated batch conversion strategy, 9OHAD production was improved to 7.23 g L−1 with a conversion rate of 90.4 % and productivity of 0.45 g (L h)−1. The results demonstrated that the NADH regeneration system of recombinant B. subtilis strain can be used in 9OHAD production. However, the low solubility of the steroid substrates in aqueous conversion system limits extremely the bioconversion rate. Thus, aqueous-organic two-phase systems and cloud point systems will be considered to be applied in the future research to improve the conversion rate of AD to 9OHAD (Wang et al. 2005).
References
Anagnostopoulos C, Spizizen J (1961) Requirements for transformation in Bacillus subtilis. J Bacteriol 81(5):741–746
Andor A, Jekkel A, Hopwood DA, Jeanplong F, Ilkoy E, Konya A, Kurucz I, Ambrus G (2006) Generation of useful insertionally blocked sterol degradation pathway mutants of fast-growing mycobacteria and cloning, characterization, and expression of the terminal oxygenase of the 3-ketosteroid 9 alpha-hydroxylase in Mycobacterium smegmatis mc(2)155. Appl Environ Microbiol 72(10):6554–6559
Angelova B, Mutafov S, Avramova T, Dimova I, Boyadjieva L (1996) 9Alpha-hydroxylation of 4-androstene-3,17-dione by resting Rhodococcus sp. cells. Process Biochem 31(2):179–184
Arnell R, Johannisson R, Lindholm J, Fornstedt T, Ersson B, Ballagi A, Caldwell K (2007) Biotechnological approach to the synthesis of 9 alpha-hydroxylated steroids. Prep Biochem Biotechnol 37(4):309–321
Bao T, Zhang X, Zhao X, Rao Z, Yang T, Yang S (2015) Regulation of the NADH pool and NADH/NADPH ratio redistributes acetoin and 2,3-butanediol proportion in Bacillus subtilis. Biotechnol J. doi:10.1002/biot.201400577
Brzostek A, Sliwinski T, Rumijowska-Galewicz A, Korycka-Machala M, Dziadek J (2005) Identification and targeted disruption of the gene encoding the main 3-ketosteroid dehydrogenase in Mycobacterium smegmatis. Microbiology 151(Pt 7):2393–2402
Capyk JK, D’Angelo I, Strynadka NC, Eltis LD (2009) Characterization of 3-Ketosteroid 9 alpha-hydroxylase, a rieske oxygenase in the cholesterol degradation pathway of Mycobacterium tuberculosis. J Biol Chem 284(15):9937–9946
Datcheva VK, Voishvillo NE, Kamernitskii AV, Vlahov RJ, Reshetova IG (1989) Synthesis of 9 alpha-hydroxysteroids by a Rhodococcus sp. Steroids 54(3):271–286
Donova MV (2007) Transformation of steroids by actinobacteria: a review. Prikl Biokhim Mikrobiol 43(1):5–18
Donova MV, Egorova OV (2012) Microbial steroid transformations: current state and prospects. Appl Microbiol Biotechnol 94(6):1423–1447
Donova MV, Gulevskaya SA, Dovbnya DV, Puntus IF (2005) Mycobacterium sp mutant strain producing 9 alpha-hydroxyandrostenedione from sitosterol. Appl Microbiol Biotechnol 67(5):671–678
Dutta RK, Roy MK, Singh HD (1992) Role of plasmid pjl1 of Arthrobacter oxydans 317 in the degradation of beta-sitosterol. J Basic Microbiol 32(5):317–324
Fernandes P, Cruz A, Angelova B, Pinheiro HM, Cabral JMS (2003) Microbial conversion of steroid compounds: recent developments. Enzyme Microb Technol 32(6):688–705
Hilt W, Pfleiderer G, Fortnagel P (1991) Glucose dehydrogenase from Bacillus subtilis expressed in Escherichia coli. I: purification, characterization and comparison with glucose dehydrogenase from Bacillus megaterium. Biochim Biophys Acta 1076(2):298–304
Kieslich K (1985) Microbial side-chain degradation of sterols. J Basic Microbiol 25(7):461–474
Martin CK (1977) Microbial cleavage of sterol side chains. Adv Appl Microbiol 22:29–58
Petrusma M, Dijkhuizen L, van der Geize R (2009) Rhodococcus rhodochrous DSM 43269 3-ketosteroid 9alpha-hydroxylase, a two-component iron-sulfur-containing monooxygenase with subtle steroid substrate specificity. Appl Environ Microbiol 75(16):5300–5307
Schallmey M, Singh A, Ward OP (2004) Developments in the use of Bacillus species for industrial production. Can J Microbiol 50(1):1–17
Seidel L, Horhold C (1992) Selection and characterization of new microorganisms for the manufacture of 9-OH-AD from sterols. J Basic Microbiol 32(1):49–55
Shao ML, Zhang X, Rao ZM, Xu MJ, Yang TW, Li H, Xu ZH (2015a) Enhanced production of androst-1,4-diene-3,17-dione by Mycobacterium neoaurum JC-12 using three-stage fermentation strategy. PLoS ONE 10(9):e0137658
Shao ML, Rao ZM, Zhang X, Xu MJ, Yang TW, Li H, Xu ZH, Yang ST (2015b) Bioconversion of cholesterol to 4-cholesten-3-one by recombinant Bacillus subtilis expressing choM gene encoding cholesterol oxidase from Mycobacterium neoaurum JC-12. J Chem Technol Biotechnol 90(10):1811–1820
Shao M, Zhang X, Rao Z, Xu M, Yang T, Li H, Xu Z, Yang S (2016a) A mutant form of 3-ketosteroid-delta-dehydrogenase gives altered androst-1,4-diene-3, 17-dione/androst-4-ene-3,17-dione molar ratios in steroid biotransformations by Mycobacterium neoaurum ST-095. J Ind Microbiol Biotechnol. doi:10.1007/s10295-016-1743-9
Shao M, Zhang X, Zhiming R, Meijuan X, Yang T, Li H, Zhenghong X, Shangtian Y (2016b) Efficient testosterone production by engineered Pichia pastoris co-expressing human 17β-hydroxysteroid dehydrogenase type 3 and Saccharomyces cerevisiae glucose 6-phosphate dehydrogenase with NADPH regeneration. Green Chem 18:1774–1784
Strijewski A (1982) The steroid-9 alpha-hydroxylation system from Nocardia species. Eur J Biochem 128(1):125–135
Van der Geize R, Hessels GI, van Gerwen R, Vrijbloed JW, van der Meijden P, Dijkhuizen L (2000) Targeted disruption of the kstD gene encoding a 3-ketosteroid Delta(1)-dehydrogenase isoenzyme of Rhodococcus erythropolis strain SQ1. Appl Environ Microbiol 66(5):2029–2036
Van der Geize R, Hessels GI, van Gerwen R, van der Meijden P, Dijkhuizen L (2002) Molecular and functional characterization of kshA and kshB, encoding two components of 3-ketosteroid 9alpha-hydroxylase, a class IA monooxygenase, in Rhodococcus erythropolis strain SQ1. Mol Microbiol 45(4):1007–1018
Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci USA 104(6):1947–1952
Wang ZL, Zhao FS, Chen DJ, Li DT (2005) Cloud point system as a tool to improve the efficiency of biotransformation. Enzyme Microb Technol 36(4):589–594
Wovcha MG, Antosz FJ, Knight JC, Kominek LA, Pyke TR (1978) Bioconversion of sitosterol to useful steroidal intermediates by mutants of Mycobacterium fortuitum. Biochim Biophys Acta 531(3):308–321
Yao K, Xu LQ, Wang FQ, Wei DZ (2014) Characterization and engineering of 3-ketosteroid-∆1-dehydrogenase and 3-ketosteroid-9alpha-hydroxylase in Mycobacterium neoaurum ATCC 25795 to produce 9alpha-hydroxy-4-androstene-3,17-dione through the catabolism of sterols. Metab Eng 24:181–191
Yuan J, Chen G, Cheng S, Ge F, Qiong W, Li W, Li J (2015) Accumulation of 9alpha-hydroxy-4-androstene-3,17-dione by co-expressing kshA and kshB encoding component of 3-ketosteroid-9alpha-hydroxylase in Mycobacterium sp. NRRL B-3805. Sheng Wu Gong Cheng Xue Bao 31(4):523–533
Zhang W, Shao M, Rao Z, Xu M, Zhang X, Yang T, Li H, Xu Z (2013a) Bioconversion of 4-androstene-3,17-dione to androst-1,4-diene-3,17-dione by recombinant Bacillus subtilis expressing ksdd gene encoding 3-ketosteroid-delta1-dehydrogenase from Mycobacterium neoaurum JC-12. J Steroid Biochem Mol Biol 135:36–42
Zhang X, Zhang R, Bao T, Yang T, Xu M, Li H, Xu Z, Rao Z (2013b) Moderate expression of the transcriptional regulator ALsR enhances acetoin production by Bacillus subtilis. J Ind Microbiol Biotechnol 40(9):1067–1076
Zhang X, Zhang RZ, Bao T, Rao ZM, Yang TW, Xu MJ, Xu ZH, Li HZ, Yang ST (2014) The rebalanced pathway significantly enhances acetoin production by disruption of acetoin reductase gene and moderate-expression of a new water-forming NADH oxidase in Bacillus subtilis. Metab Eng 23:34–41
Authors’ contributions
ZX, ZL and RZ designed the study and performed the data analysis. ZX, ZL and XM carried out the laboratory work of the study. ZX, ZL and YT wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by the High-tech Research and Development Programs of China (2011AA02A211, 2015AA021004, 2014AA021304), the National Natural Science Foundation of China (31570085, 31500065), the Jiangsu Province Science Fund for Distinguished Young Scholars (BK20150002), the Natural Science Foundation of Jiangsu Province (BK20150142), the China Postdoctoral Science Foundation Funded Project (2015M570407, 2016T90421), the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 111 Project (111-2-06), the Program of the Key Laboratory of Industrial Biotechnology, Ministry of Education, China (KLIB-KF201406), the Fundamental Research Funds for the Central Universities (JUSRP11545), and the Jiangsu province “Collaborative Innovation Center for Advanced Industrial Fermentation” industry development program.
Author information
Authors and Affiliations
Corresponding author
Additional file
40064_2016_2871_MOESM1_ESM.docx
Additional file 1: Figure S1. The NADH regeneration system constructed by this work. Figure S2. Construction steps of the plasmids used in this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, X., Rao, Z., Zhang, L. et al. Efficient 9α-hydroxy-4-androstene-3,17-dione production by engineered Bacillus subtilis co-expressing Mycobacterium neoaurum 3-ketosteroid 9α-hydroxylase and B. subtilis glucose 1-dehydrogenase with NADH regeneration. SpringerPlus 5, 1207 (2016). https://doi.org/10.1186/s40064-016-2871-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-2871-4