
Abstract
Proteinopathy, defined as the abnormal accumulation of proteins that eventually leads to cell death, is one of the most significant pathological features of neurodegenerative diseases. Tauopathies, represented by Alzheimer’s disease (AD), and synucleinopathies, represented by Parkinson’s disease (PD), show similarities in multiple aspects. AD manifests extrapyramidal symptoms while dementia is also a major sign of advanced PD. We and other researchers have sequentially shown the cross-seeding phenomenon of α-synuclein (α-syn) and tau, reinforcing pathologies between synucleinopathies and tauopathies. The highly overlapping clinical and pathological features imply shared pathogenic mechanisms between the two groups of disease. The diagnostic and therapeutic strategies seemingly appropriate for one distinct neurodegenerative disease may also apply to a broader spectrum. Therefore, a clear understanding of the overlaps and divergences between tauopathy and synucleinopathy is critical for unraveling the nature of the complicated associations among neurodegenerative diseases. In this review, we discuss the shared and diverse characteristics of tauopathies and synucleinopathies from aspects of genetic causes, clinical manifestations, pathological progression and potential common therapeutic approaches targeting the pathology, in the aim to provide a timely update for setting the scheme of disease classification and provide novel insights into the therapeutic development for neurodegenerative diseases.
Similar content being viewed by others
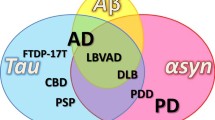

Background
Tauopathies are defined as a spectrum of neurodegenerative diseases with pathological features of intracellular (neuronal or glial) deposition of hyperphosphorylated tau, forming neurofibrillary tangles (NFTs) [1]. So far, more than 26 neurological diseases have been identified as tauopathies, including Alzheimer’s disease (AD), Pick’s disease, and progressive supranuclear palsy (PSP), manifesting symptoms of dementia and motor deficits [2]. Tauopathies can be classified as primary or secondary depending on whether other proteinopathies are involved, such as extracellular Aβ plaque formation. For example, frontotemporal dementia (FTD), in which tau is the dominant composition of protein aggregates, is a primary tauopathy [3, 4]. On the contrary, AD is a secondary tauopathy, possessing both NFTs and Aβ plaques [2, 5].
Certain series of neurodegenerative diseases can be pathologically characterized by their common “driving-force” proteins, such as tauopathies by tau [6]. Similarly, synucleinopathies including Parkinson’s disease (PD), Lewy body dementia (DLB) [7] and multiple system atrophy (MSA) are characterized by aggregation of α-synuclein (α-syn) [8]. Based on the cellular deposition of α-syn, synucleinopathies can be categorized into Lewy body diseases with α-syn inclusions in neurons forming Lewy bodies (LB) and Lewy neurites (LN), and MSA with α-syn aggregation in glia forming glial cytoplasmic inclusions [9]. Synucleinopathies mainly manifest motor deficits appearing as parkinsonism, such as tremor, rigidity, and bradykinesia in PD. However, in the early stages of diseases, non-motor symptoms such as rapid eye movement sleep behavior disorder are also present. Meanwhile, cognitive decline is also observed in synucleinopathies such as DLB [10].
Genome-wide association studies have revealed that synucleinopathies and tauopathies share genetic risks such as MAPT (encoding microtubule-associated protein tau) mutations [11,12,13,14,15]. Also, association between SNCA (α-syn-encoding gene) polymorphism and increased risk of AD has been reported [16]. A known risk locus of AD, apolipoprotein E (APOE), is also a genetic risk factor for DLB [17, 18]. The APOE allele status has been associated with both PD onset and its progression to Parkinson’s disease dementia (PDD) [19, 20]. Clinically, familial FTD (FTDP-17) exhibits motor symptoms of parkinsonism, while 83% of PD cases progress into PDD in the late stage [8, 21,22,23]. Pathologically, tau and α-syn have been shown to cross-seed each other in the progression of aggregation formation [24]. In summary, tauopathies and synucleinopathies have overlapping characteristics in etiology, pathology and clinical manifestations. The interacting and independent features of both pathological proteins and groups of diseases may indicate the possibility of developing differential diagnosis and pathology-targeting therapeutic interventions.
In this review, we summarize the overlapping and the divergent features of tauopathies and synucleinopathies, from perspectives of genetic risk to pathological development, with a special focus on pathology continuum between tau and α-syn aggregation. Furthermore, we discuss possible immunotherapies targeting pathology spreading that can be used for both tauopathies and synucleinopathies. A summary of examples of tauopathies and synucleinopathies and their pathological features is presented in Table 1.
Genetic overlap between tauopathies and synucleinopathies
MAPT in tauopathies
The MAPT gene is located on chromosome 17q21.31 and consists of 15 exons. MAPT has two haplotypes due to the inversion of the sequence on the 17q21 chromosome [38]. The H1 haplotype is more frequent in humans [39]. The alternative splicing of MAPT exons gives rise to six isoforms of tau protein depending on the number of microtubule-binding repeat domains (MTBD) (3R, 4R) and N-terminal inserts (0N, 1N, 2N) [40, 41] (Fig. 1). Over 50 mutations in MAPT have been identified to be related to neurodegenerative diseases, most of which are missense mutations [42, 43]. Around 30% of primary tauopathy cases are associated with pathogenic mutations of MAPT [43]. In the coding regions of MAPT, the P301L mutation on exon 10 is the most prevalent. It has been shown to cause an inherited form of FTD, and induce pathogenesis in FTD, corticobasal syndrome (CBS) and globular glial tauopathies [44, 45]. In addition to the autosomal dominant mutations, MAPT variants also serve as a risk factor for primary tauopathies. In sporadic PSP, MAPT mutations such as P301L, R5L and S285R are the strongest genetic risk factors [46,47,48]. In CBS, MAPT is the second most common genetic risk factor, only next to the progranulin-coding gene [49]. MAPT mutations mainly induce downstream MAPT mRNA splicing deficiency and structural changes in tau protein, leading to weak binding of tau to microtubule, alterations of the 4R/3R ratio and the sequential tau aggregation [50].

A schematic showing mutations of MAPT and composition of tau isoforms. The H1 and H2 haplotypes are formed by the 900 kb inversion in the q21.3 region of chromosome 17. The H1 haplotype is often the one contributing to disease initiation due to multiple missense mutations from exon 1 to 13, especially on exon 10. The tau protein can be classified into 6 isoforms depending on the number of amino inserts (0N, 1N, 2N) on exons 2 and 3 and the number of microtubule-binding domains (3R, 4R). Exon 10 encodes the R2 domain, and its alternative splicing produces 3R or 4R tau isoforms. The microtubule-binding domain contains hexapeptide motifs VQIINK in R2 and VQIVYK in R3. Interactions between the two motifs promote dimer formation of tau [1, 2, 73]
SNCA in synucleinopathy
The SNCA gene is mapped to chromosome 4q22.1 and consists of 6 exons with the last 5 translatable to α-syn protein, which is pathologically responsible for synucleinopathies [51]. The SNCA gene plays a prominent role in the onset of synucleinopathies especially PD, both as a causative gene and as risk variants [52]. Similar to MAPT, exon splicing of SNCA gives rise to alternative transcripts. Both missense mutation and gene multiplication of SNCA play important roles in pathology initiation. In the 1990s, the A53T autosomal dominant mutation was found to be related to the early onset of PD in the Contursi Kindred [53]. SNCA genetic variants such as E46K [54], H50Q [55], G51D [56] and A30P [57], have been revealed to be associated with the familial forms of synucleinopathies, especially PD. Point mutations such as G51D and multiplication of SNCA can promote formation of the amyloid structure of α-syn and facilitate the progression of pathology in synucleinopathies [54, 56, 58, 59]. Thus, gene coding dosage mutations play an important role in the progress of synucleinopathies. SNCA triplication leads to early-onset PD with symptoms of dementia [59], while SNCA duplication is associated with late-onset PD [58].
MAPT and SNCA correlate in the onset of proteinopathies
MAPT and SNCA gene variants have been shown to be correlated with each other in the etiology of various proteinopathies. First, synucleinopathy and tauopathy have been detected with overlapping presence of mutations of the two genes. Meta-analyses of genome-wide association studies have repeatedly spotted MAPT H1 haplotype as a risk locus for PD onset [60]. Although not among the entire spectrum of diseases, certain tauopathies and synucleinopathies have been shown to share genetic risks. AD and DLB, while both presenting dementia as the main clinical symptom, share APOE and BIN1 (bridging integrator-1) as risk genetic loci [18]. Genome-wide analysis revealed that the global genetic correlation between AD and DLB is as high as 0.578, while that between PD and AD is 0.08 [61]. The correlation between genetic risks of AD and ALS is also significant, with shared locus of TSPOAP1-AS1 [62]. The MAPT inversion polymorphism increases the risk of PD and is strongly associated with the development of dementia among PD patients [63]. Carriers of LRRK2 (leucine-rich repeat kinase 2) mutations, which are the most common cause of familial PD, show abundant tau deposition [64]. There are also cases of MAPT mutation working independently of SNCA to cause Parkinsonism, showing the possibility that some tauopathies may share pathogenic pathways with synucleinopathies [65, 66].
Besides the direct overlaps between MAPT and SNCA, tauopathies and synucleinopathies also share common pathogenic pathways [67]. SNCA gene point mutations are associated with impairment of key cellular functions, such as tubulin binding, as some point mutations of SNCA map to the putative tubulin-binding site, paving the way to tauopathies [68]. In addition, two genes, MAPT and HLA (human leukocyte antigen), and 10 pathways such as proteolytic signatures, overlap between AD and PD [69, 70]. Studies have shown that the tauopathy-related MAPT variants and the synucleinopathy-associated SNCA mutation may make neurons more vulnerable to cellular dysfunction such as mitochondrial deficits, setting similar mechanistic paths towards neurodegenerative cell death [71].
Structural basis for the formation of tau and α-syn pathology
Structural basis for tau aggregation
The six tau protein isoforms produced by alternative mRNA splicing of MAPT range from 352 to 441 amino acids [72]. Tau has physiological functions of stabilizing microtubules and maintaining axonal transport in highly polarized neurons [73]. The N-terminus of tau directly binds to the microtubules, laying the structural basis for tau physiological function [74]. The central domain of tau is highly disordered and rich in proline [75]. The C-terminus of tau contains three (3R) or four (4R) MTBD due to the splicing of MAPT on exon 10. The number of MTBDs affects the microtubule-binding affinity and the propensity of tau to aggregate [76]. Under physiological conditions, the protein structure of tau possesses a large range of variation. A previous study showed that the soluble native tau has multiple conformations in cytoplasm [77]. The initiation of tau aggregation lies in its own structure, i.e., the two hexapeptide sequences in the second and third MTBDs, laying the foundation for fibrilization [78].
Pathological assembly of soluble tau and post-translational modification (PTMs) of tau may result in the formation of insoluble NFTs, leading to neuronal dysfunction such as synaptic dysfunction and altered mitochondrial trafficking [43, 79]. The starting point of tau aggregation is the dimerization through interactions between the two hexapeptide motifs VQIVYK (located at the beginning of the third MTBD, thus present in both 3R and 4R isoforms) and VQIINK (located at the beginning of the second MTBD, thus present only in 4R isoforms) [78, 80]. Dimerization of tau serves as the core for sequential nucleation and elongation [81]. Elongated oligomers further serve as the template for amyloid formation with β-sheet structure, which eventually forms paired helical filaments (PHFs), giving rise to highly structured polymorphs seen in NFTs [82]. Genetic mutations such as P301L [83], and PTMs such as multi-site hyperphosphorylation [84, 85], are believed to facilitate tau aggregation. For example, the P301L tau mutation results in alterations of the hexapeptides, making tau more susceptible to aggregation [83]. A diagram of mutations in MAPT and tau isoforms is illustrated in Fig. 1.
Structural basis for α-syn aggregation
α-Syn contains 140 amino acids encoded by the SNCA gene [86]. Little is known about the physiological functions of the protein. α-Syn has been shown to play a role in regulating synaptic neurotransmitter release [87]. The protein contains a N-terminal amphipathic region, a central amyloidal region and a C-terminal acidic domain (Fig. 2). The N-terminus of α-syn has lipid-binding properties and facilitates the initiation of aggregation. The central region is also named as the non-amyloid-β component core, which contains an amyloidogenic domain (61-95) [88]. The C-terminus is the structural basis for the unfolded nature of the protein [89]. α-Syn aggregation is partly due to its own natively unfolded structure and various pathogenetic SNCA mutations, as mentioned above [90]. Physiologically, the protein executes its neurological function in the cytoplasm in the forms of monomers and oligomers. Genetic instability, introduction of external amyloid seeds, dys-homeostasis of membrane structure, etc., can disrupt the equilibrium of α-syn monomers and oligomers, induce abnormal membrane binding and cause initiation of aggregation [91]. During the process of α-syn aggregate formation, PTMs play important roles (Fig. 2). Phosphorylation of α-syn at serine 129 is considered as a pathological marker of LB formation. About 90% of the α-syn extracted from synucleinopathy patient brains is phosphorylated at serine 129 [92]. Under pathological conditions, α-syn converts from the monomeric or oligomeric forms to the β-sheet structures. The β-sheets serve as the template and seed soluble monomers to generate protofibrils and eventually amyloid fibrils [91, 93]. The processes of tau and α-syn aggregation are illustrated in Fig. 3.

Structure, mutations and PTMs of α-syn protein. α-Syn has three domains: the amphipathic N-terminus, the non-amyloid-β component (NAC) which is prone to aggregate, and the acidic C-terminus. Common point mutations of α-syn are A30P, E46K, H50Q, G51D, A53T and A53E. The PTMs of α-syn mainly include phosphorylation, nitration, acetylation, ubiquitination and SUMOylation [43, 162]

Processes of tau and α-syn aggregation. Under pathological conditions, α-syn accumulates and forms the β-sheet structure in the cytoplasm, which then becomes an amyloid fibrillary structure, the main component of LBs. On the other hand, tau monomers become hyperphosphorylated and form numeric molecules, which then aggregate into protofilaments and form PHFs, eventually becoming NFTs
Tau and α-syn pathology spreading
Postmortem studies of AD and PD have revealed the possibility of intercellular spreading of tau and α-syn, either to adjacent cells or between brain regions and peripheral organs [94,95,96]. Pathological staging using AD patient brain tissues has outlined a path of NFT deposition, beginning in the locus coeruleus and spreading to the entorhinal region and hippocampus, and eventually to the neocortex. The predictable manner of tau spreading correlates with the progress of cognitive decline [97, 98]. α-Syn spreads from the periphery to the brain following the Braak staging [99]. Typically, misfolded and aggregated α-syn in the dorsal motor nucleus of vagus can spread to other brain regions. Enteric deposition of LBs in early PD and incidental LB disease (iLBD) cases has also been reported [100], suggesting the propagating pathway from the peripheral enteric nervous system to the central nervous system (CNS) via the vagal nerve [101].
The cell-to-cell transfer of both tau and α-syn are hypothesized to follow a “prion-like” manner [102,103,104,105,106,107]. The molecular mechanisms by which the external aggregated seeds from donor cells enter recipient cells are still unknown. In the recipient cells, the seeds provoke the unfolding of the native monomeric proteins and initiate sequential aggregate formation. This is rather a process of dissemination than simple endocytosis and exocytosis.
The processes of tau and α-syn release and uptake are not fully understood. It is speculated that tau mainly spreads between cells through synaptic connectivity, rather than being predicted by proximity [98, 99]. Tau and α-syn both enter and exit cells through endocytosis, exosomes [91] and tunneling nanotubes [108, 109]. In addition, receptor proteins that mediate cell-to-cell transmission of both tau and α-syn have been reported. Lipoprotein receptor-related protein 1 (LRP1) has been shown to regulate the cellular entry of α-syn monomers and oligomers. Knock-out of LRP1 in mice significantly reduces the uptake of α-syn [110]. LRP1 is also a receptor for tau endocytosis, as blocking LRP1 significantly reduces tau uptake in neuroglioma cells and stem cell-derived neurons [111]. Lymphocyte activation gene 3 (LAG3) is necessary for receptor-mediated α-syn endocytosis [112]. Meanwhile, depletion of LAG3 can also decrease the uptake of tau by primary neurons [113].
When acting on cells, both tau and α-syn induce neuroinflammation and oxidative stress, which are common pathogenic features of neurodegeneration [114, 115]. For example, in primary microglial culture, preformed tau fibrils activate microglia in an NF-κB-dependent manner [116]. In addition, P301S tau transgenic mice show activation of astrocytes [117]. In synucleinopathies, longitudinal gene profiling studies have revealed the essential role of microgliosis in the early stages of PD, preceding the course of neurodegeneration [118]. Moreover, impairment in mitochondrial function [119, 120], synaptic transmission [121] and autophagy [122, 123] has been revealed in both proteinopathies.
The intercellular propagation of tau and α-syn pathologies follow preferential routes and share mechanisms such as exosomes and receptor-mediated endocytosis [124]. The similar spreading pathways and cellular toxicity between synucleinopathy and tauopathy may serve as a reference for developing pathology-targeting therapeutic interventions.
Pathological continuum between tauopathies and synucleinopathies
Co-occurrence of tau and α-syn pathology
Both tau and α-syn have a structural basis that confers an aggregation-prone property, leading to the typical pathological progress of tauopathy and synucleinopathy [86]. Co-presence of tau and α-syn has been shown in various diseases and models of neurodegeneration. Brain deposition of α-syn is found in more than 50% of AD patients [125, 126]. α-Syn was first identified in the AD brain in 1993 by Ueda et al., which then was identified as a non-Aβ peptide fragment in the isolated Aβ plaques [127]. However, in later studies, the co-localization of α-syn was found to a greater extent with tau than with Aβ in AD brains [125]. Lippa et al. demonstrated that the insoluble forms of α-syn were present mainly in the amygdala of familial AD patients, some of which co-localized with tau NFTs [128]. In 145 sporadic AD patients examined in the study by Hamilton et al., LBs were found in the brains of more than 60% patients, predominantly in the amygdala, rarely in the substantia nigra [126]. Uchikado et al. found that in 260 AD patients 62 cases had amygdala LBs, and in most of these cases, NFTs and LBs were found in the same cells [129]. The distribution pattern of LBs in AD patients differs significantly from that in PD, with absence of LBs in the brainstem, but more in the limbic and olfactory regions [130]. Tau fibrilization was also observed in PD patients carrying the A53T mutation [131]. Colocalization of tau and α-syn epitopes has been detected in LBs in multiple studies [132, 133]. In 80% cases of AD and diffused LB disease, tau immunoreactivity, especially phosphorylated tau, was spotted in LBs in the medulla [132]. In familial PD and DLB cases, PHF tau antibody can partially label LBs in the same neuronal cell, suggesting the co-occurrence of tau and α-syn in synucleinopathy brains [133]. The co-existence of tau and α-syn indicates the overlapping of pathology formation in both groups of disease.
Tau/α-syn co-aggregates in pathology spreading
Compared to a singular administration of tau or α-syn, the existence of co-pathology of α-syn and tau has stronger effects in facilitating pathological spreading of the proteins. In mice injected with both AD patient brain lysates and α-syn fibrils, phosphorylated tau aggregation was significantly elevated in the presence of α-syn, and spread to broader brain regions such as the retrosplenial cortex and the supramammillary nucleus [134]. In vivo, transduction of tau and α-syn fibrils together induced a significant increase of insoluble tau pathology but not α-syn aggregates in neurons [134]. In addition, mice injected with tau-modified α-syn fibrils exhibited more severe α-syn pathology and faster spreading of α-syn in the striatum, compared with pure α-syn fibril injections. Knockout of tau attenuated the α-syn propagation and the accompanied mitochondrial toxicity [24]. In addition, PD patient-derived α-syn/tau oligomers administered in tau transgenic mouse brains accelerated tau oligomer formation and induced more severe neuronal loss, compared to administration of tau oligomers alone [135]. Hippocampal injection of the tau/α-syn hybrid fibrils exacerbated tau pathology transmission in a tauopathy mouse model, compared with pure tau or α-syn fibril administration. Interestingly, when tau/α-syn co-polymers were administered to synucleinopathy mouse brains, no elevation of α-syn pathology propagation was observed compared to pure α-syn injection. It seems that the effect of tau/α-syn hybrid oligomers on pathology transmission is much stronger in tauopathy models than in synucleinopathy models, suggesting a more thorough cooperation of the two proteins in the tau seeding process, reflecting a more prominent effect of α-syn amyloidogenicity compared to tau [136].
Possible mechanisms underlying the formation of tau and α-syn co-pathology
Several studies have repeatedly proven the continuum of pathology between tau and α-syn. However, how the tau and α-syn co-pathology occurs is still unclear. Here we propose three hypothesized models for the molecular mechanisms underlying the tau and α-syn co-pathology based on current literature (Fig. 4).

Mechanisms of co-pathology formation between tau and α-syn. We propose three possible models of tau and α-syn co-pathology formation in neurons. (1) Tau and α-syn monomers directly bind to each other and form co-aggregates (left), (2) tau or α-syn aggregates serve as the template and initiate elongation with monomers of the other protein, a process termed cross-seeding (middle); and (3) tau and α-syn aggregate independently from each other in the same neurons
First, direct binding between tau and α-syn monomers promotes the co-aggregation of proteins and initiates fibrilization. The proline-rich domain and the MTBD-containing region within the structure of tau protein directly interact with α-syn and presenilin-1, both regulating the formation of pathology in synucleinopathy [81, 137]. A recent study using a bimolecular fluorescence complementation system, which allows direct visualization of protein–protein interaction, revealed contacts between α-syn and tau at the molecular level, again laying the molecular basis for the formation of co-pathology [138]. Co-incubation of monomeric α-syn with tau variants promotes the amyloid formation of both tau and α-syn aggregates in vitro. NMR spectroscopy revealed that α-syn binds to tau molecules through its negatively charged C terminal [86]. Despite the direct binding, there is also a study showing co-aggregation of tau and α-syn through liquid–liquid phase co-separation, by forming electrostatic complex coacervates, explaining the mechanisms of co-pathology formation from the biophysical perspective [139].
Second, tau and α-syn form co-polymers by means of templating and cross-seeding. The cross-seeding process follows a nucleation-dependent pathway, where one type of protein aggregate serves as a template, recruiting the monomeric, mutant or oligomeric form of the other [140]. From the biophysical perspective, oppositely charged proteins are more likely to cross-seed each other and the aggregation kinetics of co-aggregation is accelerated [141]. α-Syn and tau can cross-seed each other [142]. Adding modified polymorphic α-syn oligomeric strains to tau aggregates results in co-polymers with stronger propensity in pathology spreading [143]. Cryo-EM and solid-state NMR evidence from the study of Hojjatian et al. revealed that the α-syn aggregation in the presence of tau cofactor is related to specific polymorphs in the core region [144]. Meanwhile, in in vitro studies, α-syn fibrillary seeds induced intracellular inclusion formation of phosphorylated tau [145]. Studies in both neuronal culture and mice showed that distinct strains of α-syn preformed fibrils seed tau aggregation differently, suggesting the close association between templating and the sequential co-aggregate formation [24, 86, 146].
Third, in the post-mortem study by Ishizawa et al., phosphorylated tau was present at the periphery of LBs instead of in an intertwining distribution pattern, in the brains of patients with Lewy body disease [132]. Nevertheless, Uchikado et al. showed that α-syn formed a central core surrounded by tau tangles in AD patient brains. Electron microscopy examination showed tightly packed tau filaments separated from the α-syn granule-patterned aggregates [129]. The finding of the co-pathology of tau and α-syn without cross-seeding of the two proteins suggests that α-syn and tau can aggregate independently in a same neuron.
Perspectives
Despite the overlapping genetic risk factors and clinical manifestations, the co-pathology of protein aggregation, and the mutual facilitation of pathology spreading and cellular toxicity, there are divergent features between tauopathies and synucleinopathies. Intracellularly, tau is distributed predominantly in neuronal axons associated with microtubule, while α-syn is rather restricted to synaptic regions under physiological conditions [147, 148]. Moreover, transcriptomic studies have revealed that tau and α-syn variants have distinct cell type affinities. α-Syn is mainly present in neurons of the dopamine system, related to cellular functions such as synaptic transmission. Tau is predominantly located in the hippocampus and entorhinal cortex, related to cellular functions such as Ca2+ homeostasis [149, 150]. The propagation of tau aggregates is more limited to the CNS in contrast to the spreading pattern of α-syn from the periphery to the CNS [151]. However, studies by Beach et al. debated on the theory that α-syn pathology originates from peripheral organs. In their post-mortem analyses of synucleinopathy in iLBD, PD and normal elderly subjects, they found no presence of peripheral α-syn in subjects without brain α-syn pathology. This revoked the “body-first” hypothesis of α-syn aggregation [152, 153]. Unlike α-syn, it is hypothesized that the tau protein is present in the peripheral nervous system more as the “Big tau”, with a higher molecular weight due to the additional exon 4a, which makes the protein more strongly bind the cytoskeleton and less likely to undergo conformational changes and aggregation [154,155,156].
Based on the pathological continuum of tauopathies and synucleinopathies, common therapeutic interventions may be applied to both groups of diseases. Tau and α-syn share similar mechanisms of cell-to-cell transmission, including exosome release, receptor-mediated endocytosis, etc. Blocking the propagation process of both amyloid proteins could be a potential way to halt pathology progression and delay the development of diseases. Immunotherapies targeting extracellular tau and α-syn have been reported to be effective in clinical trials for disease modification, such as the passive-immunization antibody PRX002 [157] for α-syn and BIIB092 for tau [158, 159]. Preclinical studies have also reported some immunotherapeutic antibodies and active peptides that can effectively decrease protein aggregation and protect neurons [160, 161].
Conclusion
We have summarized the clinical symptoms, genetic risks and pathological features of tauopathies and synucleinopathies. More importantly, we have discussed recent findings on the aggregation, propagation and toxicity of tau and α-syn, with a special focus on the pathological continuum of the two proteins and the two groups of proteinopathies. Tau and α-syn can cross-seed each other in the process of aggregate formation and spreading. The present review provides an update on disease classification of neurodegeneration and suggests niches of pathology intervention at early stages of tauopathies and synucleinopathies.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- APOE:
-
Apolipoprotein E
- CBS:
-
Corticobasal syndrome
- CNS:
-
Central nervous system
- DLB:
-
Lewy body dementia
- FTD:
-
Frontotemporal dementia
- iLBD:
-
Incidental Lewy body disease
- LB:
-
Lewy body
- LN:
-
Lewy neurite
- MAPT:
-
Microtubule-associated protein tau
- MSA:
-
Multiple system atrophy
- NFT:
-
Neurofibrillary tangle
- PD:
-
Parkinson’s disease
- PDD:
-
Parkinson’s disease dementia
- PHF:
-
Paired helical filament
- PSP:
-
Progressive supranuclear palsy
- PTM:
-
Post-translational modifications
- α-Syn:
-
α-Synuclein
References
Gotz J, Halliday G, Nisbet RM. Molecular pathogenesis of the tauopathies. Annu Rev Pathol. 2019;14:239–61.
Zhang Y, Wu KM, Yang L, Dong Q, Yu JT. Tauopathies: new perspectives and challenges. Mol Neurodegener. 2022;17(1):28.
Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A. 1997;94(8):4113–8.
Chung DC, Roemer S, Petrucelli L, Dickson DW. Cellular and pathological heterogeneity of primary tauopathies. Mol Neurodegener. 2021;16(1):57.
Kovacs GG, Ghetti B, Goedert M. Classification of diseases with accumulation of Tau protein. Neuropathol Appl Neurobiol. 2022;48(3): e12792.
Goedert M, Jakes R, Spillantini MG. The synucleinopathies: twenty years on. J Parkinsons Dis. 2017;7(s1):S51–69.
Outeiro TF, Koss DJ, Erskine D, Walker L, Kurzawa-Akanbi M, Burn D, et al. Dementia with Lewy bodies: an update and outlook. Mol Neurodegener. 2019;14(1):5.
Koga S, Sekiya H, Kondru N, Ross OA, Dickson DW. Neuropathology and molecular diagnosis of synucleinopathies. Mol Neurodegener. 2021;16(1):83.
Goedert M, Spillantini MG. Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol Psychiatry. 1998;3(6):462–5.
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30(12):1591–601.
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–12.
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303–7.
de Rojas I, Moreno-Grau S, Tesi N, Grenier-Boley B, Andrade V, Jansen IE, et al. Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat Commun. 2021;12(1):3417.
Jun G, Ibrahim-Verbaas CA, Vronskaya M, Lambert JC, Chung J, Naj AC, et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry. 2016;21(1):108–17.
Chen J, Yu JT, Wojta K, Wang HF, Zetterberg H, Blennow K, et al. Genome-wide association study identifies MAPT locus influencing human plasma tau levels. Neurology. 2017;88(7):669–76.
Wang Q, Tian Q, Song X, Liu Y, Li W. SNCA gene polymorphism may contribute to an increased risk of Alzheimer’s disease. J Clin Lab Anal. 2016;30(6):1092–9.
Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol. 2018;17(1):64–74.
Chia R, Sabir MS, Bandres-Ciga S, Saez-Atienzar S, Reynolds RH, Gustavsson E, et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet. 2021;53(3):294–303.
Li YJ, Hauser MA, Scott WK, Martin ER, Booze MW, Qin XJ, et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology. 2004;62(11):2005–9.
Real R, Martinez-Carrasco A, Reynolds RH, Lawton MA, Tan MMX, Shoai M, et al. Association between the LRP1B and APOE loci and the development of Parkinson’s disease dementia. Brain. 2023;146(5):1873–87.
Szeto JYY, Walton CC, Rizos A, Martinez-Martin P, Halliday GM, Naismith SL, et al. Dementia in long-term Parkinson’s disease patients: a multicentre retrospective study. NPJ Parkinsons Dis. 2020;6:2.
Ganguly J, Jog M. Tauopathy and movement disorders-unveiling the chameleons and mimics. Front Neurol. 2020;11: 599384.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95(13):7737–41.
Pan L, Li C, Meng L, Tian Y, He M, Yuan X, et al. Tau accelerates alpha-synuclein aggregation and spreading in Parkinson’s disease. Brain. 2022;145(10):3454–71.
Abeliovich A, Gitler AD. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature. 2016;539(7628):207–16.
Kon T, Tomiyama M, Wakabayashi K. Neuropathology of Lewy body disease: Clinicopathological crosstalk between typical and atypical cases. Neuropathology. 2020;40(1):30–9.
Hishikawa N, Hashizume Y, Yoshida M, Sobue G. Clinical and neuropathological correlates of Lewy body disease. Acta Neuropathol. 2003;105(4):341–50.
Monzio Compagnoni G, Di Fonzo A. Understanding the pathogenesis of multiple system atrophy: state of the art and future perspectives. Acta Neuropathol Commun. 2019;7(1):113.
Jellinger KA. Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov Disord. 2014;29(14):1720–41.
Wang XJ, Ma MM, Zhou LB, Jiang XY, Hao MM, Teng RKF, et al. Autonomic ganglionic injection of alpha-synuclein fibrils as a model of pure autonomic failure alpha-synucleinopathy. Nat Commun. 2020;11(1):934.
Valentino RR, Koga S, Walton RL, Soto-Beasley AI, Kouri N, DeTure MA, et al. MAPT subhaplotypes in corticobasal degeneration: assessing associations with disease risk, severity of tau pathology, and clinical features. Acta Neuropathol Commun. 2020;8(1):218.
Tamvaka N, Manne S, Kondru N, Ross OA. Pick’s disease, seeding an answer to the clinical diagnosis conundrum. Biomedicines. 2023;11(6):16464.
Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: The Movement Disorder Society Criteria. Mov Disord. 2017;32(6):853–64.
Bieniek KF, Ross OA, Cormier KA, Walton RL, Soto-Ortolaza A, Johnston AE, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 2015;130(6):877–89.
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(Pt 1):43–64.
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755–66.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1): a006189.
Zody MC, Jiang Z, Fung HC, Antonacci F, Hillier LW, Cardone MF, et al. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet. 2008;40(9):1076–83.
Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. 2005;42(11):837–46.
Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. 2016;126(Pt 3):238–92.
Zhang CC, Xing A, Tan MS, Tan L, Yu JT. The role of MAPT in neurodegenerative diseases: genetics, mechanisms and therapy. Mol Neurobiol. 2016;53(7):4893–904.
Wolfe MS. Tau mutations in neurodegenerative diseases. J Biol Chem. 2009;284(10):6021–5.
Leveille E, Ross OA, Gan-Or Z. Tau and MAPT genetics in tauopathies and synucleinopathies. Parkinsonism Relat Disord. 2021;90:142–54.
Llado A, Ezquerra M, Sanchez-Valle R, Rami L, Tolosa E, Molinuevo JL. A novel MAPT mutation (P301T) associated with familial frontotemporal dementia. Eur J Neurol. 2007;14(8):e9-10.
Erro ME, Zelaya MV, Mendioroz M, Larumbe R, Ortega-Cubero S, Lanciego JL, et al. Globular glial tauopathy caused by MAPT P301T mutation: clinical and neuropathological findings. J Neurol. 2019;266(10):2396–405.
Bird TD, Nochlin D, Poorkaj P, Cherrier M, Kaye J, Payami H, et al. A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain. 1999;122(Pt 4):741–56.
Ogaki K, Li Y, Takanashi M, Ishikawa K, Kobayashi T, Nonaka T, et al. Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord. 2013;19(1):15–20.
Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52(4):511–6.
Arienti F, Lazzeri G, Vizziello M, Monfrini E, Bresolin N, Saetti MC, et al. Unravelling genetic factors underlying corticobasal syndrome: a systematic review. Cells. 2021;10(1):171.
Xia Y, Sorrentino ZA, Kim JD, Strang KH, Riffe CJ, Giasson BI. Impaired tau-microtubule interactions are prevalent among pathogenic tau variants arising from missense mutations. J Biol Chem. 2019;294(48):18488–503.
Pihlstrom L, Toft M. Genetic variability in SNCA and Parkinson’s disease. Neurogenetics. 2011;12(4):283–93.
Kasten M, Klein C. The many faces of alpha-synuclein mutations. Mov Disord. 2013;28(6):697–701.
Golbe LI, Di Iorio G, Bonavita V, Miller DC, Duvoisin RC. A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol. 1990;27(3):276–82.
Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, et al. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem. 2005;280(9):7800–7.
Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier AL, Ruggeri FS, et al. The H50Q mutation enhances alpha-synuclein aggregation, secretion, and toxicity. J Biol Chem. 2014;289(32):21856–76.
Rutherford NJ, Moore BD, Golde TE, Giasson BI. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J Neurochem. 2014;131(6):859–67.
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18(2):106–8.
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364(9440):1167–9.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science (New York, NY). 2003;302(5646):841.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–102.
Guerreiro R, Escott-Price V, Darwent L, Parkkinen L, Ansorge O, Hernandez DG, et al. Genome-wide analysis of genetic correlation in dementia with Lewy bodies, Parkinson’s and Alzheimer’s diseases. Neurobiol Aging. 2016;38(214):e7–10.
van Rheenen W, van der Spek RAA, Bakker MK, van Vugt J, Hop PJ, Zwamborn RAJ, et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet. 2021;53(12):1636–48.
Goris A, Williams-Gray CH, Clark GR, Foltynie T, Lewis SJ, Brown J, et al. Tau and alpha-synuclein in susceptibility to, and dementia in Parkinson’s disease. Ann Neurol. 2007;62(2):145–53.
Henderson MX, Sengupta M, Trojanowski JQ, Lee VMY. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol Commun. 2019;7(1):183.
Lee VM, Giasson BI, Trojanowski JQ. More than just two peas in a pod: common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci. 2004;27(3):129–34.
Jellinger KA. Absence of alpha-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol. 2009;118(3):371–9.
Moussaud S, Jones DR, Moussaud-Lamodiere EL, Delenclos M, Ross OA, McLean PJ. Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener. 2014;9:43.
Cartelli D, Cappelletti G. Microtubule destabilization paves the way to Parkinson’s disease. Mol Neurobiol. 2017;54(9):6762–74.
Stolp Andersen M, Tan M, Holtman IR, Hardy J, International Parkinson’s Disease Genomics C, Pihlstrom L. Dissecting the limited genetic overlap of Parkinson’s and Alzheimer’s disease. Ann Clin Transl Neurol. 2022;9(8):1289–95.
Arneson D, Zhang Y, Yang X, Narayanan M. Shared mechanisms among neurodegenerative diseases: from genetic factors to gene networks. J Genet. 2018;97(3):795–806.
Korn L, Speicher AM, Schroeter CB, Gola L, Kaehne T, Engler A, et al. MAPT genotype-dependent mitochondrial aberration and ROS production trigger dysfunction and death in cortical neurons of patients with hereditary FTLD. Redox Biol. 2023;59: 102597.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3(4):519–26.
Brunello CA, Merezhko M, Uronen RL, Huttunen HJ. Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci. 2020;77(9):1721–44.
Chhatwal JP, Schultz AP, Dang Y, Ostaszewski B, Liu L, Yang HS, et al. Plasma N-terminal tau fragment levels predict future cognitive decline and neurodegeneration in healthy elderly individuals. Nat Commun. 2020;11(1):6024.
Zhang X, Vigers M, McCarty J, Rauch JN, Fredrickson GH, Wilson MZ, et al. The proline-rich domain promotes Tau liquid-liquid phase separation in cells. J Cell Biol. 2020;219(11):e202006054.
Hurtle BT, Xie L, Donnelly CJ. Disrupting pathologic phase transitions in neurodegeneration. J Clin Invest. 2023;133(13):e168549.
Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife. 2018;7:e36584.
Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133(5):665–704.
Tracy TE, Madero-Perez J, Swaney DL, Chang TS, Moritz M, Konrad C, et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell. 2022;185(4):712-28 e14.
Kellogg EH, Hejab NMA, Poepsel S, Downing KH, DiMaio F, Nogales E. Near-atomic model of microtubule-tau interactions. Science (New York, NY). 2018;360(6394):1242–6.
Goedert M, Spillantini MG. Propagation of Tau aggregates. Mol. Brain. 2017;10(1):18.
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547(7662):185–90.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–5.
Despres C, Byrne C, Qi H, Cantrelle FX, Huvent I, Chambraud B, et al. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci U S A. 2017;114(34):9080–5.
Xia Y, Prokop S, Gorion KM, Kim JD, Sorrentino ZA, Bell BM, et al. Tau Ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies. Acta Neuropathol Commun. 2020;8(1):88.
Lu J, Zhang S, Ma X, Jia C, Liu Z, Huang C, et al. Structural basis of the interplay between alpha-synuclein and Tau in regulating pathological amyloid aggregation. J Biol Chem. 2020;295(21):7470–80.
Sulzer D, Edwards RH. The physiological role of alpha-synuclein and its relationship to Parkinson’s disease. J Neurochem. 2019;150(5):475–86.
Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z, et al. Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res. 2018;28(9):897–903.
Sorrentino ZA, Giasson BI. The emerging role of alpha-synuclein truncation in aggregation and disease. J Biol Chem. 2020;295(30):10224–44.
Cremades N, Dobson CM. The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol Dis. 2018;109(Pt B):178–90.
Hijaz BA, Volpicelli-Daley LA. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol Neurodegener. 2020;15(1):19.
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160–4.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–40.
Li W, Englund E, Widner H, Mattsson B, van Westen D, Latt J, et al. Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci U S A. 2016;113(23):6544–9.
Li J-Y, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. J Park Dis. 2008;14(5):501–3.
Gibbons GS, Lee VMY, Trojanowski JQ. Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol. 2019;76(1):101–8.
Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121(2):171–81.
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389–404.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211.
Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396(1):67–72.
Chen QQ, Haikal C, Li W, Li MT, Wang ZY, Li JY. Age-dependent alpha-synuclein accumulation and aggregation in the colon of a transgenic mouse model of Parkinson’s disease. Transl Neurodegener. 2018;7:13.
Goedert M, Masuda-Suzukake M, Falcon B. Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain. 2017;140(2):266–78.
Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121(2):715–25.
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–13.
Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57–71.
Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106(47):20051–6.
Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845–52.
Tardivel M, Begard S, Bousset L, Dujardin S, Coens A, Melki R, et al. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun. 2016;4(1):117.
Chakraborty R, Nonaka T, Hasegawa M, Zurzolo C. Tunnelling nanotubes between neuronal and microglial cells allow bi-directional transfer of alpha-Synuclein and mitochondria. Cell Death Dis. 2023;14(5):329.
Chen K, Martens YA, Meneses A, Ryu DH, Lu W, Raulin AC, et al. LRP1 is a neuronal receptor for alpha-synuclein uptake and spread. Mol Neurodegener. 2022;17(1):57.
Rauch JN, Luna G, Guzman E, Audouard M, Challis C, Sibih YE, et al. LRP1 is a master regulator of tau uptake and spread. Nature. 2020;580(7803):381–5.
Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science (New York, NY). 2016;353(6307):aah3374.
Chen C, Kumbhar R, Wang H, Yang X, Gadhave K, Rastegar C, et al. Pathological tau transmission initiated by binding lymphocyte-activation gene 3. bioRxiv. 2023. https://doi.org/10.1101/2023.05.16.541015.
Patani R, Hardingham GE, Liddelow SA. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat Rev Neurol. 2023;19(7):395–409.
Liu YJ, Ding Y, Yin YQ, Xiao H, Hu G, Zhou JW. Cspg4(high) microglia contribute to microgliosis during neurodegeneration. Proc Natl Acad Sci U S A. 2023;120(8): e2210643120.
Dutta D, Jana M, Paidi RK, Majumder M, Raha S, Dasarathy S, et al. Tau fibrils induce glial inflammation and neuropathology via TLR2 in Alzheimer’s disease-related mouse models. J Clin Invest. 2023;133(18):e161987.
Sun Y, Guo Y, Feng X, Jia M, Ai N, Dong Y, et al. The behavioural and neuropathologic sexual dimorphism and absence of MIP-3alpha in tau P301S mouse model of Alzheimer’s disease. J Neuroinflammation. 2020;17(1):72.
Garcia P, Jurgens-Wemheuer W, Uriarte Huarte O, Michelucci A, Masuch A, Brioschi S, et al. Neurodegeneration and neuroinflammation are linked, but independent of alpha-synuclein inclusions, in a seeding/spreading mouse model of Parkinson’s disease. Glia. 2022;70(5):935–60.
Cheng Y, Bai F. The association of tau with mitochondrial dysfunction in alzheimer’s disease. Front Neurosci. 2018;12:163.
Risiglione P, Zinghirino F, Di Rosa MC, Magri A, Messina A. Alpha-synuclein and mitochondrial dysfunction in Parkinson’s disease: the emerging role of VDAC. Biomolecules. 2021;11(5):718.
Roy B, Jackson GR. Interactions between Tau and alpha-synuclein augment neurotoxicity in a Drosophila model of Parkinson’s disease. Hum Mol Genet. 2014;23(11):3008–23.
Hamano T, Enomoto S, Shirafuji N, Ikawa M, Yamamura O, Yen SH, et al. Autophagy and tau protein. Int J Mol Sci. 2021;22(14).
Tang Q, Gao P, Arzberger T, Hollerhage M, Herms J, Hoglinger G, et al. Alpha-Synuclein defects autophagy by impairing SNAP29-mediated autophagosome-lysosome fusion. Cell Death Dis. 2021;12(10):854.
Uemura N, Uemura MT, Luk KC, Lee VM, Trojanowski JQ. Cell-to-cell transmission of tau and alpha-synuclein. Trends Mol Med. 2020;26(10):936–52.
Twohig D, Nielsen HM. Alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):23.
Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10(3):378–84.
Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(23):11282–6.
Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153(5):1365–70.
Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65(7):685–97.
Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, et al. Pathological alpha-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol. 2016;131(3):393–409.
Kotzbauer PT, Giasson BI, Kravitz AV, Golbe LI, Mark MH, Trojanowski JQ, et al. Fibrillization of alpha-synuclein and tau in familial Parkinson’s disease caused by the A53T alpha-synuclein mutation. Exp Neurol. 2004;187(2):279–88.
Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. 2003;62(4):389–97.
Arima K, Mizutani T, Alim MA, Tonozuka-Uehara H, Izumiyama Y, Hirai S, et al. NACP/alpha-synuclein and tau constitute two distinctive subsets of filaments in the same neuronal inclusions in brains from a family of parkinsonism and dementia with Lewy bodies: double-immunolabeling fluorescence and electron microscopic studies. Acta Neuropathol. 2000;100(2):115–21.
Bassil F, Meymand ES, Brown HJ, Xu H, Cox TO, Pattabhiraman S, et al. Alpha-synuclein modulates tau spreading in mouse brains. J Exp Med. 2021;218(1):e20192193.
Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Gerson JE, Kayed R. Alpha-synuclein oligomers induce a unique toxic tau strain. Biol Psychiatry. 2018;84(7):499–508.
Williams T, Sorrentino Z, Weinrich M, Giasson BI, Chakrabarty P. Differential cross-seeding properties of tau and alpha-synuclein in mouse models of tauopathy and synucleinopathy. Brain Commun. 2020;2(2):fcaa090.
Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R. Alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem. 1999;274(36):25481–9.
Torres-Garcia L, JM PD, Brandi E, Haikal C, Mudannayake JM, Bras IC, et al. Monitoring the interactions between alpha-synuclein and Tau in vitro and in vivo using bimolecular fluorescence complementation. Sci Rep. 2022;12(1):2987.
Gracia P, Polanco D, Tarancon-Diez J, Serra I, Bracci M, Oroz J, et al. Molecular mechanism for the synchronized electrostatic coacervation and co-aggregation of alpha-synuclein and tau. Nat Commun. 2022;13(1):4586.
Subedi S, Sasidharan S, Nag N, Saudagar P, Tripathi T. Amyloid cross-seeding: mechanism, implication, and inhibition. Molecules. 2022;27(6).
Oki S, Iwashita K, Kimura M, Kano H, Shiraki K. Mechanism of co-aggregation in a protein mixture with small additives. Int J Biol Macromol. 2018;107(Pt B):1428–37.
Sengupta U, Kayed R. Amyloid beta, Tau, and alpha-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog Neurobiol. 2022;214: 102270.
Sengupta U, Puangmalai N, Bhatt N, Garcia S, Zhao Y, Kayed R. Polymorphic alpha-synuclein strains modified by dopamine and docosahexaenoic acid interact differentially with tau protein. Mol Neurobiol. 2020;57(6):2741–65.
Hojjatian A, Dasari AKR, Sengupta U, Taylor D, Daneshparvar N, Yeganeh FA, et al. Tau induces formation of alpha-synuclein filaments with distinct molecular conformations. Biochem Biophys Res Commun. 2021;554:145–50.
Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31(21):7604–18.
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154(1):103–17.
Vacchi E, Kaelin-Lang A, Melli G. Tau and alpha synuclein synergistic effect in neurodegenerative diseases: when the periphery is the core. Int J Mol Sci. 2020;21(14):5030.
Iwata M, Watanabe S, Yamane A, Miyasaka T, Misonou H. Regulatory mechanisms for the axonal localization of tau protein in neurons. Mol Biol Cell. 2019;30(19):2441–57.
Praschberger R, Kuenen S, Schoovaerts N, Kaempf N, Singh J, Janssens J, et al. Neuronal identity defines alpha-synuclein and tau toxicity. Neuron. 2023;111(10):1577–9011.
Henrich MT, Geibl FF, Lakshminarasimhan H, Stegmann A, Giasson BI, Mao X, et al. Determinants of seeding and spreading of alpha-synuclein pathology in the brain. Sci Adv. 2022;6(46):1eabc2487.
Vasili E, Dominguez-Meijide A, Outeiro TF. Spreading of alpha-synuclein and tau: a systematic comparison of the mechanisms involved. Front Mol Neurosci. 2019;12:107.
Beach TG, Adler CH, Sue LI, Shill HA, Driver-Dunckley E, Mehta SH, et al. Vagus nerve and stomach synucleinopathy in Parkinson’s disease, incidental lewy body disease, and normal elderly subjects: evidence against the “body-first” hypothesis. J Parkinsons Dis. 2021;11(4):1833–43.
Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119(6):689–702.
Couchie D, Mavilia C, Georgieff IS, Liem RK, Shelanski ML, Nunez J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc Natl Acad Sci U S A. 1992;89(10):4378–81.
Himmler A, Drechsel D, Kirschner MW, Martin DW Jr. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9(4):1381–8.
Derkinderen P, Rolli-Derkinderen M, Chapelet G, Neunlist M, Noble W. Tau in the gut, does it really matter? J Neurochem. 2021;158(2):94–104.
Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. 2018;75(10):1206–14.
Qureshi IA, Tirucherai G, Ahlijanian MK, Kolaitis G, Bechtold C, Grundman M. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement (N Y). 2018;4:746–55.
Shin J, Kim HJ, Jeon B. Immunotherapy targeting neurodegenerative proteinopathies: alpha-synucleinopathies and tauopathies. J Mov Disord. 2020;13(1):11–9.
Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci. 2014;34(28):9441–54.
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80(2):402–14.
Iyer A, Claessens M. Disruptive membrane interactions of alpha-synuclein aggregates. Biochim Biophys Acta Proteins Proteom. 2019;1867(5):468–82.
Acknowledgements
We thank Hao-wen Dou and Cong Feng for the intellectual inputs.
Funding
Open access funding provided by Lund University. This review was supported by the National Natural Science Foundation of China (82371273, 81430025, and U1801681 to J-Y.L. and W.L.), the Key Field Research Development Program of Guangdong Province (2018B030337001), the Swedish Research Council (2019-01551, 2023-02216), ParkinsonFonden (1351/21, 1421/22,1494/23), the Strategic Research Area Multipark (Multidisciplinary research in Parkinson’s disease at Lund University), Hjarnfonden (FO2022-0282, FO2023-0397, PS2018-0062) and Svenska Sällskapet för Medicinsk Forskning (SSMF, P18-0194).
Author information
Authors and Affiliations
Contributions
JYL and WL wrote the paper and have both read and approved the manuscript. Jia-Yi Li and Wen Li contributed equally to the conception and writing of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, W., Li, JY. Overlaps and divergences between tauopathies and synucleinopathies: a duet of neurodegeneration. Transl Neurodegener 13, 16 (2024). https://doi.org/10.1186/s40035-024-00407-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-024-00407-y